
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAn element through the looking glass: exploring the Au–C, Au–H and Au–O energy landscape
Dragoş-Adrian
Roşca
ab,
Joseph A.
Wright
a and
Manfred
Bochmann
*a
aSchool of Chemistry, University of East Anglia, Norwich, NR4 7TJ, UK. E-mail: m.bochmann@uea.ac.uk; Tel: +44 (0)16035 92044
bMax-Planck-Institut für Kohlenforschung, D-45470 Mülheim/Ruhr, Germany
First published on 13th November 2015
Abstract
Gold, the archetypal “noble metal”, used to be considered of little interest in catalysis. It is now clear that this was a misconception, and a multitude of gold-catalysed transformations has been reported. However, one consequence of the long-held view of gold as inert metal is that its organometallic chemistry contains many “unknowns”, and catalytic cycles devised to explain gold's reactivity draw largely on analogies with other transition metals. How realistic are such mechanistic assumptions? In the last few years a number of key compound classes have been discovered that can provide some answers. This Perspective attempts to summarise these developments, with particular emphasis on recently discovered gold(III) complexes with bonds to hydrogen, oxygen, alkenes and CO ligands.
 Dragoş-Adrian Roşca | Dragoş Roşca obtained his PhD degree from the University of East Anglia in 2014 working on gold pincer complexes under the supervision of Professor Manfred Bochmann. He had earlier graduated with an M.Sc from Babeş-Bolyai University, Romania, on organoselenium chemistry (2009) and worked at the University of Rennes 1 on alkali-metal catalyzed ring opening polymerisations (2009–10, with Dr Y. Sarazin and Prof. J.-F. Carpentier). He is now a postdoctoral research associate in the group of Prof. Alois Fürstner at the MPI für Kohlenforschung in Mülheim/Ruhr. |
 Joseph A. Wright | Joseph Wright obtained his PhD degree from the University of Cambridge in 2003 working on the mechanism of the Wacker reaction under the supervision of Dr Jonathan Spencer. He then undertook postdoctoral positions at the University of Southampton (2003–2004 in the group of Dr Andreas Danopoulos) and at UEA (2005–2008 with Professor Manfred Bochmann and 2008–2012 with Professor Christopher Pickett). In 2012 he was appointed as Lecture in Energy Materials at the University of East Anglia, and has interests in organometallic chemistry, reaction mechanism and DFT modelling. |
 Manfred Bochmann | Manfred Bochmann received his Diploma degree from the University of Marburg in 1977, followed by a PhD at Imperial College London in 1979 with Professor Sir Geoffrey Wilkinson. He worked in ICI central research 1980–83 before moving to a lectureship in inorganic chemistry at the University of East Anglia. He was promoted to a personal chair in 1994. From 1995–2000 he worked at the University of Leeds as Head of Inorganic and Structural Chemistry. On his return to UEA he served as Head of the School of Chemical Sciences and Pharmacy 2006–9 and as Head of the School of Chemistry until 2011. He was awarded the RSC Medal for Organometallic Chemistry in 2003, a Leverhulme Senior Research fellowship in 2011 and an ERC Advanced Grant in 2014. He has published over 280 research articles, several books and numerous patents, mainly on Group 4 metallocene-based olefin polymerisation catalysts. In the last few years his research has concentrated on the organometallic chemistry of gold. |
Introduction
Gold catalysts, both homogeneous and heterogeneous, have seen a meteoric rise in importance in the last three decades, and interest continues unabated.1 This has been amply documented, for example, in special editions of major review journals.2,3 For organic transformations, gold in the oxidation state +I is overwhelmingly used as the catalyst, as documented in recent reviews concerning synthetic applications.4–8 Important aspects in homogeneous gold(I) catalysis include, inter alia, the role of oxidation states, silver activators, ligand structure and the role of couteranions,4 the formation of heterocycles,9,10 the nature of gold carbene intermediates,11 the influence of aurophilic interactions12 on Au(I) catalysis,13 silver-free gold catalysts,14 as well as the important role of counterions and their effect on chemo- and regioselectivity15 and of chiral Brønsted acids16 in gold catalysis. Gold(III) catalysts have also found widespread applications in synthetic methodology, most commonly in the form of simple gold salts such as AuCl3 as catalysts or catalyst precursors.17,18 While such catalysts can be fast and efficient, as Schmidbaur pointed out in a recent review,18 the nature and oxidation state of the catalyst is in most cases difficult to determine and remains a matter of conjecture.Gold occupies a unique position in the Periodic Table. It is the most electronegative metallic element, with a Pauling electronegativity value close to that of carbon and hydrogen (Au: 2.54; C: 2.55; H: 2.20). As gold(I), with d10 configuration, it is akin to a main group element, while as gold(III) it behaves as a transition metal and displays a coordination chemistry which mirrors that of other heavy-metal d8 ions. The 3rd row elements platinum, gold and mercury are strongly affected by relativistic effects,19 which cause an expansion of the 5d orbitals and a contraction of the 6s shell and reach their maximum for gold. There are certainly many chemical and structural similarities between the d8 systems platinum(II) and gold(III), which have led to platinum and gold catalysts often being compared side-by-side.20,21 Numerous reaction mechanisms and intermediates proposed for gold catalysed reactions have been drawn up in analogy to organometallic palladium or platinum catalysts.
However, there are also significant differences between those two neighbours in the Periodic Table. Most prominent are the redox potentials, which are very much more positive for gold than for platinum (values under standard conditions, 1 M aqueous acid, 25 °C):
| Au3+/Au0 1.52 V |
| Au3+/Au+ 1.36 V |
| Au+/Au0 1.83 V |
| Pt2+/Pt0 1.19 V |
Gold(III) is therefore very much more readily reduced than platinum(II), to either Au(I) or Au(0), and the formation of purple gold nanoparticles during the course of unsuccessful reactions is only too familiar to practitioners of gold chemistry. The redox chemistry of gold is illustrated, for example, by the ability of “naked” solvated Au+ ions to oxidatively cleave benzylic C–H bonds of arenes with an oxidation potential of up to 1.82 V, and even the C–C bonds of diarylmethanes, to give carbocation intermediates, with metallic gold being generated in the process.22 Oxidation catalysts based on mixtures of AuCl and the diketiminate anion [HC(MeC![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) NC6H3Pri2)2]− have been reported,23 the assumption being that under such conditions Au(I) diketiminato chelate complexes are generated in situ as catalytically active species.24 However, it was shown subsequently that even at 0 °C Au(I) and [HC(MeCNAr)2]− anions lead to the immediate reduction to metallic gold,25 which would seem to suggest that the observed oxidation catalysis is in all probability effected by gold nanoparticles. In other organic transformations, reduction of Au(III) vinyl intermediates to Au(I) could be demonstrated, while the formation of gold nanoparticles could be excluded by X-ray absorption spectroscopy (XAS) methods.26 On the other hand, there are also notable cases where Au(III) vinyl reaction intermediates of the type [(vinyl)AlCl3]− and [(vinyl)2AuCl2]−, could be isolated and structurally characterised.27
NC6H3Pri2)2]− have been reported,23 the assumption being that under such conditions Au(I) diketiminato chelate complexes are generated in situ as catalytically active species.24 However, it was shown subsequently that even at 0 °C Au(I) and [HC(MeCNAr)2]− anions lead to the immediate reduction to metallic gold,25 which would seem to suggest that the observed oxidation catalysis is in all probability effected by gold nanoparticles. In other organic transformations, reduction of Au(III) vinyl intermediates to Au(I) could be demonstrated, while the formation of gold nanoparticles could be excluded by X-ray absorption spectroscopy (XAS) methods.26 On the other hand, there are also notable cases where Au(III) vinyl reaction intermediates of the type [(vinyl)AlCl3]− and [(vinyl)2AuCl2]−, could be isolated and structurally characterised.27
The particular bonding and redox characteristics of gold are of course reflected in the reactivity of the types of intermediates that, by analogy to other metal-catalysed reactions, are assumed to be involved in catalysis: complexes with alkenes and alkynes, CO, metal hydrides and alkyls are all quite reasonable proposals, but, in the case of gold, how much is really known about such compounds? For which ligands or ligand combinations, and for which gold oxidation states, is it realistic to assume the existence, or indeed non-existence, of such species?
Gold, as the embodiment of a noble metal, had long been regarded as unreactive and chemically uninteresting; it is therefore a late-comer to catalysis. This means that many aspects of fundamental organometallic chemistry, which after all forms the basis of our understanding of catalytic mechanisms, remained unexplored for gold. The most dramatic example of this are olefin complexes: whereas platinum(II) provided the first alkene complex of any transition metal, Zeise's salt K[PtCl3(η2-C2H4)], reported in 1827,28,29 ethylene complexes of the isoelectronic Au(III) ion are conspicuous by their absence (Scheme 1). Similarly, hydride complexes of platinum(II) have been known for over 50 years,30 whereas those of Au(III) remained unknown.
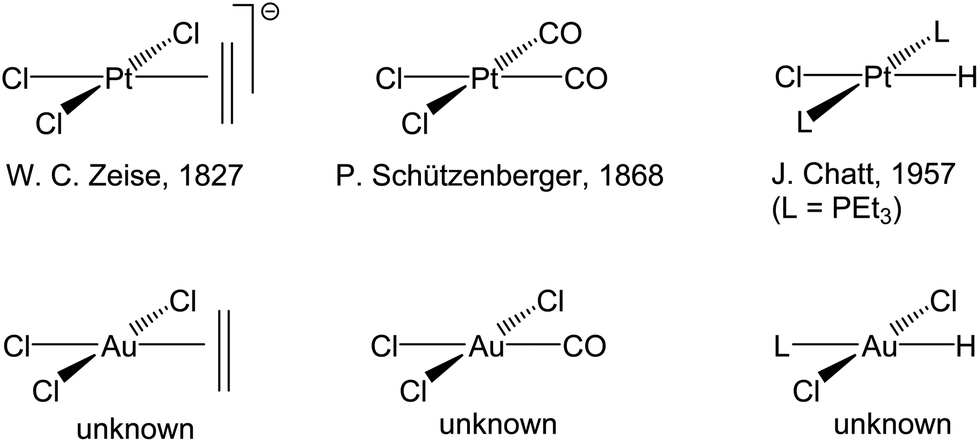 | ||
| Scheme 1 Classical platinum(II) complex types and their hypothetical gold(III) analogues. | ||
This brief Perspective presents some recent results on the identification and characteristics of some key types of catalysis-relevant gold complexes and tries to highlight the similarities and differences in bonding and reactivity of species with Au–C, Au–H and Au–O bonds. As will be seen, in some cases these gold complexes display a reactivity pattern that is rather different from that of most other transition metal compounds.
The Au–H bond
