 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceOptimizing GC-ICP-MS for ultra-trace quantification of PBDEs in natural water samples using species-specific isotope dilution†
Adriana
González-Gago
,
Daniel
Pröfrock
* and
Andreas
Prange
Helmholtz Zentrum Geesthacht – Zentrum für Material und Küstenforschung, Institute for Coastal Research, Department Marine Bioanalytical Chemistry, Max-Planck Str. 1, 21502 Geesthacht, Germany. E-mail: daniel.proefrock@hzg.de
First published on 26th June 2014
Abstract
Due to the increased interest regarding the fate, transport and toxic effects of polybrominated diphenyl ethers (PBDEs) within the aquatic environment a variety of different methods have been reported, trying to fulfil the requirements of the European Water Framework Directive (WFD, Directive 2000/60/EC). For PBDEs the WFD specifies Environmental Quality Standards (EQS) at very low trace levels, which correspond to 0.5 ng L−1 for 6 priority brominated flame retardants. The reliable quantification of such low concentration levels in environmental sample matrices requires the development of new analytical approaches. Current standard GC-MS methods, which are frequently used for PBDE analysis, often do not provide sufficient sensitivity for their reliable analysis at ultra-trace levels. Within this context a new GC-ICP-MS method has been developed and optimized allowing the ultra-trace determination of the selected priority congeners at sub ng L−1 levels. The quantification approach is based on the application of 81Br labelled PBDEs as internal standards for species-specific isotope dilution analysis. Natural water samples were analysed to demonstrate the potential of this new detection and quantification approach.
Introduction
Polybrominated diphenyl ethers (PBDEs) are frequently used as additive flame retardants for polymers used as plastics or in consumer products such as computers, furniture or textiles. In addition to their proved suitability as flame retardants, over the last two decades they have gained also great interest in the environmental health field due to their high persistence and lipophilic characteristics, which result in their ubiquitous presence as contaminants in all important environmental compartments (e.g. soil, sediment, water, air, biota) as well as wildlife and humans.1 Although there may be more than 200 possible PBDE congeners, only some of them have been found in environmental samples, mainly due to the extensive use of the three most important commercial mixtures (c-pentaBDE, c-octaBDE and c-decaBDE). However, in some cases the initial congener distribution of the applied technical mixtures can change, presumably due to naturally occurring processes such as photolytic debromination,2 biologically mediated debromination reactions3–5 or metabolic reactions in higher organisms,6,7 showing a trend towards the formation of lower brominated congeners. Since the observed adverse effects in living organisms are more pronounced for congeners with bromination degrees below seven,8 the technical mixtures c-pentaBDE and c-octaBDE have been banned in Europe and several states of the USA since 2004 while the major congeners present in these mixtures have been considered as target substances by different institutions.Within the context of the European Water Framework Directive (WFD, Directive 2000/60/EC) the European Union has included congeners 28, 47, 99, 100, 153 and 154 in the list of priority substances that need to be measured and monitored in all surface waters covered by the WFD, to finally achieve the objective of the WFD, namely good quality of surface, ground and coastal waters in the EU by 2015 with targeted Environmental Quality Standards (EQS), equivalent to an annual average concentration below 0.5 ng L−1.
For reliable measurements of contaminants at the EQS level, the WFD daughter directive 2009/90/EC demands analytical methods for test laboratories which have a limit of quantification (LOQ) equal to or lower than 30% of the EQS combined with a measurement uncertainty (95% confidence) of 50% or less at the EQS level. Therefore, primary methods of measurement, which should form the reference for measurements at test laboratories, should have a considerably lower LOQ; unfortunately, such methods are not available yet.
Mass spectrometric detection techniques represent the state of the art regarding the sensitive determination of PBDEs in environmental samples. The most commonly used techniques for PBDE analysis are GC-EI-MS or GC-NCI-MS. High resolution (HR) mass spectrometers, mainly used in EI ionization mode, show a high specificity towards PBDEs and allow their sensitive quantification. However, these instruments have not been used very often, as they are expensive and only experienced users can operate them. When working at low resolution (LR) in EI mode, the sensitivity decreases significantly in comparison with HR, being in some cases insufficient to ensure the reliable quantification of PBDEs, which are usually present at ultra-trace levels in real samples. For this reason, GC-NCI-MS has been often the technique of choice, despite showing lower specificity than GC-EI-MS, it provides much higher sensitivity for the detection of PBDEs.9
The overall low concentrations of the target analytes in water, derived from their high lipophilicity, together with the challenging requirements defined by the WFD for methods that are applicable to the determination of priority pollutants, requires the development of suitable methodologies combining sensitive detection approaches with accurate and precise analytical techniques in terms of sample preparation, calibration and quantification.10,11
Besides the mentioned techniques, typically used for the analysis of PBDEs, as well as some other semivolatile halogenated organic pollutants e.g. PCBs, hyphenated approaches based on Inductively Coupled Plasma Mass Spectrometry (ICP-MS), commonly considered only suitable for the determination of heavy metals, have recently gained a lot of interest also as sensitive detection methods for some heteroatoms such as phosphorus, sulphur, chlorine, bromine or iodine. The detection of bromine using ICP-MS is a priori challenging as it shows low ionization efficiency,12 due to its high first ionization potential, and some polyatomic ions (78Se1H+ or 40Ar21H+) can interfere with its detection, but despite these challenges, GC-ICP-MS still represents an interesting alternative approach for the analysis of PBDEs,13,14 as it has been demonstrated to provide even better instrumental limits of detection than the GC-NCI-MS,14 being very helpful to deal with the high analytical requirements specified in the EU WFD.
Here we describe for the first time the in deep optimization and application of GC-ICP-MS for the ultra-trace determination of PBDEs in water samples. A special focus lies, in particular, on the optimization of bromine detection using GC-ICP-MS and the benefits of the dry plasma conditions obtained when using GC as a sample introduction technique. A species-specific isotope dilution quantification methodology has been developed and applied to the accurate determination of the priority BDE congeners in natural water samples.
Experimental
Chemicals and standards
A standard reference material, SRM 2257 PBDE congeners in 2,2,4-trimethylpentane,15 was obtained from NIST (National Institute of Standards and Materials, Gaithersburg, USA). The SRM, including the congeners BDE 17, BDE 25, BDE 28, BDE 30, BDE 47, BDE 49, BDE 66, BDE 71, BDE 74, BDE 75, BDE 85, BDE 97, BDE 118, BDE 99, BDE 100, BDE 101, BDE 116, BDE 119, BDE 138, BDE 139, BDE 153, BDE 154, BDE 155, BDE 156, BDE 173, BDE 190, BDE 181, BDE 182, BDE 183, BDE 185, BDE 191, BDE 196, BDE 197, BDE 204, BDE 198, BDE 203, BDE 206 and BDE 208, at different concentrations, has been used for the GC-ICP-MS optimization experiments.Another standard (EPA method 1614 native PAR stock solution) containing a mixture of 8 PBDEs (BDE 28, BDE 47, BDE 99, BDE 100, BDE 153, BDE 154, BDE 183 and BDE 209) in nonane was obtained from Cambridge Isotope Laboratories Inc. (Andover, MA, USA) and was used for the optimisation of the GC column position inside the GC-ICP-MS coupling interface and for the water sample fortification experiments.
