
Self-supported composites of thin Pt–Sn crosslinked nanowires for the highly chemoselective hydrogenation of cinnamaldehyde under ambient conditions†
Lin-Xiu
Dai
,
Wei
Zhu
,
Mu
Lin
,
Zhi-Ping
Zhang
,
Jun
Gu
,
Yu-Hao
Wang
and
Ya-Wen
Zhang
*
Beijing National Laboratory for Molecular Sciences, State Key Laboratory of Rare Earth Materials Chemistry and Applications, PKU-HKU Joint Laboratory in Rare Earth Materials and Bioinorganic Chemistry, College of Chemistry and Molecular Engineering, Peking University, Beijing 100871, China. E-mail: ywzhang@pku.edu.cn; Fax: +86-10-62756787; Tel: +86-10-62756787
First published on 31st August 2015
Abstract
Alloying a second metal in a parent metal is an efficient way to improve the catalytic performances of monometallic nanocatalysts, thus the understanding of the alloying effects on the catalytic properties of bimetallic nanocatalysts is of great significance. Herein, we demonstrate the synthesis of self-supported composites of thin (ca. 3.8 nm) Pt–Sn crosslinked nanowires (PtSn/SnO2 CNs) with a Pt![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Sn molar ratio of 3–4:1 via a one-pot hydrothermal method. The growth mechanism of the CNs consisted of spontaneous hydrolysis and reduction of metal ions in water solution and the subsequent oriented attachment of M(OH)x (M = Pt, Sn) particulate species under reduction by poly(vinylpyrrolidone) during hydrothermal processing. The nature of metal precursors, capping reagents and reduction rate has shown great influences on the shaping of the nanocatalysts into CNs. The PtSn/SnO2 catalyst exhibited 87% selectivity (superior over pure Pt nanoparticles (NPs) and physical mixtures of Pt and SnO2 NPs) under ambient conditions for the hydrogenation of cinnamaldehyde, owing to the formation of a Pt–Sn alloy in the CNs. The PtSn/SnO2 catalyst also showed good recyclability and stability in chemoselectivity for cycle experiments of the catalysts.
:Sn molar ratio of 3–4:1 via a one-pot hydrothermal method. The growth mechanism of the CNs consisted of spontaneous hydrolysis and reduction of metal ions in water solution and the subsequent oriented attachment of M(OH)x (M = Pt, Sn) particulate species under reduction by poly(vinylpyrrolidone) during hydrothermal processing. The nature of metal precursors, capping reagents and reduction rate has shown great influences on the shaping of the nanocatalysts into CNs. The PtSn/SnO2 catalyst exhibited 87% selectivity (superior over pure Pt nanoparticles (NPs) and physical mixtures of Pt and SnO2 NPs) under ambient conditions for the hydrogenation of cinnamaldehyde, owing to the formation of a Pt–Sn alloy in the CNs. The PtSn/SnO2 catalyst also showed good recyclability and stability in chemoselectivity for cycle experiments of the catalysts.
Introduction
Noble metal catalysts play an important role in the areas of energy conversion and storage, environmental improvement, organic catalysis and sensors, etc.1 In order to minimize the usage of noble metals and promote the catalytic performance (activity, selectivity and durability), bimetallic catalysts have attracted tremendous attention from chemists to material scientists. It has been demonstrated that the bi-function effect, surface atom arrangement and electronic structure essentially affect the catalytic performance of bimetallic catalysts. With the control over the surface chemistry (e.g., element ratio, oxidation state, exposed facets, lattice strain, proportion of corner/edge atoms, etc.) of bimetallic catalysts, which can be realized via various synthetic protocols of nanomaterials, elevated catalytic properties of bimetallic catalysts are achieved as a result of the optimized adsorptions and desorptions of reactants and products.2–14Based on the universal growth mechanism of nanocrystals, well-developed wet chemistry synthetic methods offer a valid way for the preparation of uniform bimetallic nanocrystals with well-defined structures. The prompt development of shape-controlled synthetic strategies of nanocatalysts boosts the fundamental studies of catalysis as well as the constructions of highly efficient and highly durable nanocatalysts with novel structures. The reaction sites that determine the catalytic properties of a catalyst can be identified when shape-controlled nanocrystals are used as catalysts. Till now, many solution based synthetic approaches have been used to prepare bimetallic NPs with various morphologies, including nanowires, nanoplates, nanodendrites, cubes, polyhedrons, etc.15–23 Previous reports have pointed that nanowires possess excellent structure stability, high density of structural defects and good recyclability, resulting in significant improvement of performances in heterogeneous catalysis (e.g., ethanol electro-oxidation, formic acid oxidation and hydrogenation reactions of cinnamaldehyde (CAL), nitrobenzene or styrene, etc.).16,22
Selective hydrogenation of α,β-unsaturated aldehydes to unsaturated alcohols is a commercially important industrial process of fragrance and pharmaceutical fields.24,25 Designing catalysts for both high activity and selectivity is still a crucial challenge in heterogeneous catalysis research, particularly for the selective hydrogenation of α,β-unsaturated aldehydes. Pt-based nanomaterials are the most wildly studied catalysts for fuel cell,1 three-way catalysis26 and catalytic hydrogenation.27 Pt serves as the main active site for hydrogenation. Smaller Pt nanoparticles with low coordination sites favor the hydrogenation of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond,28 and the selective hydrogenation does not occur on the clean surface of Pt (111).29 Thus, monometallic Pt NPs showed high hydrogenation activity but low selectivity to unsaturated alcohols for the selective hydrogenation of α,β-unsaturated aldehydes. Therefore, it is necessary to improve the selectivity by modifying the Pt-based surface of the catalysts.
C bond,28 and the selective hydrogenation does not occur on the clean surface of Pt (111).29 Thus, monometallic Pt NPs showed high hydrogenation activity but low selectivity to unsaturated alcohols for the selective hydrogenation of α,β-unsaturated aldehydes. Therefore, it is necessary to improve the selectivity by modifying the Pt-based surface of the catalysts.
