
Meinwald-type rearrangement of monosubstituted epoxides to methyl ketones using an [Al porphyrin]+[Co(CO)4]− catalyst†
Jessica R.
Lamb
,
Yukyung
Jung
and
Geoffrey W.
Coates
*
Department of Chemistry and Chemical Biology, Baker Laboratory, Cornell University, Ithaca, NY 14853-1301, USA. E-mail: gc39@cornell.edu; Fax: +1 607-255-4137; Tel: +1 607-255-5447
First published on 16th February 2015
Abstract
A Meinwald-type rearrangement of monosubstituted epoxides to methyl ketones using a well-defined aluminum porphyrin catalyst is reported. This reaction shows good functional group tolerance under mild reaction conditions to selectively give methyl ketones in excellent yields.
Epoxides are versatile intermediates in organic chemistry due to their inherent high reactivity and synthetic availability. The oxirane polarity and ring strain enable these moieties to undergo a variety of useful reactions, including nucleophilic ring opening, deoxygenation, reduction, and a variety of rearrangements.1 One reaction that has attracted a great deal of attention due to its high efficiency and synthetic potential is the Lewis-acid induced isomerization of epoxides to carbonyl compounds, known as the Meinwald rearrangement (Fig. 1).1,2
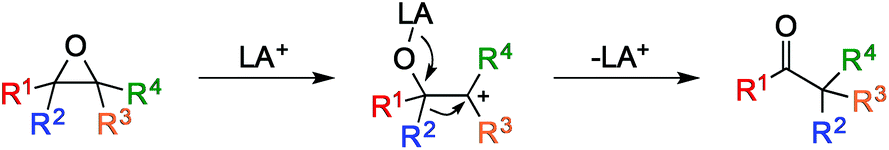 | ||
| Fig. 1 Mechanism of the Meinwald rearrangement of epoxides activated by a Lewis acid. | ||
Despite previous work to make this transformation synthetically useful, multiple products are commonly observed due to unselective ring opening and substituent migration.3 While the migratory aptitude of various substituents can often be tuned by the Lewis acid and conditions employed,4 selective ring opening using Lewis acids generally relies on substrate bias to form a single, stable tertiary (Fig. 1, R3 and R4 ≠ H) or benzylic (R3 or R4 = aryl) carbocation.5 Similarly, terminal epoxides generally yield aldehyde products because the instability of primary carbocations (R3 = R4 = H) disfavors ketone formation.6
Methyl ketones have been observed as the major product of the isomerization of monosubstituted epoxides when lithium iodide7 or transition metals—such as Fe,8 Ru,9 Co,10 Rh,11 or Pd12—are used to alter the mechanism of ring opening. While important progress has been made in this field, existing systems still have drawbacks, including limited substrate scope, modest yields, high reaction temperature, and high catalyst loading.
During our group's investigation of carbonylative ring expansion of epoxides to β-lactones using catalysts of the form [Lewis Acid]+[Co(CO)4]−,13 we observed concurrent ketone formation, especially at low pressures of carbon monoxide (Fig. 2).14 We decided to explore this reaction in more detail and selected the porphyrin-based catalyst15 [pClTPPAl(THF)2]+[Co(CO)4]− (1) because of its straightforward synthesis and known functional group tolerance.16 Herein we report a mild, high yielding, and chemo- and regioselective rearrangement of monosubstituted epoxides to methyl ketones.
