
Cu- or Fe-catalyzed C–H/C–C bond nitrogenation reactions for the direct synthesis of N-containing compounds
Yujie
Liang
,
Yu-Feng
Liang
and
Ning
Jiao
*
State Key Laboratory of Natural and Biomimetic Drugs, School of Pharmaceutical Sciences, Peking University, Xue Yuan Rd. 38, Beijing 100191, China. E-mail: jiaoning@pku.edu.cn
First published on 5th February 2015
Abstract
Nitrogen-containing compounds are widely present in both natural products and synthetic compounds, for example, they show up within functional materials, top-selling drugs, as well as bioactive molecules. Thus, organic chemists have paid considerable attention in developing novel methodologies for their preparation. To synthesize these compounds in a green and sustainable way, researchers have focused on the direct functionalization of hydrocarbons via C–H and/or C–C bond cleavage. Although significant progress has been made in the direct functionalization of simple hydrocarbons, direct incorporation of N-atoms into simple substrates via C–H and/or C–C bond cleavage remains challenging due to the inert chemical bonds and the unstable character of some N-sources under oxidative conditions. Azide reagents are frequently used as nitrogen source in incorporating nitrogen into carbon skeletons. Although the exact reaction pathway remains unclear, detailed mechanistic studies revealed that the carbon cation containing the azido group may exist as the key intermediate which would undergo Schmidt-type rearrangement to afford nitriles, tetrazoles, arylamines, or other kinds of nitrogen-containing compounds. Considering the high cost and toxicity of heavy metals, copper and iron, as inexpensive, readily accessible metals have already shown their unique utilities. This account attempts to focus on C–H/C–C bond nitrogenation reactions via Cu and Fe catalysis, as well as their applications in synthetic chemistry.
 Yujie Liang | Yujie Liang was born in 1992 in Hainan, China. He received his BS degree at Nankai University in 2014. Then he joined Prof. Ning Jiao's group in State Key Laboratory of Natural and Biomimetic Drugs, Peking University. His research interests include developing highly efficient synthesis methods for nitrogen containing compounds via inert bond activations. |
 Yu-Feng Liang | Yu-Feng Liang was born in 1986 in Jiangxi, China. He received his B.S. and M.S. degrees in 2009 and 2012 with Prof. Hua-Jian Xu at Hefei University of Technology. Then he joined Prof. Ning Jiao's group at Peking University, and is currently a third-year doctoral candidate. His research interests include developing efficient aerobic oxidation as well as oxygenation reactions with economical and environmentally friendly oxidants. |
 Ning Jiao | Ning Jiao received his Ph.D. degree (2004) (with Prof. Shengming Ma) at Shanghai Institute of Organic Chemistry, CAS. He spent 2004–2006 as an Alexander von Humboldt postdoctoral fellow with Prof. Manfred T. Reetz at Max Planck Institute für Kohlenforschung. In 2007, he joined the faculty at Peking University as an Associate Professor, and was promoted to Full Professor in 2010. His current research efforts are focused on: (1) to develop green and efficient synthetic methodologies through the Single Electron Transfer (SET) process; (2) aerobic oxidation, oxygenation and nitrogenation reactions; and (3) the activation of inert chemical bonds. |
Introduction
Nitrogen-containing compounds are of great importance in medicinal chemistry because they exhibit interesting and diverse biological activities.1 For example (Fig. 1), the COX-2 inhibitor and the celecoxib derivative exhibit valuable biological activities,1f valsartan acts as a blockade for angiotensin-II receptors,1g atorvastatin, one of the top selling drugs worldwide since 2003, blocks the production of cholesterol,1h and rizatriptan is a triptan drug used for the treatment of migraine headaches.1i Moreover, in materials sciences, the presence of nitrogen in the structures of polymers can have a profound effect on their electronic, physical, or surface properties.1 The construction of the C–N bond is significant as it offers the chance to incorporate nitrogen atoms into organic molecules. In general, the methods for C–N bond formation include substitution reactions (both electrophilic and nucleophilic amination), condensation, cycloaddition, rearrangement and cross coupling reactions.2 Among them, the advent of catalytic cross coupling reactions provides efficient approaches to N-containing compounds. In the few past decades, some direct C–H aminations have been significantly developed.3 Despite significant advancements in this area, the direct construction of the C–N bond via C–H and/or C–C bond cleavage is still a challenging task, because it requires: (1) to find suitable conditions for the generation of highly active catalytic species; (2) to control the regio- and chemo-selective C–H and/or C–C bond cleavage; (3) to form stable N-partners under oxidative conditions.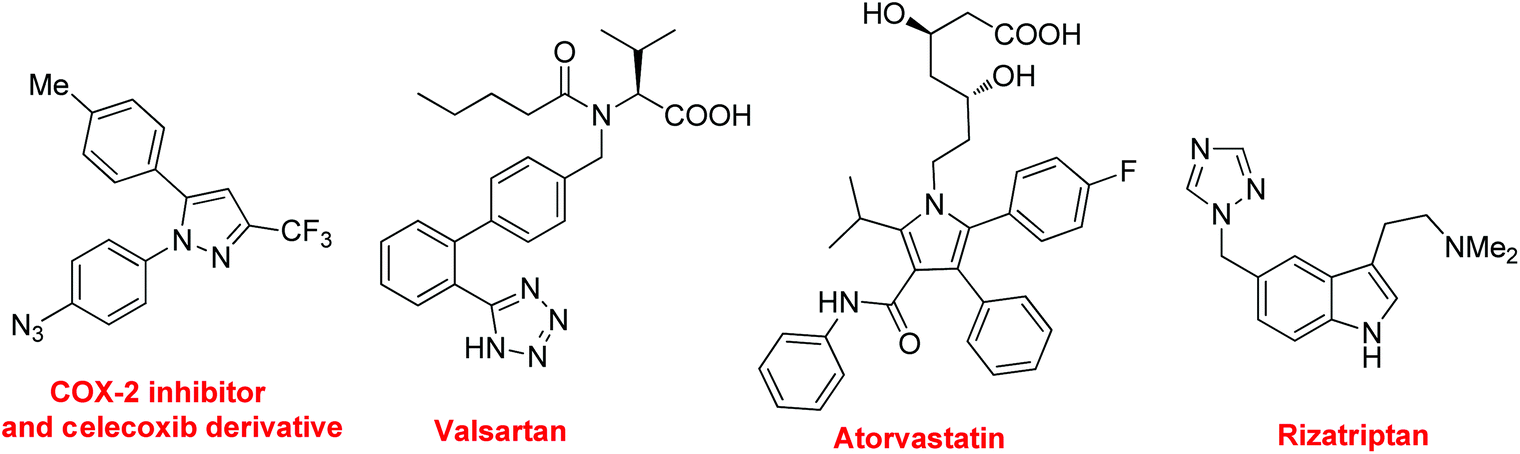 | ||
| Fig. 1 Typical N-containing molecules in pharmaceuticals. | ||
The C–H bond and the C–C bond are the most widespread and fundamental bonds existing in organic compounds. The transformation of these bonds plays an important role in modern scientific research. Recently, we have developed some novel approaches to ortho-azido anilines, nitriles, amides, arylamines, and tetrazole derivatives via C–H and/or C–C bond cleavage from some readily available substrates by using simple nitrogen sources under mild conditions.
Copper-catalyzed C–H azidation of anilines
Organic azides are widely used in organic synthesis as valuable intermediates and building blocks, particularly in the synthesis of nitrogen-containing heterocycles, in peptide chemistry, materials science, polymer chemistry and drug discovery.4 Moreover, aryl azides have found their biological and industrial use as photoaffinity labeling agents.4 Thus, a direct synthesis method of aryl azides is highly desirable.Recently, Yu and coworkers reported a Cu-catalyzed aerobic oxidative aryl C–H bond functionalization via a single electron transfer (SET) process.5 The amine group of aniline substrate assisted vinyl C–H bond activation and alkenylation was individually disclosed by the groups of You6a,b and Zhang.6c We found that the ortho-azidation of aniline derivatives directed by the amino group could be achieved by Cu-catalysis under mild conditions (eqn (1)).7a It is noteworthy that Wang and coworkers developed a Cu(ClO4)2-catalyzed aerobic oxidative direct azidation of arenes7b using sodium azide as an azide source and air as a terminal oxidant under mild conditions through a significantly proved Cu(III) involved mechanism.7c,d Recently, Fan et al. reported a successful Cu(OAc)2-catalyzed azidation of anilines using azidobenziodoxolone as an azidating agent.7e
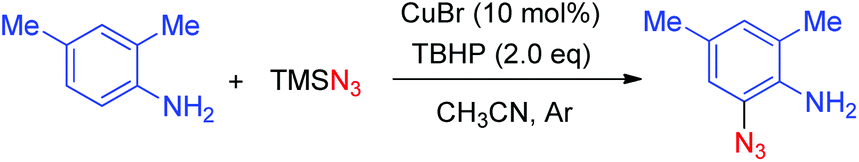 | (1) |
The reaction was performed in the presence of TMSN3 and TBHP using CuBr as the catalyst at 30 °C in CH3CN by using 2,4-dimethylaniline as a model substrate, the desired 2-azido-4,6-dimethylaniline was obtained in 68% yield. The catalytic system displayed outstanding chemoselectivity, which was illustrated by a remarkable functional-group tolerance. Moreover, both primary as well as secondary amines proved to be suitable substrates. However, there are still some limitations of the abovementioned method: (1) when ortho-unsubstituted anilines were employed as the substrates, both mono- and di-azidated products were obtained; (2) when a secondary amine was employed, a large amount of starting material was remained, thus leading to a relatively low yield; (3) the reaction of arylamines with a tertiary amino group did not work under the standard conditions, indicating that a free N–H bond is required for this transformation (Scheme 1).
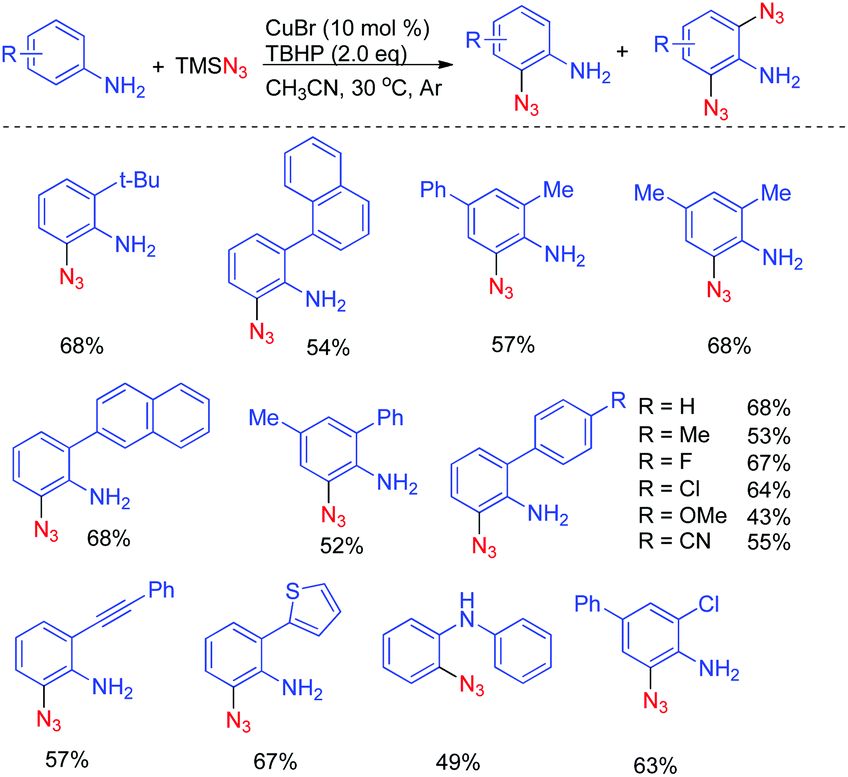 | ||
| Scheme 1 Cu(I)-catalyzed direct azidation of anilines. | ||
The power of the amino-directed C–H bond azidation approach was elegantly illustrated by the subsequent diversification of the thus obtained ortho-azido anilines (Scheme 2).7a
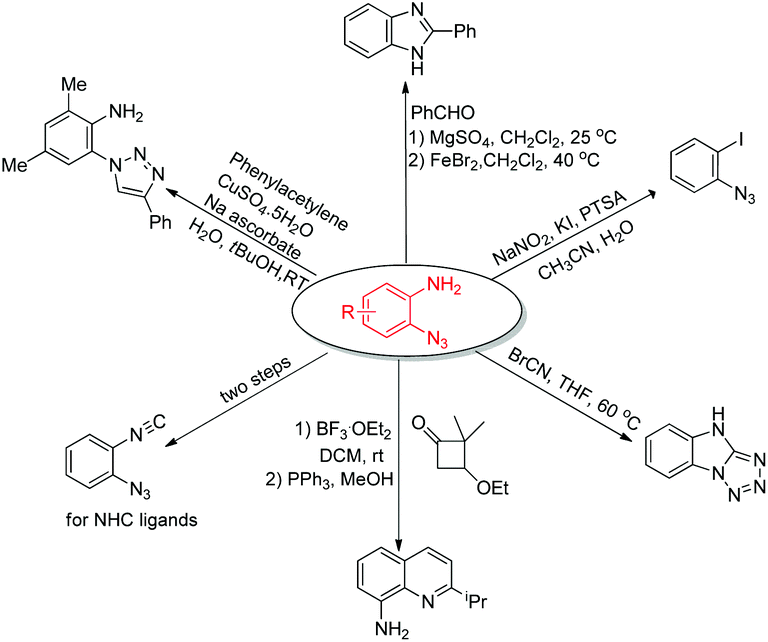 | ||
| Scheme 2 Diversification of ortho-azido anilines. | ||
To further understand the mechanism, radical inhibitor 2,2,6,6-tetramethylpiperidine 1-oxyl (TEMPO) or hydroquinone (HQ) was added to the reaction, and the formation of 2a was completely inhibited, thus indicating that a radical process may be involved (Scheme 3).