Individual 81Br enriched PBDEs for the six priority congeners (BDE 28, BDE 47, BDE 99, BDE 100, BDE 153 and BDE 154), obtained from ISC Science (Oviedo, Spain), were used to spike the water samples in the isotope dilution experiments. Table 1 summarises the concentrations and isotopic composition of the spikes obtained during their initial characterisation.
| Congener | Label | Enrichment (At%) | Concentration μg g−1 |
|---|---|---|---|
| BDE 28 | 81Br3 | 99.33 ± 0.06 | 3.08 ± 0.24 |
| BDE 47 | 81Br4 | 99.54 ± 0.02 | 4.18 ± 0.33 |
| BDE 99 | 81Br5 | 99.14 ± 0.03 | 3.24 ± 0.19 |
| BDE 100 | 81Br2 | 68.84 ± 0.73 | 8.3 ± 1.0 |
| BDE 153 | 81Br6 | 99.21 ± 0.12 | 2.91 ± 0.13 |
| BDE 154 | 81Br3 | 73.68 ± 0.37 | 2.15 ± 0.13 |
Dark glass bottled natural still mineral water (from Fürst Bismark source, Aumühle, Germany) was bought in a local supermarket and has been used for the different experiments. River water samples were collected from the River Elbe.
Ultrapure water (18 MΩ cm) was obtained from a Millipore Elix 3/Milli-Q Element water purification system (Millipore, Milford, MA, USA).
All solvents used in this work were specified for organic trace analysis. Isooctane (Suprasolv® from Merck, Darmstadt, Germany) was used to prepare standard working solutions from the SRM 2257 and as a keeper for the water sample extracts. 2-Propanol (Suprasolv® from Merck, Darmstadt, Germany) was used to prepare dilute solutions of the mixtures of PBDEs, unlabelled (EPA method 1614 native PAR stock solution) for the fortification of the water samples as well as the 81Br-labelled PBDE mix used to spike the samples for isotope dilution quantification. Hexane (Unisolv® from Merck, Darmstadt, Germany) and dichloromethane (Picograde® from LGC Promochem, Wesel, Germany) were used as solvents to extract the water samples. Standard working solutions and water sample extracts were stored in the dark at 4 °C until their use.
All the reusable glassware (amber bottles, separatory funnels and pre-concentration tubes) was cleaned with detergent in a laboratory glassware dish washer, rinsed with ultrapure water and baked for 10 h at 250 °C. Hexane and acetone (both Picograde® from LGC Promochem, Wesel, Germany) were used to rinse all the glassware after and before use. Disposable glassware was also rinsed with hexane and acetone and baked prior to use.
Helium 5.0 (99.999% purity) was used as a GC carrier gas, as a collision gas inside the octopole reaction system as well as an additional plasma gas for sensitivity enhancement during GC-ICP-MS analysis. Nitrogen 5.0 (99.999% purity) was also used as an additional plasma gas. A mixture of 10% xenon 4.8 (99.998% purity) in 90% argon 5.0 (99.999% purity) was used for general tuning of the GC-ICP-MS setup. Argon 5.0 (99.999% purity) was used as a plasma gas and a carrier gas for the GC interface. All gases were obtained from Messer Griesheim (Messer Griesheim, Krefeld, Germany).
Instrumentation
An Agilent 6890 gas chromatography system (Agilent Technologies, Waldbronn, Germany) equipped with an Agilent 7683 series auto-sampler was used during all experiments. To reduce the potential for thermal degradation of the analysed PBDEs and discrimination effects, as known from split/splitless injectors, a cool on column (COC) injection port and a DB5MS column (J&W Scientific, Folsom, CA, USA) have been used in this work.16 The GC was also equipped with an additional 3 way gas-flow controller which allows the precise mixing of different gases to the ICP-MS controlled carrier-gas. Details of the chromatographic conditions are given in Table 2.| GC parameters | |
|---|---|
| Column | Pre-column: 3 m, Siltek deactivated fused silica |
| Column: Agilent DB5MS, 15 m, i.d. 0.25 mm, film thickness 0.1 μm, 5% phenyl, 95% methyl-polysiloxane | |
| Carrier gas | Helium |
| Carrier gas flow rate | 2.6 mL min−1 |
| Injection volume/mode | 1 μL, cool on column |
| Injector temperature | Oven track |
| Temperature program | Initial temperature 100 °C hold for 2 min, ramp of 30 °C min−1 to 300 °C hold for 11 min |
| GC-ICP-MS Interface parameters | |
|---|---|
| Transfer line temperature | 300 °C |
| Injector temperature | 300 °C |
| ICP-MS parameters | |
|---|---|
| RF power | 750–850 W |
| Carrier gas flow rate | 0.95 L min−1 |
| Sampling depth | 6 mm |
| Cones | Nickel |
| Cell gas flow rate | 2 mL min−1 helium |
| Extraction lens | −100 V to −125 V |
| Octopole bias | −16 V |
| Quadrupole bias | −14 V |
| Measured isotopes/dwell time | 79Br (0.1 s), 81Br (0.1 s) |
| Optional plasma gas | N2 at 55 psi controlled by the GC |
An Agilent GC-ICP-MS interface which features a temperature controlled Silcosteel® transfer line as well as a heated stainless steel injector tip was used for the hyphenation of GC and ICP-MS (Agilent Technologies, Tokyo, Japan). Details on the interface could be found elsewhere.17 The selected interface settings are also summarized in Table 2.
An Agilent 7700 ICP-MS system (Agilent Technologies, Tokyo, Japan), that features a new on-axis octopole ion guide which operates in an RF-only mode, a modified ions lens system and shield torch technology was used as element-specific detector. The ions were extracted by an omega lens-system, consisting of a dual extraction lens and an omega lens assembly with an off-axis design for increased sensitivity and an overall low background. The off-axis design prevents photons and neutral compounds from entering the on-axis reaction cell chamber.18,19Fig. 1 gives a schematic overview of the setup used. Details concerning the ICP-MS settings are given in Table 2.