The strategy of introducing second metals such as Fe, Co, Cu, Ni and Sn into Pt is widely used to increase hydrogenation selectivity to unsaturated alcohols.30–36 The second metals were utilized to change the electronic structure and the surface atomic arrangement of Pt-based alloys. As an example of the Pt–Sn alloy, experimental and computational investigations have demonstrated that the adsorption energy of α,β-unsaturated aldehydes on the (111) facet of Pt–Sn alloys is significantly weaker than that on the (111) facet of Pt.37–39 On the alloy surfaces, an oxygen–tin interaction is more stable than the CO bonding with a Pt surface. Density functional theory (DFT) has proved that the addition of tin in Pt led to the atop adsorption mode of α,β-unsaturated aldehydes, thus favoring the hydrogenation of the CO bond.40 Several researches have explored the composition-dependent catalytic performances of Pt-based catalysts. On doping a second metal (Ti, Mn, Fe, Co, Ni, Cu, Zn, Sn, Pb) to Pt, the selectivity presented a volcano trend, suggesting changes in the electronic structure of Pt.32 Actually, the controlling of particle size, shape and atomic segregation changed the alloying effects to different degrees, thus resulting in the improvement of selectivity.41,42 Hence, rationally regulating electronic and geometrical structures to activate certain functionalities of α,β-unsaturated aldehydes could overcome thermodynamically favorable CC hydrogenation over CO hydrogenation. Moreover, support effects can be utilized to enhance the stability and activity of main catalysts.43–45 Recent research studies have manifested that bimetallic catalysts achieved 99% selectivity to unsaturated alcohol for CAL hydrogenation. Nevertheless, the high catalytic selectivity was acquired under the conditions of high temperature and high H2 pressure.32 The harsh reaction conditions normally have a high demand for the structure stability of catalysts.
Herein, we report on the synthesis of self-supported composites of thin Pt–Sn crosslinked nanowires by a facile one-pot hydrothermal method. With the as-prepared PtSn/SnO2 CNs, high chemoselectivity to unsaturated alcohols under ambient reaction conditions for CAL hydrogenation has been achieved. The PtSn/SnO2 composites exhibited 87% selectivity to cinnamyl alcohol (COL) without significant loss in activity. It was deduced that the formation of a Pt–Sn alloy in the CNs probably changed the electronic structure of Pt, thus favoring the activation of the CO bond during the CAL hydrogenation.
Experimental section
Chemicals
H2PtCl6·6H2O (A.R., Sinopharm Chemical Reagent Co. Ltd, China), K2PtCl4 (A.R., Beijing Tongguang Fine Chemicals Company), SnCl2·2H2O (A.R., Xilong Chemical Co. Ltd, China), SnCl4·5H2O (A.R., Beijing Chemical Works, China), poly(vinylpyrrolidone) (PVP: Mw = 29000 and 55000, Sigma-Aldrich), ethylene glycol (extra pure, J&K), cinnamaldehyde (Shanghai Shuangxiang Chemical Works, China), naphthalene (A.R., Beijing Chemical Works, China), ethyl ether (A.R., Beijing Tongguang Fine Chemicals Company), HCl solution (A.R., Xilong Chemical Co. Ltd, China), ethanol (A.R.), acetone (A.R.), cyclohexane (A.R.), and deionized water (Millipore, 18.2 MΩ cm).
Synthesis of PtSn/SnO2 CNs
The precursors, 0.05 mmol of H2PtCl6, 0.07 mmol of SnCl2·2H2O, 100 mg of PVP (Mw = 55000), and 0.3 mmol of HCl (1.0 mol L−1) were dissolved in 15 mL of deionized water with stirring for 20 min. The solution was transferred to a 25 mL Teflon-lined stainless steel autoclave and heated at 180 °C for 12 h. After the mixture was cooled, the black products were centrifuged by mixing 40 mL of acetone and washed with ethanol/acetone twice, and lastly dispersed in ethanol for further characterization.
Synthesis of SnO2 NPs
The synthesis of SnO2 NPs was similar to that of PtSn/SnO2 CNs except for the absence of H2PtCl6.Synthesis of Pt NPs
0.06 mmol of K2PtCl4 and 100 mg of PVP (Mw = 29000) were mixed in 15 mL of ethylene glycol with stirring for 20 min. The solution was transferred to a 25 mL Teflon-lined stainless steel autoclave and held at 180 °C for 24 h. The obtained black products were centrifuged and washed with the addition of 40 mL of acetone and 3 mL cyclohexane several times, and lastly dispersed in ethanol for further characterization.
Cinnamaldehyde hydrogenation reaction studies
CAL hydrogenation was run in a reaction tube (2.5 cm in diameter) with a solution of 0.5 mmol of cinnamaldehyde, 2.5 mL of ethanol and catalysts (0.0025 mmol, 0.5 mol%). After being purged with H2, the reaction mixture was stirred at 25 °C under a H2 atmosphere (1 bar). The as-obtained products were quantitatively determined by GC with 0.5 mmol of naphthalene as the internal standard.Instrumentation
Transmission electron microscopy (TEM), high-resolution transmission electron microscopy (HRTEM), energy dispersive X-ray spectroscopy (EDS), and high-angle annular dark-field scanning transmission electron microscopy (HAADF-STEM) EDS spot scan analysis were conducted on a FEG-TEM (JEM-2100F, JEOL, Japan) operated at 200 kV. The samples for TEM characterization were prepared by dropping the colloid solution on copper grids coated with amorphous carbon and drying naturally. Powder X-ray diffraction (PXRD) analysis was performed on a Rigaku D/MAX-2000 diffractometer (Japan). Inductively coupled plasma atomic emission spectroscopy (ICP-AES) analysis was conducted on a Profile Spec ICP-AES spectrometer (Leeman, U.S.A.). X-ray photoelectron spectroscopy (XPS) analysis was performed on an Axi Ultra imaging photoelectron spectrometer (Kratos, UK). All gas chromatography spectrometry experiments were carried out and recorded by GC (GC 7890A, Agilent Technologies).Results and discussion
Hydrothermal synthesis and structural characterization of PtSn/SnO2 CNs
The PtSn/SnO2 CNs were prepared via hydrothermal synthesis at 180 °C. The redox process occurred between H2PtCl6 and SnCl2 solution. PVP served as the capping agent to obtain PtSn/SnO2 CNs. The TEM image in Fig. 1a shows that the samples appeared in the shape of cross-linked nanowires. The width of the CNs was 3.8 ± 1.0 nm. HAADF-STEM-EDS was used to analyze the elemental distribution of Pt and Sn in a single nanowire. Fig. 1b shows that the atom ratio of Pt and Sn was about 3–4:1 for PtSn/SnO2 CNs. Uniform Pt-rich elemental distribution was found on both the branches and the nodes of nanowires. In addition, the PXRD pattern of the as-prepared composites in Fig. 1c was attributed to the co-existing fcc Pt phase (JCPDS no. 04-0802) and the tetragonal SnO2 phase (JCPDS no. 41-1445). The PXRD data were further quantitatively analysed by Rietveld refinement in TOPAS 3.0 (Table S1 and Fig. S1 in ESI†), indicating a weight ratio of 3:2 for Pt9Sn and SnO2. The (111) peak position (39.19°) of the fcc-structured portion of composites fell in between that of Pt (39.76°) and that of Pt3Sn (38.94°), suggesting the formation of a Pt–Sn alloy with a Pt:Sn molar ratio of more than 3:1 in the CNs.