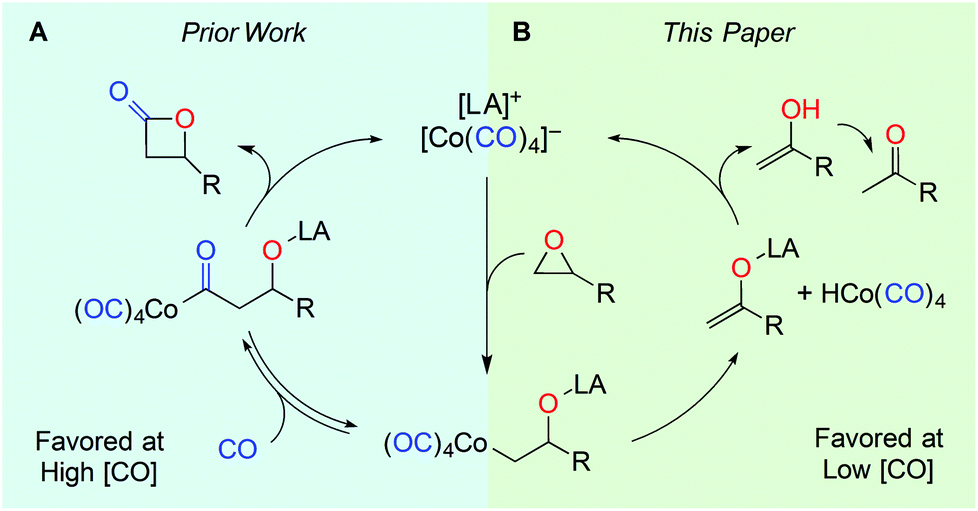 | ||
| Fig. 2 Catalytic (A) carbonylation and (B) isomerization of mono-substituted epoxides. | ||
Optimization of the reaction conditions with 1-hexene oxide (2a, Table 1) showed that the best solvent for this transformation was THF (entries 1–6). A concentration screen revealed 1.0 M as the most effective, with both higher and lower concentrations resulting in lower conversions (entries 6–11). We initially hypothesized that at higher concentrations the polarity of the ketone product might affect the rate of the reaction; however, adding 1–2 equivalents of acetone at the beginning of the reaction did not appreciably affect the conversion.17 Increasing the catalyst loading from 1 to 2 mol% resulted in full conversion of epoxide at room temperature (entry 12).
|
|
||||
|---|---|---|---|---|
| Entry | Mol% catalyst 1 | Solvent | Concentration 2a (M) | Conv.a (%) |
| a Determined by 1H NMR spectroscopy of the crude reaction mixture. | ||||
| 1 | 1 | 1,4-Dioxane | 0.5 | 21 |
| 2 | 1 | Benzene | 0.5 | 27 |
| 3 | 1 | Toluene | 0.5 | 31 |
| 4 | 1 | Ether | 0.5 | 36 |
| 5 | 1 | Hexanes | 0.5 | 28 |
| 6 | 1 | THF | 0.5 | 45 |
| 7 | 1 | THF | 1.0 | 82 |
| 8 | 1 | THF | 1.5 | 72 |
| 9 | 1 | THF | 2.0 | 67 |
| 10 | 1 | THF | 4.0 | 71 |
| 11 | 1 | Neat | 8.7 | 38 |
| 12 | 2 | THF | 1.0 | >99 |
Once viable reaction conditions were found, the substrate scope of the isomerization was explored (Chart 1). Aliphatic epoxides, including those containing substantial α-branching, were rearranged in excellent yields (≥89%, 3a–i), and no aldehyde or alcohol side products were observed. Capillary GC analysis of the crude reaction mixtures revealed trace amounts of β-lactone formation, which would result in partial deactivation of 1via loss of a CO ligand bound to the cobaltate counterion.17
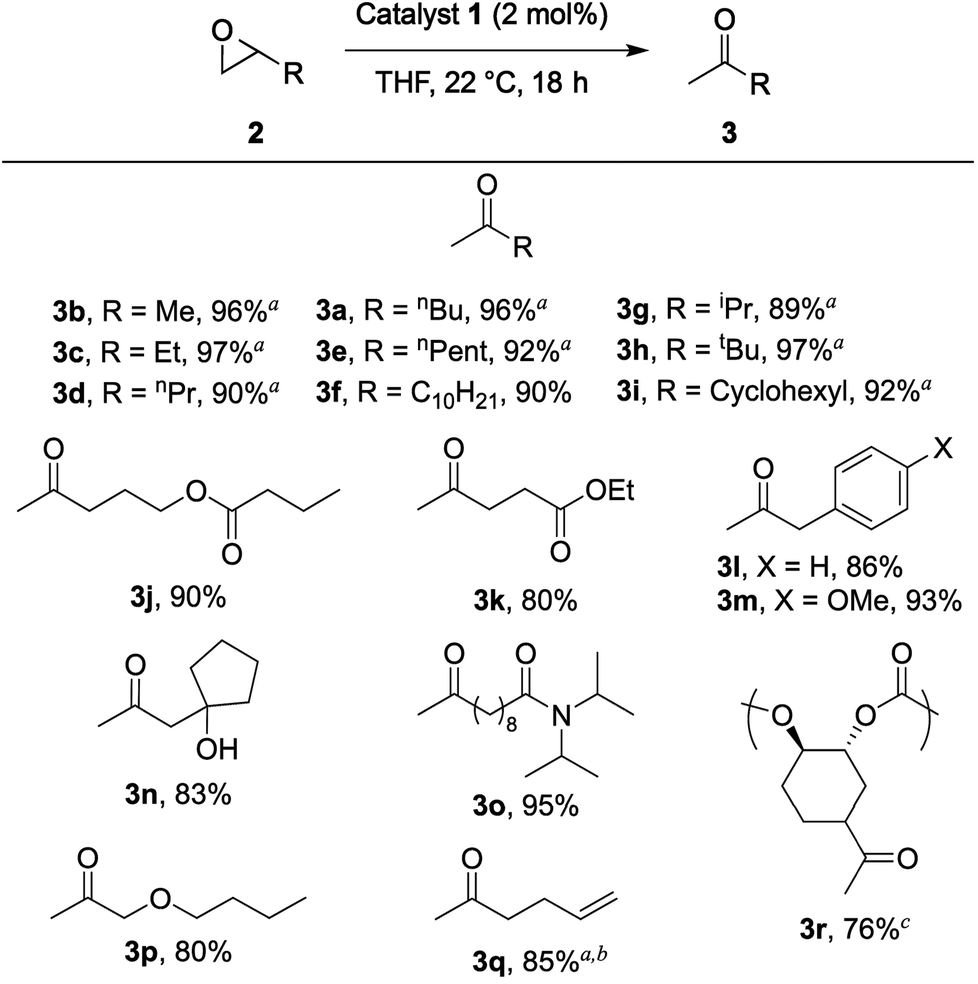 | ||
Chart 1 Isolated yields and substrate scope for the rearrangement of monosubstituted epoxides catalyzed by 1. a![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) Quantitative GC yield versus dodecane internal standard. b5 mol% 1 used. c2 mol% 1 per epoxide moiety used based on the molecular weight of the repeat unit. Quantitative GC yield versus dodecane internal standard. b5 mol% 1 used. c2 mol% 1 per epoxide moiety used based on the molecular weight of the repeat unit. | ||