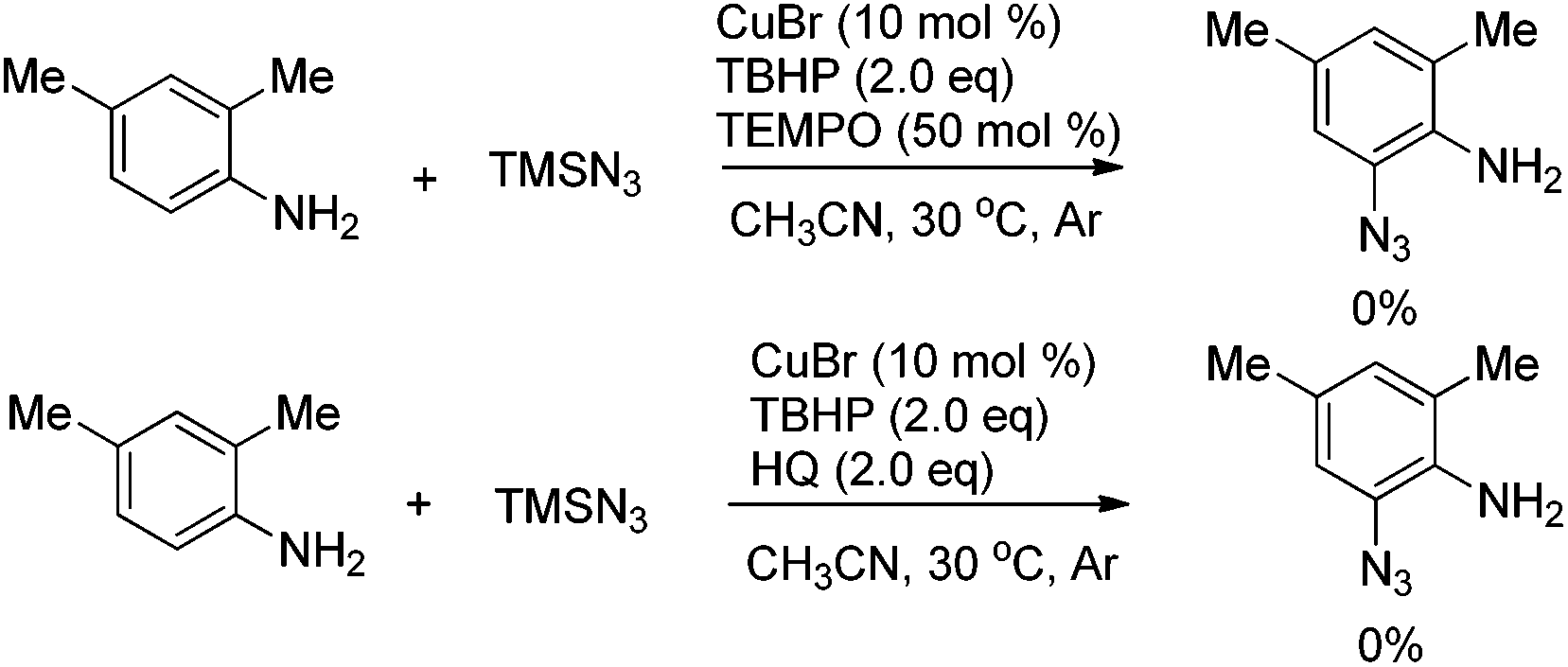 | ||
| Scheme 3 Mechanism studies. | ||
Although the mechanism of this transformation is not completely clear yet, on the basis of the mechanistic studies, we proposed that intermediate 1 is initially generated by the coordination of copper to the substrate.8 Subsequently, intermediate 1 combines with an azide radical generated in situ from TMSN3 and TBHP to form intermediate 2. A single electron transfer (SET) from the aryl ring to the metal center (from 2 to 3) may be involved in this process.5 Then, azido group transfer into the aryl ring with the release of CuBr forms intermediate 4, which undergoes deprotonation via the SET process leading to the product (Scheme 4).
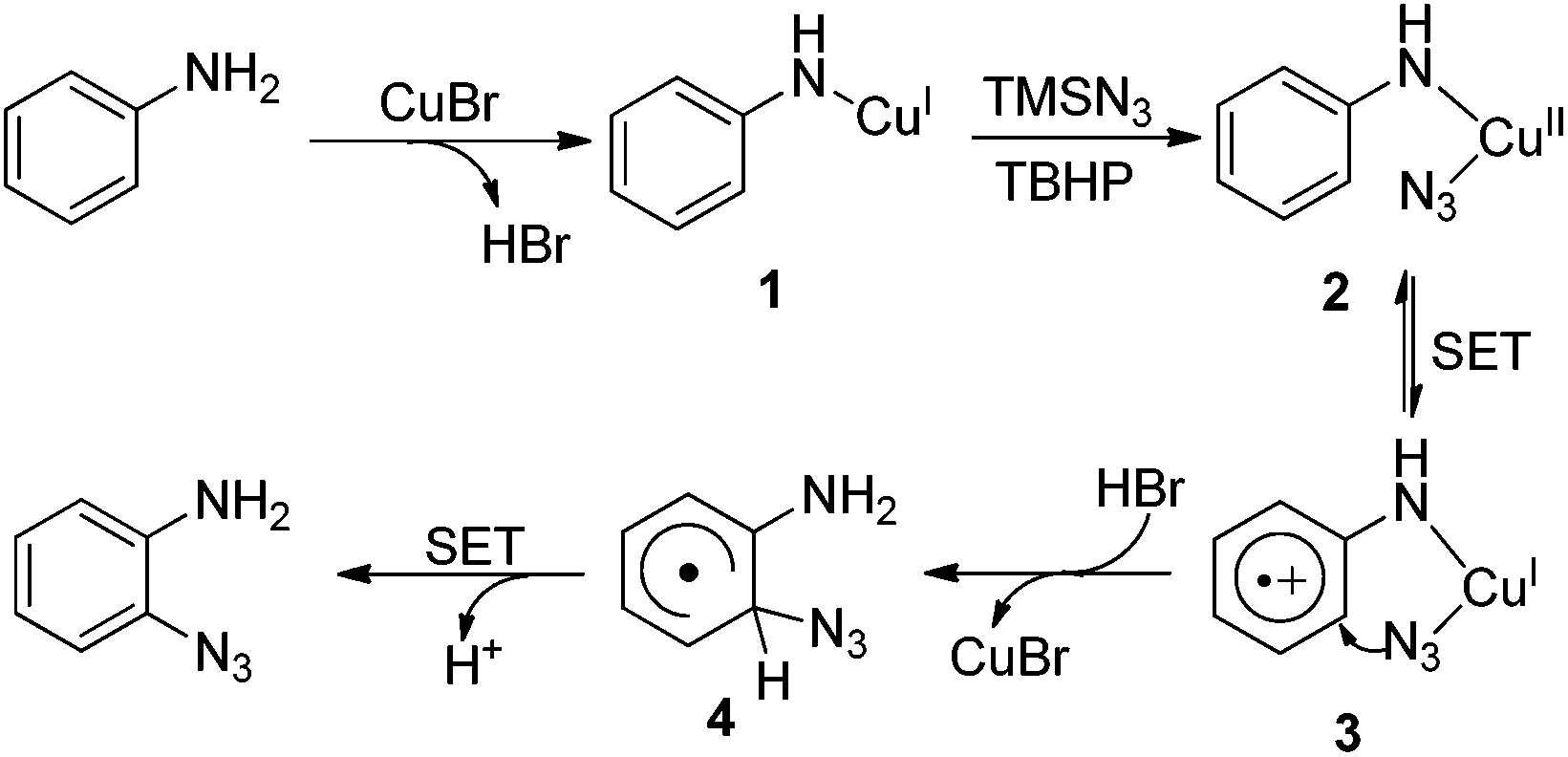 | ||
| Scheme 4 Proposed mechanism of Cu(I)-catalyzed direct azidation of anilines. | ||
The present chemistry demonstrates the first example of copper catalyzed direct ortho C–H azidation of arenes. This reaction uses an inexpensive copper catalyst under remarkably mild conditions to enable site selective direct C–N bond formations on readily available aniline derivatives with a broad substrate scope. Moreover, the removable amino group could be converted into H, Cl, Br, I, CN, and OH by the Sandmeyer reaction.
Cu-catalyzed aerobic oxidative C–C bond cleavage for C–N bond formation
Carbon–carbon (C–C) bonds are ubiquitous in organic compounds. Compared with highly developed C–C bond forming reactions, chemoselective cleavage of the C–C bond is always on the cutting edge in organic chemistry, biodegradation and industry applications. Among them, the cleavage of the C–C single bond is the most challenging issue and draws great attention due to its thermodynamic stability and uncontrollable selectivity.9 Recently, some elegant examples of C–C bond cleavage of ketone substrates with the carbonyl functional fragment as the leaving group have been reported.10,11 Using 1,3-dione substrates, we developed the Cu-catalyzed aerobic oxidative esterification reaction for the synthesis of α-ketoesters combining C–C(CO) σ-bond cleavage, dioxygen activation and oxidative C–H bond functionalization processes.11a The group of Bi and Liu11b reported a copper-catalyzed C(CO)–C(methyl) bond cleavage under an oxygen atmosphere, which terminated at the aldehyde stage without being overoxidized. With our continuing interest in the construction of the C–N bond, we designed a novel copper-catalyzed aerobic oxidative C–C bond cleavage to directly convert aryl alkyl ketones to benzamides (eqn (2)).12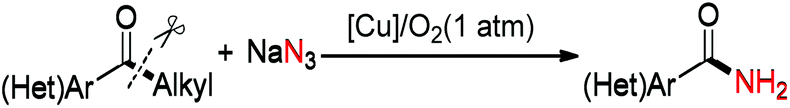 | (2) |
The conditions optimization indicated that the nitrogenation of 4′-phenylacetophenone with sodium azide afforded 4-phenylbenzamide in 84% yield when the reaction was catalyzed with CuCl2 in DMF at 120 °C in the presence of TEMPO and O2 (1 atm). Under the standard conditions, various substituted acetophenones can be smoothly converted to benzamides. Aryl ketones with electron-donating as well as electron-withdrawing substituents at the aryl ring can be tolerated well. Besides, the aryl methyl ketones could be expanded to heteroaryl ketones (Scheme 5).
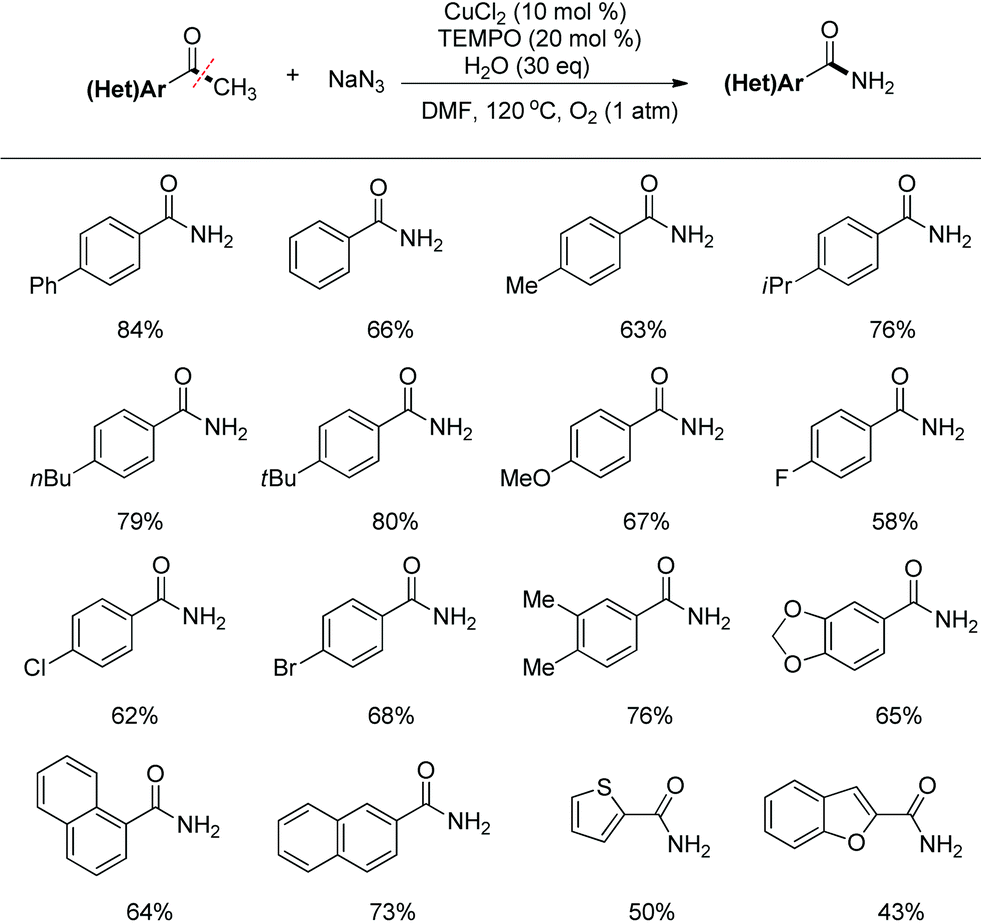 | ||
| Scheme 5 Transformation of aryl methyl ketones to benzamides. | ||
It is noteworthy that relatively inactive aryl ketone substrates with long-chain alkyl groups which were not tolerated in both the reported α-ketoester11a and aldehyde11b formation reactions via C(CO)–C bond cleavage can be successfully converted into the corresponding benzamides by our developed protocol (Scheme 6).
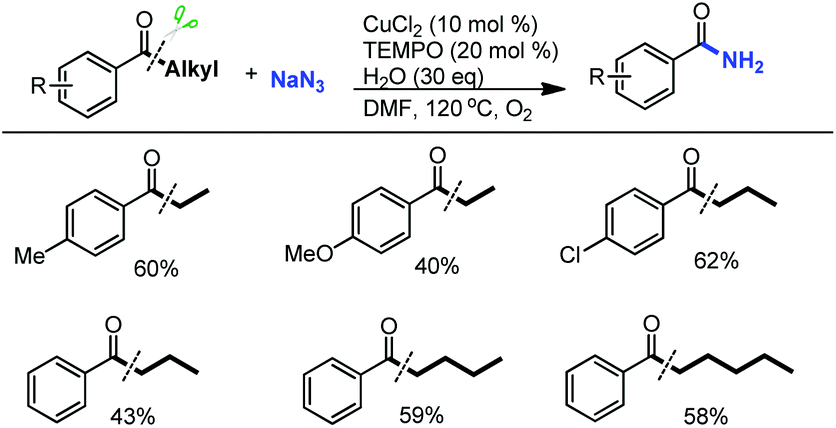 | ||
| Scheme 6 Transformation of aryl ketone substrates with long-chain alkyl groups. | ||
To elucidate the reaction pathway, some potential intermediates such as the corresponding aldehyde, α-hydroxy ketone, α-ketoaldehyde, α-keto carboxylic acid, and amide species were prepared and tested under the standard conditions (Scheme 7). However, none of them afforded the desired product (Scheme 7). These results indicated that the ketone substrate may preferentially react with an azide nucleophile, then undergoes an oxidation process under the Cu/O2 system. Furthermore, N-methylbenzamide showed no efficiency under standard conditions which ruled out the Schmidt type reaction of acetophenone and sodium azide in this system. In addition, (1-azidovinyl)benzene was completely unreactive under standard conditions.