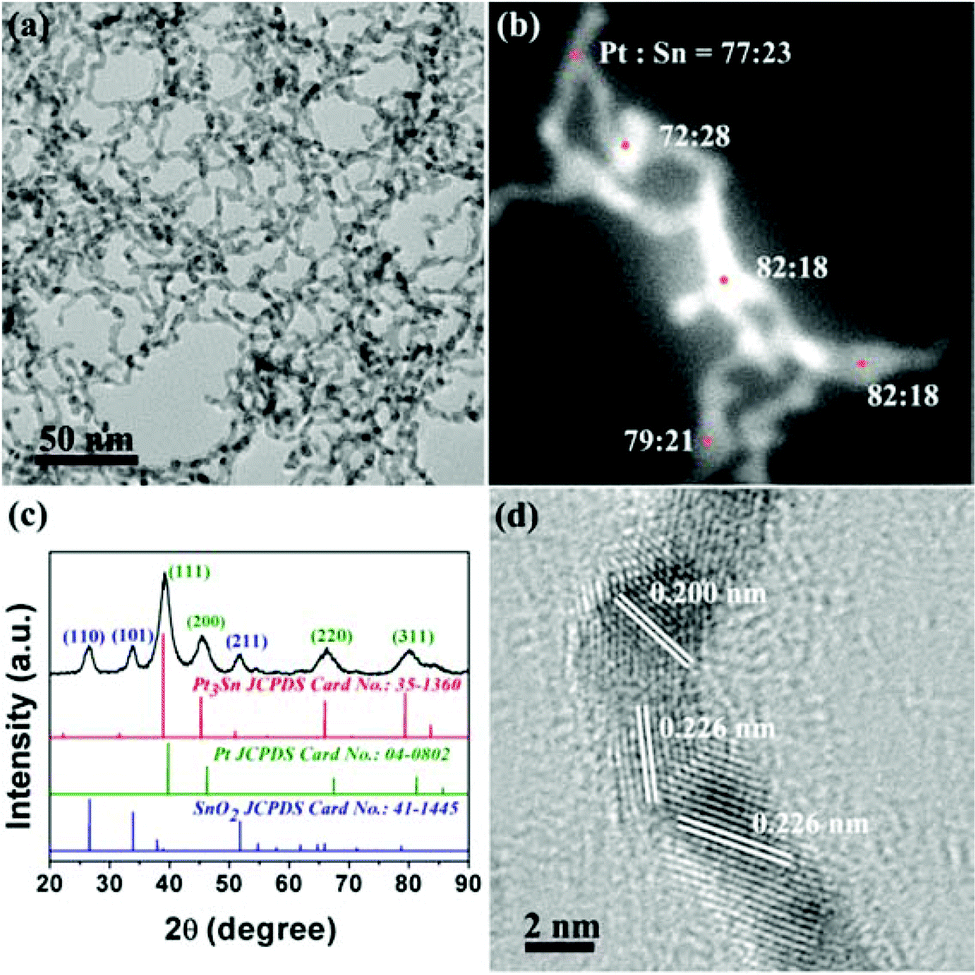 | ||
| Fig. 1 TEM image (a), EDS spot scan image (b), PXRD pattern (c) and HRTEM image (d) of the as-obtained composites. For (c), marked lattice facets in dark blue belong to SnO2, green ones belong to Pt. | ||
As shown in the HRTEM image of CNs in Fig. 1d, the CNs were crystallized to some extent. The lattice fringes of SnO2 were hardly found on the nanowires because of the segregation of SnO2 from the nanoaggregates in the products. The nanoaggregates with a Pt:Sn molar ratio of 1:4 were found in TEM images (Fig. S2 in ESI†), as proved by the observable lattice fringes for {110}, {101} and {211} of tetragonal SnO2.
XPS spectra (Fig. 2a) show that Pt4+ ions in precursors were reduced to Pt0 and Pt2+ with a ratio of 60:40 in the CN composites. As illustrated in Fig. 2b, the XPS Sn 3d5/2 peaks at 486.9 eV suggested that Sn was mainly in the oxidized state. However, according to the literature data,46 the peaks assignable to tin(II) and tin(IV) overlap. It is difficult to assign the peaks to Sn2+ or Sn4+ only on the basis of the observed binding energy value. Considering the absence of SnO in the PXRD pattern of Fig. 1c, the oxidized tin ought to be Sn4+.47 The result of 12% Sn0 observed at a binding energy of 485.2 eV implied the formation of a Pt–Sn alloy in the CNs, consistent with the negative shift of main peaks in PXRD data. Hence, the above characterization results clarified that the obtained products consisted of PtSn/SnO2 CNs with a Pt:Sn molar ratio of 3–4:1 and nanoaggregates.
 | ||
| Fig. 2 XPS spectra of the as-obtained products for Pt 4f (a) and Sn 3d5/2 (b). | ||
The growth mechanism of PtSn/SnO2 CNs
To further explore the formation mechanism of PtSn/SnO2 CNs, we carried out a battery of condition experiments with the help of TEM characterization. The results demonstrated that the growth mechanism of PtSn/SnO2 CNs was closely related to reaction time as well as starting materials such as metal precursors, raw material ratio, reductants and capping reagents. In order to design a bimetallic catalyst with strong interaction between two metals on the atomic scale, Pt4+ (H2PtCl6) and Sn2+ (SnCl2) were used as the preferred metal precursors in view of the redox process of Pt4+ and Sn2+. From the standard electrode potential of Pt4+ and Sn2+ (φ0Pt(IV)/Pt(0) = 0.93 V, φ0Sn(IV)/Sn(II) = 0.15 V), a redox reaction (Pt4+ + Sn2+ → Pt + Sn4+) should take place. When SnCl2·2H2O was added into H2PtCl6 solution, the light yellow solution became brick red, and the centrifuged products were further characterized by XPS. Fig. 3a shows that the ratio of Pt2+ and Pt4+ is 79:21 (without Pt0), indicating that incomplete reduction occurred at room temperature. From the XPS spectrum of Sn 3d5/2, no metallic Sn0 was found in Fig. 3b, indicating that the addition of PVP and the high temperature reduction (180 °C) played important roles in the reduction of tin ions and the formation of a Pt–Sn alloy at later periods.