As previously reported,16 the aluminum porphyrin catalyst 1 tolerates a variety of functionality under the same mild reaction conditions while maintaining high isolated yields (3j–p). Notably, this system tolerates a terminal olefin (3q), which demonstrates its potential over other ketone-forming reactions such as the Wacker oxidation. Even though this substrate required 5 mol% catalyst to achieve full conversion of epoxide, we were able to access the methyl ketone in the presence of a terminal olefin by utilizing the difference in reactivity of an epoxide and a double bond. Conversely, the Wacker oxidation would result in a diketone from the corresponding diene.18
In addition to small molecule substrates, this transformation works cleanly on polycarbonate 2r using the same reaction conditions with 2 mol% 1 per epoxide moiety. This substrate is prepared via a Zn-catalyzed copolymerization of vinylcyclohexene dioxide and CO2.19 As expected, the molecular weight and molecular weight distribution of the polymer did not change during this transformation, but a clear change in the NMR and IR spectra indicate clean conversion to the ketone-functionalized polymer (3r).17
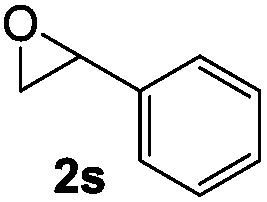
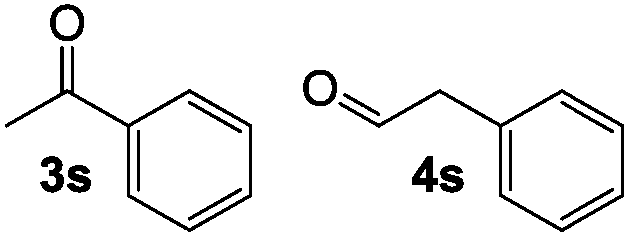
A limitation of this method was revealed during our attempt to rearrange terminal epoxides with aromatic substituents. Styrene oxide resulted in a 1:1.6 mixture of acetophenone and phenylacetaldehyde due to competing SN2 attack at the sterically unhindered methylene and the adjacent benzylic methine (Table 2, entry 1). While a variety of catalysts are known to selectively isomerize styrene oxide to either acetophenone20 or phenylacetaldehyde,5c,d,21 cobaltate-based catalysts have been reported to give a mixture of products with this substrate.22 Attempts to rearrange 2,3-disubstituted epoxides with 1 led to low conversions under standard reaction conditions (entry 2).