The chemistry of gold hydrides and compounds with hydrogen bonds to gold have recently been the subject of a comprehensive review.31 Early attempts to generate gold hydride complexes and to stabilise gold(III) with complex anions like BH4− and AlH4− at −120 °C were reported by Wiberg; the result in all cases was reduction to metallic gold.32 The formation of gold hydrides and their stability is of course particularly interesting in connection with catalytic cycles since H-transfer, β-H elimination or M–C bond hydrogenolysis are common reaction steps in transition metal catalysis. The first examples of isolable gold hydrido species were binuclear metal complexes (CO)5M(μ-H)AuPPh3 (M = Cr, Mo, W) and LAu(μ-H)MLn [MLn = IrH2(PPh3)3 or Pt(C6Cl5)(PEt3)2];33,34 all these bimetallic compounds show strongly shielded hydride NMR resonances between δ −2 and −6 ppm (in contrast to terminal AuI–H, vide infra). Hydrides of gold in oxidation states I, II and III could be identified by vibrational spectroscopy in the reactions of laser-ablated gold atoms condensed into frozen gas matrices at 5 K or below, including (H2)AuH, (H2)AuH3, AuH2, AuH4− and AuH2−.35 Evidence for more thermally stable hydride complexes was provided by mass spectrometry, to give ions of type 1,36 and by NMR studies of the reactions of (P^P)AuCl with PhMe2SiH, which gave binuclear H-bridged compounds 2 and 3 (Scheme 2); such hydrides are thought to be involved in gold-catalysed alcohol dehydrosilylation. The 1H NMR hydride signal of 2 was found at δ 8.29 (JPH 49.5 Hz), and while the 1H signal of 3 could not be detected, the 2H resonance occurs at δ 7.0 ppm (JPD 8.4 Hz).37,38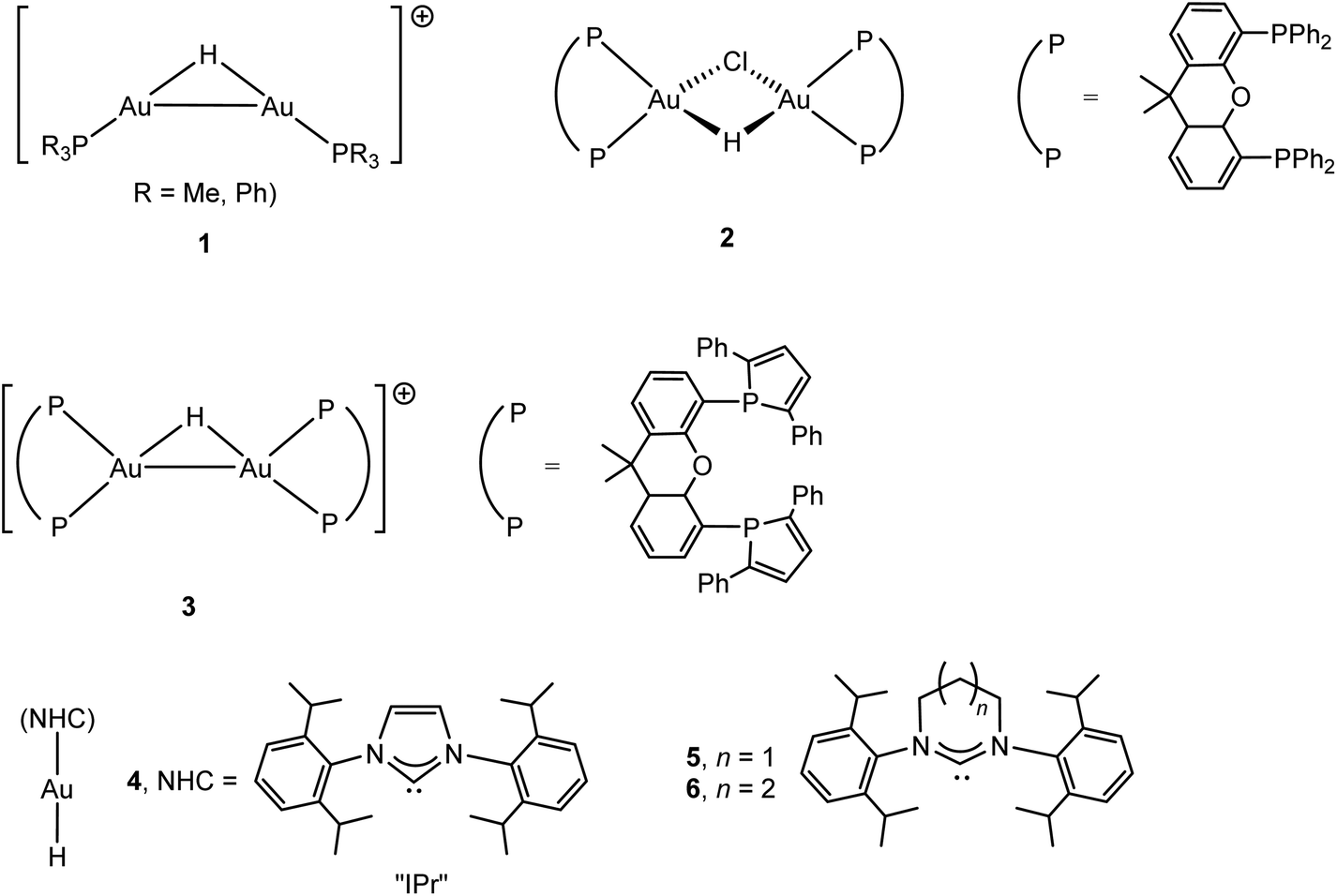 | ||
| Scheme 2 Types of gold(I) hydride complexes. | ||
The first example of a mononuclear gold(I) hydride was isolated by Sadighi in 2008, (IPr)AuH (4), either by the reaction of (IPr)AuCl with LiHBEt3, or by treating (IPr)AuOBut with HSi(OMe)3 (IPr = 1,3-bis(2,6-diisopropylphenyl)imidazole-2-ylidene). The hydride ligand gives rise to a signal with positive chemical shift, at δ 5.11 in C6D6 (δ 3.38 in CD2Cl2), with an Au–H IR stretching frequency of 1976 cm−1 (νAu-D 1407 cm−1).39 The same compound was also made from (IPr)AuOH.40 This family of gold hydrides stabilised by N-heterocyclic carbene (NHC) ligands was recently increased by ring-expanded carbenes to give 5 and 6 (δ1H 3.57 and 3.15, respectively, in C6D6) (Scheme 2). Protonation of 5 or 6 with [H(OEt2][B{C6H3(CF3)2}4] at −80 °C gave μ-H cations of type 1, indicated by a high-field shift of the hydride 1H NMR signal by 4–5 ppm; the suspected intermediate gold dihydrogen complexes [(NHC)Au(H2)]+ could not be detected.41
Given the very similar electronegativities of gold and hydrogen, the Au–H bond can be expected to show little “hydridic” character. And indeed, solid 4 is stable to air and moisture at ambient conditions, does not react with protic solvents like t-butanol, and reacts only very slowly with benzoic acid. It also does not react with 3-hexyne or diphenylacetylene. On the other hand, dimethyl acetylenedicarboxylate gives a trans-insertion product, and ethyldiazoacetate reacts under C–H activation (Scheme 3). The hydride ligand in 4 can however be abstracted by CPh3+ to give a binuclear hydride-bridged cation of type 1, indicated by the high-field shift of the 1H NMR signal to δ 0.42.39
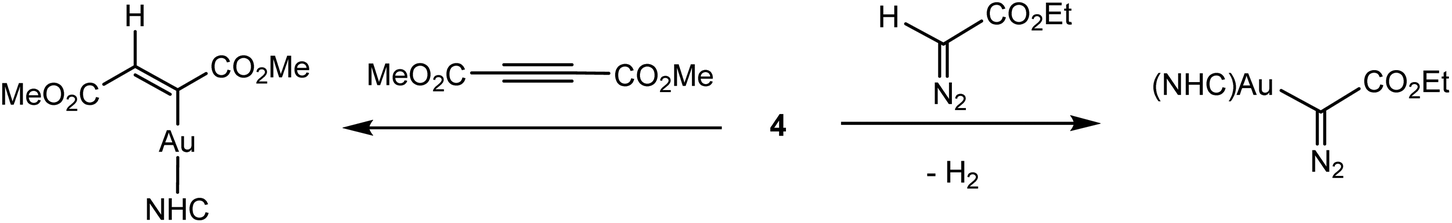 | ||
| Scheme 3 Reactions of (IPr)AuH. | ||
The mesityl-substituted analogue of 4, (IMes)AuH, shows low reactivity in the hydrodefluorination of perfluoroarenes, but the reaction is accelerated by p-dimethylaminopyridine (DMAP) and involves a spectroscopically detectable π-stacked intermediate (Scheme 4).42 Under 1–9 bar O2, complex 4 undergoes slow dioxygen insertion into the Au–H bond to give the hydroperoxide 7. The reaction is 1st order in both [O2] and [Au]. Product 7 readily decomposes to (IPr)AuOH (8), followed by condensation of 7 with 8 to give the structurally characterised peroxide 9 (Scheme 4).43
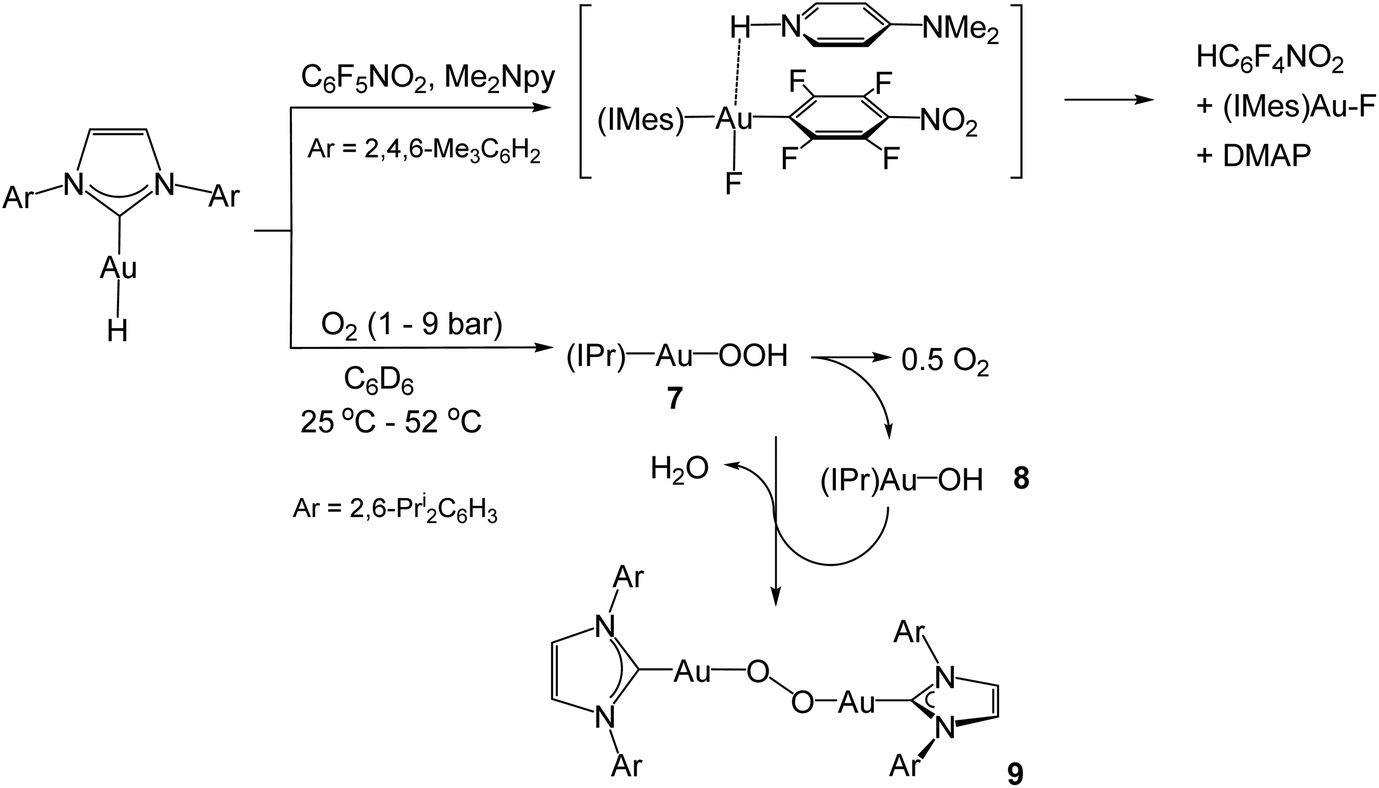 | ||
| Scheme 4 | ||
The reaction of 4 with stable radicals was used to estimate the Au–H bond energy. Whereas 4 does not react with TEMPO (= 2,2,6,6-tetramethyl-1-piperidyl-N-oxide), the hydride ligand is abstracted by galvinoxyl, with formation of the crystallographically characterised galvinoxide salt 10 (Scheme 5). Since the O–H bond dissociation energies (BDEs) of TEMPOH and galvinoxyl-H are 291 kJ mol−1 and 329 kJ mol−1, respectively, this provides an estimate of the Au–H bond energy between these two values.43
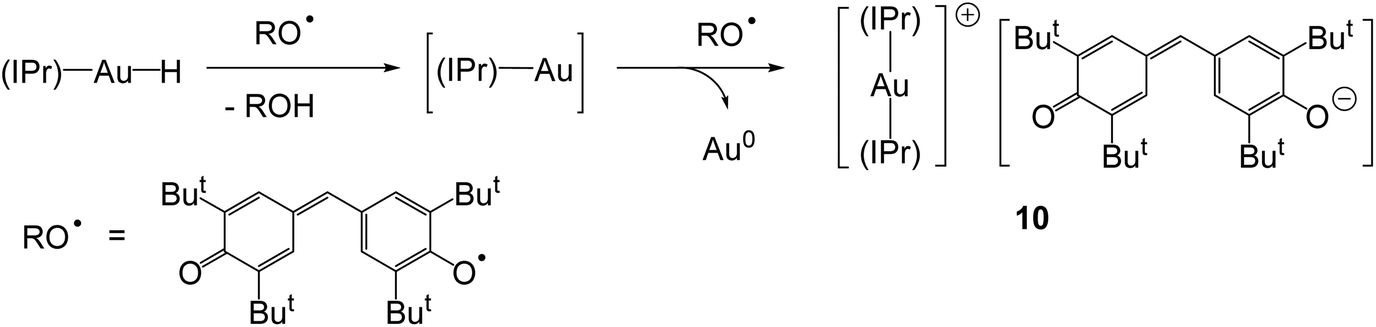 | ||
| Scheme 5 | ||
Gold(I) hydrides could, in principle, also be formed by β-H elimination of gold(I) alkyls, and conversely, the hydrogenation of unsaturated substrates is usually thought of as involving the β-insertion of a coordinated alkene or alkyne into a metal–H bond. Gold(I) is of course a d10 system, without suitable energetically accessible vacant orbitals for interacting with a β-C–H bond, nor for the coordination of an olefinic substrate. It is therefore consistent that for (IPr)AuEt a high barrier for β-H elimination was found, such that this pathway becomes kinetically competitive only above the decomposition temperature of the complex (>200 °C).44 As Bertrand has shown, both β-H and, much more slowly, α-H abstractions from gold alkyls are possible, but only by using an external electrophile such as CPh3+, not however by intramolecular H-transfer reactions.45 In line with this, calculations have suggested that for alkene hydrogenations with homogeneous gold(I) catalysts in polar solvents like ethanol, a heterolytic, solvent-assisted H2 activation pathway prevails over a σ-bond metathesis-type Au–C bond cleavage by an H2 molecule (Scheme 6). Alkene hydrogenation is thought to follow an ionic mechanism, in which a proton is delivered from the solvent to the alkene. The rate of alkene hydrogenation increases therefore as the stability of the resulting carbocation increases, e.g. cyclohexene > ethene. The hydride is delivered from the gold species. Whatever the detailed energetics of these solvent-assisted mechanisms, it is clear that for gold catalysts the ionic reaction profiles are always energetically favoured over the homolytic pathway.46
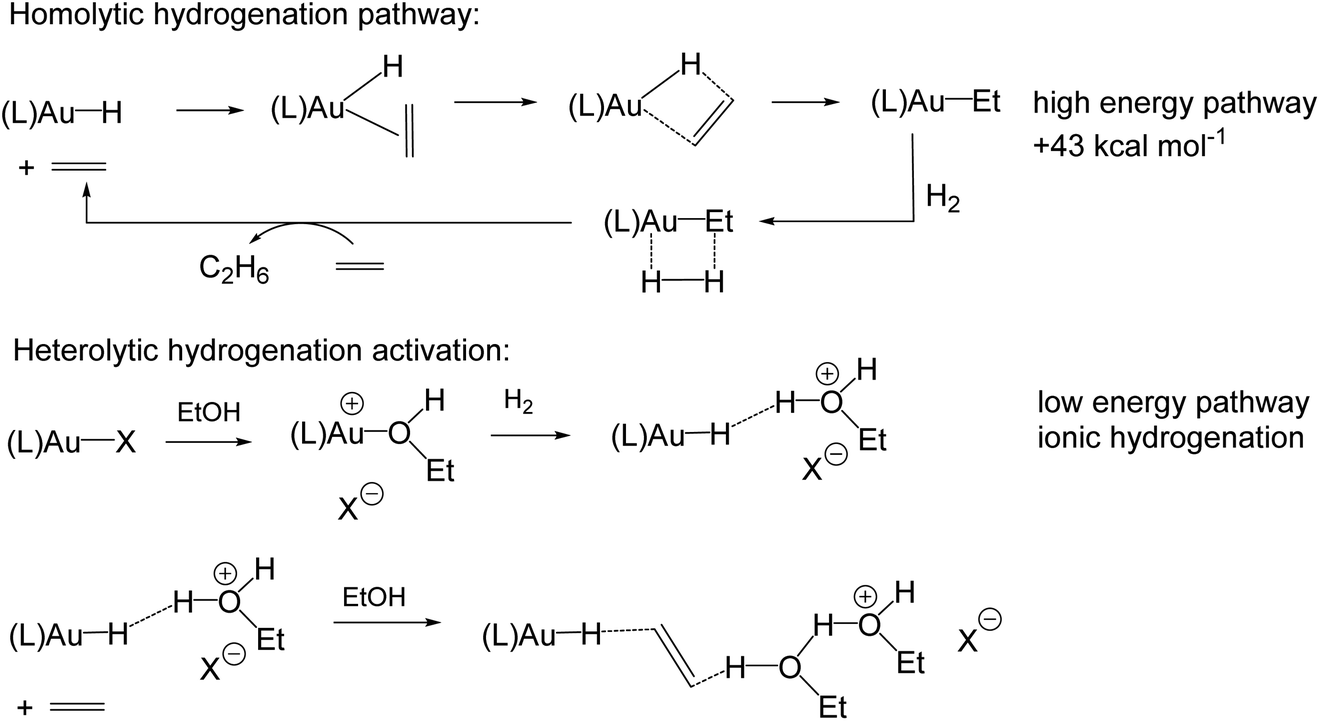 | ||
| Scheme 6 Computational modelling of Au(I) mediated hydrogenation steps (L = IPr). | ||
In addition to gold(I) catalysts, gold(III) catalyst precursors are also frequently employed in organic transformations. However, in the large majority of cases the fate of the gold species, the oxidation state and the coordination environment of the resulting gold species under such conditions are a matter of conjecture. The facile reduction of Au(III) to Au(I) suggests that the active species formed in situ are most probably gold(I), particularly if catalysts with labile ligands are used, such as AuCl3.18,47,48 The inadvertent formation of catalytically active gold nanoparticles is also not easy to rule out. Hydride complexes such as HAuCl2 are occasionally invoked in catalytic cycles,49 but as yet no such species has actually been identified. Schiff-base gold(III) complexes, supported and unsupported, proved to be potent catalysts for the hydrogenation of diethyl itaconate, comparable in activity to palladium catalysts. The process of hydrogen activation (heterolytic, ethanol-assisted) and the likely intermediacy of Au(III) hydride species was explored by computational methods. Square-planar Au(III) hydride and hydrido-ethylene complexes are energetically favoured; the alternative formation of a 5-coordinate [(O^N^N)AuH(C2H4)]+ species was disfavoured by an increased energy barrier (Scheme 7).50 This confirms the general picture that, although Au(III) complexes with coordination numbers 5 and 6 are not unknown,51 the square planar geometry is strongly preferred. There was, however, no experimental evidence that the postulated species could actually be detected.
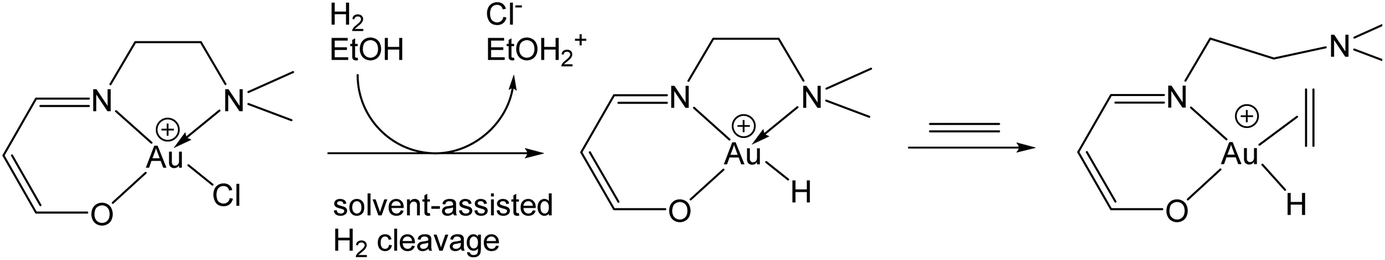 | ||
| Scheme 7 Computational models for proposed intermediates in alkene hydrogenation catalysed by Au(III) Schiff base complexes.50 | ||
Polydentate ligands offer the possibility of stabilising complexes kinetically and may also shift the redox potentials sufficiently to allow isolation and characterisation of such elusive hydride species. Cyclometallated mono- and diarylpyridine ligands provide particular stability; e.g. 2,6-diphenylpyridine derivatives form C^N^C bonded pincer complexes with Au(III). An example is the gold hydroxide 11 which has proved to be a highly versatile starting material; for example, it undergoes aryl transfer reactions with boronic acids in toluene under neutral conditions and C–H activates fluoroarenes, to give photoluminescent gold aryl complexes in essentially quantitative yields.52 Surprisingly, the reaction of 11 with “superhydride”, LiHBEt3, at −78 °C followed by stirring at room temperature proceeds without reduction of the metal centre and gives the gold hydride 12 as a yellow crystalline complex, the first example of an isolable Au(III) hydrido complex (Scheme 8).53
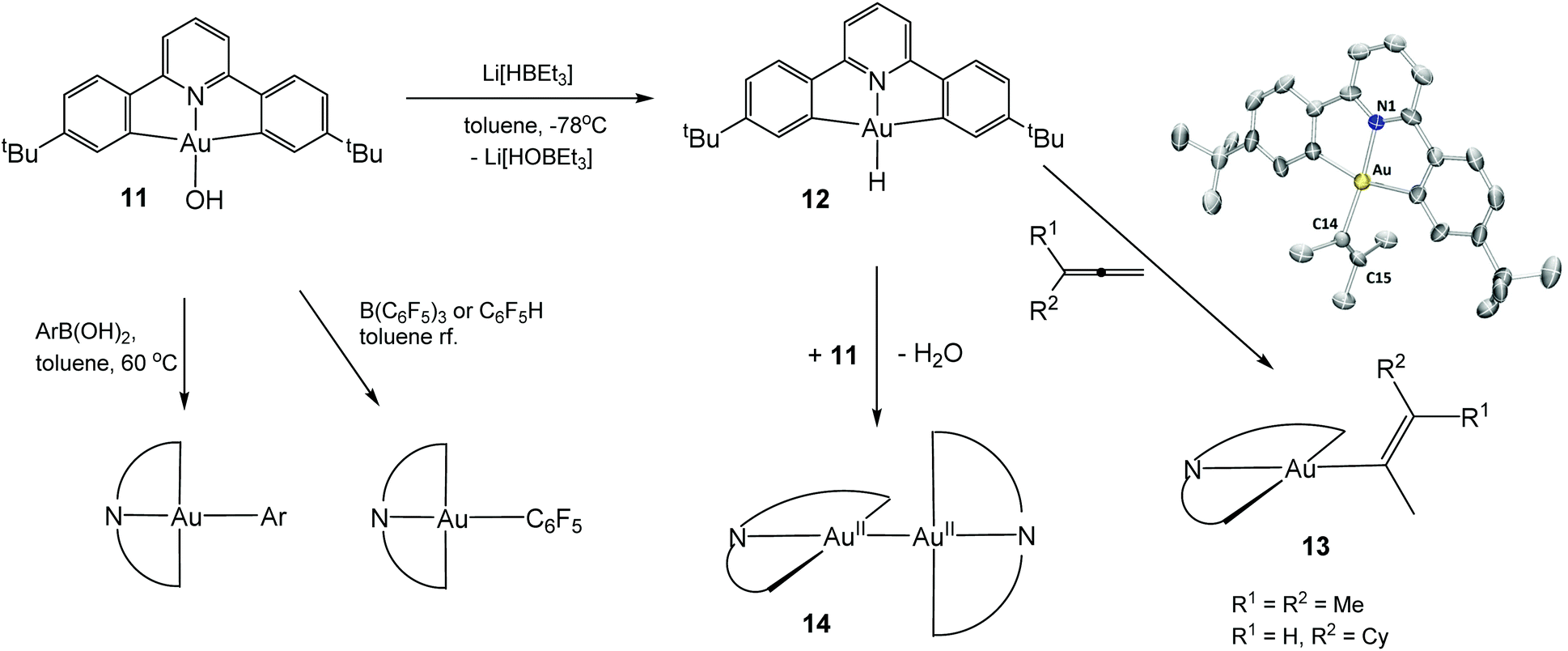 | ||
| Scheme 8 Formation and reactivity of (C^N^C)AuIIIH. | ||
The complex was crystallographically characterized and the gold-bound H atom was located in the electron density map. The infrared spectrum of the compound shows a sharp νAu–H band at 2188 cm−1. This is higher than the value for (IPr)AuH (1976 cm−1) but comparable to the frequencies assigned to AuH and (H2)AuH in an argon matrix (2226.6 and 2173.6 cm−1, respectively). In the 1H NMR spectrum the hydride ligand gives rise to a broad singlet at δ −6.51 in CD2Cl2 and at δ −5.73 in C6D6. The deuteride shows a 2H NMR resonance at δ −6.58 (CD2Cl2).53
The observed Au–H 1H NMR chemical shift for 12 is in line with theoretical predictions (taking relativistic effects into account), which show that systems with fully filled d-shell are dominated by deshielding spin–orbit coupling and signals are therefore found at low field, (e.g. the Au(I) complexes 4–6), whereas for d8 systems the spin–orbit coupling is strongly shielding, resulting in hydride resonances high-field of TMS,54 as is indeed observed. The chemical shift therefore does not allow any conclusions to be drawn concerning the polarity or “hydridic” character of the Au–H bond.
The Au(III) hydride 12 is stable to air and moisture at ambient conditions, an indication of the highly covalent character of the Au–H bond. The compound was found to be moderately light sensitive, and exposing CH2Cl2 solutions to sunlight for a few hours leads to chloride abstraction from the solvent, with formation of (C^N^C)AuCl. Complex 12 does not reduce CO2 or benzaldehyde in the dark or under photolytic conditions, and attempted insertion reactions with ethylene, 3-hexyne, phenylacetylene, and even dimethyl acetylenedicarboxylate were unsuccessful. However, it does react with allenes under 1,2-insertion to give isolable Au(III) vinyl complexes.
Hydride 12 is stable to acetic acid. However, treating dichloromethane or toluene solutions with the strong Brønsted acid [H(OEt2)2][H2N{B(C6F5)3}2]55 at −70 °C led to the disappearance of the hydride resonance, together with signals that resembled the product of cleavage of one of the Au–phenyl bonds, [as for example in (HC–N^C)Au(C6H4F)(O2CCF3)56]. Resonances that could be assigned to H2 were not observed. Similarly, upon reacting the deuteride 12-D with [H(OEt2)2][H2N{B(C6F5)3}2] or trifluoroacetic acid at −70 °C gave no evidence of the characteristic triplet of HD. The major product on warming to room temperature was the free 2,6-diphenylpyridine ligand.57 It is evident that H+ attack takes place on the Au–C(phenyl) bond in preference to the hydride, in agreement with the covalent nature of the Au–H bond in 12.