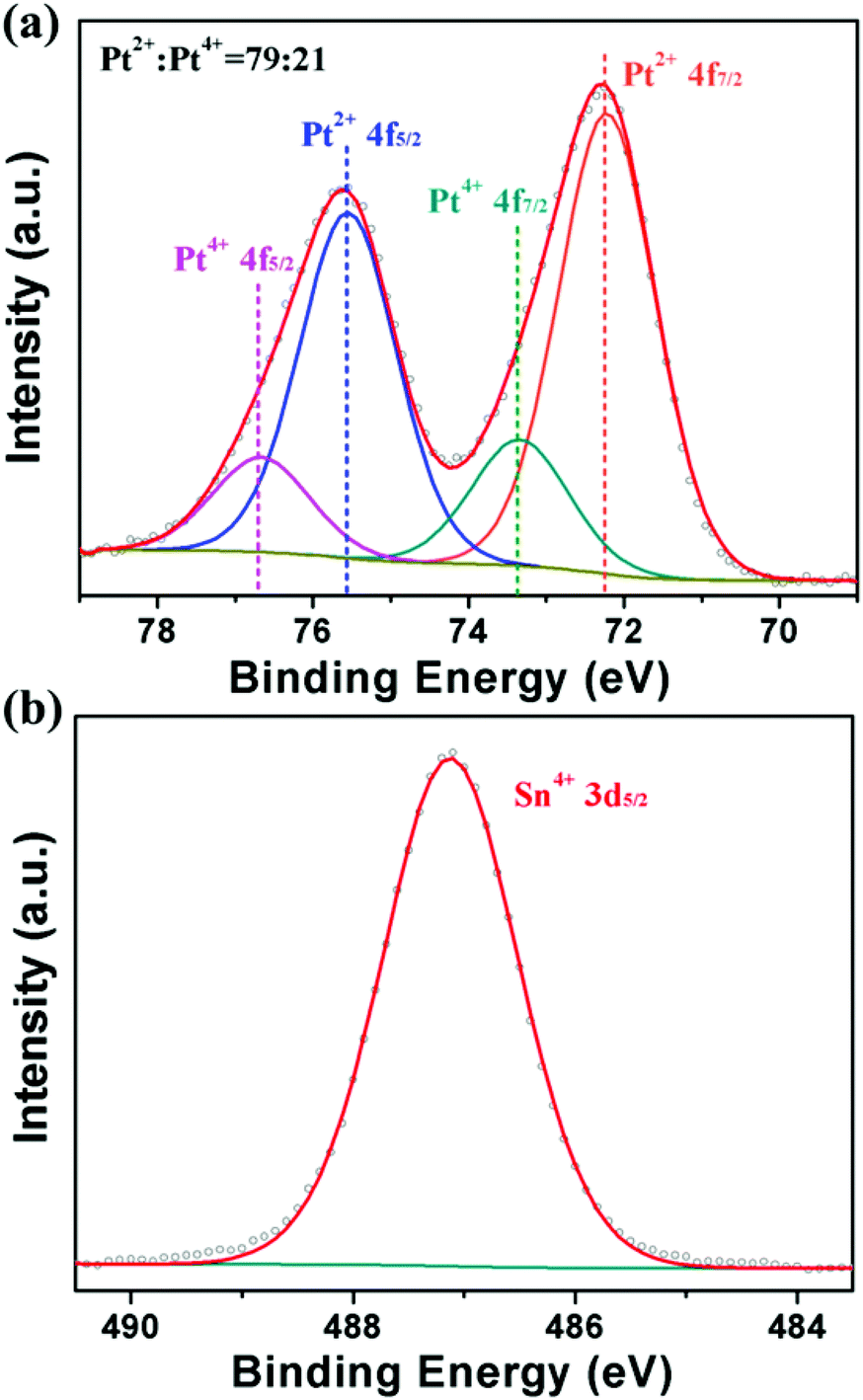 | ||
| Fig. 3 XPS spectra of the as-obtained products for Pt 4f (a) and Sn 3d5/2 (b) after mixing two metal precursors with stirring for 20 min. | ||
Fig. 4 shows different morphologies of the products resulting from using different combination of metal precursors in the synthesis. The adoption of H2PtCl6/SnCl4, K2PtCl4/SnCl4 and K2PtCl4/SnCl2 did not produce any nanowires within 12 h of reaction (Fig. 4a–c). When the reaction time was prolonged to 24 h, nanowires appeared only with using H2PtCl6/SnCl4 or K2PtCl4/SnCl4 as the metal precursors, as shown in Fig. S3a and S3b in the ESI.† However, the combination of H2PtCl6/SnCl2 could produce CNs after the reactions from 12 h (Fig. 4d) to 24 h (Fig. S3d in the ESI†). On taking K2PtCl4 and SnCl2 as metal precursors, the Pt2+ ions were rapidly reduced and grew into isolated NPs after the initial reaction (Fig. S3c in the ESI†). Then, tin ions were reduced to Sn0 in the later stage, resulting in the appearance of dispersed NPs. Therefore, it was deduced that the formation of nanowires highly depended on the generation of more platinum and tin hydroxyl species.
 | ||
| Fig. 4 TEM images of the products using H2PtCl6 and SnCl4 (a), K2PtCl4 and SnCl4 (b), K2PtCl4 and SnCl2 (c) and H2PtCl6 and SnCl2 (d) as metal precursors after 12 h reaction. | ||
In addition, the influence of the ratio of the two metal precursors, H2PtCl6 and SnCl2, was also studied. Only when the molar ratio of Pt4+ and Sn2+ in precursors was in the range of 1:1 to 2:1, CNs could be synthesized (Fig. 5b and c). When the molar ratio was more than 2, the reduction of excess Pt4+ ions was mainly assisted by PVP. The probably weakened interaction between Pt and Sn caused the formation of isolated particles (Fig. 5d). On the other hand, more SnO2 NPs appeared if excess Sn2+ ions in precursors were introduced. Less attachment between platinum and tin hydroxyl species produced more SnO2 NPs to form SnO2-supported Pt NPs.
 | ||
| Fig. 5 TEM images of the products using different ratios of H2PtCl6 and SnCl2 as metal precursors: (a) 1:2, (b) 1.5:1, (c) 2:1 and (d) 3:1. | ||
Then, we tried to use PVP and ascorbic acid (AA) as the alternative reductants to elaborate the influence of the reduction rate. As shown in Fig. 6a, only large Pt NPs appeared without the addition of PVP due to the lack of capping agents to prevent the aggregation of the NPs, while no significant influence on the nanowire structures was observed with the addition of excessive PVP. AA has stronger reducibility than PVP, probably resulting in a faster reduction rate. As illustrated in Fig. 6b, the addition of AA led to the formation of small NPs. It was because more nuclei resulted from the faster reduction rate of the metal precursors, which led to the prompt aggregation of the nuclei, and thus retarded the formation of CNs. Hence, the kinetic-controlled formation of nanowire structures required a modest reduction rate of metal precursors with PVP serving as both the capping reagent and the reductant.