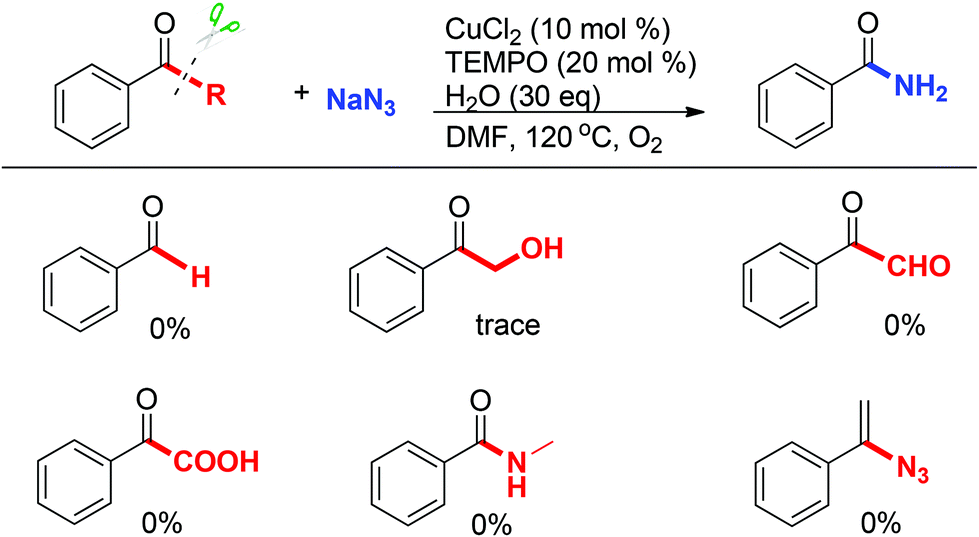 | ||
| Scheme 7 Mechanistic studies. | ||
Based on the above results, a plausible mechanism was proposed. The substrate initially undergoes a reversible equilibrium with intermediate 5. The subsequent aerobic oxidation of intermediate 5 occurs to generate α-hydroxylated intermediate 6 in the Cu/O2 oxidative system.11,13 The subsequent rearrangement of intermediate 6via its resonance structure 7 produces intermediate 8 through C–C bond cleavage with the release of molecular nitrogen and aldehyde as byproducts. Finally, tautomerization of 8 affords the desired amide product (Scheme 8).
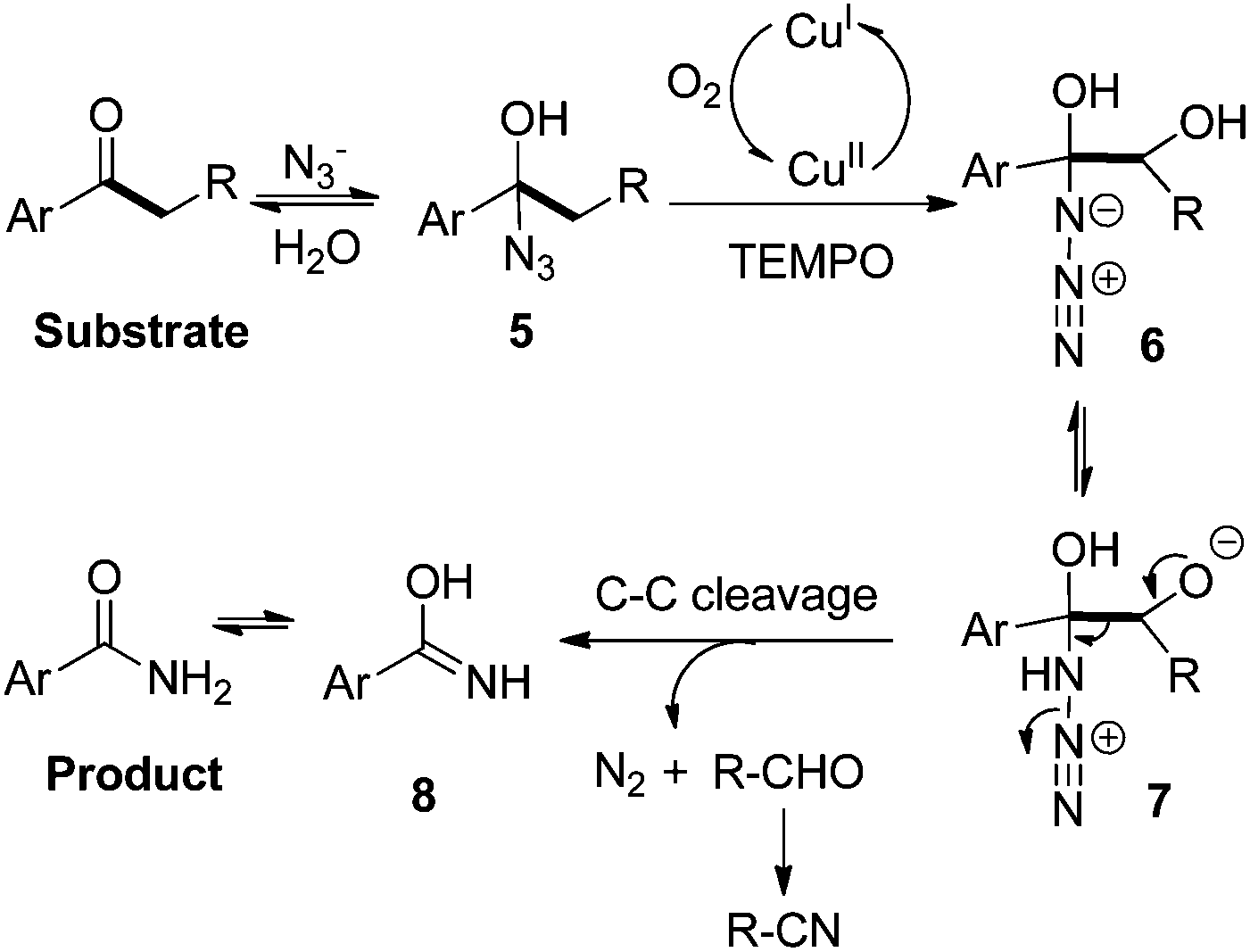 | ||
| Scheme 8 Proposed mechanism. | ||
Cu- or Fe-promoted direct synthesis of aryl nitriles and alkenyl nitriles via Csp3–H nitrogenation
Nitriles are building blocks widely used in the synthesis of pharmaceuticals, agrochemicals, and natural products.14 Recently, we reported a direct transformation of methyl arenes to aryl nitriles via three benzylic C–H bond cleavages employing phenyliodonium diacetate (PIDA) as an oxidant and sodium azide (NaN3) as the nitrogenation source. The efficiency of this transformation could be significantly improved by the addition of a catalytic amount of CuSO4·5H2O salt. As a limitation, a heteroatom or aryl substituent at the para-position of the methyl group of the toluene substrate is required for this direct transformation (Scheme 9).15 Significantly, Wang and co-workers developed a Pd-catalyzed direct transformation of methyl arenes into aromatic nitriles using tert-butyl nitrite (TBN) as the nitrogen source and oxidant,16 in which the substrate scope is much broader.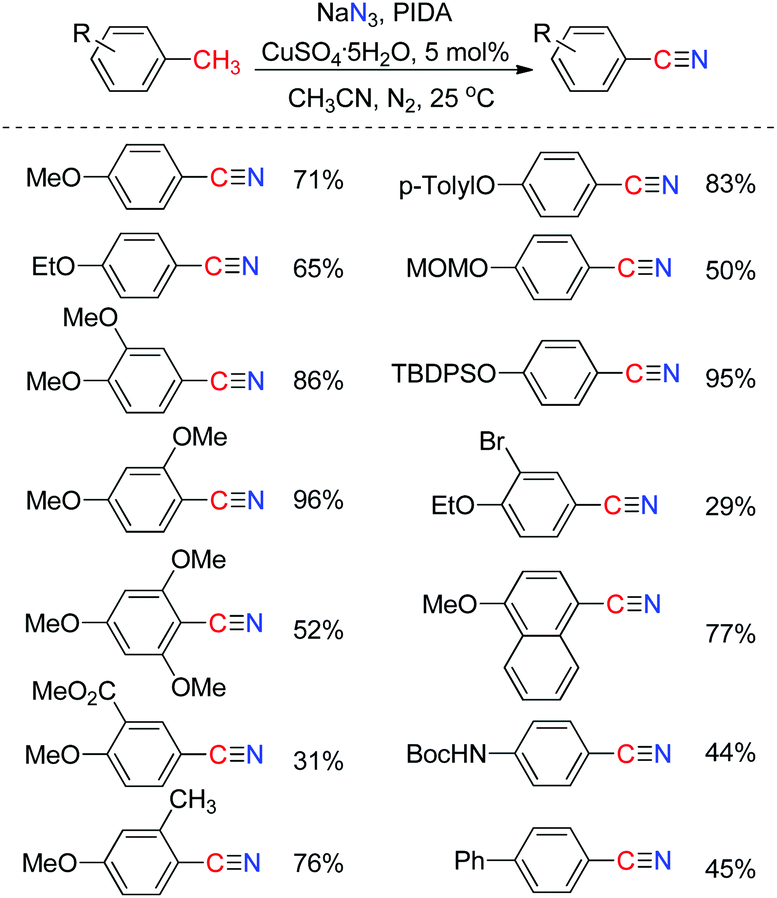 | ||
| Scheme 9 Direct transformation of methyl arenes to aryl nitriles. | ||
Benzylic azide was proposed to be the key intermediate to afford aryl nitrile. The azide radical can be produced by combining PIDA and NaN3. The hydrogen abstraction of methyl arenes affords benzylic radical 9, followed by the formation of benzylic azide 10, which is further abstracted a hydrogen to form radical 11. The radical species 11 is then further oxidized to benzylic cation 12 through single electron transfer (SET). Eventually, benzylic cation 12 undergoes a Schmidt type rearrangement17 to afford the desired aryl nitriles. The cation can be stabilized by the para-substituent on the aromatic ring (Scheme 10). A similar strategy was also applied in the synthesis of α-iminonitriles.18 Besides, by employing this oxidative strategy, the transformation of some organo azides to their corresponding nitriles has been developed.19
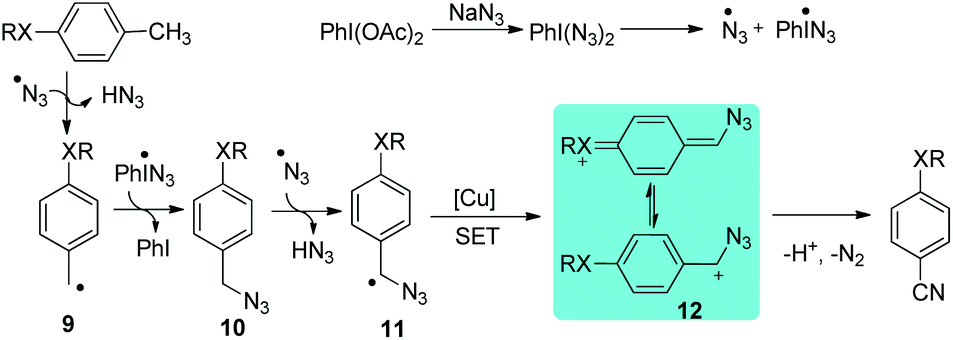 | ||
| Scheme 10 Proposed mechanism of the direct transformation of methyl arenes to aryl nitriles. | ||
Inspired by this work, we envisioned that the tandem Csp3–H azidation and oxidative rearrangement of allyl arenes would afford the corresponding alkenyl nitriles. Although fascinating examples of allylic amination by palladium catalysis have been disclosed,20 the direct transformation of allyl arenes via Csp3–H activation remains challenging.21 Therefore, the designed tandem C–H azidation and oxidative rearrangement of allyl arenes was then investigated. When DDQ was employed as the oxidant, 1-allylbenzene gave the desired product (E)-3-phenyl-2-propenenitrile in 19% yield under the reaction conditions (eqn (3)).22 To our delight, the yield of the desired product was promoted to 95% by using 10 mol% FeCl2 as the catalyst.
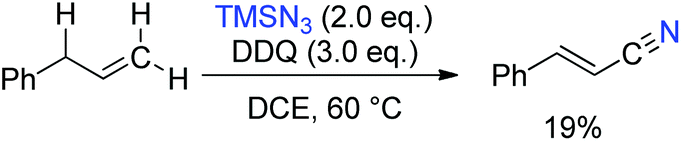 | (3) |
Various allyl substrates were investigated under the iron catalytic oxidative conditions. The allylarenes bearing different substituents were compatible in this transformation. Substituents at different positions of the aryl ring (para-, meta-, and ortho-position) did not affect the efficiencies. Notably, a heteroaryl substituted propene, such as 1-allyl-2-thiophene provided the corresponding nitrile in 77% yield. A simple aliphatic (E)-deca-1,4,9-triene was tested too, but no desired product was detected. Both (E)- and (Z)-propenylbenzene can provide high stereoselectivity, indicating that a π-allyl species may be involved in the reaction (Scheme 11).
 | ||
| Scheme 11 Transformation of allylarenes or alkenes to alkenyl nitriles. | ||
The allyl Csp3–H bond activation was proposed to be the rate-determining step. Both intra- and intermolecular kinetic isotopic effects were evident (kH/kD = 3.5 and 4.6, respectively). Besides, no azide substitution product was observed by reducing the amount of DDQ (Scheme 12).