A more unusual reaction of 12 occurs if excess (C^N^C)AuOH is present: water is eliminated in a slow reductive condensation reaction, to give the gold–gold bonded dimeric Au(II) complex 14, a rare example of a thermally stable Au(II) complex with an unsupported Au–Au bond. For comparison, the recently reported dimer [AuII(CF3)2(py)]2 follows the thermodynamic trend and spontaneously disproportionates in solution to give Au(I) and Au(III) products.58 By contrast, 14 can be heated to 120 °C without decomposition, is stable to air and moisture in the solid state, and in some reactions turns out to be the unreactive end product. On UV irradiation it shows green photoluminescence. No reaction was observed between 14 and elemental sulfur in toluene (60 °C, 12 h) or with O2 under 9 bar pressure, even though calculations suggest that O2 insertion into the gold–gold bond is exothermic by 136 kJ mol−1.59 The lack of reactivity is most probably a reflection of the steric shielding provided by the four t-butyl substituents that envelope the Au–Au bond. On the other hand, iodine cleaves the Au–Au bond cleanly to give (C^N^C)AuI.
The reduction of the hydride 12 to the Au(II) dimer 14 was studied by cyclic voltammetry. 12 shows three reduction waves with peak potentials at −2.00 V (corresponding to a metal-centred Au(III → II) reduction), −2.25 V and −2.60 V vs. Fc+/0, respectively. The −2.25 V wave was tentatively assigned to the reduction of a hydride-bridged species, such as [(C^N^C)AuII–H–AuIII(C^N^C)], formed from the reaction of 12 with a (C^N^C)AuII radical (Scheme 9). The reduction wave at −2.60 V corresponds to ligand based multi-electron processes. The reduction process of 12 differs from that of the hydroxide 11, where no such binuclear intermediate could be identified, which suggested that under these conditions the Au–O bond is more readily cleaved than the Au–H bond.59
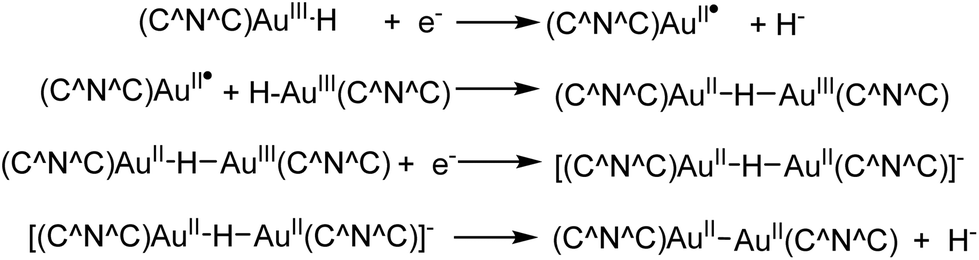 | ||
| Scheme 9 Electrochemical processes in the reduction of the gold(III) hydride 12 to the Au(II) dimer 14.59 | ||
The results also provided an estimate of the bond enthalpy difference between Au–OH and Au–H bonds in 11 and 12 (neglecting differences in solvation between hydride and hydroxide), which showed that the AuIII–H bond is more stable than Au–OH by ca. 19 kJ mol−1. This agrees qualitatively with the bond strength trend determined by DFT calculations.60 This energy difference has a number of consequences and makes gold chemistry distinctive from other metals (vide infra).
The oxidation of the dimer 14 (+0.59 V vs. Fc0/+) was found to be irreversible at all voltage scan rates. This seems to suggest that the 1-electron oxidation product of the Au(II) dimer is unstable and likely undergoes rapid follow–up chemistry. There was no evidence for detectable amounts of a mixed-valence radical cation intermediate [(C^N^C)AuII–AuIII(C^N^C)]+ in the oxidation process.
Electrochemistry also allowed the determination of the AuII–AuII bond energy of 14. Neglecting entropic contributions, the AuII–AuII bond enthalpy was estimated as 198 ± 1 kJ mol−1, in reasonably good agreement with DFT calculations (225 kJ mol−1).59 For comparison, Pyykkö described the AuII–AuII bond as a 6s6pz5dxy hybrid and calculated a bond energy of 255 kJ mol−1 for [AuII(CF3)2(py)]2.61 This suggests that the stability differences between this complex and 14 is due to kinetic rather than thermodynamic factors.
The Au–O bond
Heterogeneous gold catalysts are involved in a number of oxidation reactions, and the reaction pathways and the surface species that are likely to be involved have been the subject of numerous experimental and computational studies. Selective oxidations and the mode of O2 activation on oxide-supported gold catalysts for CO oxidation have been reviewed.62,63 A number of studies are concerned with oxygen activation and the role of Au(μ-O)Au species in CO oxidation and at the Au/oxide support interface.64 Models including Au–O–Au and Au–O–O–Au moieties have been proposed as key species in O2 activation of supported gold catalyst supported on a variety of metal oxides.65 Gold hydroperoxides are likely involved in the catalytic synthesis of H2O2,66 as gold surface species67 and in catalytic methanol oxidation.68 Evidence has also been provided for peroxide and superoxide adsorbed on anionic69 and cationic70 gold clusters, and a variety of species including hydrides, hydroxides, peroxides and hydroperoxides are thought to be involved in propene oxidation.71 With few exceptions these proposed mechanisms and surface intermediates are based on DFT modelling. Key to rationalising all these processes is a detailed understanding of the gold–oxygen bond, including possible π-bonding contributions and preferred coordination geometries.The nature of the metal–oxide bond has been the subject of considerable debate, notably the possibility (or impossibility) of forming terminal oxo species LnMO with metals in groups 9–11 (the “oxo wall”). Indeed, the only structurally confirmed example of a noble metal oxo complex is Wilkinson's OIr(mes)3 (15, d4) (Scheme 10),72 and there is indirect evidence for PtIVO (d6).73 Earlier claims of an octahedral Au(III) (d8) oxo species with an AuO double bond proved to be based on error.74
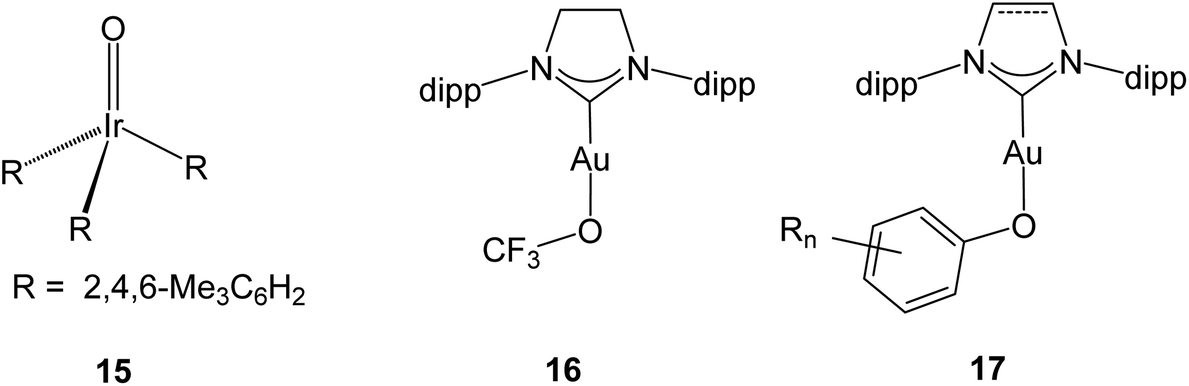 | ||
| Scheme 10 Examples of metal oxo and gold(I) alkoxide complexes (dipp = 2,6-Pri2C6H3). | ||
The gold–oxygen bond in Au(I) complexes has been the subject of some detailed theoretical investigations, e.g. (NHC)Au–OCF3 (16) which, with an Au–O–C angle of 118.3(4)°, shows little evidence of π-overlap.75 For (NHC)Au–O–aryl complexes of type 17 it has been argued that there is interaction between the phenolate lone electron pairs and the Au–C antibonding orbital. The Au–O–C(aryl) angles are indeed much wider than expected for sp3 hybridisation [in the range of 121.2–126(3)°], but significant steric repulsion between the bulky NHC and aryloxide ligands must be taken into account.76
In contrast to gold(I), which prefers coordination to “soft” donors like phosphorus and sulfur, gold(III) has long been known as a “hard”, Lewis acidic metal ion. This is probably best exemplified by the square-planar tetrahydroxoaurate anion, [Au(OH)4]− (18) which forms numerous salts,77 as well as by the classical work of Tobias concerning the identification of dimethylgold aquo ions [Me2Au(OH2)2]+, Na[Me2Au(OH)2] and dimethylgold hydroxide 19 (Scheme 11). The latter is a dimer in solution but forms a tetrameric structure in the crystal, based on square-planar Me2Au(μ-OH)2 building blocks.78 Vicente showed that gold complexes with aryl ligands form similar compounds with Au–O bonds, including phenolate (20) and hydroxide (21) derivatives.79 The structure of the hydroxide 22 has also been determined.80
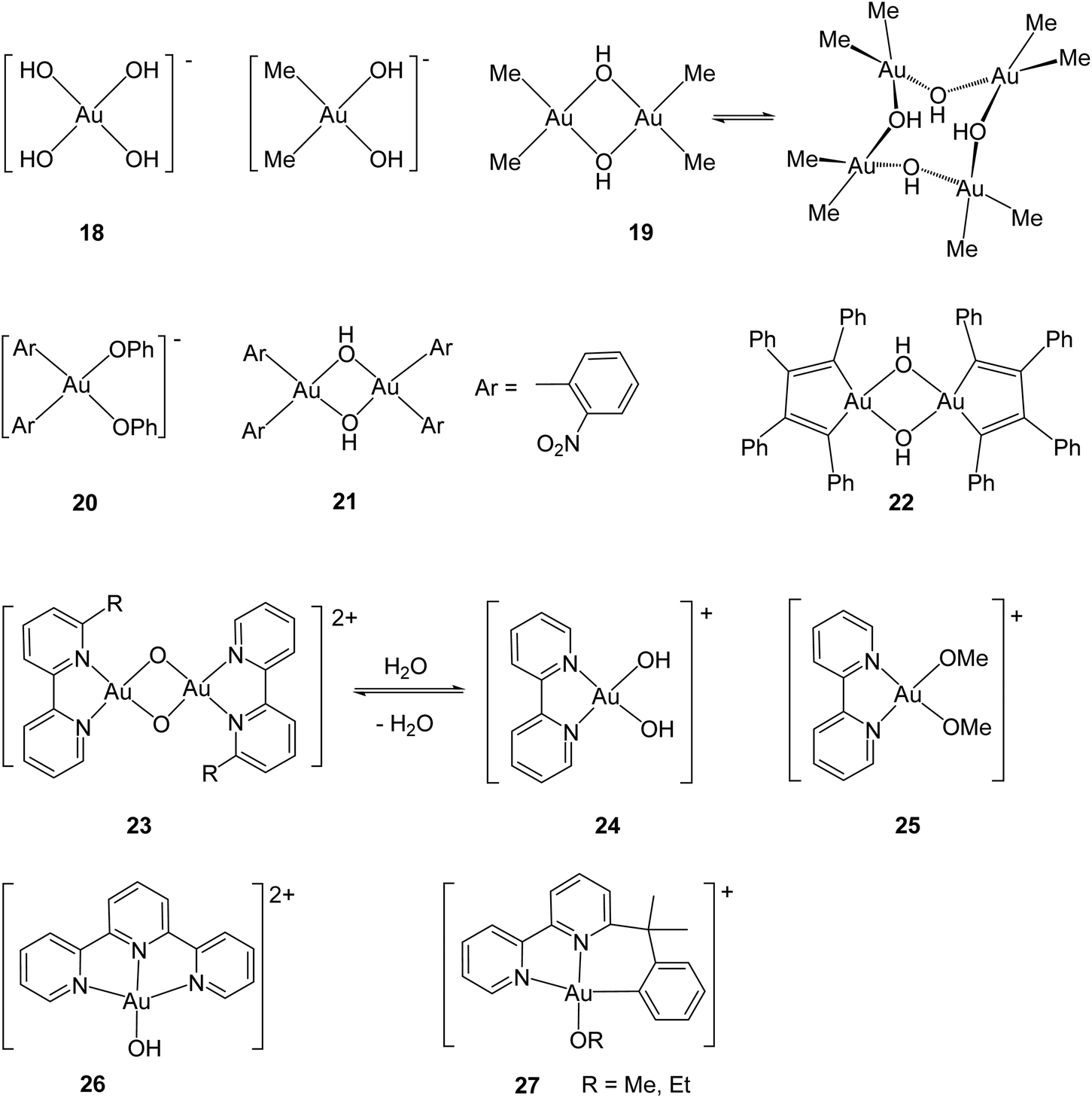 | ||
| Scheme 11 Classical examples of gold(III) oxo, hydroxo and alkoxo complexes. | ||
Like other Lewis acids, gold(III) readily forms adducts with pyridine and related N-donors, such as 2,2′-bipyridyl and terpyridyl. The formation of cyclometallated ortho-arylpyridine derivatives has proved a particularly successful strategy for the preparation of gold(III) complexes with oxo and hydroxo ligands. Scheme 11 shows a number of representative examples. While terminal AuO complexes are unknown, gold(III) compounds with bridging oxo ligands are well established. In 23, for example, the square-planar metal centres form part of a planar Au2O2 core, with Au–O single-bond distances of 1.976(3) and 1.961(3) Å (for R = CH2But).81 The oxide 23 is in equilibrium with the hydroxo complex 24.82 Analogous alkoxides have also been prepared and structurally characterised, such as 25.82 Tridentate ligands form more stable pincer complexes and have been useful in providing dicationic terpyridyl-ligated gold hydroxides such as 26,83 as well as the cyclometallated N^N^C-bonded complexes 27.84
However, as of 2012 there were no reports of isolable gold complexes containing the types of O-ligands that were being postulated in heterogeneous gold-catalysed oxidations, such as superoxides Au-OO˙, peroxides Au–OO–Au, and hydroperoxides Au–OOH. We were therefore interested in the synthesis and characterisation of such compounds.
Given that the hydroxide (C^N^C)AuOH (11) had proved an ideal starting material for hydride synthesis (Scheme 8), we chose this compound as the starting point for reactions with peroxides. The reaction of 11 with t-BuOOH readily affords the alkylperoxide 28. Similarly, treatment of 11 with H2O2 resulted in the rather delicate hydroperoxide 29, which has a tendency to condense with excess 11 to give the peroxide 30. This water elimination is reversible. Such a dehydration reaction also became apparent when 11 was stirred in dry solvents (e.g. dry acetone), which led to the formation of the μ-oxide 31 (Scheme 12). The identity of all these species was confirmed by X-ray crystallography. Oxide and hydroxide are in equilibrium, as are peroxide and hydroperoxides.60 Subsequently a gold(I) t-butylhydroperoxide has also been prepared by an analogous route.85
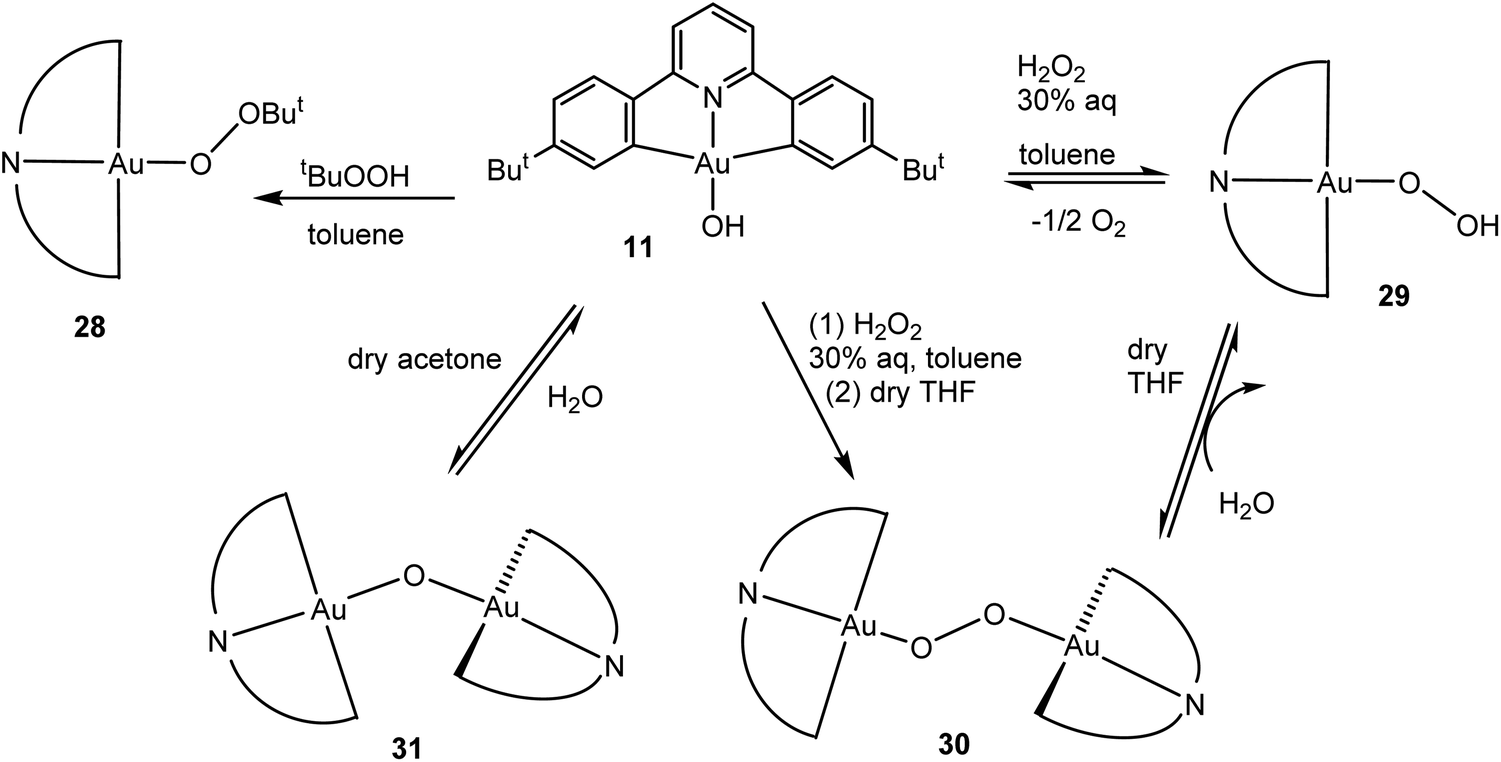 | ||
| Scheme 12 Generation and interconversion of gold(III) oxide and peroxide complexes.60 | ||
The crystal structure of the μ-oxide 31 also answers the question concerning possible π-bonding contributions to the Au–O interaction. With less electron-rich metals, M–O π-bonding leads to a widening of the M–O–M angle up to 180° and M–O bonds with partial double-bond character. The Au–O–Au angle in 31, on the other hand, is 113.5°, only slightly less acute than the tetrahedral angle of 109.5°, a rather small steric effect given the bulkiness of the two (C^N^C)Au fragments. There is therefore no evidence for any interaction between the Au(III) centres and the lone pairs on the bridging O ligand. This is further borne out by DFT calculations on 11, which show that the lobes of the gold(III) d-orbitals and of the oxygen lone pairs in the relevant MO (here HOMO−1) are out of phase (Fig. 1). The same result was found for the hydroperoxide 29.60
 | ||
| Fig. 1 HOMO−1 orbital of (C^N^C)AuOH (11), showing the relative phases of Au orbitals and the lone pairs of the OH ligand. | ||
Attempts to generate these Au peroxide species directly from O2 failed: neither the hydride 12 nor the Au(II) dimer 14 insert O2, even under forcing conditions, in spite of the calculated exothermic character of the reaction. The reactivity of these (C^N^C)Au(III) pincer complexes differs therefore from the behaviour of palladium and platinum, where O2 insertions into M–H and Pd–Pd bonds proceed with ease.86,87
The reactivity of these gold(III) hydride and oxide species is in keeping with the Au–X bond energies. Pertinent values are given in Table 1.
| Compounda | BDE | On Au(111) surfaceb | Diatomic compound | BDEc |
|---|---|---|---|---|
| a L = C^N^C pincer ligand. b Estimates based on single-crystal absorption calorimetry, values from ref. 65c. c Data from: J. A. Martinho Simoes and J. L. Beauchamp, Chem. Rev., 1990, 90, 629. | ||||
| LAu–OH (11) | 279 | 251 | ||
| LAu–OAuL (31) | 206 | Au–O | 223 ± 21 | |
| LAu–OOH (29) | 168 | 108 | ||
| LAu–OOAuL (30) | 126 | |||
| LAu–H (12) | 317 | Au–H | 292 ± 8 | |
| LAu–Me | 262 | |||
| LAu–OMe | 209 | |||
| (C^N^N)Pt–H | 350 | Pt–H | 352 ± 38 | |
| (C^N^N)Pt–OH | 367 | Pt–O | 391 ± 42 | |
| (C^N^N)Pt–Me | 297 | |||
The gold(III) hydroperoxides, peroxide and oxide species are interconvertible. Treatment of the hydroperoxides with a phosphine PAr3 led to selective and stepwise O-atom abstraction, with formation of the phosphine oxide (Ar = 4-C6H4R; R = H, F, Me, OMe) (Scheme 13). Electron-donating R groups increased the reaction rate. O-abstraction from the oxide 31 generated the Au(II) dimer 14. Remarkably, even the hydroxide 11 could be deoxygenated, to give the hydride 12.