 | ||
| Fig. 6 TEM images of the products using no PVP (a) and 100 mg PVP + 176 mg AA (b). The synthetic conditions are the same as the synthesis of PtSn/SnO2 CNs. | ||
As shown in the FTIR spectra (Fig. S4 in the ESI†), the appearance of the vibration peak of CO at 1642 cm−1 for the composites indicated the existence of a trace amount of PVP on the surfaces of the CNs. A red shift of ca. 24 cm−1 in the vibration peak of CO compared with that of pure PVP was attributed to the coordination of PVP molecules with PtSn/SnO2 CNs by their O atoms.48
To further understand the growth mechanism of PtSn/SnO2 CNs, the time of the hydrothermal reaction was varied to observe the morphology evolution of the CNs in the synthesis process. Fig. 7 shows the TEM images of the time-sequence synthesis process. In the first 0.5 h, only a number of small NPs appeared. As the reaction time went on, these small NPs were attached to form short nanowires. After 12 h reaction, PtSn/SnO2 CNs were obtained. By further extending the reaction time to 24 h, we did not observe a morphological change of CNs. The growth mechanism of the CNs was speculated to be similar to the sol–gel process.49 Redox reaction and spontaneous hydrolysis of Pt ions and Sn ions took place to form metal hydroxyl species at the beginning, then, these metal hydroxyl species proceed towards the oriented attachment of surface hydroxyl groups to produce PtSn/SnO2 CNs under the reduction by PVP at 180 °C, residual oxidized tin constituted nanoaggregates. As shown in Scheme 1, the growth mechanism is the integration of the redox reaction and the self-organization process.
 | ||
| Fig. 7 TEM images of PtSn/SnO2 CNs synthesized at 180 °C for 0.5 h (a), 2 h (b), 4 h (c) and 24 h (d). | ||
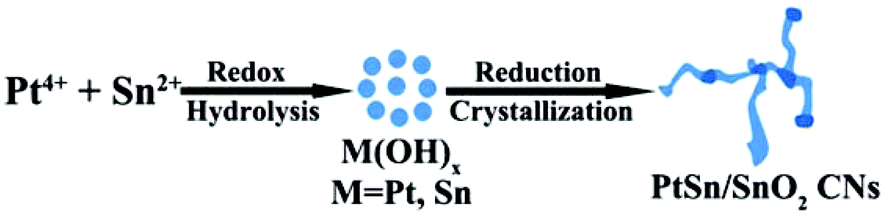 | ||
| Scheme 1 The growth mechanism of PtSn/SnO2 CNs. | ||
Catalytic performance in the hydrogenation reaction
The selective hydrogenation of cinnamaldehyde was employed to evaluate the catalytic performances of PtSn/SnO2 CNs (Scheme 2). In the catalytic tests, Pt NPs in the size of 3.1 ± 0.5 nm, and SnO2 NPs in the size of 7.8 ± 2.0 nm, and physical mixtures of Pt and SnO2 (Pt + SnO2 NPs) prepared via a similar hydrothermal method, were used as references (Fig. S5 in the ESI†).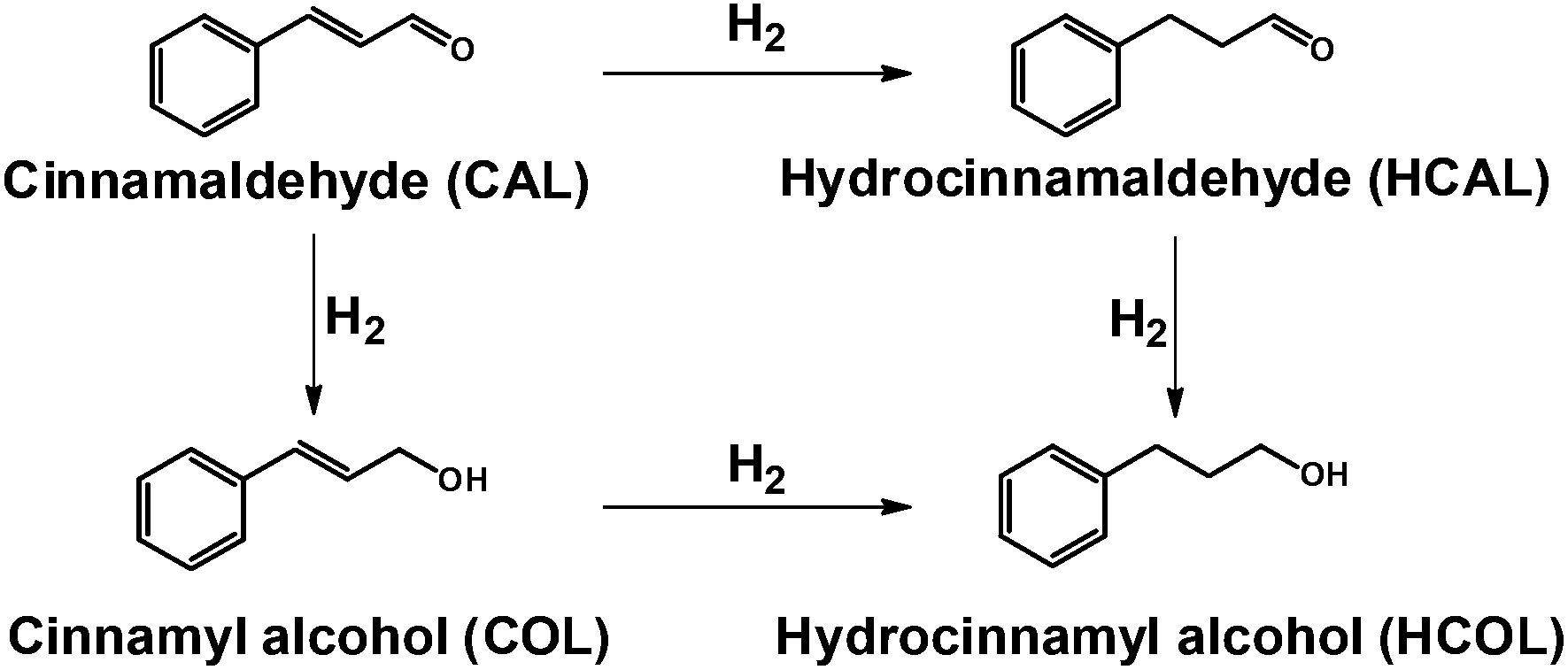 | ||
| Scheme 2 Reaction paths for the hydrogenation of cinnamaldehyde. | ||
In this reaction, hydrogenation on the CO bond and the CC bond generally led to a parallel and consecutive hydrogenation reduction to obtain different catalytic products. The formation of the HCAL is favored over the COL because of thermodynamics.50 The distribution of the three hydrogenation products depends on the structure and composition of catalysts.51
From the results of experiments, CAL was completely converted by Pt NPs, PtSn/SnO2 CNs and Pt + SnO2 NPs with the addition of the same amount of Pt atoms (determined by ICP-AES) in 12 h. No hydrogenation activity was observed for SnO2 NPs, demonstrating the role of Pt as the main active site. For the PtSn/SnO2 CNs, SnO2 as the supporter improved the physical stability of the catalyst, but contributed little to the overall hydrogenation activity. As shown in Table 1, we calculated the mass activity of the three catalysts, the mass activity was defined as below:
| Mass activity = mmolCAL converted min−1 mmolPt−1 |
| Catalysts | Time [min] | Conversion [%] | n Pt [μmol] | Mass activity [min−1] |
|---|---|---|---|---|
| PtSn/SnO2 CNs | 4 | 10 | 2.5 | 4.7 |
| Pt NPs | 4 | 15 | 2.5 | 6.4 |
| Pt + SnO2 NPs | 4 | 12 | 2.5 | 5.7 |
From Table 1, it is seen that the mass activities of the three catalysts were comparable.