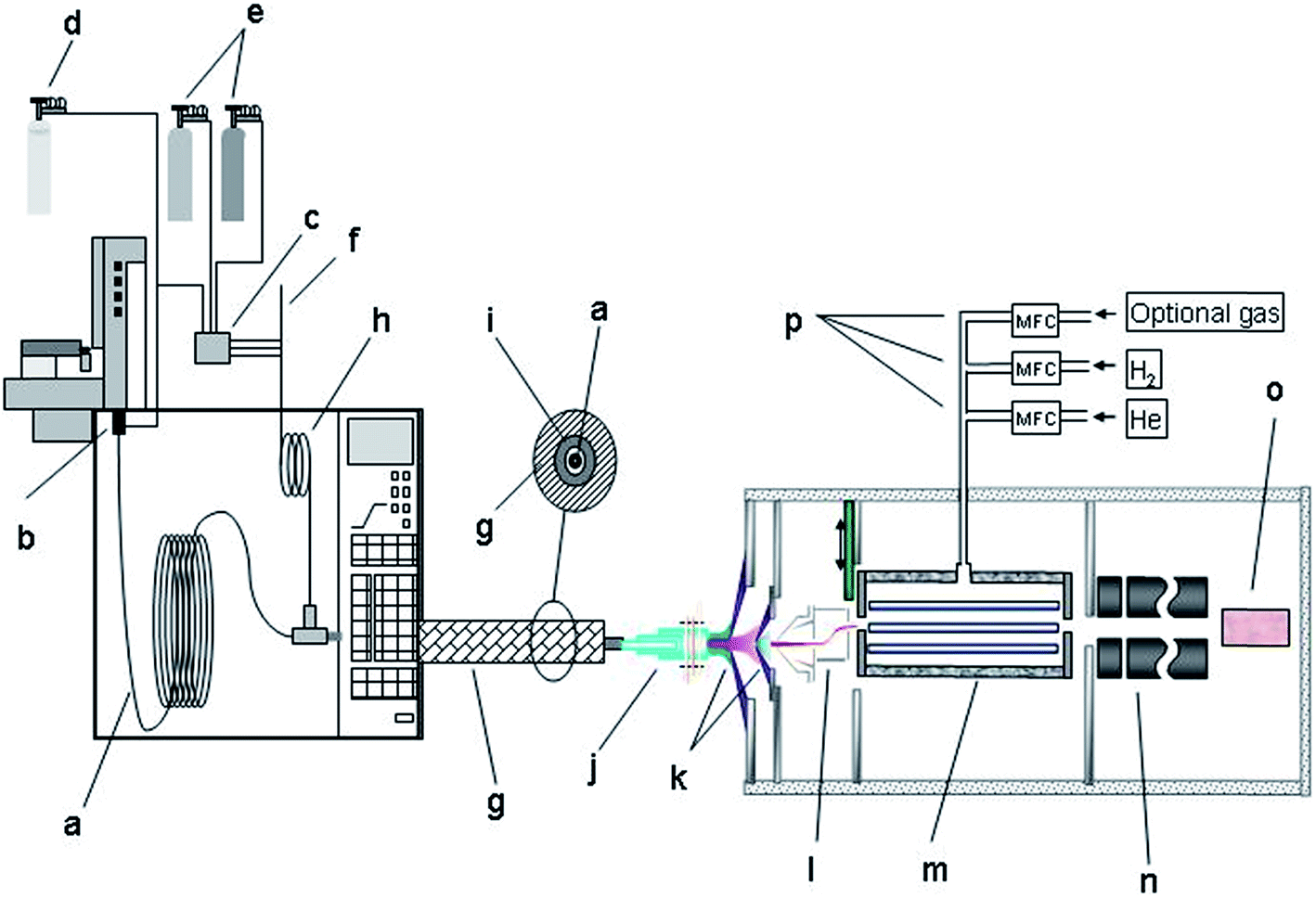 | ||
| Fig. 1 Schematic overview of the used instrumental setup: (a) fused silica capillary, (b) injector, (c) optional 3 way gas controller, (d) He, (e) optional gases, (f) Ar carrier gas/ICP-MS controlled, (g) heated transfer-line, (h) carrier gas pre-heater coil, (i) Silcosteel® transfer-line, (j) ICP torch with Pt shield, (k) cones Ni, (l) dual extraction and omega lens assembly (off-axis design, s-lens), (m) in-line octopole reaction system, (n) quadrupole mass analyser, (o) detector and (p) mass-flow controller. | ||
Sample preparation
All working standards (natural and labelled), used in the isotope dilution experiments, were prepared gravimetrically using an analytical balance (AT261 Delta Range from Mettler-Toledo, Greifensee, Switzerland).The samples were placed into 1 L amber glass bottles and spiked with a mixture of six 81Br enriched PBDEs in isopropanol. After equilibration the samples were extracted three times with a mixture of hexane and dichloromethane (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1). The extracts were pre-concentrated, first by rotary evaporation and afterwards under a nitrogen flow using a heated gas flow laboratory evaporator (Flowtherm Optocontrol from Barkey, Leopoldshoehe, Germany), to get a final volume between 50 and 75 μL. Finally the extracts were injected into the GC-ICP-MS system. A mineral water sample was run in parallel to check the procedural blanks. Recovery experiments were carried out by fortifying mineral and river water samples with a mixture of native PBDEs in isopropanol resulting in concentration levels of approximately 0.2–0.4 ng L−1 for each of the six priority congeners. The fortified samples were analysed as described above.
:1). The extracts were pre-concentrated, first by rotary evaporation and afterwards under a nitrogen flow using a heated gas flow laboratory evaporator (Flowtherm Optocontrol from Barkey, Leopoldshoehe, Germany), to get a final volume between 50 and 75 μL. Finally the extracts were injected into the GC-ICP-MS system. A mineral water sample was run in parallel to check the procedural blanks. Recovery experiments were carried out by fortifying mineral and river water samples with a mixture of native PBDEs in isopropanol resulting in concentration levels of approximately 0.2–0.4 ng L−1 for each of the six priority congeners. The fortified samples were analysed as described above.
Results and discussion
Optimisation of the instrumental settings
Since the application of GC as a sample introduction system for ICP-MS results in a dry and nearly matrix free plasma, common problems known from liquid sample introduction such as interference or ionization suppression effects are more or less negligible. However, under such conditions the optimal instrumental parameters differ also strongly, in terms of plasma power as well as lens and gas flow settings, from the values commonly used for wet plasma applications. For the optimization of the GC-ICP-MS coupling, instrumental settings adapted from previous work devoted to the simultaneous determination of phosphorus, sulphur, chlorine, bromine and iodine containing pesticides using an Agilent 7500 cs ICP-MS system have been used as a starting point.20 Unfortunately these conditions showed only a poor sensitivity for the new instrumental system. The differences observed in the optimal instrumental settings may be related to the different properties shown by this instrument, which can be due to the use of a new lens and collision cell system as well as the new RF generator design. Therefore, different parameters, which influence the sensitivity of the setup, have been optimized using a standard reference material (NIST 2257, 38 PBDE congeners in 2,2,4-trimethylpentane) after a 100 fold dilution with isooctane. To optimize the bromine specific detection of the various congeners present in the test sample, 1 μL of the solution was injected several times. The instrumental response was analysed in terms of signal to noise ratio (S/N), which was calculated using the Agilent MassHunter software supplied with the instrument, for each separated compound under different working conditions. In comparison with the optimization of the instrumental conditions by monitoring the signal of Xe, the proposed method allows the direct bromine specific optimization as well as the consideration of the influence of changing settings on the Br background. To improve the readability of the following figures only signal to noise ratios for the congeners 28, 47, 99, 100, 153, 154, 183, 208, 206 will be presented with the aim to cover the most important ones. However, during the experiments all congeners and both isotopes showed the same trend for the different changed parameters. The following section will focus on the optimization of different instrumental parameters for dry plasma conditions, which in sum have a strong influence on the overall sensitivity of the instrumental setup, which is of high importance in particular when aiming on ultra-trace analysis at sub ng L−1 levels. Due to the overall high robustness of the instrumental setup in terms of instrumental drifts over time no internal standardization was necessary during the different optimization procedures. This is in particular a result of the very small amount of matrix, that enters the ICP-MS when using GC as a sample introduction system. The main matrix that reaches the ICP-MS is caused by the solvent peak that appears at the beginning of each separation, which is caused by the used solvent (0.5 to 1 μL per injection). Typical peak area and peak height RSDs during long term measurements over 15 to 20 hours are below 3%, so the instrumental fluctuations have only minimal influence on the observed trends. Xe isotopes (126Xe or 132Xe), which can be found as impurities in the argon gas or which can be actively mixed via the GC into the Ar carrier gas flow controlled by the ICP-MS have been used to monitor possible long term drifts of the setup.RF power. Due to the high 1st ionization potential of Br as well as its limited ionization efficiency12 when using an argon based inductively coupled plasma, optimization of the RF power is mandatory. Due to the dry plasma conditions, slightly different power settings are necessary to obtain the best sensitivity during bromine specific detection. As indicated in Fig. 2 the best sensitivity and signal to noise ratios for all congeners evaluated were obtained under low RF power conditions, comparable to those used for cool plasma applications. Based on these results all further experiments were carried out under low plasma RF power conditions of 750–850 W.