We propose that 1 reacts via a similar mechanism as that reported by Eisenmann10a and Kagan,10c in which Co2(CO)8 was combined with MeOH to form [Co(MeOH)6]+2[Co(CO)4]2−in situ. Kagan's proposed catalytic cycle involves ring opening via a Lewis acid-assisted SN2 attack of the cobaltate at the least substituted carbon followed by classical β-hydride elimination from cobalt (Fig. 2B). This proposal was based on the observed epoxide reactivities and product distributions, but the full mechanism has yet to be thoroughly studied.
We utilized our well-defined complexes to probe the synergistic nature of this two-component catalyst system by replacing either the aluminum porphyrin with the non-Lewis acidic sodium cation or the cobaltate with the non-nucleophilic tetraphenyl borate anion. Neither of these variants was able to promote the isomerization of 2a (Table 3, entries 2 and 3). Similarly, pClTPPAlCl, the precursor to catalyst 1, does not lead to any conversion to ketone (entry 4). This conclusively shows that both the Lewis acid cation and nucleophilic anion are necessary for this reaction, which supports the first step in Kagan's proposed mechanism and parallels the ring-opening step of carbonylation (Fig. 2A),23 which catalyst 1 is also known to do. The fact that cobalt is required to effect β-hydride elimination clearly distinguishes this mechanism from the Lewis acid pathway depicted in Fig. 1.
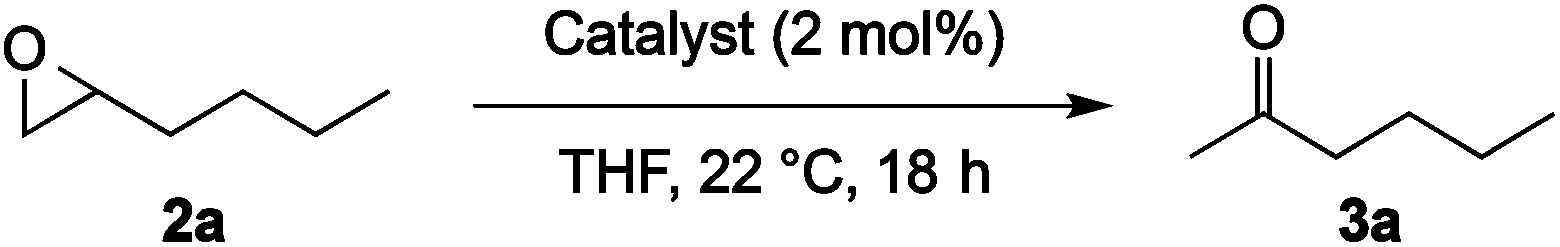
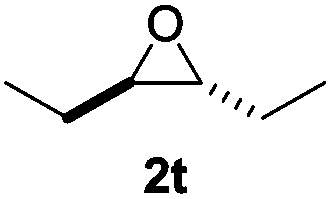
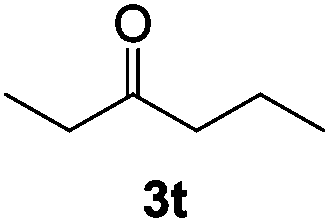
The β-hydride elimination step was investigated by subjecting isobutylene oxide to standard reaction conditions.17 This 2,2-disubstituted epoxide results in a tertiary alkoxide upon SN2 ring opening that cannot undergo β-hydride elimination. As expected, no methyl shift to form a ketone was observed. Less than 2% conversion to isobutyraldehyde occurred, which could arise from either an SN1-type attack of cobaltate24 or the classic Lewis-acid mediated mechanism (Fig. 1).25
We have presented a new application of a readily available, well-defined catalyst for the isomerization of monosubstituted epoxides to methyl ketones in excellent yields. Compared to previous catalyst systems, 1 is effective under mild conditions, low catalyst loading, and has good functional group tolerance. Current work focuses on applying more recent, selective catalysts to the rearrangement of 2,3-disubstituted epoxides.