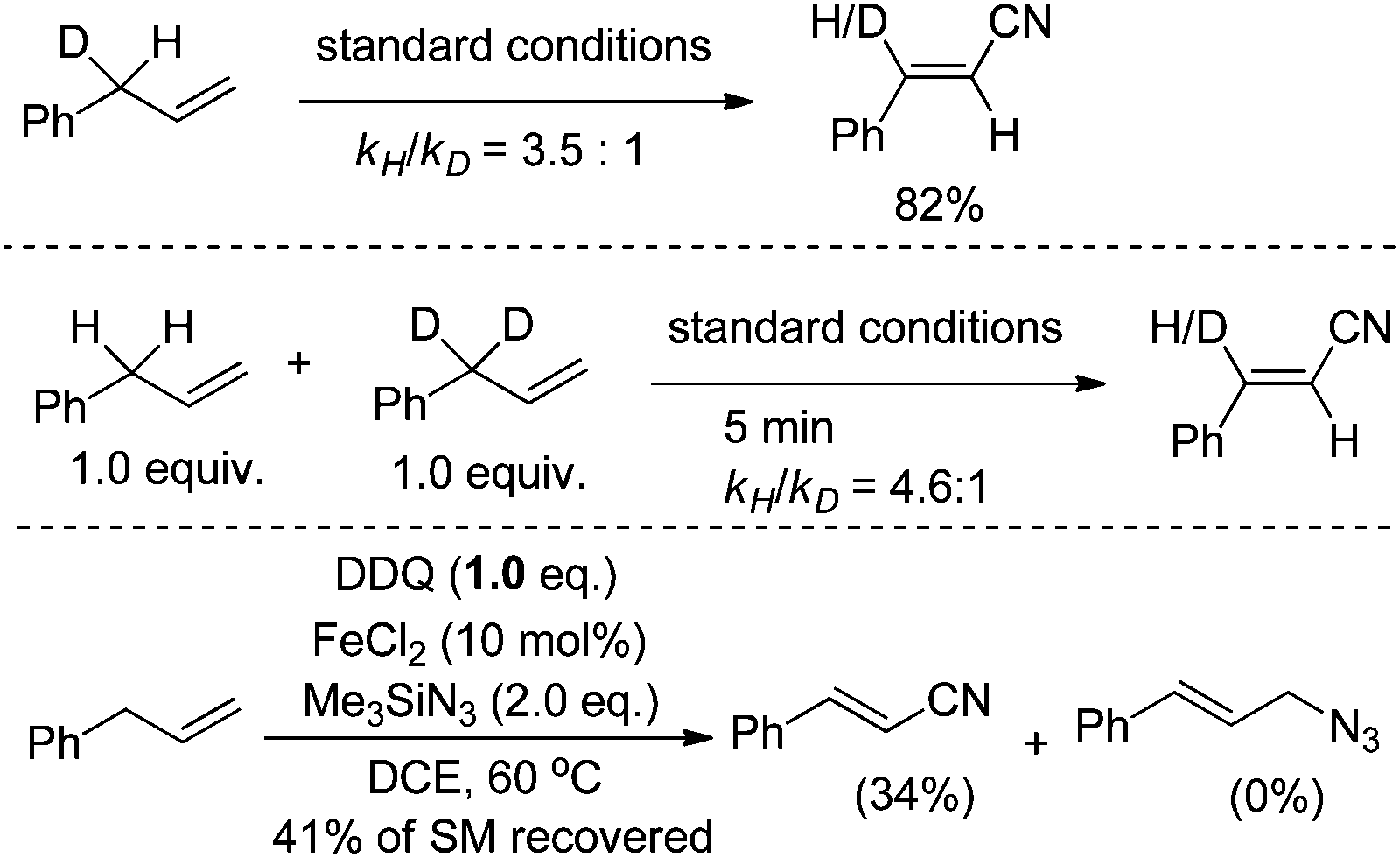 | ||
| Scheme 12 Mechanism studies. | ||
A SET oxidation mechanism is proposed for this reaction. Initially, allyl arene undergoes successive iron-assisted single-electron oxidation to produce the corresponding allyl cation 14. Subsequently, 14 experiences nucleophilic attack to yield allyl azide 15 and 15′, which would exist as an equilibrating mixture by [3,3]-sigmatropic rearrangement. Allyl azide 15 is further oxidized to generate allyl azide cation 16, which undergoes Schmidt-type rearrangement to afford the desired nitrile (Scheme 13).
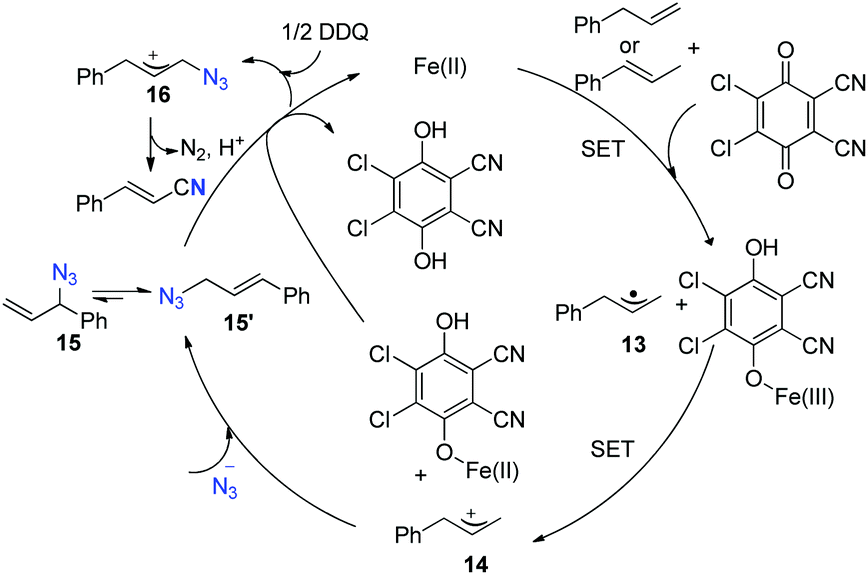 | ||
| Scheme 13 Proposed mechanism for this alkenyl nitrile synthesis method. | ||
Iron-catalyzed nitrogenation of simple hydrocarbons through C–H and C–C bond cleavage for the synthesis of amides
The development of a more atom-efficient, novel catalytic method for the preparation of amides under mild conditions has been one of the hot topics in organic synthesis, because they are important moieties in organic chemistry, biology and pharmaceutical molecules.23 Although the functionalized compounds prepared by direct C–H or C–C bond activation have been the focus of significant recent interest, the amide bond formation through C–H and/or C–C bond cleavage remains challenging.24 Inspired by our previous direct aryl and alkenyl nitrile synthesis methods and to further enhance the utility of azide in synthesizing nitrogen-containing molecules, we demonstrated a novel iron-catalyzed transformation of benzyl hydrocarbons to their corresponding amides through C–H and C–C bond cleavage under mild conditions. The substrate scope is broad and the ring expansion can be significantly achieved using this present strategy to generate lactams. In addition, under the optimized reaction conditions, (E)-1,3-diphenylpropenes can be converted into the corresponding acrylamides in good to excellent yields, the high chemoselectivity indicates that the aryl groups have a better migration ability to the nitrogen atom than alkenyl groups in the rearrangement process (Scheme 14).25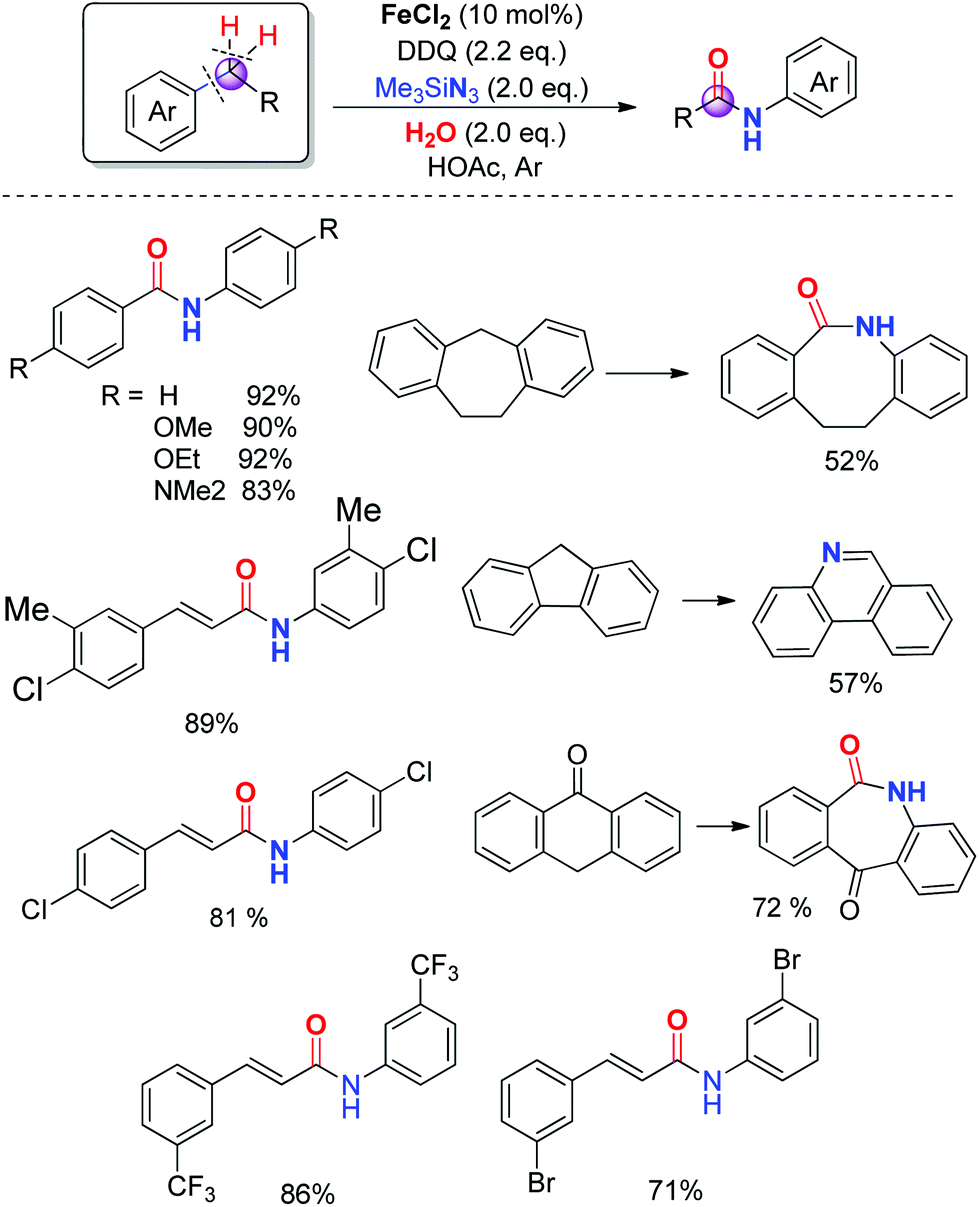 | ||
| Scheme 14 Direct transformation of benzyl hydrocarbons and 1,3-diphenylpropenes to amides. | ||
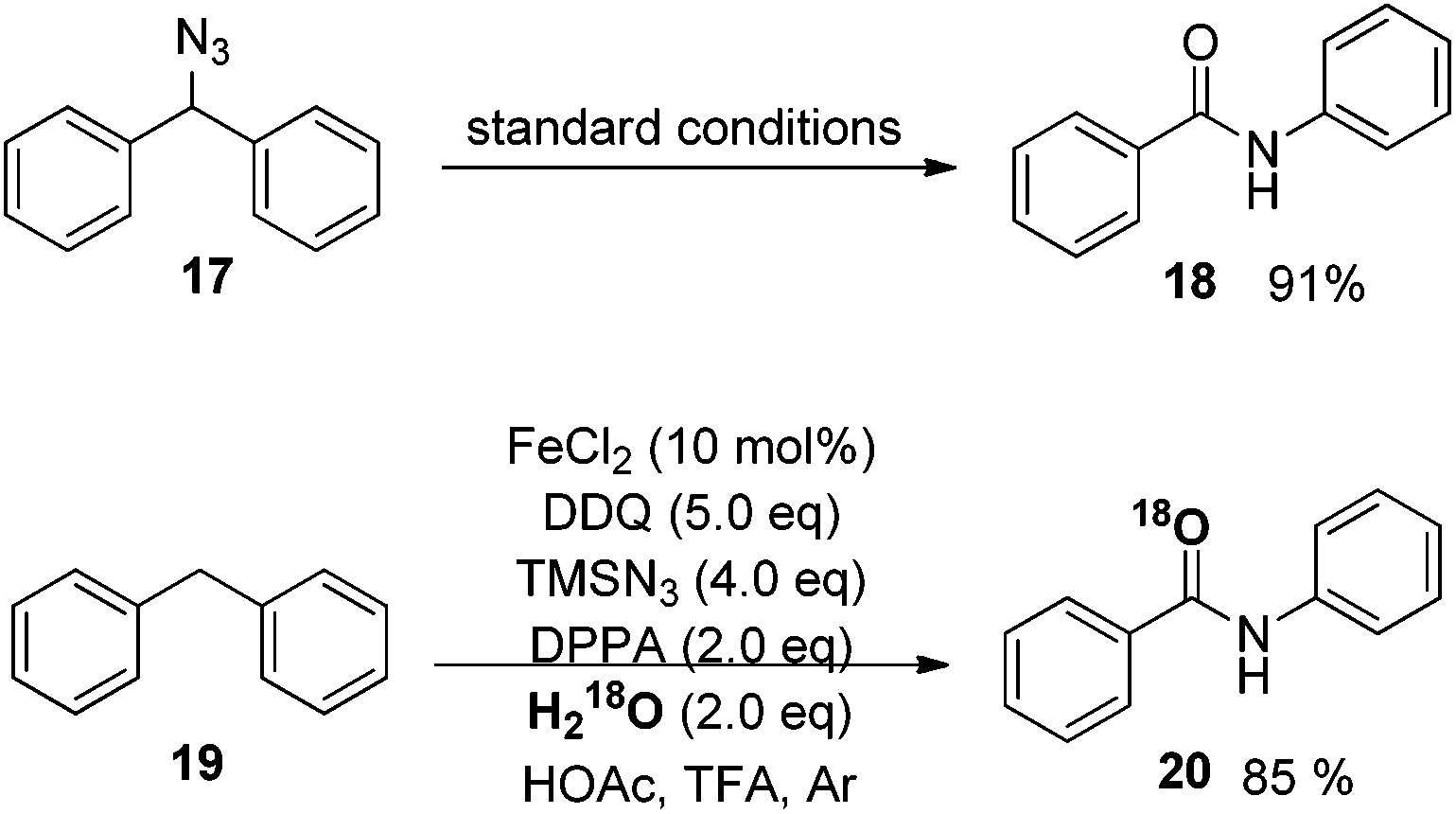
To better understand the mechanism, several possible intermediates have been investigated. All of these results suggest that the reaction does not undergo the process of oxidation of diphenylmethane to benzophenone followed by the Schmidt reaction. The possible intermediate (azidomethylene)dibenzene 17 was synthesized and subjected to standard conditions, affording amide product 18 in 91% yield. The reaction conducted in the presence of H218O afforded the desired isotope labeled product 20 as expected (Scheme 15).25
 | ||
| Scheme 15 Mechanism studies. | ||
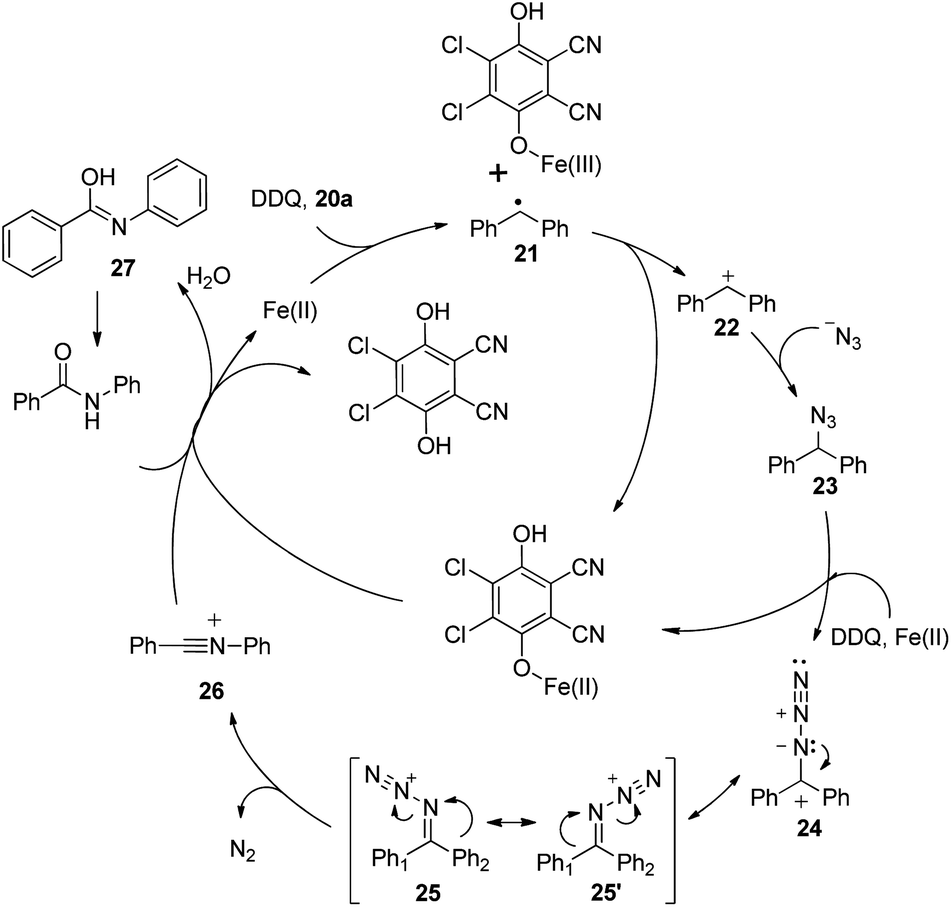
A SET oxidation mechanism is proposed for the reaction. Initially, the substrate undergoes the iron-assisted SET oxidation with DDQ22 to produce the corresponding diphenylmethyl radical 21, which could be further oxidized to diphenylmethyl cation 22. Then diphenylmethyl cation 22 is attacked by an azide anion to afford 23, which would be oxidized to diphenylmethyl azide cation 24 by the iron assisted DDQ oxidative system. Subsequent isomerization of diphenylmethyl azide cation 24 leads to intermediates 25 and 25′. For the unsymmetrical substrate, two isomeric iminodiazonium ions (25 and 25′) would be produced in this step. As in the Beckmann rearrangement, the trans group migrates from carbon to nitrogen to generate intermediate 26. A subsequent nucleophilic attack by H2O leads to the desired amide product via isomerization of intermediate 27 (Scheme 16).