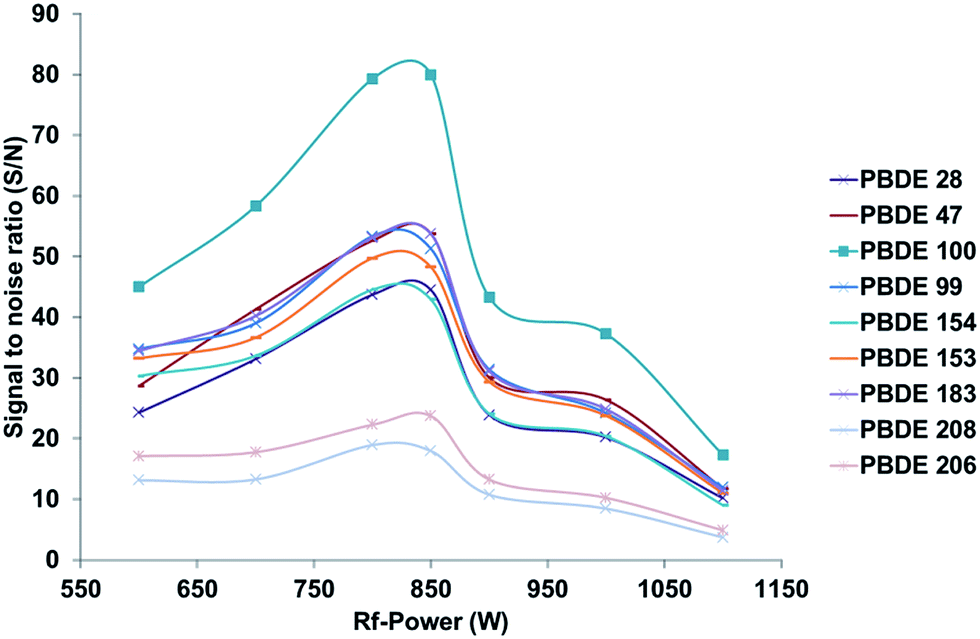 | ||
| Fig. 2 Effect of the RF power settings on the signal to noise ratio of the selected PBDE congeners when using GC-ICP-MS conditions. Calculations were based on the 79Br signal. | ||
Fig. S3(b) shows the effect of the addition of nitrogen to the plasma for the same congeners as shown before. The addition of N2 also decreases the background, especially for m/z = 81, which helps a lot to improve S/N. In the context of this paper this is very important since for IDMS both Br isotopes have to be measured and a high background for one of the measured isotopes results in a worse precision (one of the main advantages of using IDMS for quantification).
GC column position inside the interface. For most applications, the outlet of the GC capillary column is placed inside the Silcosteel® line 2–3 mm behind the end of the heated injector tubing. The tangential flowing, pre-heated Ar carrier gas supplied by the ICP provides rapid transfer of the eluting analytes directly into the plasma. First separations of a PBDE test mixture (EPA method 1614 native PAR stock solution) using the described instrumental settings indicated a good separation of all congeners except PBDE 209, which could not be eluted from the column (Fig. 3(a)). This might be, in particular, a result of the constant high interface temperature (300 °C), which represents also the constant temperature level of the last, at least 1 m of the GC column. During its passage through the last meter of the column the compound is still retained by the stationary phase. Keeping in mind the long retention time of this congener, it results in a prolonged exposure time of this compound to the constant high temperature of the interface. This may lead to its degradation and potentially interference with the quantification of lower brominated congeners.
 | ||
| Fig. 3 Effect of the GC capillary position inside the interface placed (a) 0.2 cm before the injector tip exit and (b) 10 cm beyond the Ar carrier gas inlet to the interface inside the GC oven. | ||
To overcome this problem the contact time of the analyte with the 300 °C interface environment was minimized by moving the GC capillary outlet back into the interface to a position approximately 10 cm beyond the carrier gas inlet inside the oven. Under such conditions the contact time is minimized, since the high carrier gas volume (0.95 L min−1), which has to pass the low i.d. (500 μm) 1/16′′ Silcosteel® line, immediately transports the eluting compounds to the plasma. Under such adapted conditions the previously observed degradation effects could be minimized as shown in Fig. 3(b).
Analytical figures of merit
Once the instrumental system was optimized for the sensitive detection of the target PBDEs by GC-ICP-MS, a measurement procedure based on the accurate and precise quantification of these substances by isotope dilution mass spectrometry was proposed. To evaluate the capabilities of this instrumental system to meet the requirements of the current legislation it was characterized in terms of instrumental limits of detection and precision.To estimate the instrumental limits of detection, standard solutions containing a mixture of 8 PBDEs at different concentration levels, from 0.05 to 10 μg L−1, were injected (1 μL) into the GC-ICP-MS system. The instrumental response for each congener was obtained from the signals measured for both bromine ions (79Br+ and 81Br+). A linear response was observed for the 6 priority congeners within the studied concentration range. The instrumental limits of detection (ILODs) were calculated for each BDE congener from both bromine isotopes as the lowest detected concentration level, obtained from a calibration curve, plus three times its standard deviation for five injections. Results are summarized in Table 3 in comparison with other published studies dealing also with the detection of PBDEs by GC-ICP-MS. Instrumental limits of detection were between 60–100 fg on the column (1 μL injected) for the six priority congeners and for both m/z monitored. As shown in Table 3, careful optimisation of the GC-ICP-MS instrumental setup provides improved ILODs (from 2 to 5 times lower than previously published values), which is a good basis for the reliable ultra-trace analysis of PBDEs. Taking into account the achieved instrumental limits of detection, an enrichment factor of 500 or higher would be sufficient to allow the detection of the six priority PBDEs when they are present in water at the required LOQ level.
However, the required enrichment factor should also ensure the compliance with the uncertainty requirements of the WFD. The precision obtained in the measurements was also evaluated, as it will eventually affect the expanded uncertainty associated with the results. Since the quantification of PBDEs is going to be carried out by IDMS, isotope ratios were measured, together with their standard deviations, from the natural abundance standard solutions injected to obtain the calibration curve for the ILOD calculations. The experimental bromine isotope ratios were obtained at different concentration levels of the six priority PBDEs and compared with the theoretical value24 to assess the accuracy. Precisions expressed as %RSD were obtained from the standard deviations of five repeated isotope ratio measurements. Fig. 4 summarizes the results obtained for the GC-ICP-MS setup when working under the optimized working conditions. As can be observed concentration levels above 0.5 μg L−1 provide acceptable accuracy (relative errors between 99–107%) and precision (%RSDs below 5%) due to the improvement of the statistics when measuring high ion counting rates. Therefore, concentration levels of the analytes in the final extract above 0.5 μg L−1 (equivalent to peak areas above 4000) would be convenient to minimise the uncertainty in the results when working at the optimum isotope ratio according to the error magnification factor, aiming to achieve uncertainties below 50% in the quantification of PBDEs at the EQS level, as set by the WFD. Enrichment factors above 1000 would be necessary to achieve concentration levels in the final extract in this range, which can be obtained by using different extraction techniques which allow the extraction of 1 L sample and afterwards pre-concentrating the extracts to a final volume below 1 mL.