We are grateful to the Department of Energy (DE-FG02-05ER15687) and the National Science Foundation (DGE-1144153, fellowship to J.R.L.) for funding.
Notes and references
- For reviews see: (a) J. G. Smith, Synthesis, 1984, 629 CrossRef CAS; (b) C.-Y. Huang and A. G. Doyle, Chem. Rev., 2014, 114, 8153 CrossRef CAS PubMed; (c) S. C. Bergmeier and D. J. Lapinsky, Prog. Heterocycl. Chem., 2013, 25, 47 CAS.
- (a) J. Meinwald, S. S. Labana and M. S. Chadha, J. Am. Chem. Soc., 1963, 85, 582 CrossRef CAS; For a review see: (b) B. Rickborn, in Comprehensive Organic Synthesis, ed. B. M. Trost, I. Fleming and G. Pattenden, Pergamon, Oxford, 1991, vol. 3, 733 Search PubMed.
- (a) H. O. House, J. Am. Chem. Soc., 1955, 77, 3070 CrossRef CAS; (b) H. O. House, J. Am. Chem. Soc., 1955, 77, 5083 CrossRef CAS; (c) B. Rickborn and R. M. Gerkin, J. Am. Chem. Soc., 1971, 93, 1693 CrossRef CAS; (d) A. Miyashita, T. Shimada, A. Sugawara and H. Nohira, Chem. Lett., 1986, 1323 CrossRef CAS.
- (a) K. Maruoka, N. Murase, R. Bureau, T. Ooi and H. Yamamoto, Tetrahedron, 1994, 50, 3663 CrossRef CAS; (b) F. Martínez, C. del Campo and E. F. Llama, J. Chem. Soc., Perkin Trans. 1, 2000, 1749 RSC.
- For selected examples see: (a) R. Sudha, K. M. Narasimhan, V. Saraswathy and S. Sankararaman, J. Org. Chem., 1996, 61, 1877 CrossRef CAS PubMed; (b) K. Suda, S. Nakajima, Y. Satoh and T. Takanami, Chem. Commun., 2009, 1255 RSC; (c) E. Ertürk, M. Göllü and A. S. Demir, Tetrahedron, 2010, 66, 2373 CrossRef; (d) V. Gudla and R. Balamurugan, Tetrahedron Lett., 2012, 53, 5243 CrossRef CAS; (e) D. J. Vyas, E. Larionov, C. Besnard, L. Guénée and C. Mazet, J. Am. Chem. Soc., 2013, 135, 6177 CrossRef CAS PubMed; (f) N. Humbert, D. J. Vyas, C. Besnard and C. Mazet, Chem. Commun., 2014, 50, 10592 RSC.
- (a) A. Yanagisawa, K. Yasue and H. Yamamoto, J. Chem. Soc., Chem. Commun., 1994, 2103 RSC; (b) K. Suda, K. Baba, S. Nakajima and T. Takanami, Tetrahedron Lett., 1999, 40, 7243 CrossRef CAS; (c) A. Procopio, R. Dalpozzo, A. De Nino, M. Nardi, G. Sindona and A. Tagarelli, Synlett, 2004, 2633 CrossRef CAS.
- Z. An, R. D'Aloisio and C. Venturello, Synthesis, 1992, 1229 CrossRef CAS.
- T. Kauffmann, C. Neiteler and G. Neiteler, Chem. Ber., 1994, 127, 659 CrossRef CAS.
- C.-L. Chang, M. P. Kumar and R.-S. Liu, J. Org. Chem., 2004, 69, 2793 CrossRef CAS PubMed.
- (a) J. L. Eisenmann, J. Org. Chem., 1962, 27, 2706 CAS; (b) J.-Y. Kim and T. Kwan, Chem. Pharm. Bull., 1970, 18, 1040 CrossRef CAS; (c) J. Prandi, J. L. Namy, G. Menoret and H. B. Kagan, J. Organomet. Chem., 1985, 285, 449 CrossRef CAS.
- D. Milstein, J. Am. Chem. Soc., 1982, 104, 5227 CrossRef CAS.