 | ||
| Scheme 16 Proposed mechanism. | ||
The mechanistic insight into this novel transformation motivates us to discover other new types of reactions via C–C bond cleavage by using other nucleophiles instead of H2O to trap intermediate 26. Moreover, we also successfully developed a direct amide synthesis method via the Csp2–Csp bond cleavage of aryl substituted alkynes, which offers a novel application of alkynes in amide synthesis (eqn (4)).26
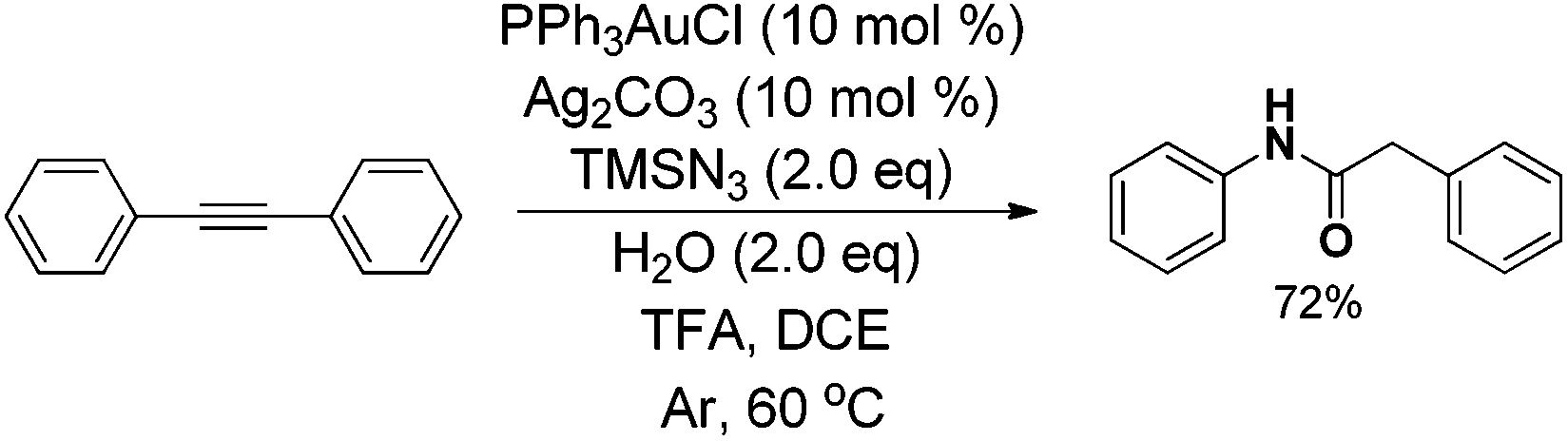 | (4) |
Cu-catalyzed nitrogenation of simple hydrocarbons through C–H and C–C bond cleavage for the synthesis of tetrazoles
Tetrazoles are of high utility in chemistry and biology, in particular 1,5-disubstituted tetrazoles. Thus, many methods have been developed for their synthesis.27 However, these approaches have some limitations, such as the use of protic acid catalysts, preactivated starting materials, and tedious workups, and hence a direct synthesis method for their access remains attractive.Inspired by the above amide construction, in this work, we envisioned that the allylic azide cation would undergo aryl migration to the proximal nitrogen with the extrusion of nitrogen gas to generate intermediate 29. We tried to trap this intermediate 29 by using excess azide as the nucleophile to produce tetrazole product 30 (Scheme 17).28
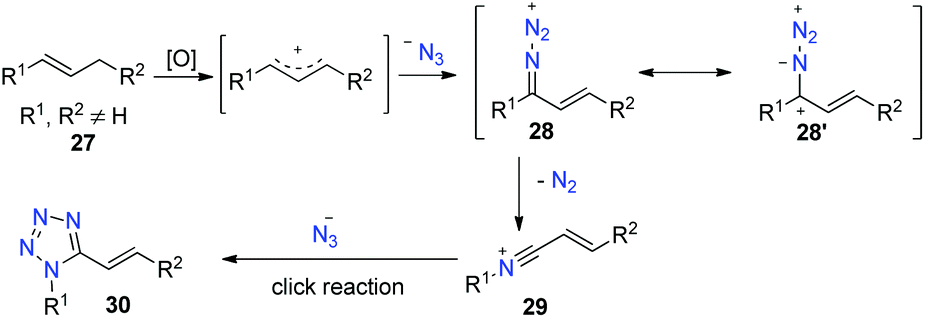 | ||
| Scheme 17 Envisioned pathway in forming tetrazoles. | ||
To test this hypothesis, we employed (E)-1,3-diphenylprop-1-ene as the substrate in the preliminary investigation. After extensive condition screening, we found that the reaction of (E)-1,3-diphenylprop-1-ene with 5.5 equivalents of TMSN3 as the nitrogen source catalyzed by CuI in the presence of a DDQ oxidant in MeCN at 80 °C worked well to afford the desired (E)-1-phenyl-5-styryl-1H-tetrazole in 88% yield. Under the standard conditions, various substituted 1,3-diarylprop-1-enes could easily be converted into the corresponding tetrazoles in good yields. Notably, bisheteroaryl methanes could be successfully converted into the corresponding 1,5-diheteroaryl tetrazoles too, albeit in moderate yields (Scheme 18).
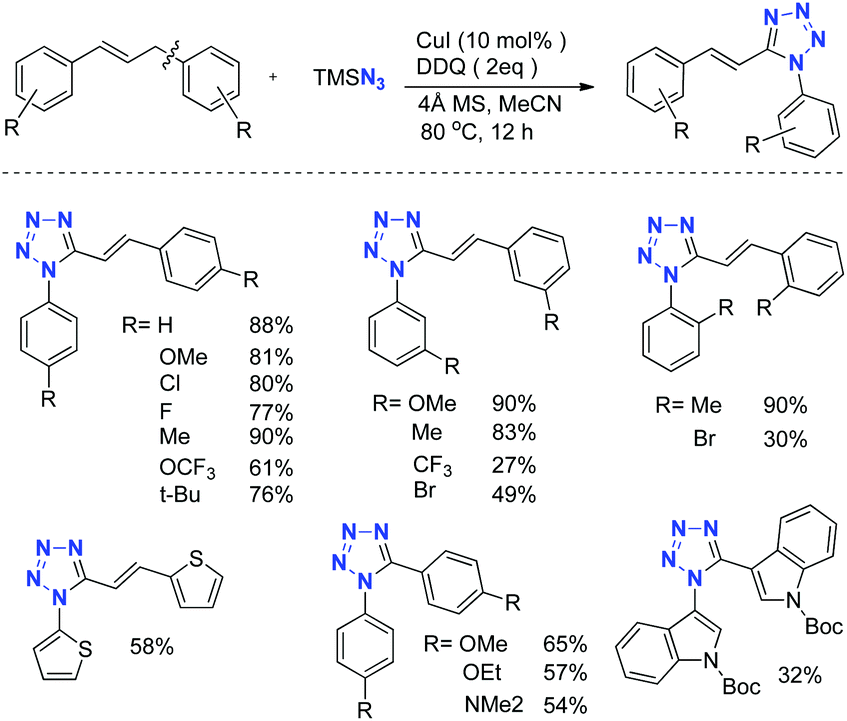 | ||
| Scheme 18 Direct synthesis of tetrazoles. | ||
This protocol demonstrates a novel and efficient Cu-promoted implanting of nitrogen into simple hydrocarbon molecules to construct 1,5-disubstituted tetrazoles through C–H and C–C bond cleavage under mild and neutral reaction conditions. It not only extends the application of azides in organic transformations, but also offers an alternative method to prepare 1,5-disubstituted tetrazoles, which are ubiquitous structural units in numerous biologically active compounds.
FeCl2-promoted arylamine synthesis from alkylarenes through C–C bond nitrogenation
Arylamines are common and fundamental industrial feedstocks. For instance, N-alkylanilines are widely used in the synthesis of important dyes, polymers, herbicides, insecticides, pharmaceuticals, plant growth agents and antiknock agents for gasoline engines.29 On the basis of the above reactions, after the generation of a carbon cation intermediate, we tried to use an alkyl azide to trap the cation intermediate and for the subsequent rearrangement via C–C bond cleavage (eqn (5)).30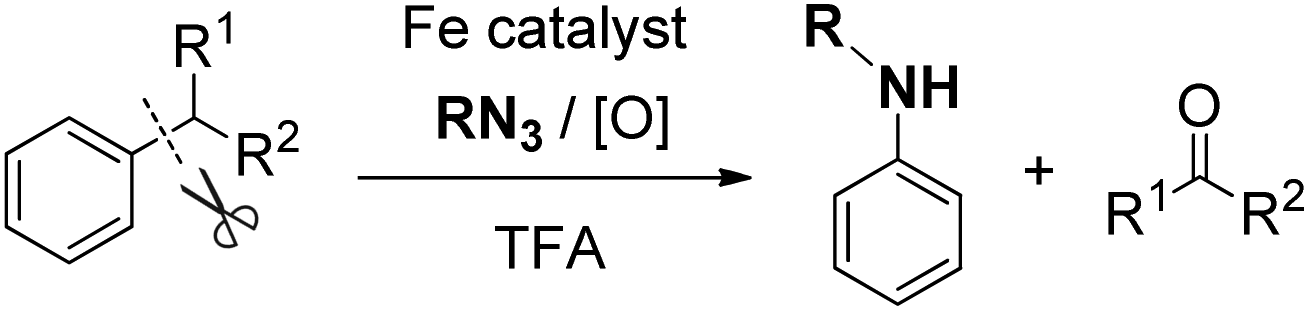 | (5) |
Interestingly, when diphenylmethane was treated with 1-azidononane, the reaction catalyzed by FeCl2 in the presence of DDQ as the oxidant in trifluoroacetic acid (TFA) afforded the desired nitrogenation product N-nonylaniline in 75% isolated yield with the generation of 72% GC yield of benzaldehyde. Under the optimized reaction conditions, various diarylmethanes and benzyl alkanes were found to be compatible with this protocol. The nitrogenation process can also be extended to inert benzyl alkanes, furnishing the desired N-alkyl aniline in good yields. The iron catalyst was found to be essential in other cases employing relatively unactivated alkylarenes as the substrates (Scheme 19).
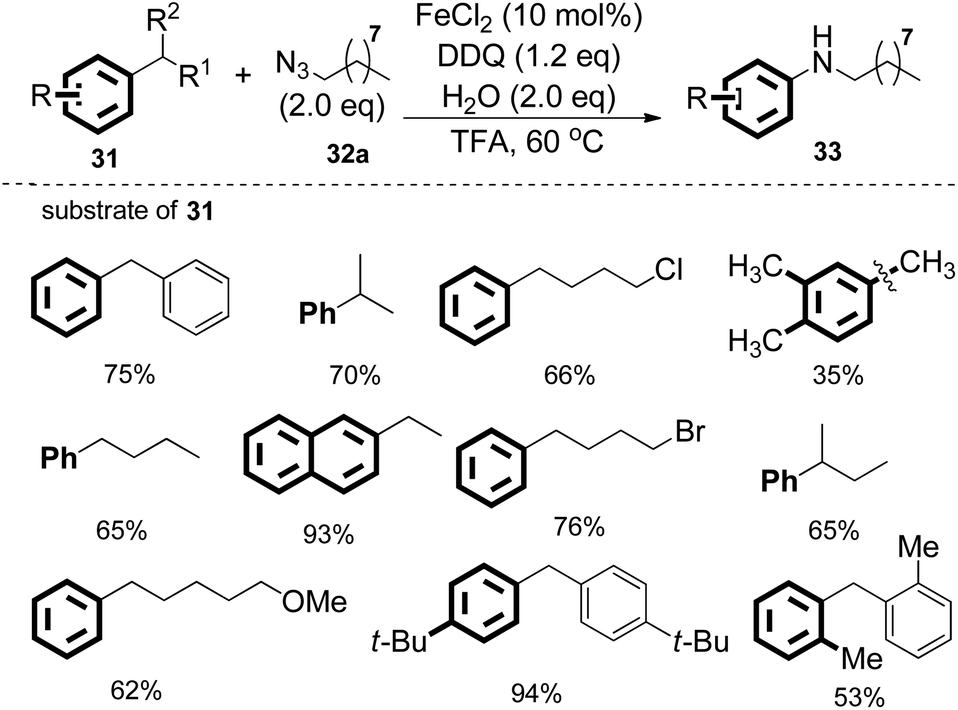 | ||
| Scheme 19 Direct synthesis of aniline from various benzyl hydrocarbons. | ||
Moreover, different alkyl azides can be applied in the reaction, and the length of alkyl chain did not affect the efficiencies, therefore extending the utility of this transformation. Alkyl azides with functional groups at the alkyl chain such as phenyl, alkenyl and ethylene glycol units were also compatible (Scheme 20).