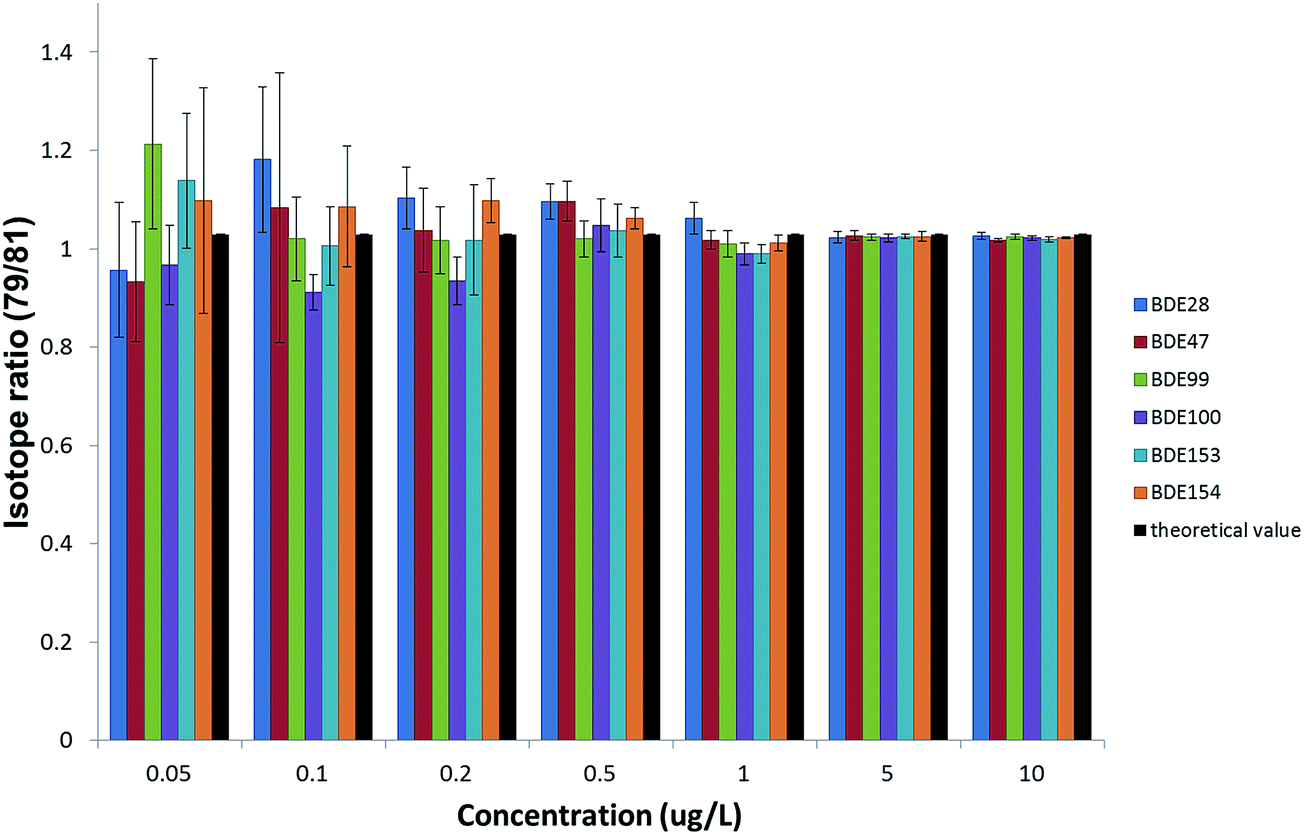 | ||
| Fig. 4 Accuracy and precision of isotope ratio measurements achieved by GC-ICP-MS under the optimized experimental working conditions. | ||
The selectivity of the proposed analytical method must be also considered for the reliable determination of the analytes. As the ICP source generates mainly elemental singly positively charged ions, all the molecular information from the analytes is lost during the ionization process and, consequently, the identification must be done based on their retention times. However, ICP-MS still shows a good selectivity towards bromine, which can help to solve possible co-elutions of PBDEs with other non-brominated pollutants, that due to their similar physicochemical properties, can be co-extracted from the sample, together with the PBDEs. Unfortunately, some brominated pollutants, such as PBBs, HBCD or TBBPA, or even some metabolites and naturally occurring compounds can be also co-extracted under the same conditions. Some of these interferents can be removed by applying different clean-up and fractionation procedures, but still some important co-elutions of the target PBDEs with some brominated substances have been reported in the literature for the DB5MS column used in this work.25,26 Thus, the proposed methodology is limited to the reliable determination of PBDEs in samples free of these potential co-eluting interferents. The use of a different GC column can be helpful to overcome the co-elution problem25 but it usually involves a higher discrimination and/or thermal degradation of the higher brominated congeners16 and consequently, higher limits of detection. So a compromise between sensitivity and selectivity must be achieved in the selection of the GC column.
When using the DB5MS column, small variations in the retention times were observed for the PBDEs of interest, so a normalization procedure was developed to facilitate their identification. To do this, a standard solution containing a mixture of 8 PBDEs was injected into the GC-ICP-MS system on different working days, covering a period of 8 months. The relative retention times (RRTs) for these congeners were calculated against the sum of the retention times of BDEs 47 and 183.25,27 Different concentration levels, from 50 ng L−1 to 10 μg L−1, were considered for the RRT calculations. As can be seen in Table 4, the stationary phase proposed for this work showed stable RRTs for most of the target congeners throughout the studied time period, so they can be used as reference values for their identification. BDE 209 showed higher variability in its RRT, which can arise from the broad peak shape observed for this congener due to its high boiling point or its thermal degradation when it is exposed to high temperatures for a long time,9,28 limiting its reliable identification, although this congener is not included in the list of priority substances of the WFD.
| Congener | 20/09/12 | 06/12/12 | 19/03/13 | 18/05/13 |
|---|---|---|---|---|
| BDE 28 | 0.4023 ± 0.0004 | 0.4024 ± 0.0002 | 0.4021 ± 0.0001 | 0.4029 ± 0.0004 |
| BDE 47 | 0.4445 ± 0.0003 | 0.4445 ± 0.0001 | 0.4445 ± 0.0003 | 0.4446 ± 0.0001 |
| BDE 99 | 0.4845 ± 0.0005 | 0.4845 ± 0.0003 | 0.4844 ± 0.0002 | 0.4845 ± 0.0002 |
| BDE 100 | 0.4748 ± 0.0005 | 0.4747 ± 0.0002 | 0.4746 ± 0.0002 | 0.4745 ± 0.0002 |
| BDE 153 | 0.5210 ± 0.0005 | 0.5211 ± 0.0002 | 0.5210 ± 0.0002 | 0.5210 ± 0.0002 |
| BDE 154 | 0.5081 ± 0.0004 | 0.5081 ± 0.0002 | 0.5081 ± 0.0002 | 0.5077 ± 0.0002 |
| BDE 183 | 0.5555 ± 0.0003 | 0.5555 ± 0.0001 | 0.5555 ± 0.0003 | 0.5554 ± 0.0001 |
| BDE 209 | 0.9302 ± 0.0019 | 0.9257 ± 0.0008 | 0.9325 ± 0.0013 | 0.9276 ± 0.0013 |
According to these results, the proposed instrumental system shows appropriate characteristics for the reliable determination of the six priority PBDEs (28, 47, 99, 100, 153 and 154) in compliance with the requirements of the WFD.