Fig. 8a shows the selectivity of the nanocatalysts for the CAL hydrogenation, and the selectivity was defined as below:
| %Selectivity = [COL]/([COL] + [HCAL] + [HCOL]) × 100 |
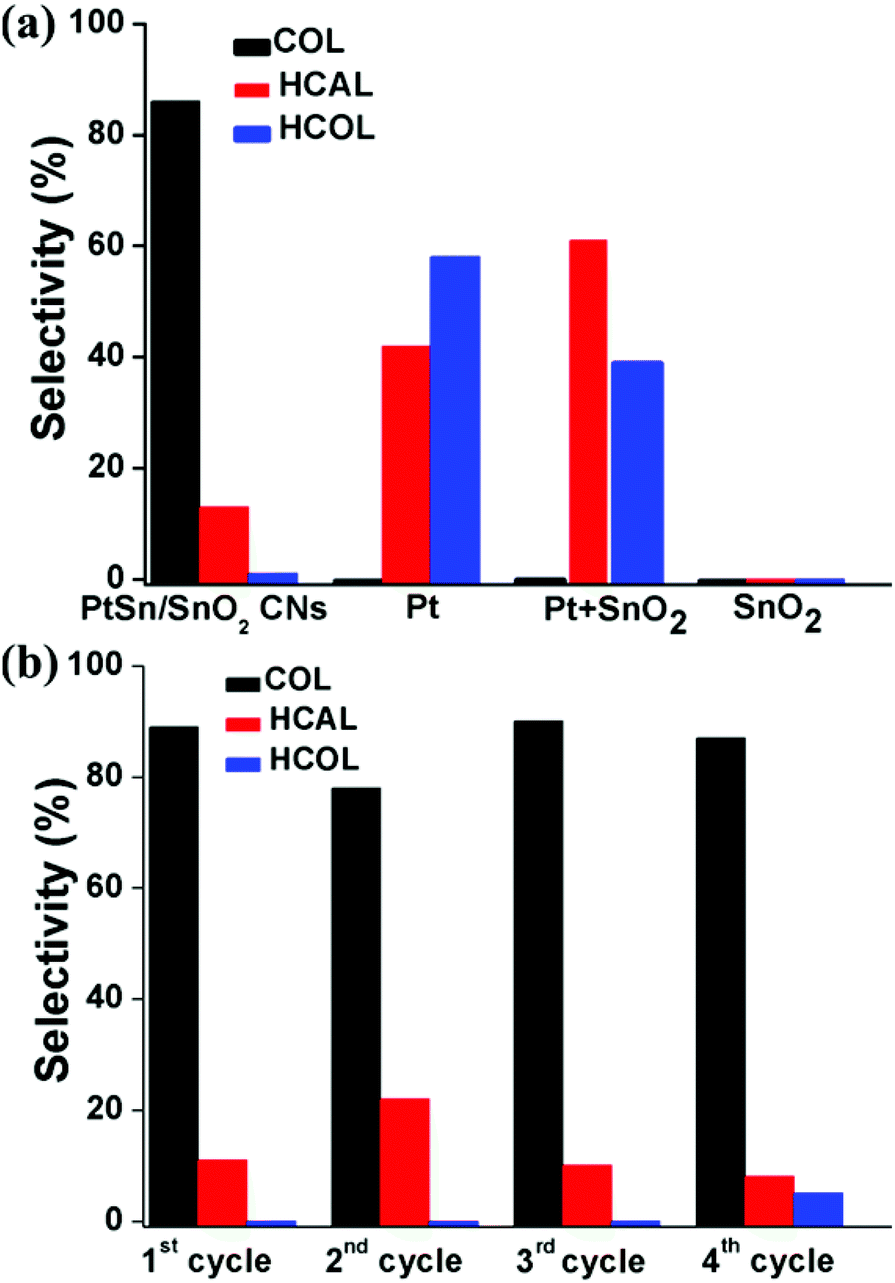 | ||
| Fig. 8 Selectivity (a) and stability (b) for cinnamaldehyde hydrogenation. | ||
PtSn/SnO2 CNs exhibited 87% selectivity to COL in ethanol at room temperature under 1 bar of H2, while the other catalysts could not catalyze the hydrogenation of CAL to COL under this condition.
As is known, molecular hydrogen is adsorbed and dissociated on surface Pt sites. The adsorption geometry of the CC bond and the CO bond is highly dependent on the surface structure of catalysts and the molecular structure of α,β-unsaturated aldehyde.52 CAL generally adopted a flat-lying co-adsorption geometry on the Pt surface.53 Pure Pt or supported Pt catalysts are well known for their activity of olefin hydrogenation, implying more favorable binding with CC than CO. So the low selectivity of Pt NPs and Pt + SnO2 NPs was caused by the strong adsorption of the CC bond and more rapid hydrogenation rate, and high desorption energy resulted in the formation of complete hydrogenation products (i.e., HCOL).52
However, the selectivity to COL of PtSn/SnO2 CNs was as high as 87%, possibly because of the formation of a Pt–Sn alloy.37–39 The introduction of electropositive metals such as Sn, could increase the electron density on Pt, which decreased the capability for CC bond adsorption and increased the interaction of the CO bond with the polar bimetallic sites.54 The oxygen of carbonyl has a stronger chemical affinity with Sn than that with Pt, thus an atop adsorption mode of α,β-unsaturated aldehydes would be more favorable, as confirmed by both experimentation and DFT studies.40 In an upright adsorption configuration, the successive hydrogenation of COL to HCOL does not take place on the alloy as it does on platinum, because no activation of CC occurs. In particular for nanowire catalysts with lots of unsaturated coordination sites, the upright adsorption geometry was more reasonable.55,56
Previous research studies have emphasized that the low desorption energy of COL on the Pt–Sn alloy is another reason for high selectivity to unsaturated alcohols, which reduces the possibility of the complete hydrogenation to HCOL.57 The physical mixtures of Pt and SnO2 showed a similar selectivity with pure Pt NPs, indicating the lack of the ensemble effect of alloying structures on the atom scale. When partial Pt atoms on the surface were replaced by Sn, the adsorption configuration was likely changed. As the number of Sn atoms increased on the surface, higher selectivity was realized.58 For PtSn/SnO2 CNs, SnO2−x is also responsible for high selectivity to COL, because the oxygen vacancy sites provided by SnO2−x could enhance the adsorption of the CO group.59 In addition, although monometallic Pt exhibited the negative effect of unsaturated coordination sites on selective hydrogenation of α,β-unsaturated aldehydes, Pt-based bimetallic catalysts with many unsaturated coordination atoms benefited the formation of COL.28
The stability of PtSn/SnO2 CNs was also investigated by injecting reactants repeatedly after complete conversion of CAL and retaining the PtSn/SnO2 CNs for the next run of the reaction. There was no obvious decrease in the conversion between the reused catalysts for the first three cycles. Full conversion of CAL required the double reaction time in the fourth cycle possibly due to the partial disassembly of nanowire structures after 3 cycles (Fig. S6 in the ESI†). Fig. S7 in the ESI† indicated that Pt NPs and Pt + SnO2 NPs also underwent significant aggregation after 1 cycle, suggesting the enhanced structural stability of the PtSn/SnO2 CNs. As shown in Fig. 8b, PtSn/SnO2 CNs still possessed about 90% selectivity for 4 cycles, possibly owing to the structural stability of the self-supported CNs under the reaction conditions.