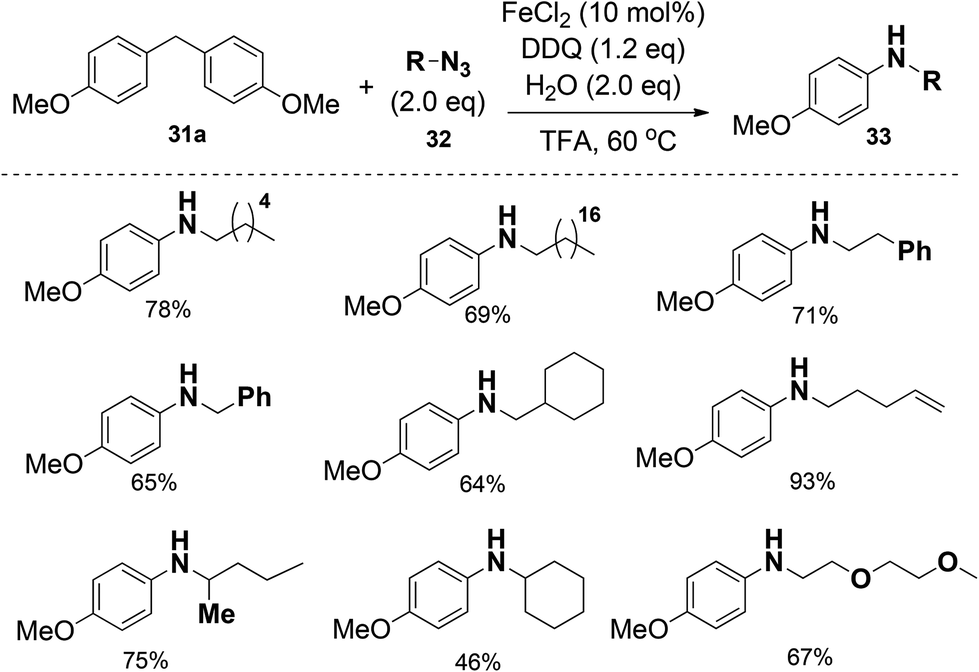 | ||
| Scheme 20 Scope of organic azide in this transformation. | ||
In addition, this protocol can be conducted on a 24 g large scale in lab (a, Scheme 21). Even the mixture of alkyl benzenes can also be smoothly converted (b, Scheme 21), which demonstrates the potential for applying this oxidative unactivated C–C bond cleavage strategy to the conversion of a crude mixture of benzyl hydrocarbons from the oil and coal industry to single N-alkyl aniline products. Moreover, the present strategy was also applicable to ring opening reactions (c, Scheme 21).
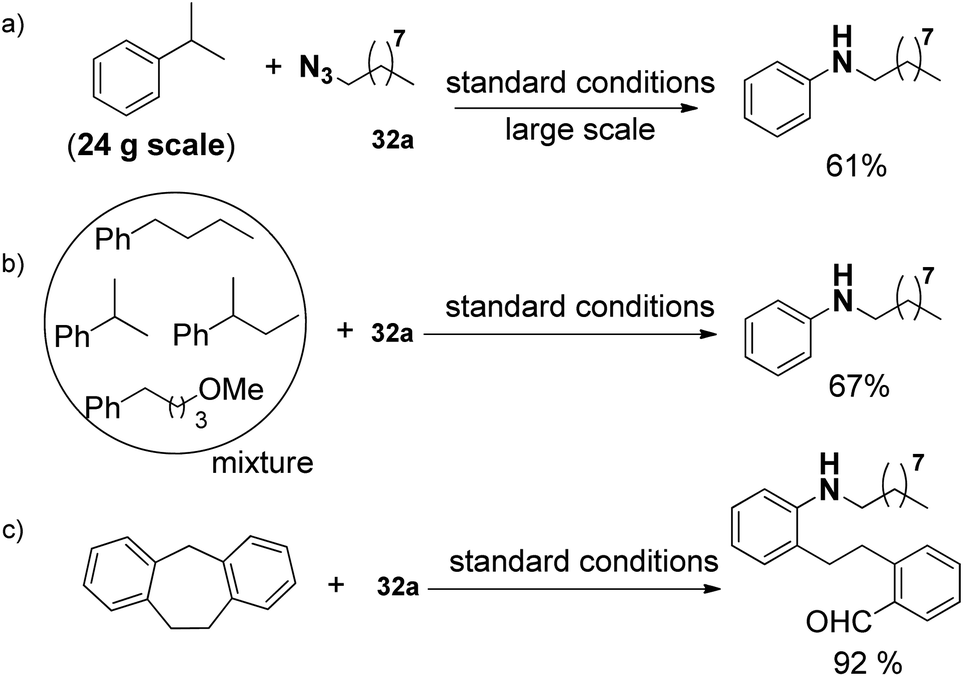 | ||
| Scheme 21 Direct transformation of various benzyl hydrocarbons into aniline. | ||
Significantly, the desired N-nonylaniline was obtained in 17% yield when polystyrene was used as the substrate. Moreover, waste polystyrene foam can also be used as a reactant to afford N-nonylaniline in 12% yield. This may provide a novel concept for polystyrene degradation and reuse (Scheme 22).
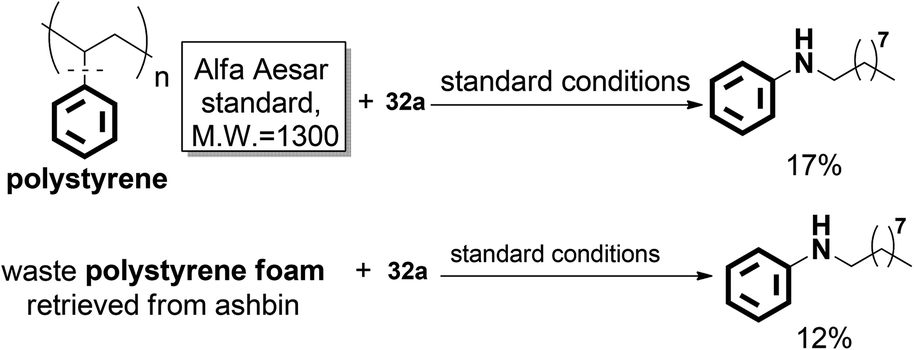 | ||
| Scheme 22 Direct transformation of polystyrene into aniline. | ||
Mechanistic studies rule out the pathway through the oxidation of diphenylmethane to benzophenone. Furthermore, deuterated substrate d2-31a was subjected to the reaction and produced the deuterated product 4-methoxybenzaldehyde 34 in 62% yield, which may exclude the pathway of the initial oxidation to ketone or the formation of imine intermediate via nitrene before the aryl shift step. Furthermore, the intra- and inter-molecular kinetic isotopic effects (kH/kD) were 5.3 and 2.6 respectively (Scheme 23).31
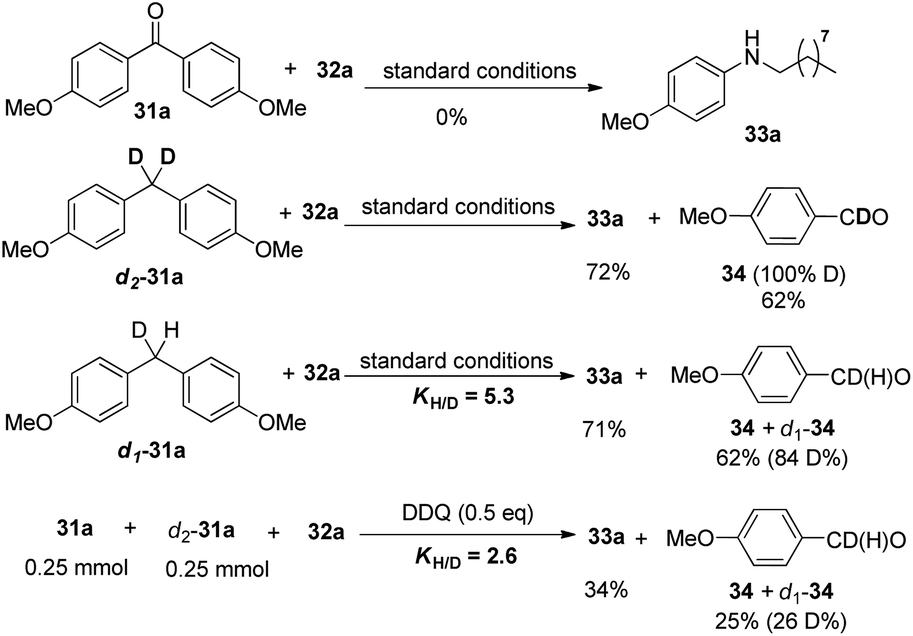 | ||
| Scheme 23 Mechanistic studies. | ||
Although the exact mechanism is not clear, a possible pathway could initiate from iron-assisted oxidation32 of the substrate to cation 35, which would be attacked by nucleophilic organic azides15 to generate intermediate 36. The following Schmidt-type rearrangement17 process via extrusion of nitrogen gas and trans migration of the aryl group R2 from carbon to the nitrogen atom generating intermediate 37. Subsequent isomerization of cation 37 leads to the iminium cation, which may exist as iminium trifluoroacetic acid salt 38 in the reaction mixture. Finally, N-alkyl anilines and benzaldehydes are generated via hydrolysis of 38 (Scheme 24).
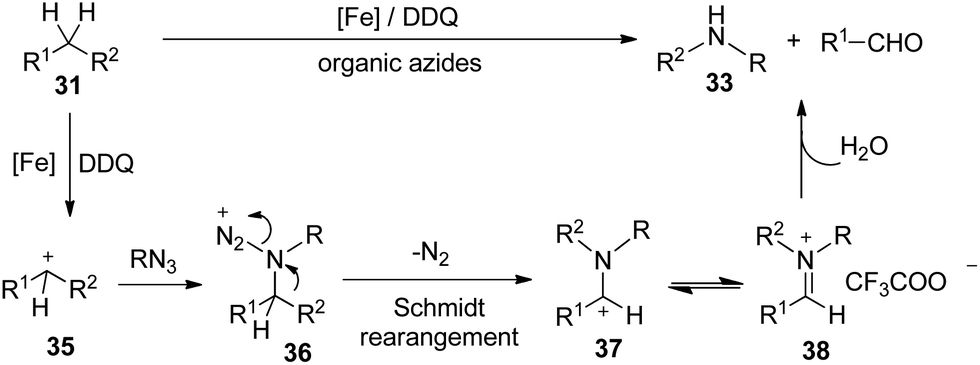 | ||
| Scheme 24 Proposed mechanism. | ||
This protocol offers a new route for aryl amide synthesis by employing benzyl hydrocarbons and very stable alkyl azides as substrates, even the unseparated alkylarene mixture and polystyrene could be employed too, which offers a new strategy for degradation and reuse of polystyrene. A Schmidt type rearrangement process is proposed for this transformation involving the release of nitrogen and migration of the aryl group from the carbon to the nitrogen atom.
Direct transformation of N,N-dimethylformamide (DMF) to –CN
Although azides exhibit powerful functions to build N-containing compounds, some features like toxicity and explosibility of these reagents limit their further applications. Traditional CN sources include metal cyanide salts and acetone cyanohydrins, which provide the cyanide unit in its entirety in metal catalyzed cyanation of aryl halides. The stoichiometric toxic cyanide salts are inevitably used in the reaction. To solve this problem, many safe and readily available cyanide sources generated in situ are developed by diverse groups in recent years.33 Interestingly, a combined cyanide source from DMF and ammonia or ammonium salts have been reported by Chang for various cyanation reactions, whereas the combination of ammonium salts and DMSO was also used as a safe cyanide source for the cyanation of indole C–H bonds. These studies revealed that DMF and DMSO are not only polar solvents, but also have been employed as versatile synthetic precursors to yield a number of functional units (Scheme 25).34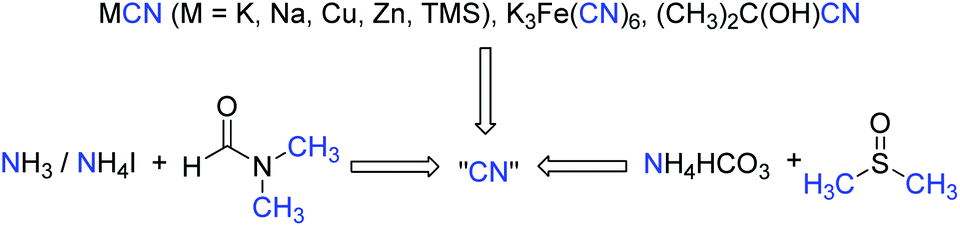 | ||
| Scheme 25 Direct transformation of N,N-dimethylformamide (DMF) to –CN. | ||
Recently, we developed a direct C–H cyanation of indoles just by using DMF as the CN source (Scheme 26).35 The control experiments indicated that CuBr2 and Pd(OAc)2 are essential to this direct cyanation reaction. FeCl2 was an efficient additive to promote the efficiency of this transformation.
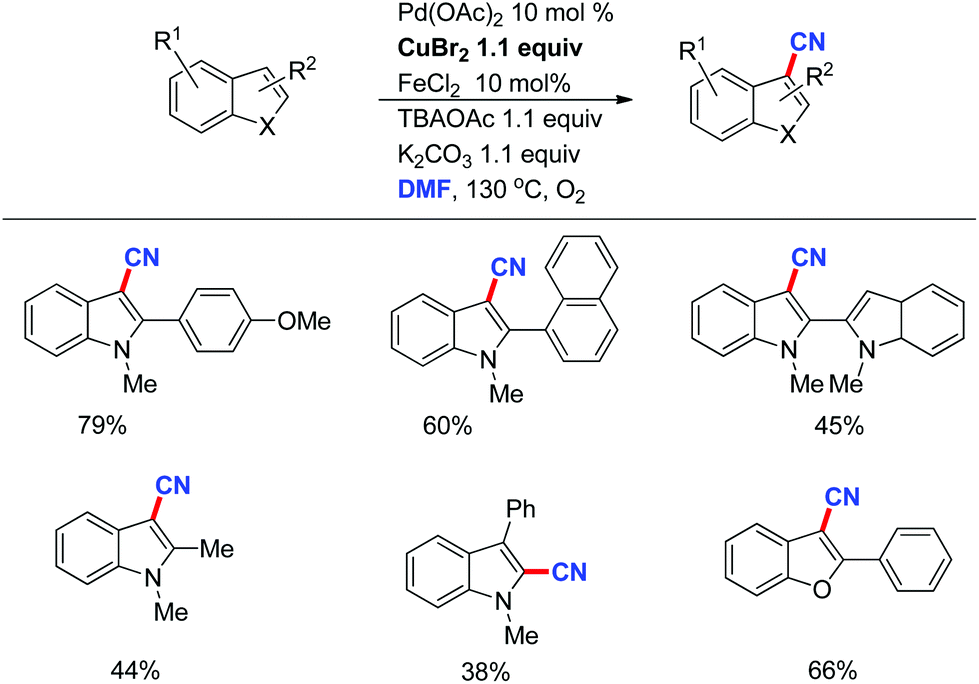 | ||
| Scheme 26 Cyanide sources in organic synthesis. | ||
To better understand the distinct role of DMF, a series of labeling reactions were carried out to determine the source of the cyanide unit (Scheme 27). The reaction of 15N-labeled DMF can pinpoint that DMF was the N-source of the cyanide group (99% incorporation, entry 1). The labeling experiment indicated that the carbon of CN and CHO hardly originated from the formyl group of DMF (<5% incorporation; entry 2); whereas the dimethylamino group of DMF became the carbon source instead. The calculation of isotope abundance of CN and CHO by inverse gated decoupling 13C NMR revealed that NMe and OMe methyl groups cleaved and exchanged with DMF in the reaction.36 The 18O labeling experiments showed that the O-source of CHO was molecular oxygen.