- (a) S. Kulasegaram and R. J. Kulawiec, J. Org. Chem., 1994, 59, 7195 CrossRef CAS; (b) S. Kulasegaram and R. J. Kulawiec, J. Org. Chem., 1997, 62, 6547 CrossRef CAS (mechanism).
- T. L. Church, Y. D. Y. L. Getzler, C. M. Byrne and G. W. Coates, Chem. Commun., 2007, 657 RSC.
- J. W. Kramer, E. B. Lobkovsky and G. W. Coates, Org. Lett., 2006, 8, 3709 CrossRef CAS PubMed.
- Iron, ruthenium, and chromium porphyrin catalysts have precedence in the Meinwald rearrangement literature, but they generally operate via a Lewis acidic mechanism which results in aldehyde products from terminal epoxides (Fig. 1). For Fe see ref. 6b and (a) T. Takanami, R. Hirabe, M. Ueno, F. Hino and K. Suda, Chem. Lett., 1996, 1031 CrossRef CAS; For Ru see: (b) J. Chen and C.-M. Che, Angew. Chem., Int. Ed., 2004, 43, 4950 CrossRef CAS PubMed; (c) G. Jiang, J. Chen, H.-Y. Thu, J.-S. Huang, N. Zhu and C.-M. Che, Angew. Chem., Int. Ed., 2008, 120, 6740 CrossRef; For Cr see: (d) K. Suda, T. Kikkawa, S. Nakajima and T. Takanami, J. Am. Chem. Soc., 2004, 126, 9554 CrossRef CAS PubMed.
- (a) J. M. Rowley, E. B. Lobkovsky and G. W. Coates, J. Am. Chem. Soc., 2007, 129, 4948 CrossRef CAS PubMed; (b) M. Mulzer, B. J. Tiegs, Y. Wang, G. W. Coates and G. A. O'Doherty, J. Am. Chem. Soc., 2014, 136, 10814 CrossRef CAS PubMed.
- See ESI† for additional information.
- For a Wacker oxidation on a similar diene substrate see: T. Mitsudome, T. Umetani, N. Nosaka, K. Mori, T. Mizugaki, K. Ebitani and K. Kaneda, Angew. Chem., Int. Ed., 2005, 45, 481 CrossRef PubMed.
- W. C. Ellis, Y. Jung, M. Mulzer, R. DiGirolamo, E. B. Lobkovsky and G. W. Coates, Chem. Sci., 2014, 5, 4004 RSC.
- E. C. H. Sykes, M. S. Tikhov and R. M. Lambert, Catal. Lett., 2002, 78, 7 CrossRef CAS.
- (a) I. Karamé, M. L. Tommasino and M. Lemaire, Tetrahedron Lett., 2003, 44, 7687 CrossRef; (b) M. W. C. Robinson, K. S. Pillinger and A. E. Graham, Tetrahedron Lett., 2006, 47, 5919 CrossRef CAS; (c) B. C. Ranu and U. Jana, J. Org. Chem., 1998, 63, 8212 CrossRef CAS.
- A. Cabrera, F. Mathé, Y. Castanet, A. Mortreux and F. Petit, J. Mol. Catal., 1991, 64, L11 CrossRef CAS.
- T. L. Church, Y. D. Y. L. Getzler and G. W. Coates, J. Am. Chem. Soc., 2006, 128, 10125 CrossRef CAS PubMed.
- Both SN2 and SN1-type attacks were observed during the carbonylation of isobutylene oxide: (a) V. Mahadevan, Y. D. Y. L. Getzler and G. W. Coates, Angew. Chem., Int. Ed., 2002, 41, 2781 CrossRef CAS; (b) Y. D. Y. L. Getzler, V. Mahadevan, E. B. Lobkovsky and G. W. Coates, J. Am. Chem. Soc., 2002, 124, 1174 CrossRef CAS PubMed.
- This alternative pathway is favored when using a non-nucleophilic anion so that conversion to isobutyraldehyde increased to 62% with [pClTPPAl(THF2)]+[BPh4]−.
Footnote |
| † Electronic supplementary information (ESI) available: Expanded Tables S1 and S2, detailed experimental procedures, characterization data. See DOI: 10.1039/c4qo00324a |
| This journal is © the Partner Organisations 2015 |