ICP-AES tests evidenced no detectable metal leaching, while control experiments verified negligible CAL conversion in the absence of either H2 or Pt, confirming the heterogeneous nature of the observed reactions and the high durability and recyclability of PtSn/SnO2 CNs during the selective hydrogenation of cinnamaldehyde.
Conclusion
In this work, we prepared self-supported composites of thin Pt–Sn CNs with a Pt:Sn molar ratio of 3–4:1, by the oriented attachment mechanism via a facile hydrothermal method. The growth of the CNs was revealed to proceed by the following two stages: spontaneous hydrolysis and reduction of metal ions in water solution and the subsequent oriented attachment of M(OH)x (M = Pt, Sn) particulate species under the reduction by poly(vinylpyrrolidone) during hydrothermal treatment. For the selective hydrogenation of CAL, the PtSn/SnO2 CNs showed the highest selectivity to unsaturated alcohols with little loss of activity under ambient conditions. It was deduced that the electronic and geometric effects of the Pt–Sn alloy benefited the activation of the CO bond in the CNs, and decreased the desorption energy of unsaturated alcohols. In addition, the observed prominent durability and recyclability could reflect the intrinsic property of the self-supported nanowire structure. Hence, the self-supported thin Pt–Sn CNs combine the merits of a bimetallic alloy with a nanowire structure having a high portion of step/edge atoms for the CAL hydrogenation reaction with high selectivity and good durability. This work shed new light on the design of high-performance bimetallic catalysts by modulating the alloying effect of bimetallics as well as dimensionality of nanostructures.
Acknowledgements
This work was supported by the National Science Foundation of China (NSFC) (Grant No. 21025101, 21271011, 21573005, and 21321001). Y.W.Z. particularly appreciates the financial aid of China National Funds for Distinguished Young Scientists from the NSFC.Notes and references
- G. A. Somorjai, Chem. Rev., 1996, 96, 1223 CrossRef CAS PubMed.
- V. R. Stamenkovic, B. S. Mun, M. Arenz, K. J. J. Mayrhofer, C. A. Lucas, G. F. Wang, P. N. Ross and N. M. Markovic, Nat. Mater., 2007, 6, 241 CrossRef CAS PubMed.
- M. S. Chen, D. Kumar, C.-W. Yi and D. W. Goodman, Science, 2005, 310, 291 CrossRef CAS PubMed.
- P. Strasser, S. Koh, T. Anniyev, J. Greeley, K. More, C. F. Yu, Z. C. Liu, S. Kaya, D. Nordlund, H. Ogasawara, M. F. Toney and A. Nilsson, Nat. Chem., 2010, 2, 454 CrossRef CAS PubMed.
- V. R. Stamenkovic, B. Fowler, B. S. Mun, G. F. Wang, P. N. Ross, C. A. Lucas and N. M. Marković, Science, 2007, 315, 493 CrossRef CAS PubMed.
- K.-Q. Sun, Y.-C. Hong, G.-R. Zhang and B.-Q. Xu, ACS Catal., 2011, 1, 1336 CrossRef CAS.
- F. Gao and D. W. Goodmanz, Chem. Soc. Rev., 2012, 41, 8009 RSC.
- H. Zhang, M. S. Jin and Y. N. Xia, Chem. Soc. Rev., 2012, 41, 8035 RSC.
- J. B. Wu, P. P. Li, Y.-T. Pan, S. Warren, X. Yin and H. Yang, Chem. Soc. Rev., 2012, 41, 8066 RSC.
- J. Gu, Y.-W. Zhang and F. Tao, Chem. Soc. Rev., 2012, 41, 8050 RSC.
- M. Sankar, N. Dimitratos, P. J. Miedziak, P. P. Wells, C. J. Kielye and G. J. Hutchings, Chem. Soc. Rev., 2012, 41, 8099 RSC.
- Y. Pei, G. B. Zhou, N. Luan, B. N. Zong, M. H. Qiao and F. Tao, Chem. Soc. Rev., 2012, 41, 8140 RSC.
- Y. W. Zhang, W. Y. Huang, S. E. Habas, J. N. Kuhn, M. E. Grass, Y. Yamada, P. D. Yang and G. A. Somorjai, J. Phys. Chem. C, 2008, 112, 12092 CAS.
- F. Tao, M. E. Grass, Y. W. Zhang, D. R. Butcher, F. Aksoy, S. Aloni, V. Altoe, S. Alayoglu, J. R. Renzas, C.-K. Tsung, Z. W. Zhu, Z. Liu, M. Salmeron and G. A. Somorjai, J. Am. Chem. Soc., 2010, 132, 8697 CrossRef CAS PubMed.
- J. Gu, G. X. Lan, Y. Y. Jiang, Y. S. Xu, W. Zhu, C. H. Jin and Y. W. Zhang, Nano Res., 2015, 8, 1480 CrossRef CAS.
- J. B. Ding, L. Z. Bu, N. Zhang, J. L. Yao, Y. Huang and X. Q. Huang, Chem. – Eur. J., 2015, 21, 3901 CrossRef CAS PubMed.
- A. Rai, M. Chaudhary, A. Ahmad, S. Bhargava and M. Sastry, Mater. Res. Bull., 2007, 42, 1212 CrossRef CAS PubMed.
- X. L. Wang, B. H. Wu, G. X. Chen, Y. Zhao, P. X. Liu, Y. Dai and N. F. Zheng, Nanoscale, 2014, 6, 6798 RSC.
- D.-Y. Wang, H.-L. Chou, C.-C. Cheng, Y.-H. Wu, C.-M. Tsai, H.-Y. Lin, Y.-L. Wang, B.-J. Hwang and C.-C. Chen, Nano Energy, 2015, 11, 631 CrossRef CAS PubMed.
- J. Zhang and J. Y. Fang, J. Am. Chem. Soc., 2009, 131, 18543 CrossRef CAS PubMed.