Application of the method for the analysis of PBDEs in water samples
An analytical method for the quantification of ultra-trace levels of PBDEs in water samples by species-specific IDMS was developed and validated. As there are no certified reference materials for the validation of the proposed methodology available to date, a test sample was prepared by spiking mineral water with a known amount (close to the EQS value) of the six priority PBDEs. A non-fortified mineral water sample was run in parallel to check the procedural blanks. Two sample replicates of the test sample and one blank were prepared according to the procedure described in the experimental section followed by the injection of the extracts into the GC-ICP-MS under optimised working conditions. Concentrations of the six priority congeners were obtained by single IDMS using the form of the equation for elemental analysis described by Rodríguez-González et al.29 Mass bias correction was carried out by the bracketing approach, injecting a natural abundance standard solution containing a mixture of PBDEs between samples and comparing the measured isotope ratios with the reference value obtained from the average isotopic abundances for bromine published by IUPAC.24The results obtained for the determination of PBDEs in a mineral water sample are summarised in Table 5. As shown, recoveries are between 88% and 106% for most congeners which means that good accuracy can be achieved in the determination of PBDEs even at concentration levels below the EQS. Recoveries were a bit higher, around 130%, for BDE 153. This result cannot be explained and further experiments would be necessary in order to assess this issue, although it could be considered an acceptable result taking into account that very low concentration levels, below 1 ppt, are being quantified. Precisions are also included in Table 5, expressed as RSDs. Repeatabilities obtained for five replicate measurements of the same sample were below 3% for the six priority congeners. The reproducibility in the concentration results obtained from the two fortified mineral water samples was below 6% in all cases.
| Congener | Spike | Concentration (ng L−1) | Recovery (%) | Precision (%RSD) | U (%) | |
|---|---|---|---|---|---|---|
| Repeatability | Reproducibility | |||||
| BDE 28 | (81Br3, 99.3%) BDE 28 | 0.417 | 97.5 | 1.3 | 1.6 | 7.8 |
| BDE 47 | (81Br4, 99.5%) BDE 47 | 0.403 | 103.5 | 1.5 | 1.7 | 8.0 |
| BDE 99 | (81Br5, 99.1%) BDE 99 | 0.405 | 88.6 | 1.3 | 2.2 | 6.2 |
| BDE 100 | (81Br2, 68.6%) BDE 100 | 0.402 | 106.3 | 2.3 | 4.4 | 16 |
| BDE 153 | (81Br6, 99.2%) BDE 153 | 0.387 | 132.7 | 0.9 | 5.9 | 5.0 |
| BDE 154 | (81Br3, 73.7%) BDE 154 | 0.416 | 96.4 | 2.6 | 2.9 | 8.3 |
Expanded uncertainties were calculated for one of the samples using the GUM uncertainty calculation software (GUM Workbench 2.4, Metrodata GmbH, Weil am Rhein, Germany) in order to check the capabilities of the proposed methodology to meet the uncertainty requirements of the WFD. The results are also summarised in Table 5. As can be seen expanded uncertainties (k = 2) were below 16% for all the congeners with the concentration of the analytes in the spike (obtained by reverse IDMS using the standard reference material SRM 2257), the isotope ratio measured for the spike and the isotope ratio measured for the sample–spike blend being the main sources of uncertainty. For all the BDE congeners the expanded uncertainty was quite below the 50% level as required by the WFD.
The limits of quantification (LOQs) of the proposed methodology were also evaluated. The results, expressed as 10 times the standard deviation from 8 replicate blank samples, are shown in Table 6 in comparison with other published methods for the determination of PBDEs in water samples. As can be observed, the LOQs obtained in this work were below the value required by the WFD (0.15 ng L−1) for the six priority congeners. In comparison to the LLE-GC-ECNI-IDMS method,10 lower LOQs could be obtained due to the improved sensitivity (leading to lower standard deviations in the measurements) achieved when using GC-ICP-MS under the optimised conditions. Table 6 shows also the LOQs obtained when using solid phase extraction and a high-resolution instrument (SPE-GC-EI-HRMS) for the determination of PBDEs in water samples, although in this case the LOQs were obtained by injecting low concentration standards of the target analytes (accounting for the enrichment factor). The LOQs obtained by LLE-GC-ICP-IDMS are at the same