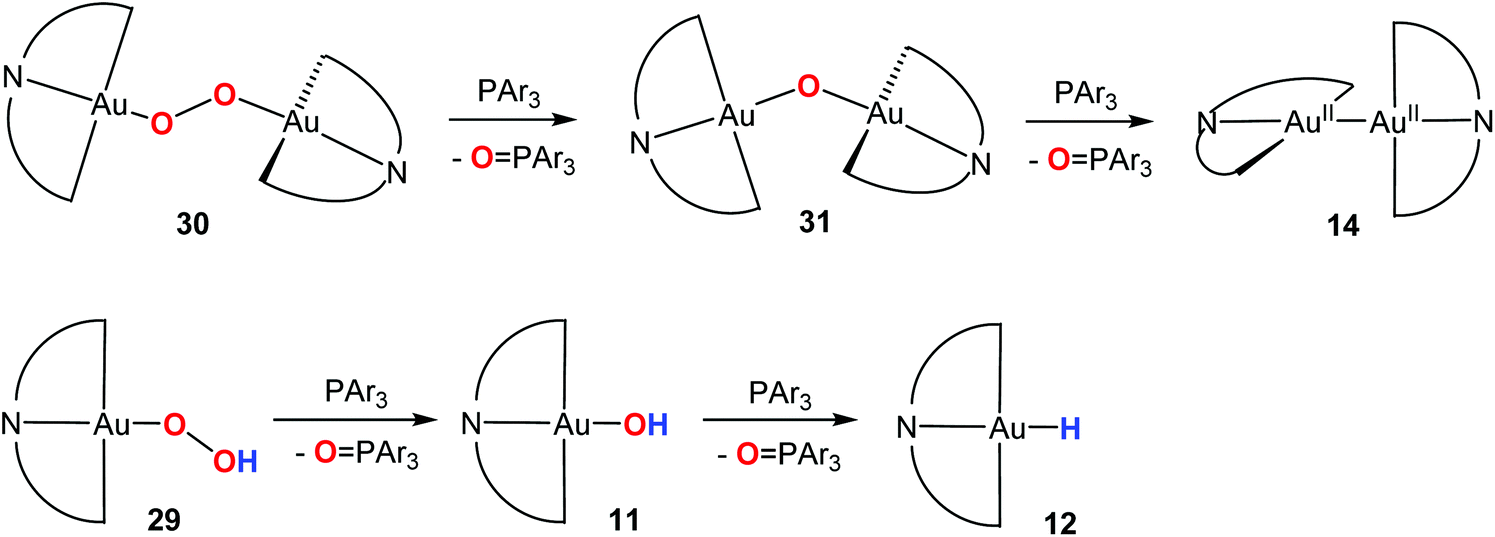 | ||
| Scheme 13 Oxygen transfer reactions of Au(III)–O complexes. | ||
These reaction sequences are in line with the bond energy trend, Au–H > Au–O. The transformation of a metal hydroxide into a metal hydride (in the absence of a hydride transfer agent) is unique to Au(III) and is not known for any other metal.
For platinum(II), the order of bond energies is reversed, Pt–H < Pt–O, and the hydroxide-to-hydride transformation is thermodynamically unfavourable. This is therefore an example of a reaction which gold can do but platinum cannot. However, the energetics are very carefully balanced: whereas the Au–OH deoxygenation works for Au(III), gold(I) hydroxides like (IPr)AuOH do not react.
The hydroxide to hydride conversion is, in principle, a key step in a water splitting cycle which does not require oxidation state changes of the metal (Scheme 14). However, so far it has not been possible to close the cycle because the gold hydride is too stable and does not react with acids under H2 elimination.
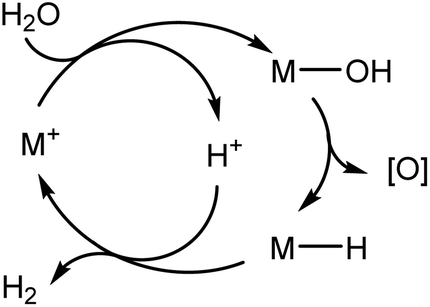 | ||
| Scheme 14 Principle of a water-splitting cycle based on M–OH deoxygenation. | ||
The oxygen abstraction by phosphines proceeds via a concerted mechanism. The kinetics (over the temperature interval −10 to −55 °C, toluene) show a secondary kinetic isotope effect kH/kD = 1.45, consistent with an Au–O–H bending motion in the rate limiting step. DFT studies confirmed that the oxygen is abstracted via an external attack by phosphine (PMe3 as model); intermediates involving a 5-coordinate phosphine adduct of gold could not be identified. 18O-labelling confirmed that the phosphine oxide formed was derived from the gold hydroxide. The reaction is 1st order in both [Au] and [PR3]; the kinetic parameters (Ea = 31.4 ± 1.7 kJ mol−1; ΔH‡ = 35.02(67) kJ mol−1; ΔS‡ = −105.9(3) J mol−1 K−1) show a strongly negative entropy of activation, in agreement with an associative mechanism. The rates for different phosphines with electron donating and withdrawing para-substituents follow a Hammett relationship (ρ = −3.15), with electron-donating groups such as Me or OMe accelerating the rates substantially. The negative ρ value (−1.05 per aryl ring) suggests that positive charge is built up on the reaction centre and is in good agreement with a zwitterionic transition state (Scheme 15).60
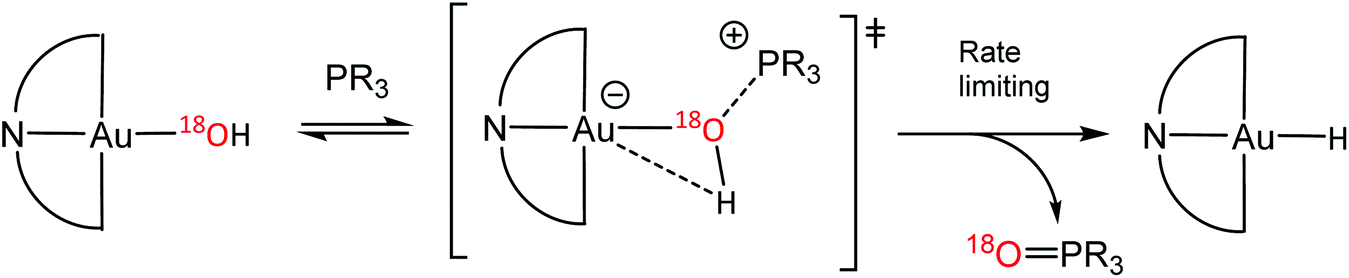 | ||
| Scheme 15 Proposed mechanism for O-abstraction from AuIII–OH by phosphines. | ||
In summary, studies in gold(III) oxide systems have confirmed the absence of double-bond character and of π-bonding contributions in the Au–O bond. The first examples of gold peroxides and hydroperoxides have been isolated. The Au–OO bond is substantially weaker than the Au–O(oxide) and Au–OH bonds, and all of these are weaker than the Au–H bond. Both Au(I) and Au(III) hydrides are highly covalent species, stable to air and moisture and surprisingly resistant to weak acids. One interesting consequence of these bond energy differences was the facile O-abstraction from a gold(III) hydroxide to give the corresponding gold hydride, a reaction that is so far unique to Au(III) and found neither for Pt(II) nor for Au(I).
Gold π-complexes of alkenes and alkynes
The chemistry of π-complexes of gold in various oxidation states with alkenes and alkynes has recently been the subject of a very detailed and comprehensive review.88 Only a brief summary of the main complex types needs to be given here. The first alkene adducts of AuCl were reported in the 1960s by Chalk89 and Hüttel;90 the latter developed this chemistry throughout the 1970s and 80s.88 The advent of very non-coordinating anions made it possible to isolate complexes where Au(I) is exclusively ligated to alkenes, as in the trigonal-planar [Au(C2H4)3]+ (32), [Au(norbornene)3]+ and tetrahedral [Au(COD)2]+ cations (COD = 1,5-cyclooctadiene) (33) (Scheme 16).91–93 The reaction of the gold(III) oxide [Au2(μ-O)2(bipy)2]2+ with alkenes proceeds with reduction to give the Au(I) bipyridyl-stabilised alkene complexes of type 34 (bipy = variously 6,6′-substituted bipyridyl), with alkene = styrene, p-methoxystyrene, α-methylstyrene, cis-stilbene; the complexes with norbornadiene, COD and dicyclopentadiene are alkene-bridged binuclear species.94 Tris(pyrazolato)borates act as bidentate N-donor ligands in the zwitterionic complex 35.95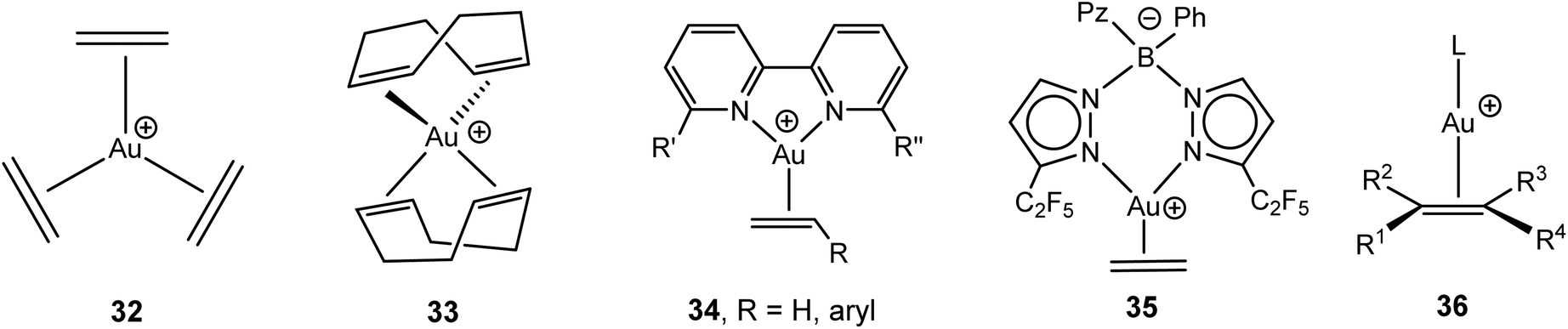 | ||
| Scheme 16 Types of Au(I) alkene complexes. | ||
Strongly donating phosphine and NHC ligands have proved useful in stabilising two-coordinate Au(I) complexes of type 36. These complexes have recently been reviewed.96 Complexes with alkenes carrying 1–4 substituents are now known, including the crystallographically characterised isobutene (L = IPr) and tetramethylethylene derivatives (L = But2PC6H4-2-Ph).97,98 Structurally similar complexes have been reported for 1,3-dienes, which are η2-coordinated, as well as for cyclic dienes, allenes,99 and vinyl ethers.100 An η2-cyclopentadiene complex of type 36 has also been characterised (L = But2PC6H4-2-Ph), and mixtures of Ph3PAuCl and AgSbF6 were found to polymerise cyclopentadiene and 1,3-cyclohexadiene. A mechanism was not suggested, although Au+ or H+ initiated carbocationic diene polymerisation seems most likely.101
The structural chemistry of gold(I) alkyne complexes follows the pattern set out for alkene adducts and was recently discussed in detail by Schmidbaur.88 The involvement of Au(I) alkyne compounds in homogeneous gold catalysed alkyne reactions has been reviewed.102 Two-coordinate adducts prevail, and complexes of cyclic alkynes tend to be the most stable due to the stronger π-interactions of strained C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C bonds with metal centres.103–105 Complexes of AuCl with noncyclic alkynes tend to be thermally sensitive. It has proved possible to crystallographically characterise the 3-hexyne complex AuCl(EtCCEt), which shows a CC bond length of 1.224(6) Å, only marginally longer than that of a typical non-coordinated alkyne (around 1.202 Å).106 In line with this very small degree of bond elongation, structural and DFT studies on Au(I) complexes with cyclic alkynes have shown that CC π-donation into the σ* orbital of the Au–L bond (L = NHC) is 3–4 times larger than d(Au) → π*(CC) back-donation. These observations explain the electrophilic activation of alkynes by LAu+ fragments in gold(I) catalysed reactions. Surprisingly, AuCl proved to be both the strongest donor and the strongest acceptor.104 With silyl alkynes, desilylation has been observed, which leads to a number of structurally characterized oligonuclear complexes (37),107 not dissimilar to the interactions postulated for di-gold activation of alkynes.108 Desilylation of bis(silyl) acetylene gave a tetranuclear dication 38 (Scheme 17).
C bonds with metal centres.103–105 Complexes of AuCl with noncyclic alkynes tend to be thermally sensitive. It has proved possible to crystallographically characterise the 3-hexyne complex AuCl(EtCCEt), which shows a CC bond length of 1.224(6) Å, only marginally longer than that of a typical non-coordinated alkyne (around 1.202 Å).106 In line with this very small degree of bond elongation, structural and DFT studies on Au(I) complexes with cyclic alkynes have shown that CC π-donation into the σ* orbital of the Au–L bond (L = NHC) is 3–4 times larger than d(Au) → π*(CC) back-donation. These observations explain the electrophilic activation of alkynes by LAu+ fragments in gold(I) catalysed reactions. Surprisingly, AuCl proved to be both the strongest donor and the strongest acceptor.104 With silyl alkynes, desilylation has been observed, which leads to a number of structurally characterized oligonuclear complexes (37),107 not dissimilar to the interactions postulated for di-gold activation of alkynes.108 Desilylation of bis(silyl) acetylene gave a tetranuclear dication 38 (Scheme 17).