- A.-X. Yin, X.-Q. Min, W. Zhu, H.-S. Wu, Y.-W. Zhang and C.-H. Yan, Chem. Commun., 2012, 48, 543 RSC.
- Q. Yuan and X. Wang, Nanoscale, 2010, 2, 2328 RSC.
- B. Lim, M. J. Jiang, P. H. C. Camargo, E. C. Cho, J. Tao, X. M. Lu, Y. M. Zhu and Y. N. Xia, Science, 2009, 324, 1302 CrossRef CAS PubMed.
- P. Gallezot and D. Richard, Catal. Rev. Sci. Eng., 1998, 40, 81 CAS.
- L. A. Saudan, Acc. Chem. Res., 2007, 40, 1309 CrossRef CAS PubMed.
- H. He, H. X. Dai, L. H. Ng, K. W. Wong and C. T. Au, J. Catal., 2002, 206, 1 CrossRef CAS.
- Y. E. Wu, S. F. Cai, D. S. Wang, W. He and Y. D. Li, J. Am. Chem. Soc., 2012, 134, 8975 CrossRef CAS PubMed.
- S. C. Tsang, N. Cailuo, W. Oduro, A. T. S. Kong, L. Clifton, K. M. K. Yu, B. Thiebaut, J. Cookson and P. Bishop, ACS Nano, 2008, 2, 2547 CrossRef CAS PubMed.
- L. E. Murilloa and J. G. G. Chen, Surf. Sci., 2008, 602, 919 CrossRef PubMed.
- W. T. Zang, G. Z. Li, L. Wang and X. W. Zhang, Catal. Sci. Technol., 2015, 5, 2532 CAS.
- T. B. L. W. Marinelli, S. Nabuurs and V. Ponec, J. Catal., 1995, 151, 431 CrossRef CAS.
- W. O. Oduro, N. Cailuo, K. M. K. Yu, H. W. Yang and S. C. Tsang, Phys. Chem. Chem. Phys., 2011, 13, 2590 RSC.
- Z. M. Rong, Z. H. Sun, Y. Wang, J. K. Lv and Y. Wang, Catal. Lett., 2014, 144, 980 CrossRef CAS.
- A. Stolle, T. Gallert, C. Schmöger and B. Ondruschka, RSC Adv., 2013, 3, 2112 RSC.
- L. E. Murilloa, C. A. Menning and J. G. G. Chen, J. Catal., 2009, 268, 335 CrossRef PubMed.
- R. Y. Zheng, M. D. Porosoff, J. L. Weiner, S. L. Lu, Y. X. Zhu and J. G. G. Chen, Appl. Catal., A, 2012, 419, 126 CrossRef PubMed.
- J. Haubrich, D. Loffreda, F. Delbecq, P. Sautet, Y. Jugnet, A. Krupski, C. Becker and K. Wandelt, J. Phys. Chem. C, 2008, 112, 3701 CAS.
- J. Haubrich, D. Loffreda, F. Delbecq, P. Sautet, A. Krupski, C. Becker and K. Wandeltt, J. Phys. Chem. C, 2009, 113, 13947 CAS.
- J. Haubrich, D. Loffreda, F. Delbecq, P. Sautet, Y. Jugnet, C. Becker and K. Wandelt, J. Phys. Chem. C, 2010, 114, 1073 CAS.
- F. Delbecq and P. Sautet, J. Catal., 2003, 220, 115 CrossRef CAS.
- J. P. Stassi, P. D. Zgolicz, S. R. de Miguel and O. A. Scelza, J. Catal., 2013, 306, 11 CrossRef CAS PubMed.
- F. Colomaa, A. Sepúlveda-Escribanoa, J. L. G. Fierrob and F. R. Reinoso, Appl. Catal., A, 1996, 136, 231 CrossRef.
- J. J. Shi, R. F. Nie, P. Chen and Z. Y. Hou, Catal. Commun., 2013, 41, 101 CrossRef CAS PubMed.
- B.-H. Zhao, J.-G. Chen, X. Liu, Z.-W. Liu, Z. P. Hao, J. L. Xiao and Z.-T. Liu, Ind. Eng. Chem. Res., 2012, 51, 11112 CrossRef CAS.
- L. J. Durndell, C. M. A. Parlett, N. S. Hondow, M. A. Isaacs, K. Wilson and A. F. Lee, Sci. Rep., 2012, 5, 9425 CrossRef PubMed.
- J. F. Moulder, W. F. Stickle, P. E. Sobol and K. D. Bomben, Handbook of X-ray photoelectron spectroscopy, Perkin-Elmer Corp., USA, 1992 Search PubMed.
- W.-Z. Hung, W.-H. Chung, D.-S. Tsai, D. P. Wilkinson and Y.-S. Huang, Electrochim. Acta, 2010, 55, 2116 CrossRef CAS PubMed.
- H. S. Wang, X. L. Qiao, J. G. Chen, X. J. Wang and S. Y. Ding, Mater. Chem. Phys., 2005, 94, 449 CrossRef CAS PubMed.
- G. Z. Cao and Y. Wang, Nanostructures and Nanomaterials: Synthesis, Properties, and Applications, Imperial College Press, China, 2004 Search PubMed.
- C. Mohr and P. Claus, Sci. Prog., 2001, 84, 311 CrossRef CAS.
- X. Q. Qi, M. R. Axet, K. Philippot, P. Lecanted and P. Serpa, Dalton Trans., 2014, 43, 9283 RSC.
- F. Delbecq and P. Sautet, J. Catal., 1995, 152, 217 CrossRef CAS.
- J. C. de Jesus and F. Zaera, J. Mol. Catal. A: Chem., 1999, 138, 237 CrossRef CAS.
- M. Englisch, V. S. Ranade and J. A. Lercher, J. Mol. Catal. A: Chem., 1997, 121, 69 CrossRef CAS.
- X.-C. Xiao, W. Shi, Z.-M. Ni, L.-Y. Zhang and J.-F. Xu, Acta Phys.-Chim. Sin., 2015, 31, 885 CAS.
- B. Eveline, P. Roel and J. A. van Bokhoven, Catal. Commun., 2007, 8, 1397 CrossRef PubMed.
- J. W. Medlin, ACS Catal., 2011, 1, 1284 CrossRef CAS.
- P. Claus, Top. Catal., 1998, 5, 51 CrossRef CAS.
- H. P. Rong, Z. Q. Niu, Y. F. Zhao, H. Cheng, Z. Li, L. Ma, J. Li, S. Q. Wei and Y. D. Li, Chem. – Eur. J., 2015, 21, 12034 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: TEM images of PtSn/SnO2 catalysts, spectra of FTIR and Rietveld refinement of PXRD. See DOI: 10.1039/c5qi00129c |
| This journal is © the Partner Organisations 2015 |