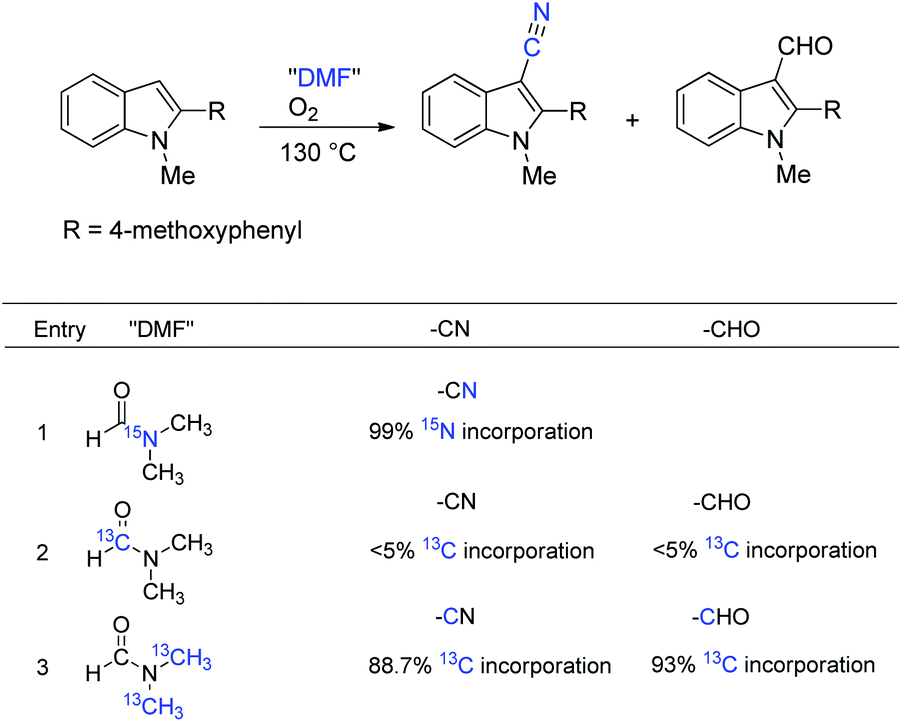 | ||
| Scheme 27 Isotope incorporation experiments. | ||
Next, various possible intermediates 39 were synthesized and investigated under optimized conditions. However, none of them could provide the desired nitrile in high yield (Scheme 28). The cyano product was produced in 28% yield in N-methylformamide instead of DMF. To investigate the possibility of isonitrile, which can be generated from DMF, as an intermediate, the control experiment was carried out using tert-butyl isonitrile or benzyl isonitrile as the solvent. However, no product was observed in these reactions. In addition, if the reaction undergoes oxidative demethylation of DMF, this species may react with copper salts to generate CuCN with the loss of “HCO”. However, control experiments excluded CuCN involved in this cyanation.
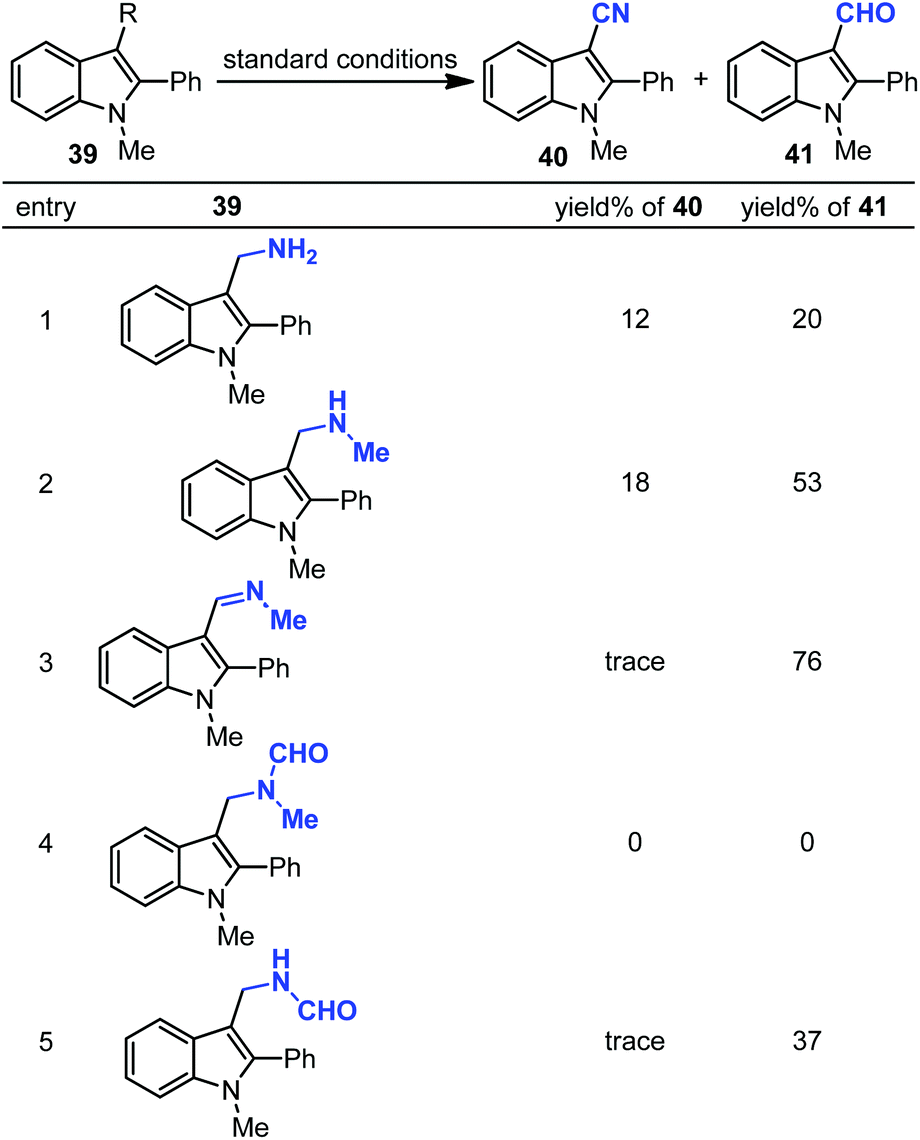 | ||
| Scheme 28 Reactions of possible intermediates. | ||
The precise mechanism is not clear yet. A proposed pathway is elucidated based on the mechanistic studies (Scheme 29). Considering that complex 44 was documented to form in DMF with copper(II) salts, we considered that it would be a possible intermediate in the reaction (as illustrated in the mechanism).37 Initially, Pd(II) intermediate 43 was afforded by electrophilic aromatic palladation. This Pd(II) species reacted with complex 44 to give 45 and/or 46via C–N cleavages. Subsequently, 39b and/or 39d were formed by reductive elimination. The cyanation product could be obtained directly from 46, whereas a pathway involving 39d cannot be excluded. Aldehyde 41 could be generated from 39b, although 41 is the major product according to the control experiment. Further mechanism studies should be conducted to explain the influencing factors of the 40![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :41 ratio.
:41 ratio.
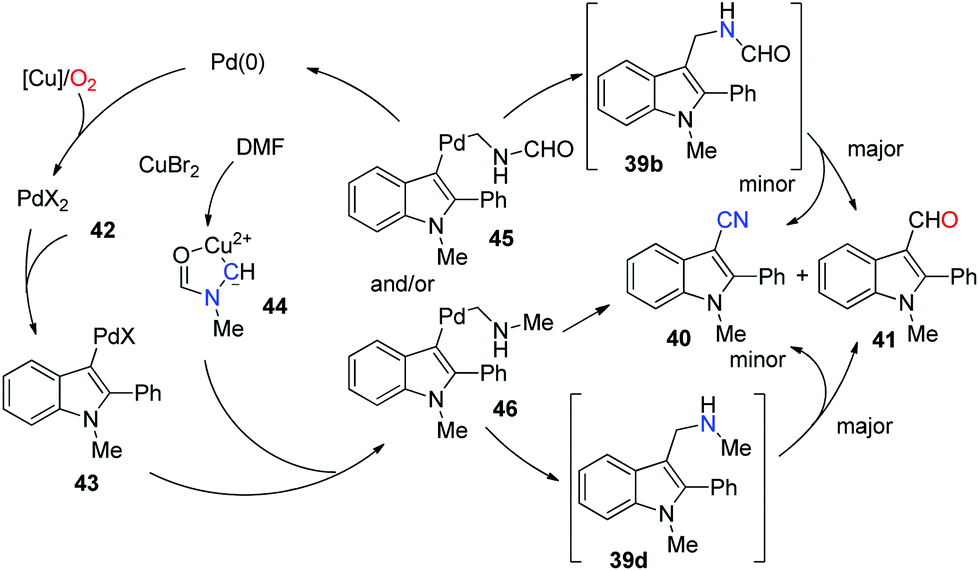 | ||
| Scheme 29 Proposed mechanism. | ||
Encouraged by this transformation, we were also interested to develop the nitrogenation reaction with some other N-containing reagents. Recently, we demonstrated a novel synthesis method for quinoxaline N-oxides through the functionalization of C(sp2)–H and C(sp3)–H bonds of readily obtainable imines by using simple and commercially available tert-butyl nitrite (TBN) as the NO source. This reaction proceeds under mild and transition metal free conditions. DFT calculation and EPR measurement suggested that this reaction involved multiple SET processes (eqn (6)).38
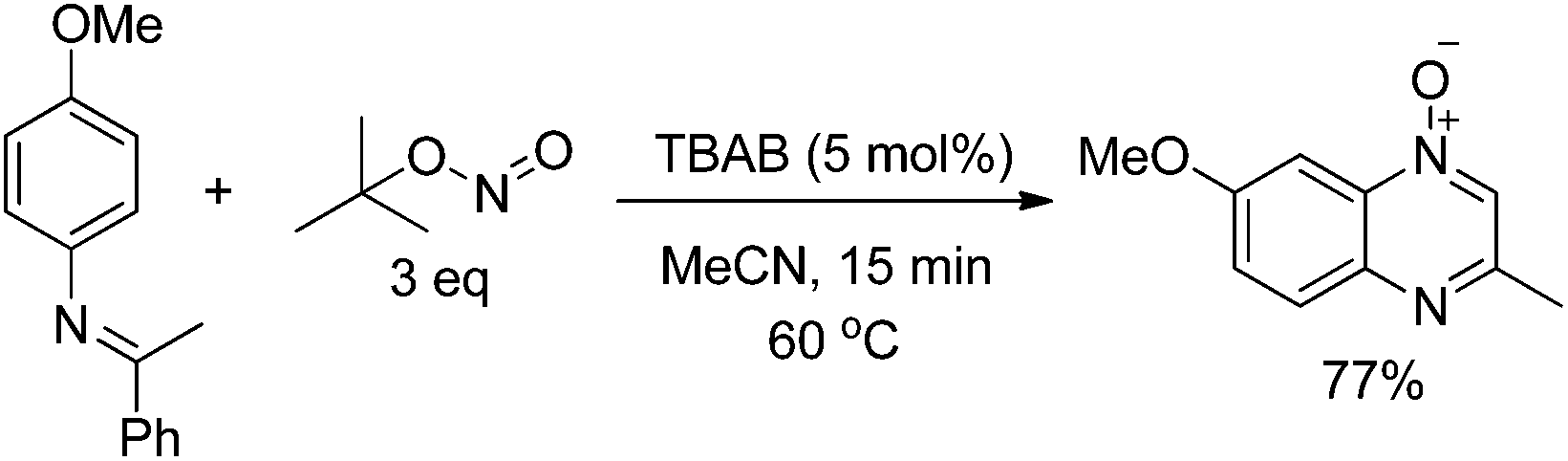 | (6) |
Conclusion
Via Cu or Fe catalyzed C–H and/or C–C bond cleavage from some readily available hydrocarbons by using nonmetal oxidants and simple nitrogen sources under mild conditions, novel synthetic approaches to ortho-azido anilines, nitriles, amides, arylamines, and tetrazoles have been developed. The azides, DMF, and TBN reagents are used as potential nitrogen sources in the C–N bond formation reaction. Mechanism studies revealed that a cation or a radical intermediate is involved in these nitrogenation processes. Despite these contributions and significant progress, many challenges still remain. To meet these, many more mechanistic studies have to be performed to shed more light on the potential intermediates involved during the reactions. A better understanding of the mechanism can provide more inspiration on new reaction design. Last but not least, the exploration of safe, efficient and readily available nitrogenation reagents is always favorable.Conflict of interest
The authors declare no competing financial interest.Acknowledgements
Financial support from the National Basic Research Program of China (973 Program) (grant no. 2015CB856600), the National Natural Science Foundation of China (no. 21325206, 21172006), and the National Young Top-notch Talent Support Program are greatly appreciated.References
- (a) G. Jones, in Comprehensive Heterocyclic Chemistry II, ed. A. R. Katritzky, C. W. Rees, E. F. V. Scriven and A. McKillop, Pergamon, Oxford, UK, 1996, vol. 5, p. 167 Search PubMed; (b) Pharmaceutical Chemistry. Drug Synthesis, ed. H. J. Roth and A. Kleemann, Prentice Hall Europe, London, 1988, vol. 1, p. 407 Search PubMed; (c) Electronic Materials: The Oligomer Approach, ed. K. Müllen and G. Wegner, Wiley-VCH, Weinheim, 1998 Search PubMed; (d) A. Kleemann, J. Engel, B. Kutscher and D. Reichert, Pharmaceutical Substances: Synthesis, Patents, Applications, Thieme, Stuttgart, Germany, 4th edn, 2001 Search PubMed; (e) N. K. Boaen and M. A. Hillmyer, Chem. Soc. Rev., 2005, 34, 267 RSC; (f) A. G. Habeeb, P. N. P. Rao and E. E. Knaus, J. Med. Chem., 2001, 44, 3039 CrossRef CAS PubMed; (g) M. de Gasparo and S. Whitebread, Regul. Pept., 1995, 59, 303 CrossRef CAS; (h) A. Graul and J. Castaner, Drugs Future, 1997, 22, 956 CAS; (i) K. Wellington and G. L. Plosker, Drugs, 2002, 62, 1539 CrossRef CAS PubMed.