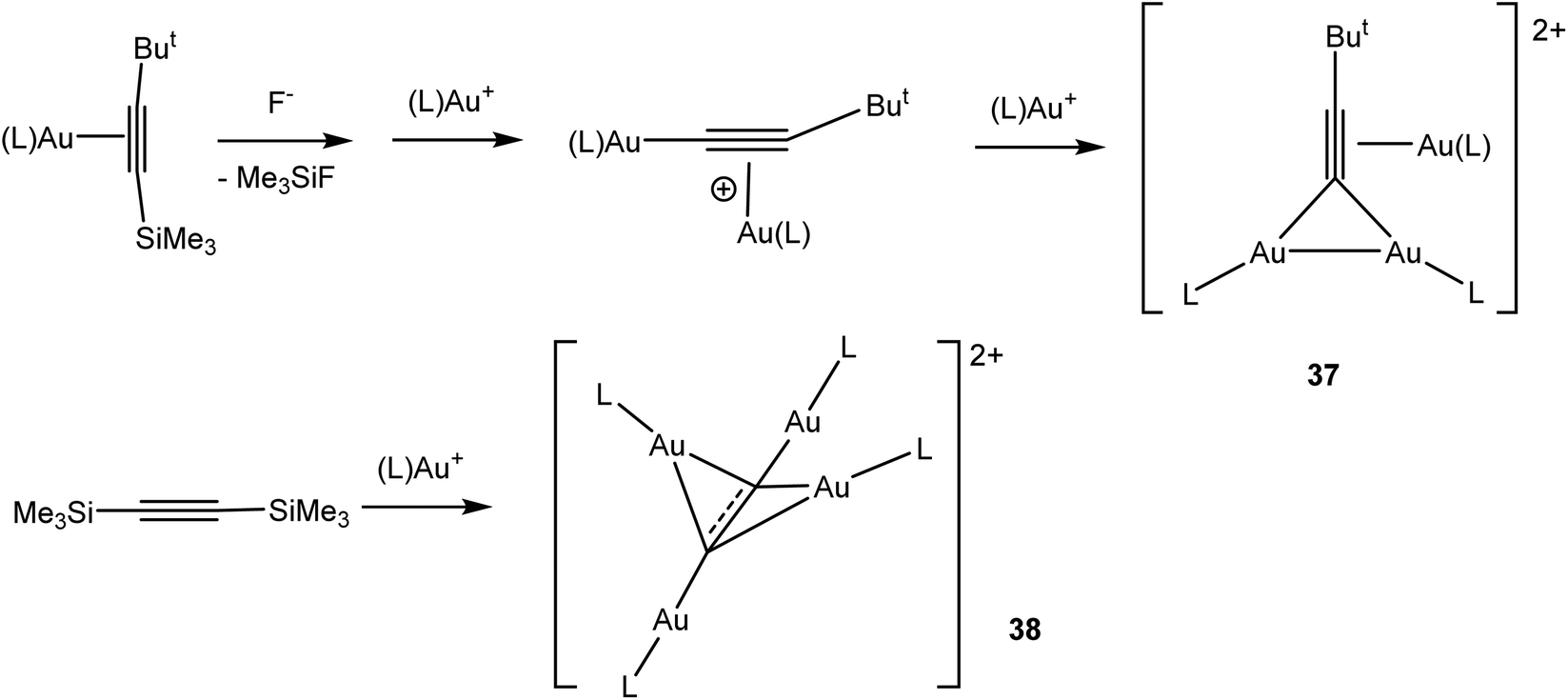 | ||
| Scheme 17 Formation of oligonuclear cationic gold alkyne complexes (L = PBut3).107 | ||
NMR spectroscopic studies (1H–19F HOESY experiments) showed that the relative cation–anion positions in gold(I) alkyne complexes [(L)Au(alkyne)]+BF4− have been found to vary, depending on the ligand structure; the anion is located close to gold in the phosphine complex 39 but has close interactions with the alkyne ligand in the NHC derivative 40 (Scheme 18). DFT studies showed that the phosphine complex in this case is the stronger π-acceptor.109 Anions also play an important role in the intermolecular alkoxylation of alkynes catalysed by carbene-based gold(I) salts, (NHC)AuX (X− = [B{C6H3(CF3)2}4]−, BF4−, OTf−, OTs−, TFA−, or OAc−). The anion is involved in alcohol deprotonation, and the intermediate basicity of the OTs− anion gave the most efficient catalysis.110
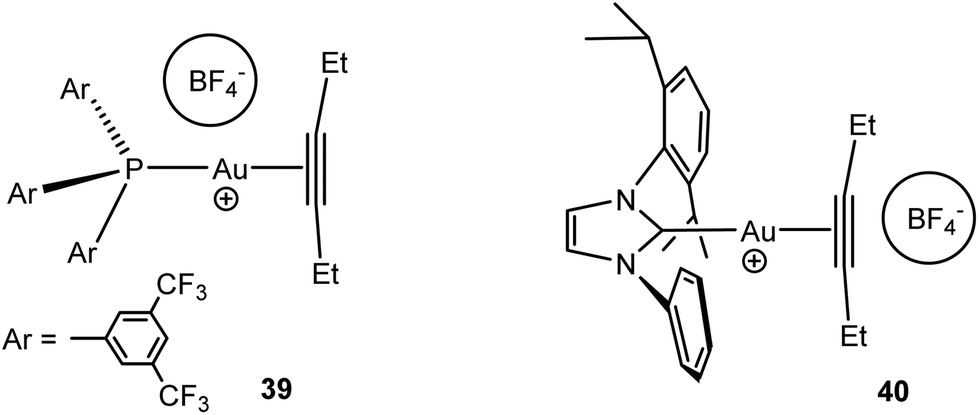 | ||
| Scheme 18 Ion pair structures of gold alkyne complexes.109 | ||
In addition to the mono-alkyne adducts of AuCl and [(L)Au]+, the bis- and tris-alkyne compounds 41–43 have also been characterised (Scheme 19). The structures are analogues of alkene adducts, but the tris-cyclooctyne complex 42 shows a slight propeller-type twist of the ring ligands, with torsion angles of 3.0, 6.3 and 43.9°. The Au–C bond lengths to the cyclooctyne with the largest twist are significantly larger than the Au–C bonds to the other, in-plane, ligands. This indicates weaker bonding of one of the alkyne ligands, and indeed on recrystallization some alkyne is lost and crystals of the bis-alkyne adduct could also be obtained. In the [Au(cyclooctyne)2]+ cation 43 the alkyne ligands are oriented almost perpendicular to one another. The CC bonds in these compounds are slightly elongated compared to the corresponding free alkynes, with the largest elongation being displayed by the neutral AuCl adduct, which can be expected to display the strongest back-donation.111
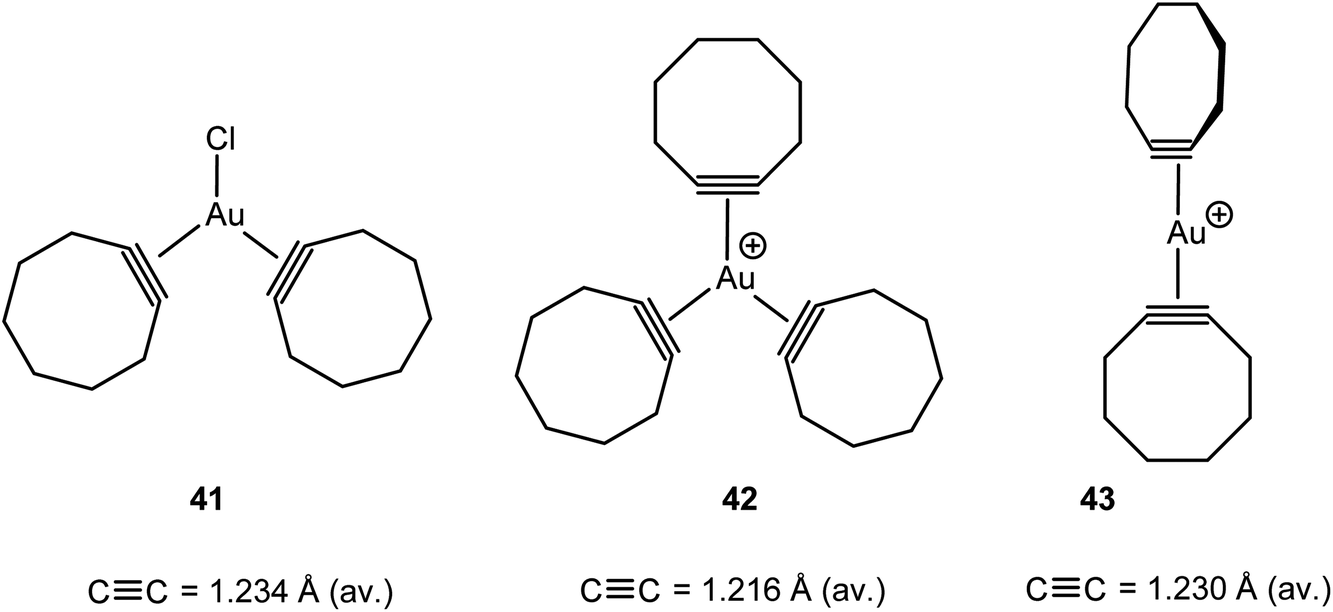 | ||
| Scheme 19 Structurally characterised bis- and tris-cyclooctyne complexes of gold(I).111 | ||
So far it has not been possible to isolate alkyne complexes of gold(III). Early attempts to react disubstituted alkynes with AuCl3 led to reduction.88 Interestingly, in NMR studies on the reaction of the thermally unstable gold(III) diaryl complex [(C6F5)2AuCl]2 at −80 to 0 °C Laguna et al. were able to detect some tantalising indications for the formation of an alkyne complex with the tentative structure (C6F5)2AuCl(η-PhCCH) (δ1H 3.76, toluene-d8). However, on removal of the solvent above −50 °C the compound decomposed to metallic gold.112
By contrast to alkynes, the first examples of alkene complexes of gold(III) have recently been reported, by two very different routes. Initial attempts seemed to confirm the susceptibility of gold(III) alkene species to reduction; for example, efforts to displace the ether ligands in Uson's bis(pentafluorophenyl)gold(III) cation113 [(C6F5)2Au(OEt2)2]+ with norbornene or 1,5-cyclooctadiene were unsuccessful, while chloride abstraction from [(C6F5)2AuCl2]− with AgSbF6 in the presence of norbornene led to reductive elimination of perfluorobiphenyl and formation of [Au(norbornene)3]+. The strategy of taking advantage of the stability of the C^N^C pincer ligand framework proved more successful. The trifluoroacetate ligand in (C^N^C)Au(OAcF) (44) is easily abstracted by B(C6F5)3 in dichloromethane, and adding alkenes to this solution at −40 °C resulted in the clean formation of the corresponding η2-alkene complexes 45–47 (Scheme 20).114
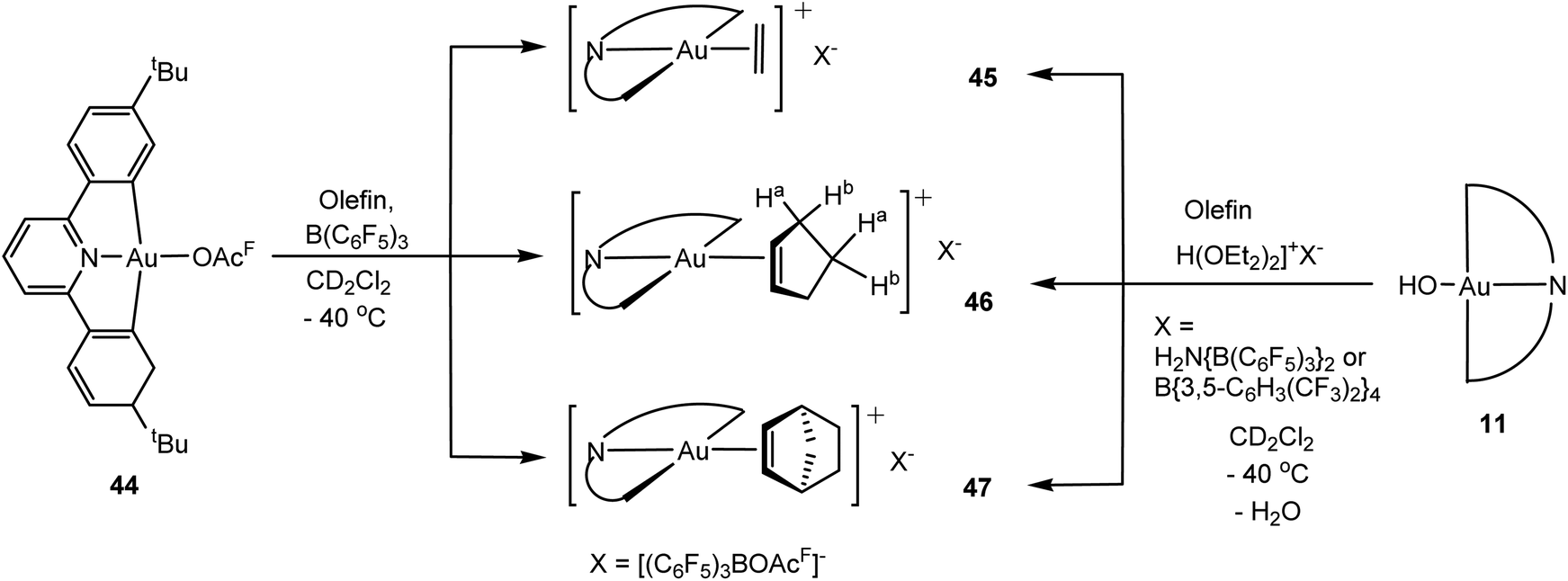 | ||
| Scheme 20 Synthesis of gold(III) alkene complexes. | ||
Dichloromethane solutions of the cyclopentene and ethylene complexes are stable up to about 0 and −20 °C, respectively, whereas solutions of the norbornene complex 47 can be handled at 20 °C. Removal of volatiles afforded the complexes as yellow powders which can be stored indefinitely under nitrogen. Once isolated, these complexes proved stable in the solid state for hours at room temperature and in air with minimal degradation.
In the absence of excess ethylene, complex 45 shows a sharp 1H NMR singlet at δ 6.29, downfield shifted from the δ 5.38 signal of free C2H4 (CD2Cl2, −40 °C), in contrast to Au(I) and Pt(II) ethylene complexes, which show an upfield shift of the ethylene resonance (Table 2), suggestive of π-electron donation which is poorly compensated by back-bonding. In the presence of excess ethylene the 1H NMR signals broaden on warming from −70 to −10 °C, as expected for ligand exchange. The 13C2H4 complex shows a 13C{1H} NMR resonance at δ 108.9, up from δ 122.8 for the free alkene; JCH increased on coordination from 156 to 166 Hz. Similar JCH values have been found for gold(I) ethylene complexes, although in those cases the changes in the 13C NMR chemical shifts Δδ (13C) are much larger, of the order of 60+ ppm, for both Au(I) and Pt(II). The Au(III)–alkene bond is evidently weaker, particularly the back-bonding contribution, than the Pt(II)–alkene bond.
| Compound | δ 1H | Δδ (H) | δ 13C | Δδ (C) | J CH [Hz] | Ref. |
|---|---|---|---|---|---|---|
| a In CD2Cl2 unless indicated otherwise. b pzCF3 = 3,5-(CF3)2pyrazolate. c In CDCl3. d bipyR = 4-(2′′,6′′-Me2C6H3)-2,2′-bipyridyl. e In 1 M methanolic HCl. The chemical shift of free C2H4 in this solvent is δ 5.37. | ||||||
| C2H4 | 5.38 | 122.8 | 156 | |||
| [Au(C2H4)3]SbF6 | 4.94 | −0.44 | 92.7 | 91a | ||
HB(pzCF3)3Au(C2H4)b,c [(bipyR)Au(C2H4)]PF6![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) d d |
3.81 | −1.6 | 63.7 | −59.8 | 165 | 95 |
| 3.09 | −2.3 | 61.6 | −55.2 | 94b | ||
| K[PtCl3(C2H4)]e | 4.83 | −0.55 | 67.1 | −55.7 | 115 and 116 | |
| [MePt(PMe2Ph)2(C2H4)]PF6 | 4.12 | −1.26 | 84.8 | −38.4 | 116 and 117 | |
| 45 | 6.29 | 0.91 | 108.9 | −13.9 | 166 | 114 |
| trans-(py)PtCl2(cyclopentene) | 6.23 | 0.49 | 118 | |||
| 46 | 6.30 | 0.56 | 114 | |||
This remarkably low Au(III)–ethylene back-bonding contribution, compared to Pt(II), is further underlined by the calculated rotational barriers. The energy difference for the in-plane and perpendicular ethylene orientations for 45 was estimated to be only 2.4 kJ mol−1, less than 10% of the value for isostructural cationic platinum pincer compounds (Scheme 21). This agrees with the donation/back-donation (d/b) ratio obtained by charge decomposition analysis, which gives a value of d/b = 3.41 for Au(III) but d/b = 1.70 for Pt(II), i.e. back-donation is much more important for platinum than for gold.119 This general picture, i.e. the remarkably small contribution of back-bonding for Au(III), has since been confirmed for other systems, including chelating olefin and CO complexes (vide infra).
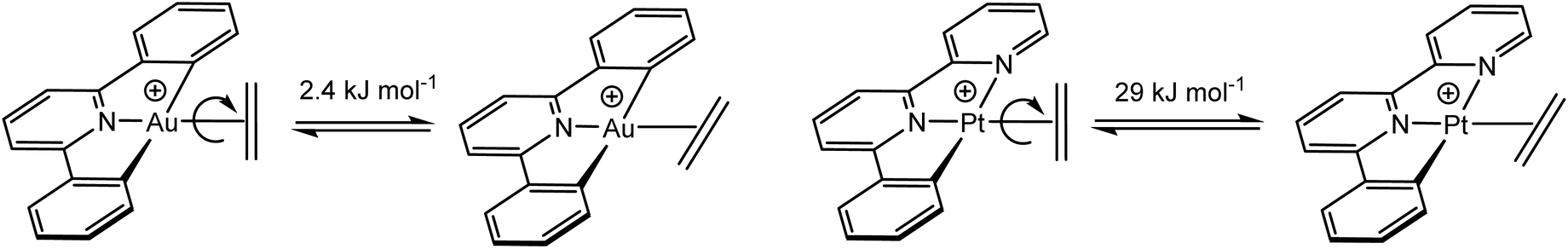 | ||
| Scheme 21 Rotational barriers in Au(III) and Pt(II) ethylene complexes. | ||
Cyclopentene coordination in 46 was indicated by a downfield shift of the olefinic proton signal from δ 5.74 to 6.3 ppm, as well as by the inequivalence of the methylene protons. There was no indication of ligand exchange, which would have resulted in the interchange of the methylene-protons Ha and Hb (Scheme 20); the CH2 grous of η2-cyclopentene are diastereotopic, and coordination is via one π-face only.
Unlike the cyclopentene and ethylene compounds, the CHCH signal of the norbornene complex 47 shows only a small upfield shift from δ 6.02 to 5.8; in this case a clearer indication of CC coordination is provided by the bridgehead CH signals, which are shifted from δ 2.84 for free norbornene to δ 3.49.114
A different route to an Au(III) alkene complex was developed by Tilset and co-workers, by reacting the cyclometallated dimethylgold complex 48 with trifluoroacetic acid at −78 to 0 °C in the presence of COD.120 Under these conditions the phenylpyridine ligand is displaced, to give cis-[Me2Au(COD)]+ (49) (Scheme 22). The same cation is formed by protonating 48 with [H(OEt2)2][B{C6H3(CF3)2}4]. Borate salts of 49+ proved suitable for X-ray diffraction. The olefinic 1H NMR signal is found at δ 6.39, 0.8 ppm higher than the signal of free COD. The Au–C bonds to the COD ligand are considerably longer than the Pt–COD bonds in the analogous platinum complex (1,5-COD)PtMe2 (average Au: 2.389; Pt: 2.228 Å), while the CC bond lengths in both complexes are identical within experimental error (average Au: 1.356(5); Pt: 1.355(17) Å); both show slight elongation compared to free 1,5-COD [CC 1.340(3) Å]. The Au–C bond length to the two C-atoms of the same CC bond differ by over 0.04 Å, a consequence of conformational strains in the COD backbone. DFT modelling showed that back-bonding from the metal dxz and dyz orbitals is substantially weaker for Au(III) than for Pt(II); this reinforces the bonding picture suggested by the spectroscopic results for compounds 45–47.
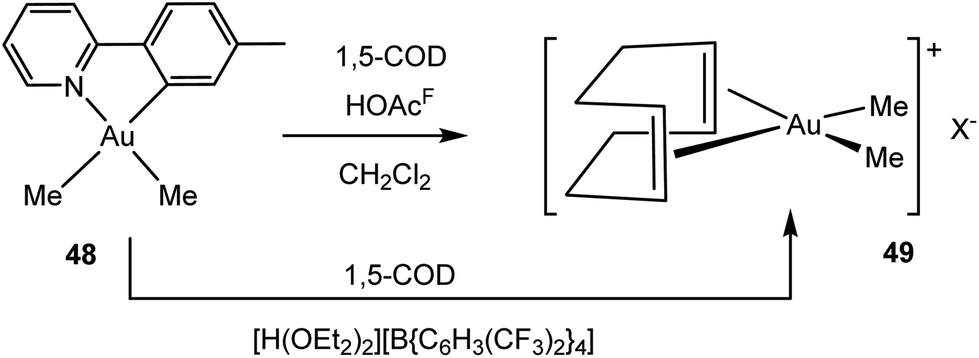 | ||
| Scheme 22 Synthesis of Au(III) COD complexes.120 | ||
The coordinated CC bonds in Au(III) complexes are very susceptible to nucleophilic attack. The norbornene complex 47 was found to react with traces of moisture or with alcohols to give the corresponding hydroxy- or alkoxyalkyl complexes 50, respectively (Scheme 23). The reaction involves the protolytic cleavage of one of the Au–phenyl bonds of the pincer ligand. The OH-derivative was structurally characterized (as SbF6− salt).114
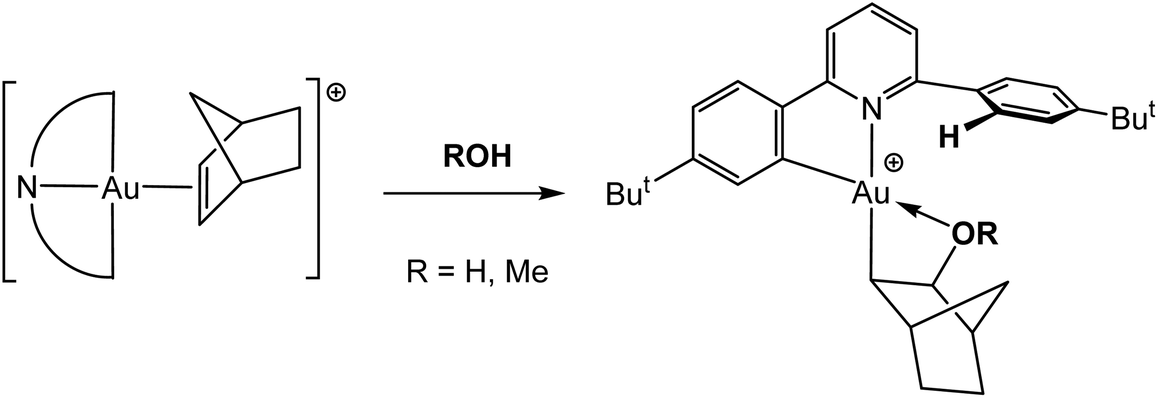 | ||
| Scheme 23 Reaction of the gold(III) norbornene complex 47 with protic reagents.114 | ||
An unusual reaction that was discovered in the course of this work is the direct insertion of ethylene into the Au-trifluoroacetate bond, to give the crystallographically characterised 2-acetatoethyl complex 51 (Scheme 24). The insertion proceeds slowly (14–72 h) at room temperature and is quantitative; it is accelerated by AgOAcF impurities which catalyse the acetate abstraction.114 The reaction is yet another expression of the thermodynamic trend in AuIII–O and AuIII–C bond strengths (cf. Table 1).