level or lower than those obtained by SPE-GC-EI-HRMS, especially for the higher brominated congeners, which usually show lower signals when using an EI source.11 Conversely, the ICP is a hard ionization source which generates only elemental ions and the expected instrumental response should be proportional to the amount of target atoms reaching the plasma and independent of the molecular structure of the analyte. This means that higher brominated congeners, which contain a higher amount of bromine atoms, should provide a higher instrumental response in ICP-MS leading to lower ILODs. However, as shown in Table 3 the ILODs were in the same range for the six priority PBDEs. This can be due to the discrimination of the higher brominated congeners through the chromatographic system (despite using the cool on column injector) because of their high boiling points. But still, the use of the elemental ICP ionization source for the detection of PBDEs can compensate to a certain extent this discrimination effect allowing the detection of all the priority congeners at very low concentration levels and improving the LOQs for the higher brominated congeners in comparison to the EI source.
| Congener | LLE-GC-ICP-IDMSa (this work) | LLE-GC-ECNI-IDMSb (ref. 10) | SPE-GC-EI-HRMSc (ref. 11) |
|---|---|---|---|
|
a Expressed as 10 times the standard deviation of 8 blank samples (preconcentration factor 10000, 1 μL injected).
b Expressed as 10 times the standard deviation of 10 individual blanks (preconcentration factor 5000, 2 μL injected).
c Expressed as the lowest concentration of the analyte for which S/N was 10 for both ions monitored (preconcentration factor 1000, 1 μL injected).
|
|||
| BDE 28 | 0.014 | 0.28 | 0.05 |
| BDE 47 | 0.067 | 0.30 | 0.05 |
| BDE 99 | 0.039 | 0.05 | 0.05 |
| BDE 100 | 0.030 | 0.05 | 0.05 |
| BDE 153 | 0.007 | 0.28 | 0.10 |
| BDE 154 | 0.023 | 0.06 | 0.10 |
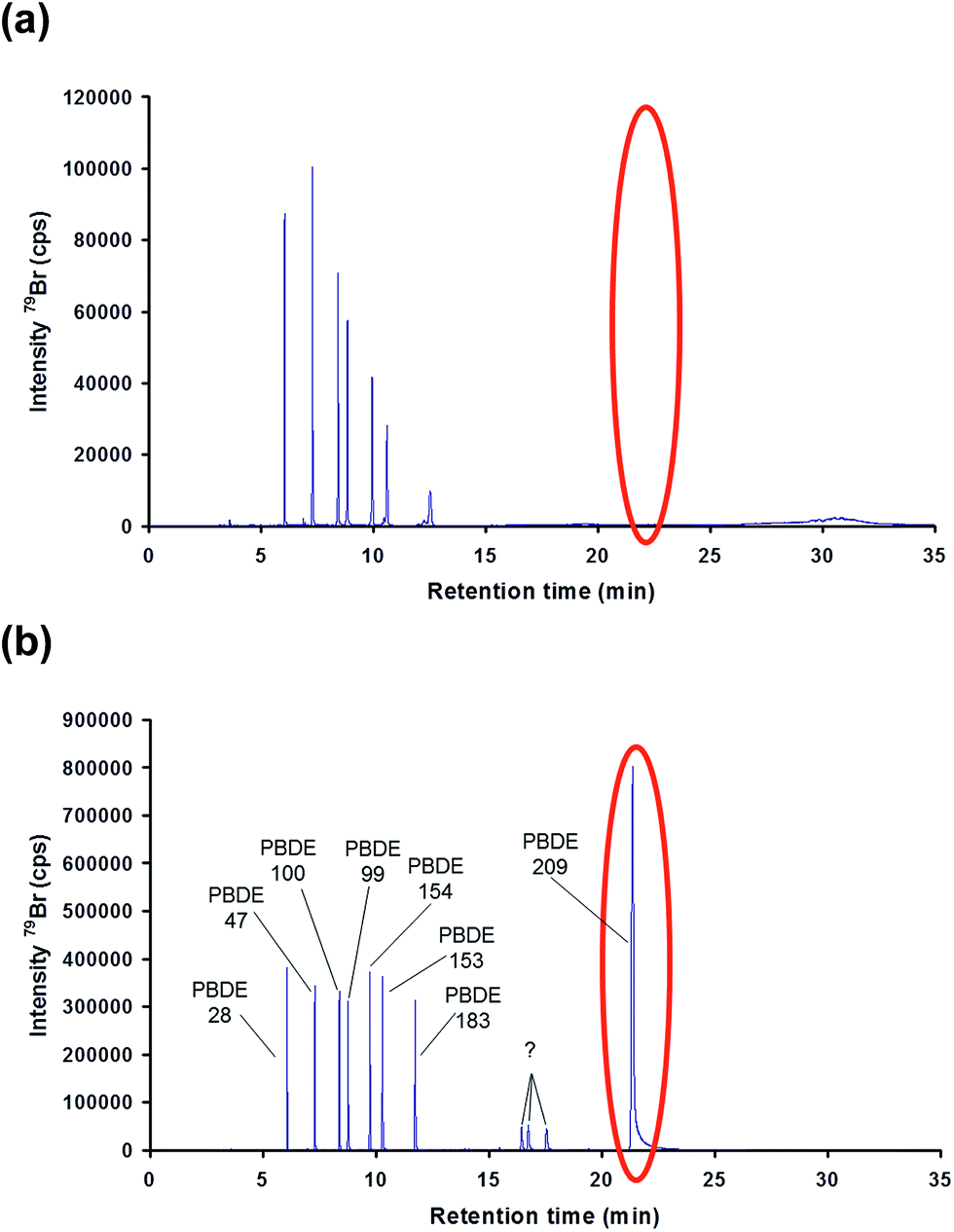
Finally, the proposed methodology was tested for the determination of PBDEs in a river water sample in order to evaluate its performance when dealing with more complex matrices. Three replicate samples and a blank (mineral water) were prepared as described before and injected into the CG-ICP-MS system. The six priority BDE congeners were not detected above the required LOQ (0.15 ng L−1) in the river water samples spiked with 81Br-labelled PBDEs. Fig. 5 shows the chromatograms of (a) a blank sample and (b) a river water sample. In order to assess the analytical performance of the proposed method in a real matrix, the river water sample was also analysed after its fortification with a mixture of native PBDEs. Three replicates and a blank sample were analysed according to the procedure described in the experimental section. The results are summarised in Table 7. Accuracy, expressed in terms of recoveries, was between 84% and 107% for the six priority congeners at concentration levels around 50% of the EQS, which demonstrates the good accuracy and selectivity provided by this method. Reproducibility, expressed in terms of %RSD for three replicate samples prepared separately, was below 8% in all cases. Compared with the results obtained for the fortified mineral water samples, the %RSDs obtained for the fortified river water samples should result in expanded uncertainties below 50% as required by the WFD.
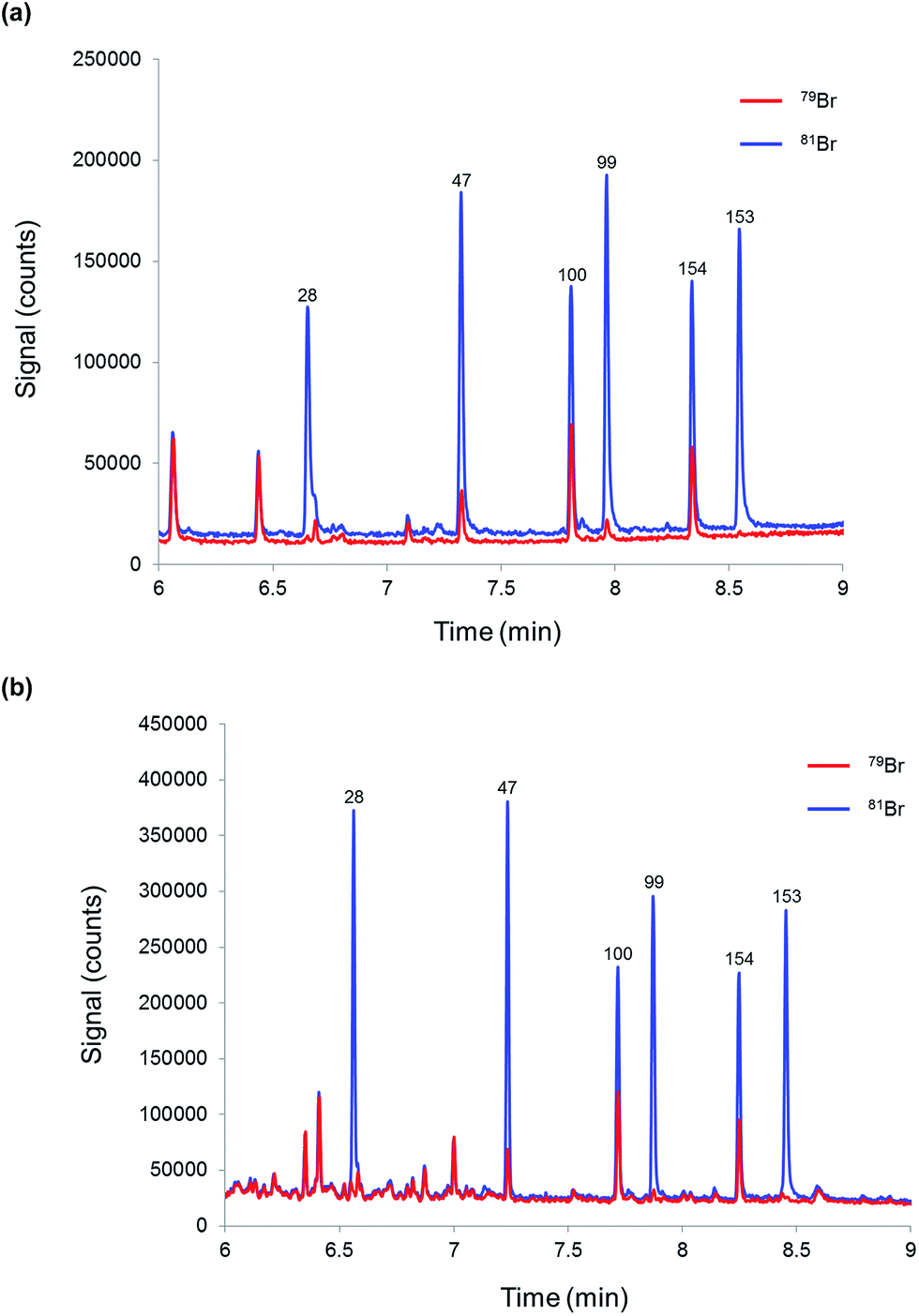 | ||
| Fig. 5 Chromatograms obtained by GC-ICP-MS for (a) mineral water (blank sample) and (b) a river water sample both spiked with 81Br-labelled PBDEs. | ||
| Congener | Concentration (ng L−1) | Recovery (%) | Reproducibility (%RSD) | ||
|---|---|---|---|---|---|
| Mineral water (blank) | River water | Fortified river water | |||
| BDE 28 | 0.0038 ± 0.0008 | 0.0030 ± 0.0003 | 0.234 ± 0.011 | 99.4 | 2.3 |
| BDE 47 | 0.0321 ± 0.0008 | 0.0412 ± 0.0010 | 0.238 ± 0.005 | 103.5 | 4.0 |
| BDE 99 | 0.0103 ± 0.0008 | 0.0143 ± 0.0011 | 0.225 ± 0.003 | 96.1 | 2.3 |
| BDE 100 | 0.0074 ± 0.0019 | 0.0060 ± 0.0017 | 0.249 ± 0.025 | 107.9 | 8.0 |
| BDE 153 | 0.0005 ± 0.0005 | 0.0015 ± 0.0006 | 0.222 ± 0.009 | 84.4 | 3.7 |
| BDE 154 | 0.0025 ± 0.0019 | 0.0041 ± 0.0017 | 0.206 ± 0.013 | 93.3 | 4.1 |
According to the results, the proposed methodology shows great potential for the determination of PBDEs in water samples meeting the requirements of the WFD. In future, this method will be tested for the determination of PBDEs in different water matrices such as coastal or estuarine waters, in order to assess also its robustness.