- (a) J. F. Hartwig, Angew. Chem., Int. Ed., 1998, 37, 2046 CrossRef CAS; (b) J. E. Hein and V. V. Fokin, Chem. Soc. Rev., 2010, 39, 1302 RSC; (c) K. B. Lipkowitz, S. Scarpone, D. McCullough and C. Barney, Tetrahedron Lett., 1979, 20, 2241 CrossRef.
- (a) S. H. Cho, J. Y. Kim, J. Kwak and S. Chang, Chem. Soc. Rev., 2011, 40, 5068 RSC; (b) Handbook of C–H Transformations, ed. G. Dyker, Wiley-VCH, Weinheim, Germany, 2005 Search PubMed; (c) C–H Activation (Topics in Current Chemistry), ed. J.-Q. Yu and Z.-J. Shi, Springer-Verlag, Berlin, 2010 Search PubMed; (d) G. Rousseau and B. Breit, Angew. Chem., Int. Ed., 2011, 50, 2450 CrossRef CAS PubMed.
- (a) E. F. V. Scriven and K. Turnbull, Chem. Rev., 1988, 88, 297 CrossRef CAS; (b) S. Bräse, C. Gil, K. Knepper and V. Zimmermann, Angew. Chem., Int. Ed., 2005, 44, 5188 CrossRef PubMed; (c) M. Minozzi, D. Nanni and P. Spagnolo, Chem. – Eur. J., 2009, 15, 7830 CrossRef CAS PubMed; (d) S. Bräse and K. Banert, Organic Azides: Syntheses and Applications, Wiley-VCH, Weinheim, 2010 Search PubMed; (e) T. G. Driver, Org. Biomol. Chem., 2010, 8, 3831 RSC; (f) G. Lapointe, A. Kapat, K. Weidner and P. Renaud, Pure Appl. Chem., 2012, 84, 1633 CrossRef CAS PubMed; (g) S. Chiba, Synlett, 2012, 21 CrossRef CAS PubMed. For its use as flexible building blocks in the partial synthesis of some very complex natural products, see: (h) B. B. Snider and J. Zhou, J. Org. Chem., 2005, 70, 1087 CrossRef CAS PubMed; (i) V. Mascitti and E. J. Corey, J. Am. Chem. Soc., 2004, 126, 15664 CrossRef CAS PubMed; (j) M. P. Cassidy, A. D. Özdemir and A. Padwa, Org. Lett., 2005, 7, 1339 CrossRef CAS PubMed. For a general review see: (k) E. M. Sletten and C. R. Bertozzi, Acc. Chem. Res., 2011, 44, 666 CrossRef CAS PubMed.
- X. Chen, X.-S. Hao, C. E. Goodhue and J.-Q. Yu, J. Am. Chem. Soc., 2006, 128, 6790 CrossRef CAS PubMed.
- (a) K.-Y. Ye, H. He, W.-B. Liu, L.-X. Dai, G. Helmchen and S.-L. You, J. Am. Chem. Soc., 2011, 133, 19006 CrossRef CAS PubMed; (b) H. He, W.-B. Liu, L.-X. Dai and S.-L. You, J. Am. Chem. Soc., 2009, 131, 8346 CrossRef CAS PubMed; (c) Z. Liang, L. Ju, Y. Xie, L. Huang and Y. Zhang, Chem. – Eur. J., 2012, 18, 15816 CrossRef CAS PubMed.
- (a) C. Tang and N. Jiao, J. Am. Chem. Soc., 2012, 134, 18924 CrossRef CAS PubMed; (b) B. Yao, Y. Liu, L. Zhao, D.-X. Wang and M.-X. Xiang, J. Org. Chem., 2014, 79, 11139 CrossRef CAS PubMed; (c) Z.-L. Wang, L. Zhao and M.-X. Wang, Org. Lett., 2011, 13, 6560 CrossRef CAS PubMed; (d) H. Zhang, L. Zhao, D.-X. Wang and M.-X. Wang, Org. Lett., 2013, 15, 3836 CrossRef CAS PubMed; (e) Y. Fan, W. Wan, G. Ma, W. Gao, H. Jiang, S. Zhu and J. Hao, Chem. Commun., 2014, 50, 5733 RSC.
- R. Giri and J. F. Hartwig, J. Am. Chem. Soc., 2010, 132, 15860 CrossRef CAS PubMed.
- (a) Y. J. Park, J.-W. Park and C.-H. Jun, Acc. Chem. Res., 2008, 41, 222 CrossRef CAS PubMed; (b) M. Tobisu and N. Chatani, Chem. Soc. Rev., 2008, 37, 300 RSC; (c) F. Chen, T. Wang and N. Jiao, Chem. Rev., 2014, 114, 8613 CrossRef CAS PubMed.
- (a) R.-J. Song, Y. Liu, R.-X. Hu, Y.-Y. Liu, J.-C. Wu, X.-H. Yang and J.-H. Li, Adv. Synth. Catal., 2011, 353, 1467 CrossRef CAS; (b) S. Chiba, L. Zhang, G. Y. Ang and B. W.-Q. Hui, Org. Lett., 2010, 12, 2052 CrossRef CAS PubMed; (c) J. Mecinovic, R. B. Hamed and C. J. Schofield, Angew. Chem., Int. Ed., 2009, 48, 2796 CrossRef CAS PubMed; (d) C. He, S. Gou, L. Huang and A. Lei, J. Am. Chem. Soc., 2010, 132, 8273 CrossRef CAS PubMed; (e) H. Liu, C. Dong, Z. Zhang, P. Wu and X. Jiang, Angew. Chem., Int. Ed., 2012, 51, 12570 CrossRef CAS PubMed; (f) C. Zhang, Z. Xu, T. Shen, G. L. Wu and N. Jiao, Org. Lett., 2012, 14, 2362 CrossRef CAS PubMed; (g) B. Tiwari, J. Zhang and Y. R. Chi, Angew. Chem., Int. Ed., 2012, 51, 1911 CrossRef CAS PubMed; (h) P. Feng, X. Sun, Y. Su, X. Li, L.-H. Zhang, X. Shi and N. Jiao, Org. Lett., 2014, 16, 3388 CrossRef CAS PubMed.
- (a) C. Zhang, P. Feng and N. Jiao, J. Am. Chem. Soc., 2013, 135, 15257 CrossRef CAS PubMed; (b) L. Zhang, X. Bi, X. Guan, X. Li, Q. Liu, B.-D. Barry and P. Liao, Angew. Chem., Int. Ed., 2013, 52, 11303 CrossRef CAS PubMed.
- C. Tang and N. Jiao, Angew. Chem., Int. Ed., 2014, 53, 6528 CrossRef CAS PubMed.
- (a) C. Zhang, C. Tang and N. Jiao, Chem. Soc. Rev., 2012, 41, 3464 RSC; (b) M. Gómez-Gallego and M. A. Sierra, Chem. Rev., 2011, 111, 4857 CrossRef PubMed.
- (a) A. J. Fatiadi, in Preparation and Synthetic Applications of Cyano Compounds, ed. S. Patai and Z. Rappaport, Wiley, New York, 1983 Search PubMed; (b) J. S. Miller and J. L. Manson, Acc. Chem. Res., 2001, 34, 563 CrossRef CAS PubMed; (c) F. F. Fleming, L. Yao, P. C. Ravikumar, L. Funk and B. C. Shook, J. Med. Chem., 2010, 53, 7902 CrossRef CAS PubMed.
- W. Zhou, L. Zhang and N. Jiao, Angew. Chem., Int. Ed., 2009, 48, 7094 CrossRef CAS PubMed.
- Z. Shu, Y. Ye, Y. Deng, Y. Zhang and J. Wang, Angew. Chem., Int. Ed., 2013, 52, 10573 CrossRef CAS PubMed.
- (a) K. F. Schmidt, Eur. J. Inorg. Chem., 1924, 57, 704 Search PubMed; (b) H. Wolff, Schmidt Reaction, Org. React., 1946, 307 CAS; (c) K. F. Z. Schmidt, Angew. Chem., 1923, 36, 511 Search PubMed.
- F. Chen, X. Huang, Y. Cui and N. Jiao, Chem. – Eur. J., 2013, 19, 11199 CrossRef CAS PubMed.
- (a) W. Zhou, J. Xu, L. Zhang and N. Jiao, Org. Lett., 2010, 12, 2888 CrossRef CAS PubMed; (b) W. Zhou, J. Xu, L. Zhang and N. Jiao, Synlett, 2011, 887 CAS; (c) T. Wang, Y. Hang and N. Jiao, Adv. Synth. Catal., 2013, 355, 1207 CrossRef CAS; (d) X. Huang and N. Jiao, Org. Biomol. Chem., 2014, 12, 4324 RSC; (e) M. Lamani and K. R. Prabhu, Angew. Chem., Int. Ed., 2010, 49, 6622 CrossRef CAS PubMed; (f) J. He, K. Yamaguchi and N. Mizuno, J. Org. Chem., 2011, 76, 4606 CrossRef CAS PubMed.
- (a) S. A. Reed and M. C. White, J. Am. Chem. Soc., 2008, 130, 3316 CrossRef CAS PubMed; (b) G. Liu, G. Yin and L. Wu, Angew. Chem., Int. Ed., 2008, 47, 4733 CrossRef CAS PubMed.
- (a) Z. Li and C.-J. Li, J. Am. Chem. Soc., 2006, 128, 56 CrossRef CAS PubMed; (b) S. Lin, C.-X. Song, G.-X. Cai, W.-H. Wang and Z.-J. Shi, J. Am. Chem. Soc., 2008, 130, 12901 CrossRef CAS PubMed.
- C. Qin and N. Jiao, J. Am. Chem. Soc., 2010, 132, 15893 CrossRef CAS PubMed.
- (a) K. Ishihara, Y. Furuya and H. Yamamoto, Angew. Chem., Int. Ed., 2002, 41, 2983 CrossRef CAS; (b) C. W. Kuo, J. L. Zhu, J. D. Wu, C. M. Chu, C. F. Yao and K. S. Shia, Chem. Commun., 2007, 301 RSC; (c) E. Choi, C. Lee, Y. Na and S. Chang, Org. Lett., 2002, 4, 2369 CrossRef CAS PubMed; (d) K. Yamaguchi, H. Fujiwara, Y. Ogasawara, M. Kotani and N. Mizuno, Angew. Chem., Int. Ed., 2007, 46, 3922 CrossRef CAS PubMed.
- For some new approaches to amide bond formation, see: (a) C. G. Espino, K. W. Fiori, M. Kim and J. Du Bois, J. Am. Chem. Soc., 2004, 126, 15378 CrossRef CAS PubMed; (b) K. W. Fiori and J. Du Bois, J. Am. Chem. Soc., 2007, 129, 562 CrossRef CAS PubMed; (c) K. Inamoto, T. Saito, M. Katsuno, T. Sakamoto and K. Hiroya, Org. Lett., 2007, 9, 2931 CrossRef CAS PubMed; (d) G. Liu, G. Yin and L. Wu, Angew. Chem., Int. Ed., 2008, 47, 4733 CrossRef CAS PubMed. For some recent Schmidt reaction of ketones via C–C bond cleavage, see: (e) D. Song, A. Rostami and F. G. West, J. Am. Chem. Soc., 2007, 129, 12019 CrossRef CAS PubMed; (f) L. Yao and J. Aubé, J. Am. Chem. Soc., 2007, 129, 2766 CrossRef CAS PubMed; (g) T. Katori, S. Itoh, M. Sato and H. Yamataka, J. Am. Chem. Soc., 2010, 132, 3413 CrossRef CAS PubMed.
- C. Qin, W. Zhou, F. Chen, Y. Ou and N. Jiao, Angew. Chem., Int. Ed., 2011, 50, 12595 CrossRef CAS PubMed.
- C. Qin, P. Feng, Y. Ou, T. Shen, T. Wang and N. Jiao, Angew. Chem., Int. Ed., 2013, 52, 7850 CrossRef CAS PubMed.
- (a) J. Quintin and G. Lewin, J. Nat. Prod., 2004, 67, 1624 CrossRef CAS PubMed; (b) A. Fettes and E. M. Carreira, Angew. Chem., Int. Ed., 2002, 41, 4098 CrossRef CAS.
- F. Chen, C. Qin, Y. Cui and N. Jiao, Angew. Chem., Int. Ed., 2011, 50, 11487 CrossRef CAS PubMed.
- (a) A. A. Doherty, in Annual Reports in Medicinal Chemistry, Academic Press, San Diego, 1999 Search PubMed; (b) M. Horie, I. Yamaguchi and T. Yamamoto, Macromolecules, 2006, 39, 7493 CrossRef CAS.
- C. Qin, T. Shen, C. Tang and N. Jiao, Angew. Chem., Int. Ed., 2012, 51, 6971 CrossRef CAS PubMed.
- E. M. Simmons and J. F. Hartwig, Angew. Chem., Int. Ed., 2012, 51, 3066 CrossRef CAS PubMed.
- (a) Y. Zhang and C.-J. Li, J. Am. Chem. Soc., 2006, 128, 4242 CrossRef CAS PubMed; (b) Y.-Z. Li, B.-J. Li, X.-Y. Lu, S. Lin and Z.-J. Shi, Angew. Chem., Int. Ed., 2009, 48, 3817 CrossRef CAS PubMed; (c) Y. Zhang and C.-J. Li, Angew. Chem., Int. Ed., 2006, 45, 1949 CrossRef CAS PubMed.
- (a) J. Kim and S. Chang, J. Am. Chem. Soc., 2010, 132, 10272 CrossRef CAS PubMed; (b) X. Ren, J. Chen, F. Chen and J. Cheng, Chem. Commun., 2011, 47, 6725 RSC; (c) S. Kim, J. Choi, K. Shin and S. Chang, J. Am. Chem. Soc., 2012, 134, 2528 CrossRef PubMed.
- S. Ding and N. Jiao, Angew. Chem., Int. Ed., 2012, 51, 9226 CrossRef CAS PubMed.
- S. Ding and N. Jiao, J. Am. Chem. Soc., 2011, 133, 12374 CrossRef CAS PubMed.
- K. E. Roege and W. L. Kelly, Org. Lett., 2009, 11, 297 CrossRef CAS PubMed.
- V. F. Plyusnin, N. M. Bazhin and O. M. Usov, Koord. Khim., 1980, 6, 856 CAS.
- F. Chen, X. Huang, X. Li, T. Shen, M. Zou and N. Jiao, Angew. Chem., Int. Ed., 2014, 53, 10495 CrossRef CAS PubMed.
| This journal is © the Partner Organisations 2015 |