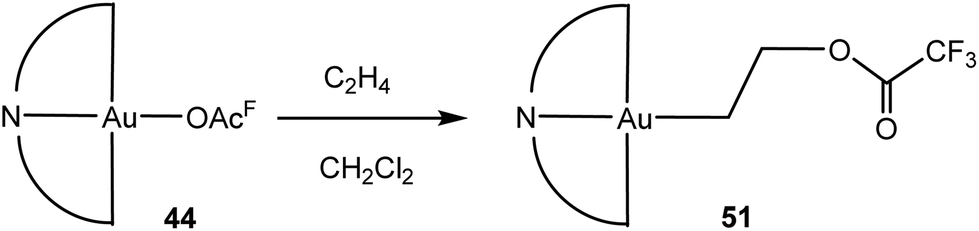 | ||
| Scheme 24 Ethylene insertion into the gold–trifluoroacetate bond.114 | ||
The mechanism of ethylene insertion into gold(III)–trifluoroacetate bonds was subsequently investigated in detail by Tilset and co-workers.121 In trifluoroacetic acid as the solvent the ethylene insertion is fast (5–30 min). There is a strong kinetic preference for insertion trans to the N-donor. The reaction is reversible, and there is ethylene exchange with C2D4. With Z-C2H2D2 the threo isomer 52-D2 is formed, arising from external anti attack by acetate on ethylene in the likely intermediate [(C^N)Au(OAcF)(η2-C2H4)]+; this ethylene complex itself could however not be detected. In trifluoroethanol as solvent the corresponding 2-alkoxyethyl complex 53 is formed (Scheme 25).
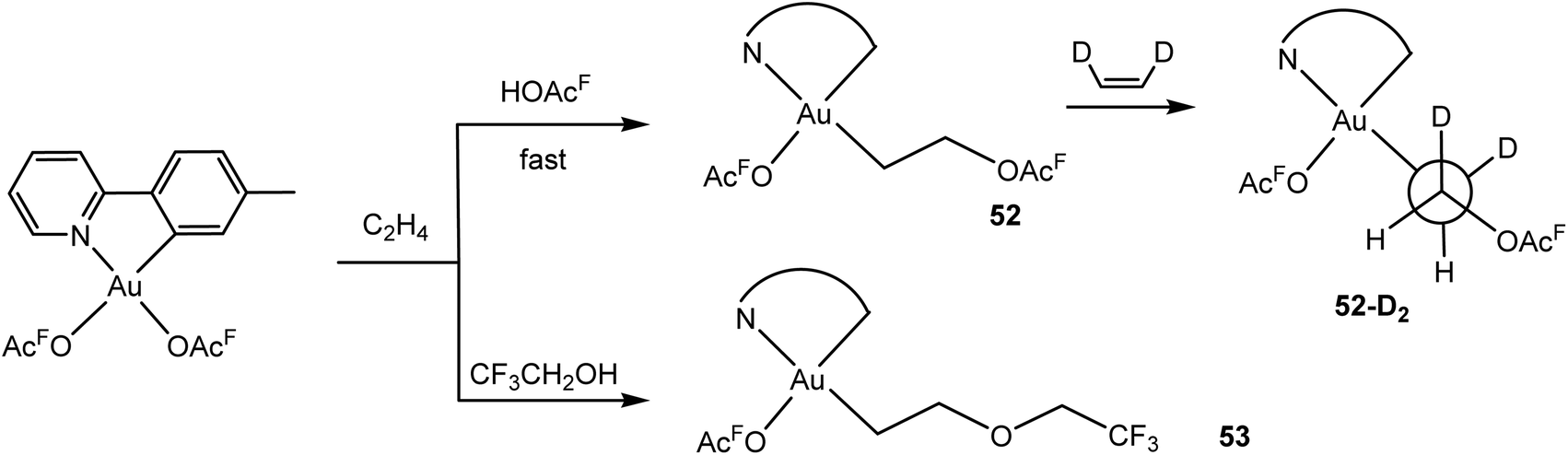 | ||
| Scheme 25 Ethylene insertion in (C^N)Au(OAcF)2 complexes.121 | ||
Whereas alkene insertions into Au–X bonds lead to functionalised alkyls, the formation of new C–C bonds requires alkene insertion into a gold–alkyl bond. These are generally not at all facile. As Amgoune and Bourissou were recently able to show, such a pathway becomes feasible if a coordinatively unsaturated, formally three-coordinate gold(III) species can be generated. This was successful in the case of the cyclometallated phosphinonaphthalene complex 54 (Scheme 26).122 While cold (−80 °C) mixtures of 54 with B(C6F5)3 as methide abstractor failed to react with ethylene or styrene and gave Au–C6F5 species on warming, the reaction with norbornene led to the first observation of an alkene insertion into a gold(III) alkyl bond. The addition of neutral N-donors allowed the isolation of 55, while chloride addition afforded the crystallographically characterised complex 56. The syn-1,2 character of the insertion process suggests an intramolecular migratory pathway via an (undetected) alkene complex [(C^P)AuMe(η2-norbornene)]+ as the intermediate.123
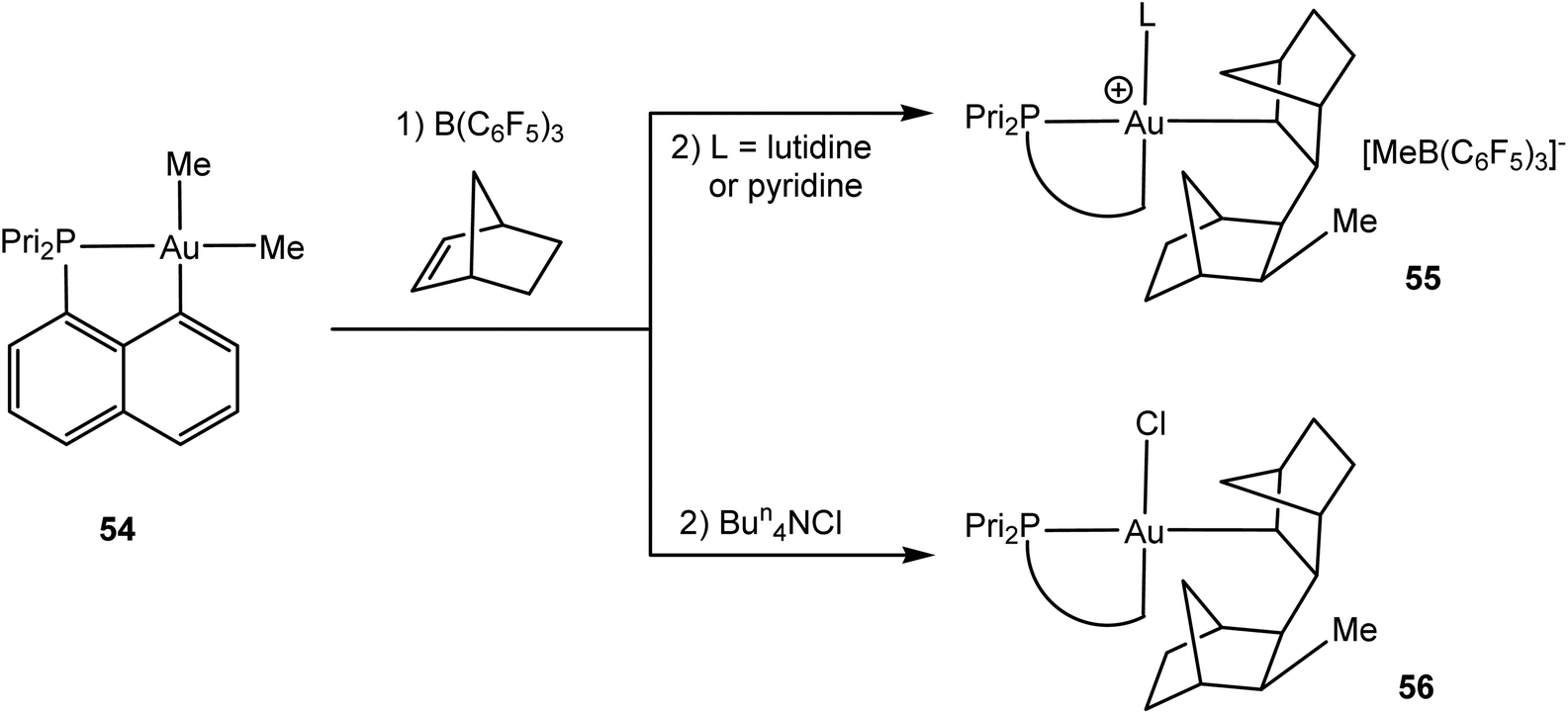 | ||
| Scheme 26 Norbornene insertion into an Au–Me bond.122 | ||
The picture that has emerged is that Au(III) alkene complexes can indeed be isolated and characterized, over 180 years after the first preparation of the famous Pt(II) analogue, Zeise's salt. They differ however substantially from the alkene complexes of both Au(I) and Pt(II) in terms of bonding and reactivity and rely almost entirely on the (CC)→Au(III) donor interaction, with very little back-bonding contribution and very low rotational barriers. This makes gold(III) alkene compounds highly susceptible to nucleophilic attack, even by traces of moisture or weak nucleophiles such as trifluoroacetate in acidic media. As yet there are no examples of isolable gold(III) alkyne adducts, most likely because alkyne complexes rely for stability even more on π-back-donation.
CO complexes
The first carbonyl complex of gold was Manchot's AuCl(CO), first reported in 1925.124,125 Since then a number of neutral126–130 and cationic128–134 CO complexes of gold(I) have been described. The bonding of CO to gold in compounds of the type [(L)AuI(CO)]+ (L = phosphine or NHC) has recently been analysed in detail.135 These complexes are linear. In gold–CO complexes the donation of the non-bonding electron pair on carbon dominates over back-donation, which leads to an increase of the CO stretching frequency to a value higher than that of free CO (2143 cm−1) and more akin to the formyl cation HCO+ (2184 cm−1), a situation fund for quite a number of highly electrophilic metal ions (often referred to as “non-classical CO complexes”,136 a term that is not without objections137). For linear gold complexes [(L)AuCO]+ it was shown that the νCO stretching frequencies do not correlate readily with the familiar Tolman electronic parameters of the L ligands; instead the νCO frequencies are mainly determined by the flux of π-electron density from the L–Au bond.135 A three-coordinate CO complex with a chelating carboranyl bis(phosphine) ligand, [(P^P)Au–CO]+ (57), has also been reported; here the increased electron density provided by the chelating phosphine increases the π-back-bonding sufficiently to give the first “classical” gold CO complex (νCO = 2113 cm−1) (Scheme 27).138 The situation resembles that in the three-coordinate styrene complex 34, which also shows stronger back-bonding than two-coordinate alkene complexes.94
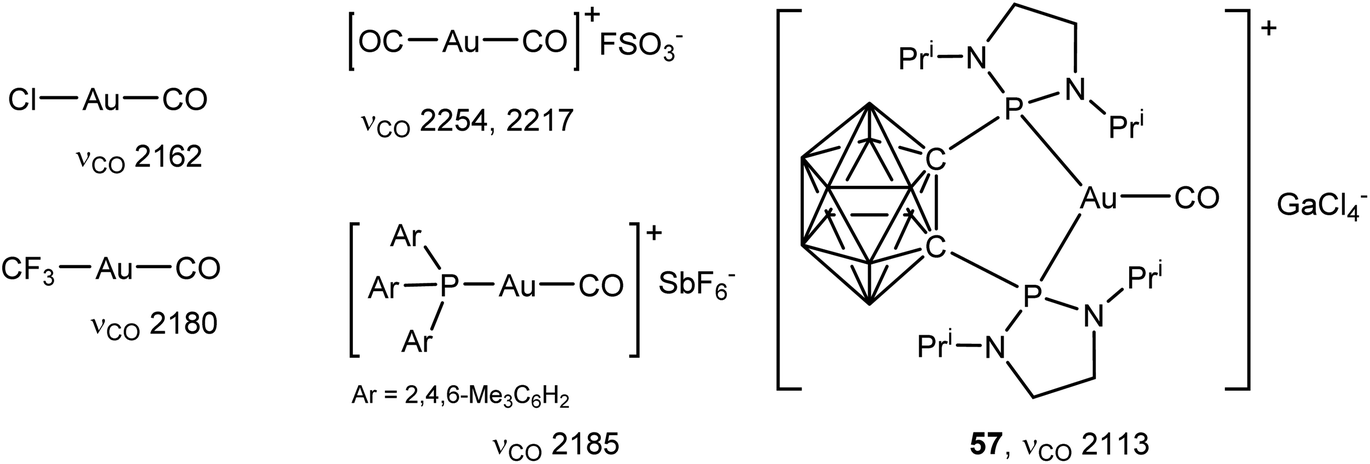 | ||
| Scheme 27 Representative types of Au(I) CO complexes, with νCO stretching frequencies (cm−1).128,129,133,138 | ||
The CO complexes of gold(I) are mainly interesting from a bonding perspective. Since the metal has a full complement of d-electrons and there are no energetically accessible orbitals cis to CO, Au(I) carbonyls do not participate in carbonylation catalysis.139 On the other hand, Au–CO adducts are likely involved in heterogeneous gold catalysis, including the much-studied catalytic oxidation of CO64,65 and of methanol.67,68 This includes surface species of TiO2-supported CO oxidation catalysts where gold is thought to be present as Au3+ ions; and CO adducts with νCO = 2158 cm−1 have been identified.140
However, isolable CO complexes of gold in the oxidation state +III were unknown. Indeed, as early as 1905 it was reported141 that treating HAuCl4 with CO leads to reduction and is in fact an excellent method for the preparation of colloidal gold nanoparticles; this method continues to be used for this purpose.142
Given that Au(III) and Pt(II) are isoelectronic and Pt(II) provided the first transition metal carbonyl complex ever made, PtCl2(CO)2 (1868),143 the lack of isolable Au(III) CO complexes is rather surprising. Redox potentials can of course be shifted by suitable ligands, and in spite of the reducing power of CO, we found recently that CO complexes of Au(III) are in fact readily accessible. Thus, treating mixtures of (C^N^C)Au(OAcF) (44) and B(C6F5)3 in dichloromethane at −30 °C with CO generates [(C^N^C)Au–CO]+[(C6F5)3BOAcF]− (58) (Scheme 28).144 The complex can be isolated by precipitation with light petroleum as a yellow microcrystalline solid. [(C^N^C)Au–13CO]+ (58-13C) was obtained similarly and gives a 13C NMR resonance at δ 167.6 (cf. δ 184 for free CO). The same product was obtained when [(C^N^C)Au(C2H4)]+[(C6F5)3BOAcF]− (45) was exposed to CO. The CO complex is thermally labile, and solutions must be kept <−10 °C. The νCO stretch of 58 was observed at 2167 cm−1 (58-13C: 2143 cm−1). This value is rather close to that of CO bound to Au3+ centres on titania-supported gold mentioned above (2158 cm−1),140 which may suggest that the CO bonding in 58 resembles that found in these supported catalysts.
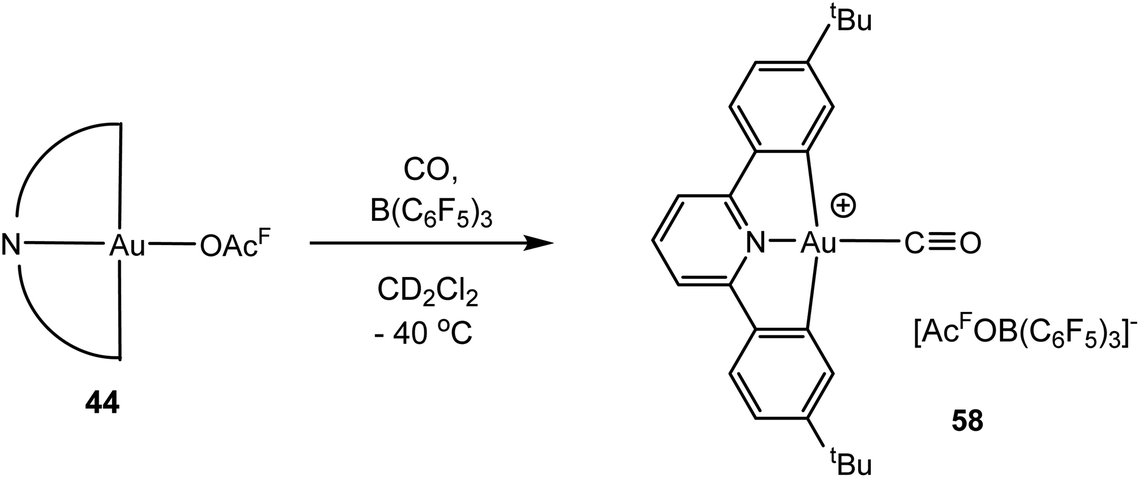 | ||
| Scheme 28 Synthesis of Au(III) CO complexes. | ||
The Au(III)–CO bonding was investigated by DFT methods. Significantly, none of the occupied molecular orbitals show any involvement in Au–CO π-interactions. These are prominent in the LUMO which, however, is separated from the HOMO by a large energy gap (Fig. 2). The Au–C interaction is a single bond, according to natural bond orbital analysis, with the major contributions from gold derived from the 6s, 6py and 5dx2–y2 molecular orbitals.
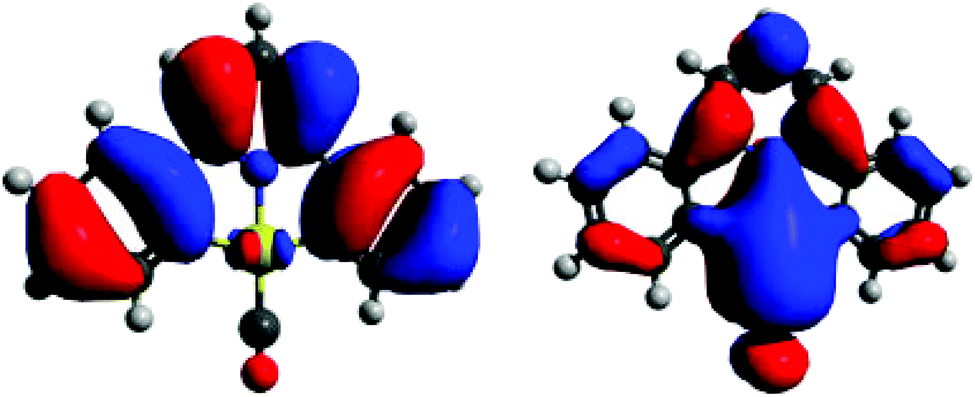 | ||
| Fig. 2 HOMO (left) and LUMO (right) of [(C^N^C)Au(CO)]+ as simulated by DFT. | ||
The low-temperature solution IR spectra of 58 and 58-13C were always accompanied by bands at 2338 cm−1 for 12CO2 and 2273 cm−1 for 13CO2, respectively (CH2Cl2). Since the CO2 must have originated from the CO complex, this observation pointed to a gold-mediated water–gas shift reaction due to the presence of traces of moisture condensation under the recording conditions (−20 °C).
The water-gas shift (WGS) reaction (CO + H2O → CO2 + H2; exothermic, ΔHr = −41.2 kJ mol−1) is catalysed by heterogeneous gold and platinum catalysts,145,146 with gold exhibiting a significantly lower activation energy than platinum.147,148 The mechanism of the heterogeneous WGS reaction is as yet poorly understood. At least three mechanisms are being discussed:149 (1) the redox mechanism assumes reduction of the oxide support, which is then re-oxidised by H+ to generate H2; (2) the formate mechanism assumes CO insertion into surface-bound Au–OH to give Au–OC(O)H, which then decomposes to give AuH + CO2, and (3) in the carboxylate mechanism CO and AuOH give Au–COOH, which also decomposes to CO2 + AuH. The nature of the catalytically active species is equally uncertain, and the debate concerns whether the active species are dispersed and mononuclear, whether they are gold nanoparticles or in fact metal ions, and if so, in which oxidation state. There are arguments in favour of nanoparticles and gold clusters,150 as well for dispersed gold ions. Very recent proposals for active sites have included models such as Au(O)x(OH)y(Na)9 (e.g. x = 6, 7; y = 2) with octahedrally coordinated gold in remarkably high oxidation states,151 with little apparent account of the coordination chemistry and ionisation potentials of gold.