Conclusion and outlook
The present work clearly indicates the potential of GC-ICP-MS for the ultra-trace analysis of priority heteroelement containing contaminants such as PBDEs. It has been shown that careful instrumental optimization is mandatory to benefit from the dry plasma conditions achieved when using GC as a sample introduction system, which results in the absence of an interfering matrix as well as 100% transport efficiency into the plasma. Instrumental limits of detection, in the low fg range without the need for any pre-concentration step, provide the basis for ultra-trace PBDE analysis in water samples. The species-specific IDMS method used for quantification provides a unique way for the accurate quantification according to the requirements of the WFD.Overall, the combination of GC-ICP-MS and as well as a species-specific isotope dilution based quantification approach has the potential to become a reference method for the ultra-trace determination of PBDEs in natural waters. Future research will also target the application of further GC columns and clean up procedures to reduce the possibility of co-elution of other bromine containing species, which could be present in natural water samples and which might interfere with the accurate quantification of the targeted priority congeners.
Acknowledgements
The research leading to these results has received funding from the European Union on the basis of Decision No 912/2009/EC within the context of an EMRP Research Grant, which was part of the EURAMET JRP ENV08 -Traceable measurements for monitoring critical pollutants under the European Water Framework Directive (WFD- 2000/60/EC).References
- F. Rahman, K. H. Langford, M. D. Scrimshaw and J. N. Lester, Sci. Total Environ., 2001, 275, 1–17, DOI:10.1016/S0048-9697(01)00852-X.
- G. Söderström, U. Sellstrom, C. A. De Wit and M. Tysklind, Environ. Sci. Technol., 2004, 38, 127–132, DOI:10.1021/es034682c.
- H. M. Stapleton, M. Alaee, R. J. Letcher and J. E. Baker, Environ. Sci. Technol., 2004, 38, 112–119, DOI:10.1021/es034746j.
- H. M. Stapleton, R. J. Letcher and J. E. Baker, Environ. Sci. Technol., 2004, 38, 1054–1061, DOI:10.1021/es0348804.
- N. Litz, J. Plant Nutr. Soil Sci., 2002, 165, 692–696 CrossRef CAS.
- R. J. Law, M. Alaee, C. R. Allchin, J. P. Boon, M. Lebeuf, P. Lepom and G. A. Stern, Environ. Int., 2003, 29, 757–770 CrossRef CAS.
- P. Lindberg, U. Sellstrom, L. Haggberg and C. A. de Wit, Environ. Sci. Technol., 2004, 38, 93–96, DOI:10.1021/es034614q.
- A. F. Lagalante and T. D. Oswald, Anal. Bioanal. Chem., 2008, 391, 2249–2256, DOI:10.1007/s00216-008-2156-z.
- A. Covaci, S. Voorspoels and J. de Boer, Environ. Int., 2003, 29, 735–756, DOI:10.1016/S0160-4120(03)00114-4.
- A. Gonzalez-Gago, S. H. Brandsma, P. E. G. Leonards, J. de Boer, J. M. Marchante-Gayon and J. I. G. Alonso, Anal. Bioanal. Chem., 2011, 401, 2639–2649, DOI:10.1007/s00216-011-5323-6.
- N. Barco-Bonilla, P. Plaza-Bolanos, N. M. V. Tarifa, R. Romero-Gonzalez, J. L. M. Vidal and A. G. Frenich, J. Sep. Sci., 2014, 37, 69–76, DOI:10.1002/jssc.201300757.
- R. S. Houk, Anal. Chem., 1986, 58, 97A–105A CrossRef CAS.
- A. P. Vonderheide, M. Montes-Bayon and J. A. Caruso, J. Anal. At. Spectrom., 2002, 17, 1480–1485, 10.1039/b207521k.
- R. F. Swarthout, J. R. Kucklick and W. C. Davis, J. Anal. At. Spectrom., 2008, 23, 1575–1580, 10.1039/b809173k.
- https://www-s.nist.gov/srmors/view_cert.cfm?srm=2257 .
- J. Björklund, P. Tollback, C. Hiarne, E. Dyremark and C. Ostman, J. Chromatogr. A, 2004, 1041, 201–210 CrossRef PubMed.
- J. Meija, M. Montes-Bayon, D. L. Le Duc, N. Terry and J. A. Caruso, Anal. Chem., 2002, 74, 5837–5844 CrossRef CAS.
- P. Leonhard, R. Pepelnik, A. Prange, N. Yamada and T. Yamada, J. Anal. At. Spectrom., 2002, 17, 189–196, 10.1039/b110180n.
- N. Yamada, J. Takahashi and K. Sakata, J. Anal. At. Spectrom., 2002, 17, 1213–1222 RSC.
- D. Pröfrock, P. Leonhard, S. Wilbur and A. Prange, J. Anal. At. Spectrom., 2004, 19, 623–631, 10.1039/b310530j.
- S. F. Durrant, Fresenius' J. Anal. Chem., 1993, 347, 389–392 CrossRef CAS.
- N. N. Sesi, A. Mackenzie, K. E. Shanks, P. Y. Yang and G. M. Hieftje, Spectrochim. Acta, Part B, 1994, 49, 1259–1282 CrossRef.
- E. H. Larsen and M. B. Ludwigsen, J. Anal. At. Spectrom., 1997, 12, 435–439 RSC.
- J. K. Bohlke, J. R. de Laeter, P. De Bievre, H. Hidaka, H. S. Peiser, K. J. R. Rosman and P. D. P. Taylor, J. Phys. Chem. Ref. Data, 2005, 34, 57–67, DOI:10.1063/1.1836764.
- P. Korytar, A. Covaci, J. de Boer, A. Gelbin and U. A. T. Brinkman, J. Chromatogr. A, 2005, 1065, 239–249, DOI:10.1016/j.chroma.2004.12.059.
- E. Eljarrat, A. de la Cal and D. Barcelo, J. Chromatogr. A, 2003, 1008, 181–192, DOI:10.1016/S0021-9673(03)00980-4.
- H. Wei, R. Q. Yang, A. Li, E. R. Christensen and K. J. Rockne, J. Chromatogr. A, 2010, 1217, 2964–2972, DOI:10.1016/j.chroma.2010.02.070.
- J. Bjorklund, P. Tollback, C. Hiarne, E. Dyremark and C. Ostman, J. Chromatogr. A, 2004, 1041, 201–210, DOI:10.1016/j.chroma.2004.04.025.
- P. Rodríguez-González, J. M. Marchante-Gayón, J. I. García Alonso and A. Sanz-Medel, Spectrochim. Acta, Part B, 2005, 60, 151–207 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4ja00112e |
| This journal is © The Royal Society of Chemistry 2015 |