The isolation of a well-defined Au(III)–CO complex 58 has now allowed the role of Au(III) in the WGS reaction to be explored. By contrast, for none of the well-known Au(I) and Pt(II) CO complexes has WGS activity been reported.
In order to monitor the reaction by NMR spectroscopy, (C^N^C)AuOH (11) was used as a surrogate for water; in which case the WGS product would be (C^N^C)Au–H (12) instead of H2, which of course has an easily recognisable NMR signature.144 Exposing 11 to CO did indeed generate 12, via a hypothetical carboxylate intermediate (C^N^C)Au–COOH (59) (Scheme 29). Reactions A, B and C are part of a WGS cycle according to the carboxylate mechanism. Calculations showed that overall steps A and B are 181 kJ mol−1 more favourable for gold than for platinum. Once again, the high stability of the hydride 12 prevented the closure of the cycle, since it does not react with H2O or acids to liberate H2.
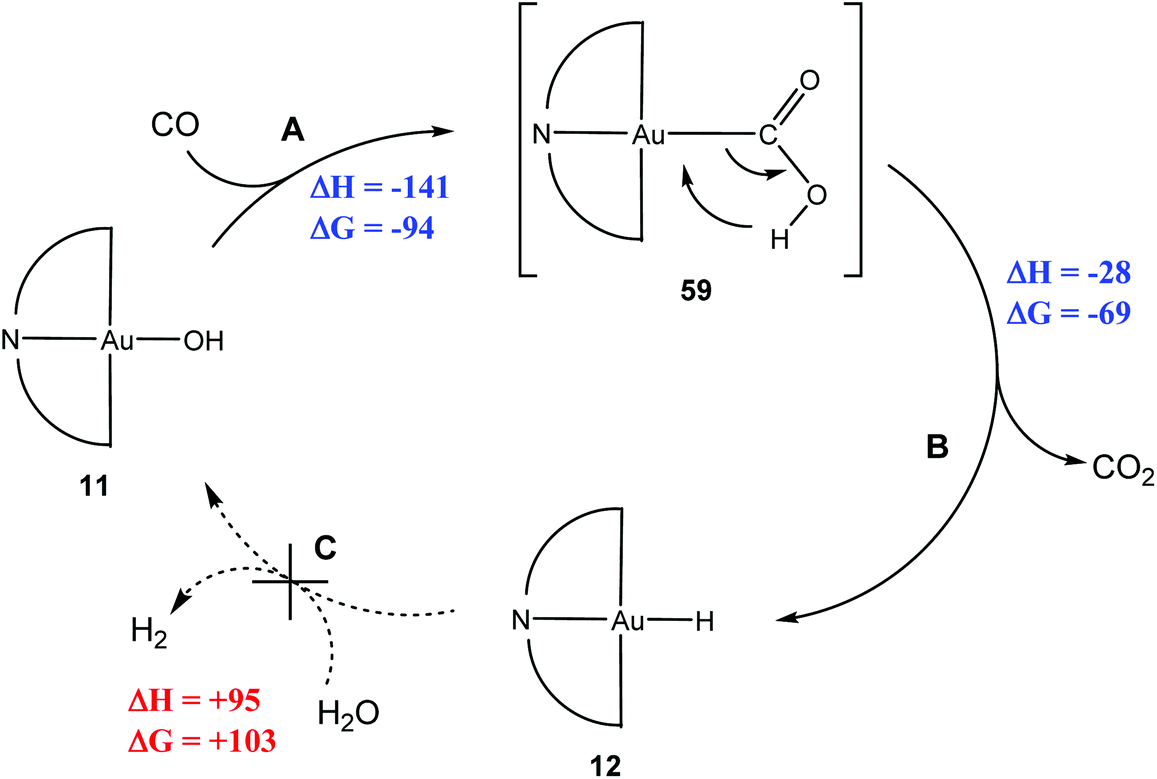 | ||
| Scheme 29 WGS-type reactions of (C^N^C)AuOH. Enthalpy and Gibbs free energy values (in kJ mol−1) for reaction steps as calculated by DFT (T = 298.15 K).144 | ||
Although intermediate 59 could not be spectroscopically identified, support for its formation comes from the analogous reaction of CO with the methoxide (C^N^C)AuOMe. The product, (C^N^C)Au–COOMe (60), is thermally stable and was isolated as white crystals (Scheme 30). In an effort to explore possible formate formation as observed in heterogeneous WGS catalysis, attempts were made to hydrogenolyse 60 under 4 bar H2. However, methylformate was not observed; the reaction was in any case calculated to be almost thermoneutral.
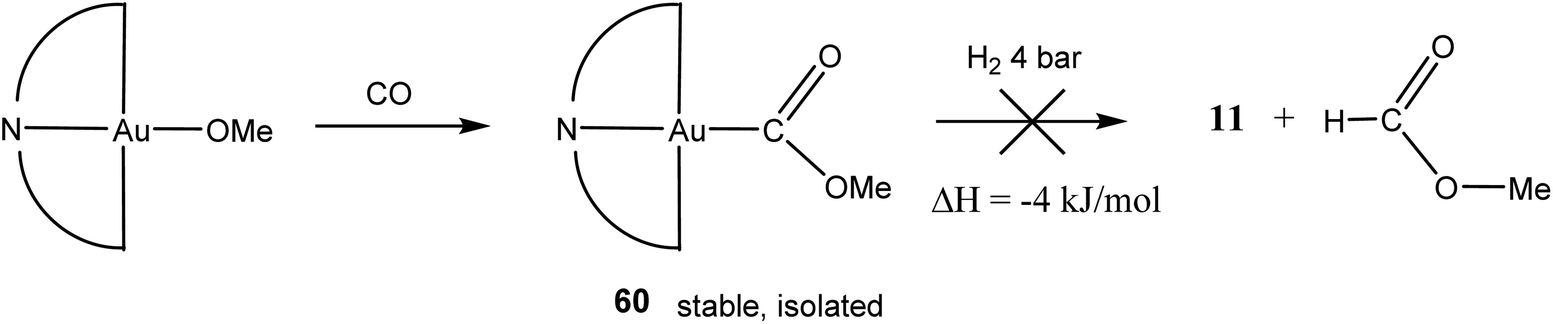 | ||
| Scheme 30 | ||
In addition, the reaction of 11 with CO also produced the CO2-bridged complex 61, which is most probably formed in an off-cycle reaction by condensation of intermediate 59 with excess hydroxide. Compound 61 was structurally characterised; it represents the first CO2 complex of gold in any oxidation state. On heating to 80–120 °C, 61 was found to release CO2 in a reductive elimination process, to give the thermally stable Au(II) complex 14 (Scheme 31).
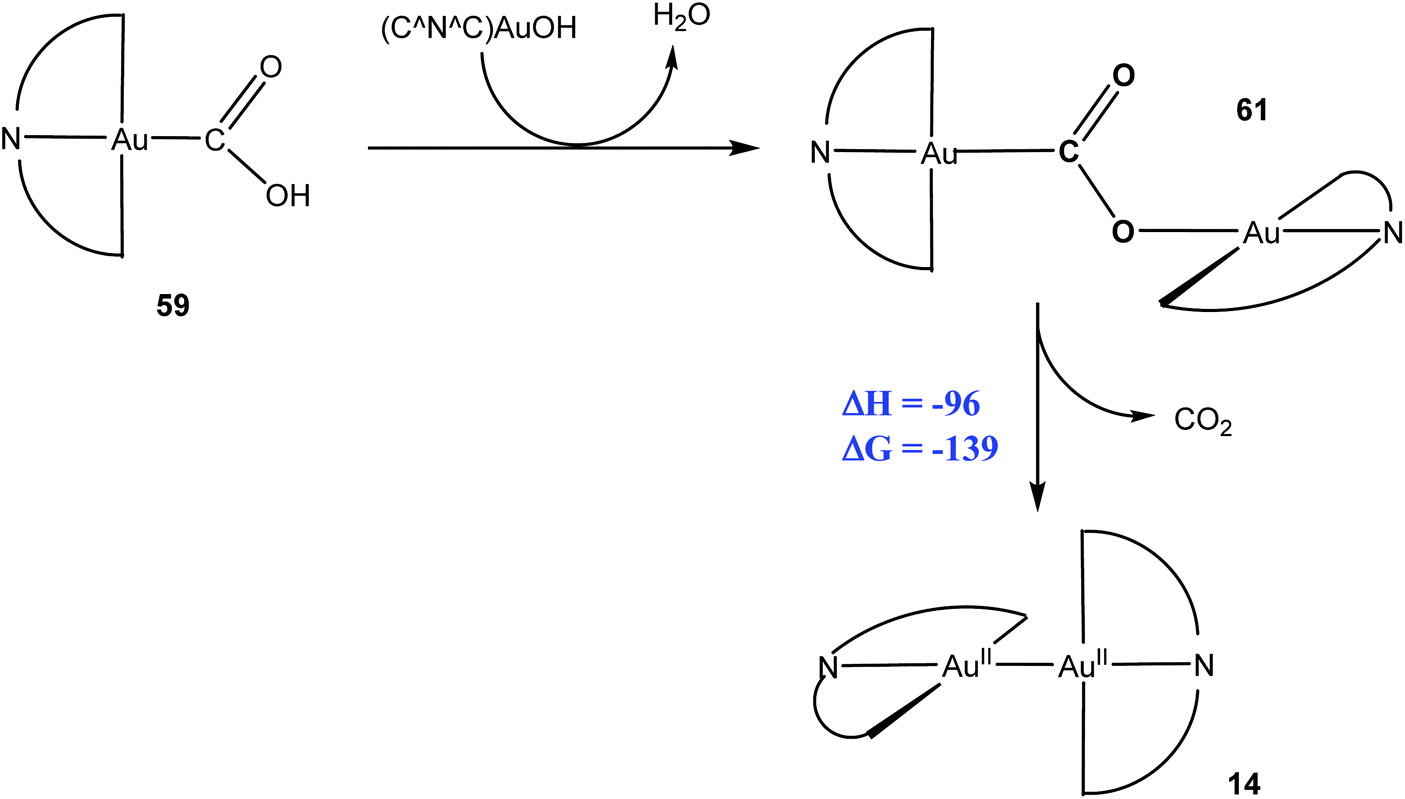 | ||
| Scheme 31 Thermally induced reductive CO2 elimination from a di-gold complex. Enthalpy and Gibbs free energy values (kJ mol−1) as calculated by DFT (T = 298.15 K). | ||
The results show that there is a very significant difference in terms of susceptibility to nucleophilic attack between CO complexes of Au(III) and Pt(II). This is in spite of the fact that 58 and, for example, cis-PtCl2(CO)2 have very similar νCO frequencies, which could be taken to mean that the C atoms in the CO ligands should possess similar degrees of electrophilicity. Drastic differences were however revealed in the donation/back-donation ratio (d/b), as estimated by charge decomposition analysis. [(C^N^C)Au(CO)]+ shows a d/b ratio of 2.26, compared to a value of only 0.65 for PtCl2(CO)2 (Scheme 32).119 Evidently back-donation, even for CO complexes with high frequencies, is much more prominent in Pt(II) than in Au(III). The study also demonstrates that it is the d/b ratio, rather than the IR frequency, which provides the more informative guide to chemical reactivity.
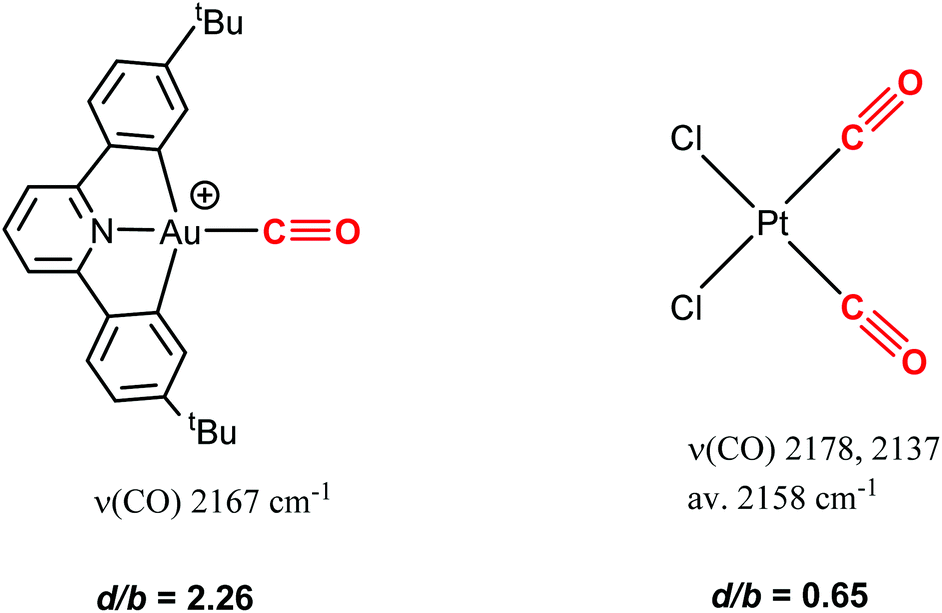 | ||
| Scheme 32 CO stretching frequencies (Au: CH2Cl2; Pt: benzene) and donation/back-donation ratios in Au and Pt carbonyls. | ||
Conclusion
In summary, this Perspective has highlighted some recent advances in gold chemistry, alongside some of its peculiarities. The characteristics that set gold apart from other metals, and even from its neighbour in the Periodic Table, platinum, are the high electronegativity, with the resulting highly covalent character of the Au–H and Au–C bonds, and the order of bond strengths: Au–H > Au–C ≥ Au–O, which runs counter to experience with other transition metals. These special characteristics need to be kept in mind in mechanistic proposals for gold catalysed reactions. The last few years have also seen the isolation of some long sought-after representatives of important compound classes; in particular, the first examples of gold(III) hydride, olefin and CO complexes. Bonding studies have highlighted the absence of π-bonding contributions to and from the metal centre, which appears to be the overriding bonding characteristic of gold(III). This applies to Au–O bonds (absence of ligand-to-metal donation) as well as to alkene and CO complexes (absence of metal-to-ligand back-bonding). In the case of CO complexes this has remarkable consequences for reactivity, and illustrates an unexpectedly drastic difference between gold and platinum.These advances notwithstanding, many types of organometallic complexes that are commonplace for other d8 metals are still missing, such as complexes with labile sites cis to H, CO or alkenes, also dihydrides, dicarbonyls and alkyne complexes, as well as cationic and anionic hydrides and carbonyls, hydrido and carbonyl phosphine complexes, and so forth. The synthesis of these much more reactive species will be both challenging and instructive, not least since these are the types of compounds that are likely to be realistic intermediates in catalytic cycles. The chemistry of gold can be expected to provide some intriguing surprises yet.
Acknowledgements
This work was supported by the European Research Council, the Leverhulme Trust and Johnson Matthey PLC. M. B. is an ERC Advanced Investigator Award holder (grant no. 338944 – GOCAT). D.A.R. thanks UEA for a Ph.D. studentship and a Katritzky scholarship.References
- H. G. Raubenheimer and H. Schmidbaur, The Late Start and Amazing Upswing in Gold Chemistry, J. Chem. Educ., 2014, 91, 2024 CrossRef CAS.
- Special issue: Chem. Rev., 2008, 108(8), 3223–3442.
- C. M. Friend and A. S. K. Hashmi (eds.), “Gold Catalysis”, Acc. Chem. Res., 2014, 47, 729–978.
- (a) A. S. K. Hashmi and R. Matthias, Chem. Soc. Rev., 2008, 37, 1766 RSC; (b) A. S. K. Hashmi, Angew. Chem., Int. Ed., 2010, 49, 5232 CrossRef CAS PubMed; (c) R. Matthias and A. S. K. Hashmi, Chem. Soc. Rev., 2012, 41, 2448 RSC; (d) I. Braun, A. M. Asiri and A. S. K. Hashmi, ACS Catal., 2013, 3, 1902 CrossRef CAS; (e) A. S. K. Hashmi, Top. Org. Chem., 2013, 44, 143 Search PubMed; (f) T. Lauterbach, A. M. Asiri and A. S. K. Hashmi, Adv. Organomet. Chem., 2014, 62, 261 CrossRef CAS.
- B. Ranieri, I. Escofet and A. M. Echavarren, Org. Biomol. Chem., 2015, 13, 7103 CAS.
- N. D. Shapiro and F. D. Toste, Synlett, 2010, 675 CAS.
- L. P. Liu and G. B. Hammond, Chem. Soc. Rev., 2012, 41, 3129 RSC.
- P. H. S. Paioti and A. Aponick, Top. Curr. Chem., 2015, 357, 63 CrossRef PubMed.
- A. S. K. Hashmi and R. Matthias, Chem. Commun., 2011, 47, 6536 RSC.
- A. Corma, A. Leyva-Pérez and M. J. Sabatier, Chem. Rev., 2011, 111, 1657 CrossRef CAS PubMed.
- Y. H. Wang, M. E. Muratore and A. M. Echavarren, Chem. – Eur. J., 2015, 21, 7332 CrossRef CAS PubMed.
- (a) H. Schmidbaur and A. Schier, Chem. Soc. Rev., 2008, 37, 1931 RSC; (b) H. Schmidbaur and A. Schier, Chem. Soc. Rev., 2012, 41, 370 RSC.
- D. Weber and M. R. Gagné, Top. Curr. Chem., 2015, 357, 167 CrossRef PubMed.
- H. Schmidbaur and A. Schier, Z. Naturforsch., B: Chem. Sci., 2011, 66, 329 CrossRef CAS.
- M. Q. Jia and M. Bandini, ACS Catal., 2015, 5, 1638 CrossRef CAS.
- S. M. Inamdar, A. Konala and N. T. Patil, Chem. Commun., 2014, 50, 15124 RSC.
- For early examples of catalysis by AuCl3 see: (a) A. S. K. Hashmi, L. Schwarz, J. H. Choi and T. M. Frost, Angew. Chem., Int. Ed., 2000, 39, 2285 CrossRef CAS; (b) A. S. K. Hashmi, T. M. Frost and J. W. Bats, J. Am. Chem. Soc., 2000, 122, 11553 CrossRef CAS.
- Review: H. Schmidbaur and A. Schier, Arabian J. Sci. Eng., 2012, 37, 1187 CrossRef CAS.
- (a) P. Pyykkö, Angew. Chem., Int. Ed., 2004, 43, 4412 CrossRef PubMed; (b) P. Pyykkö, Inorg. Chim. Acta, 2005, 358, 4113 CrossRef.
- A. Leyva-Pérez and A. Corma, Angew. Chem., Int. Ed., 2012, 51, 614 CrossRef PubMed.
- (a) A. Fürstner and P. W. Davies, Angew. Chem., Int. Ed., 2007, 46, 3410 CrossRef PubMed; (b) A. Fürstner, Chem. Soc. Rev., 2009, 38, 3208 RSC; (c) A. Fürstner, Acc. Chem. Res., 2014, 47, 925 CrossRef PubMed.
- N. Savjani, S. Bew, D. L. Hughes, S. J. Lancaster and M. Bochmann, Organometallics, 2012, 31, 2534 CrossRef CAS.
- B. Guan, D. Xing, G. Cai, X. Wan, N. Yu, Z. Fang, L. Yang and Z. Shi, J. Am. Chem. Soc., 2005, 127, 18004 CrossRef CAS PubMed.
- A. S. K. Hashmi and G. J. Hutchings, Angew. Chem., Int. Ed., 2006, 45, 7896 CrossRef PubMed.
- N. Carrera, N. Savjani, J. Simpson, D. L. Hughes and M. Bochmann, Dalton Trans., 2011, 40, 1016 RSC . By contrast, gold(I) nacnac complexes with fluoroalkyl substituents are thermally stable: N. Savjani, M. Schormann and M. Bochmann, Polyhedron, 2012, 38, 137 CrossRef CAS.
- (a) A. S. K. Hashmi, M. C. Blanco, D. Fischer and J. W. Bats, Eur. J. Org. Chem., 2006, 1387 CrossRef CAS; (b) A. S. K. Hashmi, C. Lothschütz, M. Ackermann, R. Doepp, S. Anantharaman, B. Marchetti, H. Bertagnolli and F. Rominger, Chem. – Eur. J., 2010, 16, 801 CrossRef PubMed.
- O. A. Egorova, H. Seo, Y. Kim, D. Moon, Y. M. Rhee and K. H. Ahn, Angew. Chem., Int. Ed., 2011, 50, 11446 CrossRef CAS PubMed.
- W. C. Zeise, Poggendorf's Ann. Phys., 1827, 9, 632 Search PubMed.
- The structure of Zeise's salt was determined much later: (a) J. A. Wunderlich and D. P. Mellor, Acta Crystallogr., 1954, 7, 130 CrossRef CAS; (b) G. B. Bokii and G. A. Kukina, Zh. Strukt. Khim., 1965, 5, 706 Search PubMed; (c) J. A. J. Jarvis, B. T. Kilbourn and P. G. Owsten, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1971, 27, 366 CrossRef CAS; (d) M. Black, R. H. B. Mais and P. G. Owston, Acta Crystallogr., Sect. A: Cryst. Phys., Diffr., Theor. Gen. Cryst., 1969, 25, 1753 CrossRef CAS; (e) R. A. Love, T. F. Koetzle, G. J. B. Williams, L. C. Andrews and R. Bau, Inorg. Chem., 1975, 14, 2653 CrossRef CAS.
- (a) J. Chatt, L. A. Duncanson and B. L. Shaw, Proc. Chem. Soc., 1957,(12), 343 CAS; (b) P. G. Owen, J. M. Partridge and J. M. Rowe, Acta Crystallogr., 1960, 13, 246–252 CrossRef; (c) J. Chopoorian, R. S. Nyholm and J. Lewis, Nature, 1961, 190, 528 CrossRef CAS.
- H. Schmidbaur, H. G. Raubenheimer and L. Dobrzanska, Chem. Soc. Rev., 2014, 43, 345 RSC.
- E. Wiberg and H. Neumaier, Inorg. Nucl. Chem. Lett., 1965, 1, 35 CrossRef CAS.
- M. Green, A. G. Orpen, I. D. Salter and F. G. A. Stone, J. Chem. Soc., Chem. Commun., 1982, 813 RSC.
- H. Lehner, D. Matt, P. S. Pregosin, L. M. Venanzi and A. Albinati, J. Am. Chem. Soc., 1982, 104, 6825 CrossRef CAS.
- (a) X. F. Wang and L. Andrews, J. Am. Chem. Soc., 2001, 123, 12899 CrossRef CAS PubMed; (b) X. F. Wang and L. Andrews, J. Phys. Chem. A, 2002, 106, 3744 CrossRef CAS; (c) L. Andrews and X. F. Wang, J. Am. Chem. Soc., 2003, 125, 11751 CrossRef CAS PubMed; (d) X. F. Wang, L. Andrews, L. Manceron and C. Marsden, J. Phys. Chem. A, 2003, 107, 8492 CrossRef CAS; (e) X. F. Wang and L. Andrews, Angew. Chem., Int. Ed., 2003, 42, 5201 CrossRef CAS PubMed; (f) L. Andrews, X. F. Wang, L. Manceron and K. Balasubramanian, J. Phys. Chem. A, 2004, 108, 2936 CrossRef CAS.
- N. Khairallah, R. A. J. O'Hair and M. I. Bruce, Dalton Trans., 2006, 3699 RSC.
- H. Ito, T. Saito, T. Miyahara, C. Zhong and M. Sawamura, Organometallics, 2009, 28, 4829 CrossRef CAS.
- A. Escalle, G. Mora, F. Gagosz, N. Mézailles, X. F. Le Goff, Y. Jean and P. Le Floch, Inorg. Chem., 2009, 48, 8415 CrossRef CAS PubMed.
- E. Y. Tsui, P. Müller and J. P. Sadighi, Angew. Chem., Int. Ed., 2008, 47, 8937 CrossRef CAS PubMed.
- S. Gaillard, A. M. Z. Slawin and S. P. Nolan, Chem. Commun., 2010, 46, 2742 RSC.
- N. Phillips, T. Dodson, R. Tirfoin, J. I. Bates and S. Aldridge, Chem. – Eur. J., 2014, 20, 16721 CrossRef CAS PubMed.
- H. B. Lv, J. H. Zhan, Y. B. Cai, Y. Yu, B. Wang and J. L. Zhang, J. Am. Chem. Soc., 2012, 134, 16216 CrossRef CAS PubMed.
- D.-A. Roşca, J. Fernandez-Cestau, D. L. Hughes and M. Bochmann, Organometallics, 2015, 34, 2098 CrossRef PubMed.
- G. Klatt, R. Xu, M. Pernpointner, L. Molinari, T. Q. Hung, F. Rominger, A. S. K. Hashmi and H. Köppel, Chem. – Eur. J., 2013, 19, 3954 CrossRef CAS PubMed.
- G. Ung and G. Bertrand, Angew. Chem., Int. Ed., 2013, 52, 11388 CrossRef CAS PubMed.
- A. Comas-Vives and G. Ujaque, J. Am. Chem. Soc., 2013, 135, 1295 CrossRef CAS PubMed.
- J. Xie, H. Li, J. C. Zhou, Y. Cheng and C. J. Zhu, Angew. Chem., Int. Ed., 2012, 51, 1252 CrossRef CAS PubMed.
- S. Orbisaglia, B. Jaques, P. Braunstein, D. Hueber, P. Pale, A. Blanc and P. De Frémont, Organometallics, 2013, 32, 4153 CrossRef CAS.
- B. Alcaide, P. Almendros, T. Martinez del Campo and I. Fernandez, Chem. Commun., 2011, 47, 9054 RSC.
- A. Comas-Vives, C. González-Arellano, A. Corma, M. Iglesias, F. Sánchez and G. Ujaque, J. Am. Chem. Soc., 2006, 128, 4756 CrossRef CAS PubMed.
- (a) M. C. Gimeno and A. Laguna, in Comprehensive Coordination Chemistry II, ed. J. A. McCleverty and T. J. Meyer, Elsevier, 2003, vol. 6, p. 996 Search PubMed; (b) J. Vicente, M. T. Chicote, M. D. Bermudez, P. G. Jones, C. Fittschen and G. M. Sheldrick, J. Chem. Soc., Dalton Trans., 1986, 2361 RSC.
- D.-A. Roşca, D. A. Smith and M. Bochmann, Chem. Commun., 2012, 48, 7247 RSC.
- D.-A. Roşca, D. A. Smith, D. L. Hughes and M. Bochmann, Angew. Chem., Int. Ed., 2012, 51, 10643 CrossRef PubMed.
- P. Hrobárik, V. Hrobáriková, F. Meier, M. Repiský, S. Komorovský and M. Kaupp, J. Phys. Chem. A, 2011, 115, 5654 CrossRef PubMed.
- S. J. Lancaster, A. Rodriguez, A. Lara-Sanchez, M. D. Hannant, D. A. Walker, D. L. Hughes and M. Bochmann, Organometallics, 2002, 21, 451 CrossRef CAS.
- D. A. Smith, D.-A. Roşca and M. Bochmann, Organometallics, 2012, 31, 5988 CrossRef.
- D.-A. Roşca, PhD Thesis, University of East Anglia, 2014.
- D. Zopes, C. Hegemann, W. Tyrra and S. Mathur, Chem. Commun., 2012, 48, 8805 RSC.
- T. Dann, D.-A. Roşca, G. G. Wildgoose, J. A. Wright and M. Bochmann, Chem. Commun., 2013, 49, 10169 RSC.
- D.-A. Roşca, J. A. Wright, D. L. Hughes and M. Bochmann, Nat. Commun., 2013, 4, 2167 Search PubMed.
- X.-G. Xiong and P. Pyykkö, Chem. Commun., 2013, 49, 2103 RSC.
- C. Della Pina, E. Falletta, L. Prati and M. Rossi, Chem. Soc. Rev., 2008, 37, 2077 RSC.
- D. Widman and R. J. Behm, Acc. Chem. Res., 2014, 47, 740 CrossRef PubMed.
- (a) C. Bürgel, N. M. Reilly, G. E. Johnson, R. Mitrić, M. L. Kimble, A. W. Castleman and V. Bonaćić-Koutecky, J. Am. Chem. Soc., 2008, 130, 1694 CrossRef PubMed; (b) I. X. Green, W. Tang, M. Neurock and J. T. Yates Jr., Science, 2011, 333, 736 CrossRef CAS PubMed.
- (a) B. T. Teng, J. J. Lang, X.-D. Wen, C. Zhang, M. Fan and H. G. Harris, J. Phys. Chem. C, 2013, 117, 18986 CrossRef CAS; (b) K. L. Howard and D. J. Willcock, Faraday Discuss., 2011, 152, 135 RSC; (c) C. T. Campbell, J. C. Sharp, Y. X. Yao, E. M. Karp and T. L. Silbaugh, Faraday Discuss., 2011, 152, 227 RSC; (d) Y. G. Wang, D. Mei, V. A. Glezakou, J. Li and R. Rousseau, Nat. Commun., 2015, 6, 6511 CrossRef CAS PubMed.
- S. J. Freakley, M. Piccinini, J. K. Edwards, E. N. Ntainjua, J. A. Moulijn and G. J. Hutchings, ACS Catal., 2013, 3, 487 CrossRef CAS.
- (a) C. Sivadinarayana, T. V. Choudhary, L. L. Daemen, J. Eckert and D. W. Goodman, J. Am. Chem. Soc., 2004, 126, 38 CrossRef CAS PubMed; (b) C. R. Chang, X. F. Yang, B. Long and J. Li, ACS Catal., 2013, 3, 1693 CrossRef CAS; (c) N. Ohta, K. Nomura and I. Yagi, J. Phys. Chem. C, 2012, 116, 14390 CrossRef CAS.
- K. Bobuatong, S. Karanjit, R. Fukuda, M. Ehara and H. Sakurai, Phys. Chem. Chem. Phys., 2012, 14, 3103 RSC.
- R. Pal, L.-M. Wang, Y. Pei, L.-S. Wang and X. C. Zeng, J. Am. Chem. Soc., 2012, 134, 9438 CrossRef CAS PubMed.
- A. P. Woodham and A. Fielicke, Angew. Chem., Int. Ed., 2014, 53, 6554 CrossRef CAS PubMed.
- (a) A. M. Joshi, W. N. Delgass and K. T. Thomson, J. Phys. Chem. B, 2006, 110, 2572 CrossRef CAS PubMed; (b) A. Pulido, M. Boronat and A. Corma, J. Phys. Chem. C, 2012, 116, 1955 Search PubMed. By contrast, the oxidation of vinyl ethers with O2 in the presence of Au(I) complexes to give alkyl hydroperoxides has been shown to proceed via a radical mechanism not involving Au–OOH species: A. S. K. Hashmi, M. C. Blanco Jaimes, A. M. Schuster and F. Rominger, J. Org. Chem., 2012, 77, 6394 CrossRef CAS PubMed.
- R. S. Hay-Motherwell, G. Wilkinson, B. Hussain-Bates and M. B. Hursthouse, Polyhedron, 1993, 12, 2009 CrossRef CAS.
- E. Poverenov, I. Efremenko, A. I. Frenkel, Y. Ben-David, L. J. W. Shimon, G. Leitus, L. Konstantinovski, J. M. L. Martin and D. Milstein, Nature, 2008, 455, 1093 CrossRef CAS.
- K. P. O'Halloran, C. Zhao, N. S. Ando, A. J. Schultz, T. F. Koetzle, P. M. B. Piccoli, B. Hedman, K. O. Hodgson, E. Bobyr, M. L. Kirk, S. Knottenbelt, E. C. Depperman, B. Stein, T. M. Anderson, R. Cao, Y. V. Geletii, K. I. Hardcastle, D. G. Musaev, W. A. Neiwert, X. Fang, K. Morokuma, S. Wu, P. Kögerler and C. L. Hill, Inorg. Chem., 2012, 51, 7025 CrossRef PubMed.
- C.-P. Zhang and D. A. Vicic, Organometallics, 2012, 31, 7812 CrossRef CAS.
- N. Ibrahim, M. H. Vilhelmsen, M. Pernpointner, F. Rominger and A. S. K. Hashmi, Organometallics, 2013, 32, 2576 CrossRef CAS.
- (a) G. Jander and G. Krien, Z. Anorg. Allg. Chem., 1960, 304, 164 CrossRef CAS; (b) P. G. Jones and G. M. Sheldrick, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1984, 40, 1776 CrossRef; (c) P. G. Jones, R. Schelbach and E. Schwarzmann, Z. Naturforsch., B: Anorg. Chem. Org. Chem., 1987, 42, 522 CAS.
- (a) M. G. Miles, G. E. Glass and R. S. Tobias, J. Am. Chem. Soc., 1966, 88, 5738 CrossRef CAS; (b) G. E. Glass, J. H. Konnert, M. G. Miles, D. Britton and R. S. Tobias, J. Am. Chem. Soc., 1968, 90, 1131 CrossRef CAS; (c) S. J. Harris and R. S. Tobias, Inorg. Chem., 1969, 8, 2259 CrossRef CAS; (d) W. M. Scovell and R. S. Tobias, Inorg. Chem., 1970, 9, 945 CrossRef CAS.
- J. Vicente, M. D. Bermudez, F. J. Carrion and P. G. Jones, J. Organomet. Chem., 1996, 508, 53 CrossRef CAS.
- (a) E. H. Braye, W. Hübel and I. Caplier, J. Am. Chem. Soc., 1961, 83, 4406 CrossRef CAS; (b) M. Peteau-Boisdenghien, J. Meunier-Piret and M. van Meerssche, Cryst. Struct. Commun., 1975, 4, 375 CAS.
- M. A. Cinellu, G. Minghetti, M. V. Pinna, S. Stroccoro, A. Zucca, M. Manassero and M. Sansoni, J. Chem. Soc., Dalton Trans., 1998, 1735 RSC.
- M. A. Cinellu, G. Minghetti, M. V. Pinna, S. Stoccoro, A. Zucca and M. Manassero, J. Chem. Soc., Dalton Trans., 2000, 1261 RSC.
- B. Pitteri, G. Marangoni, F. Visentin, T. Bobbo, V. Bertolasi and P. Gilli, J. Chem. Soc., Dalton Trans., 1999, 677 RSC.
- M. A. Cinellu, G. Minghetti, M. V. Pinna, S. Stoccoro, A. Zucca and M. Manassero, J. Chem. Soc., Dalton Trans., 1999, 2823 RSC.
- A. Collado, A. Gomez-Suarez, Y. Oonishi, A. M. Z. Slawin and S. P. Nolan, Chem. Commun., 2013, 49, 10745 RSC.
- (a) L. Boisvert and K. I. Goldberg, Acc. Chem. Res., 2012, 45, 899 CrossRef CAS PubMed; (b) D. D. Wick and K. I. Goldberg, J. Am. Chem. Soc., 1999, 121, 11900 CrossRef CAS; (c) J. M. Keith, R. P. Muller, R. A. Kemp, K. I. Goldberg, W. A. Goddard III and J. Oxgaard, Inorg. Chem., 2006, 45, 9631 CrossRef CAS PubMed.
- R. Huacuja, D. J. Graham, C. M. Fafard, C.-H. Chen, B. M. Foxman, D. E. Herbert, G. Alliger, C. M. Thomas and O. V. Ozerov, J. Am. Chem. Soc., 2011, 133, 3820 CrossRef CAS PubMed.
- H. Schmidbaur and A. Schier, Organometallics, 2010, 29, 2 CrossRef CAS.
- A. J. Chalk, J. Am. Chem. Soc., 1964, 86, 4733 CrossRef CAS.
- R. Hüttel and H. Dietl, Angew. Chem., Int. Ed. Engl., 1965, 4, 438 CrossRef.
- (a) H. V. R. Dias, M. Finchini, T. R. Cundaru and C. F. Campana, Angew. Chem., Int. Ed., 2008, 47, 556 CrossRef CAS PubMed; (b) J. Schäfer, D. Himmel and I. Krossing, Eur. J. Inorg. Chem., 2013, 2712 CrossRef.
- T. N. Hooper, C. P. Butts, M. Green, M. F. Haddow, J. E. McGrady and C. A. Russell, Chem. – Eur. J., 2009, 15, 12196 CrossRef CAS PubMed.
- M. Fianchini, H. Dai and H. V. R. Dias, Chem. Commun., 2009, 6373 RSC.
- (a) M. A. Cinellu, M. Minghetti, S. Stoccoro, A. Zucca and M. Manassero, Chem. Commun., 2004, 1618 RSC; (b) M. A. Cinellu, G. Minghetti, F. Cocco, S. Stoccoro, A. Zucca, M. Manassero and M. Arca, Dalton Trans., 2006, 5703 RSC.
- H. V. R. Dias and J. Wu, Organometallics, 2012, 31, 1511 CrossRef CAS.
- R. E. M. Brooner and R. A. Widenhoefer, Angew. Chem., Int. Ed., 2013, 52, 11714 CrossRef CAS PubMed.
- T. N. Hooper, M. Green, J. E. McGrady, J. R. Patel and C. A. Russell, Chem. Commun., 2009, 3877 RSC.
- (a) T. J. Brown, M. G. Dickens and R. A. Widenhoefer, J. Am. Chem. Soc., 2009, 131, 6350 CrossRef CAS PubMed; (b) T. J. Brown, M. G. Dickens and R. A. Widenhoefer, Chem. Commun., 2009, 6451 RSC.
- (a) R. E. M. Brooner and R. A. Widenhoefer, Organometallics, 2011, 30, 3182 CrossRef CAS; (b) T. J. Brown, B. D. Robertson and R. A. Widenhoefer, J. Organomet. Chem., 2014, 758, 25 CrossRef CAS.
- (a) R. A. Sanguramath, T. N. Hooper, C. P. Butts, M. Green, J. E. McGrady and C. A. Russell, Angew. Chem., Int. Ed., 2011, 50, 7592 CrossRef CAS PubMed; (b) Y. Zhu, C. S. Day and A. C. Jones, Organometallics, 2012, 31, 7332 CrossRef CAS.
- R. A. Sanguramath, S. K. Patra, M. Green and C. A. Russell, Chem. Commun., 2012, 48, 1060 RSC.
- H. G. Raubenheimer and H. Schmidbaur, S. Afr. J. Sci., 2011, 107, 31 CAS.
- P. Schulte and U. Behrens, Chem. Commun., 1998, 1633 RSC.
- S. Flügge, A. Anoop, R. Goddard, W. Thiel and A. Fürstner, Chem. – Eur. J., 2009, 15, 8558 CrossRef PubMed.
- M. A. Celik, C. Dash, V. A. K. Adiraju, A. Das, M. Yousufuddin, G. Frenking and H. V. R. Dias, Inorg. Chem., 2013, 52, 729 CrossRef CAS PubMed.
- J. Wu, P. Kroll and H. V. R. Dias, Inorg. Chem., 2009, 48, 423 CrossRef CAS PubMed.
- T. N. Hooper, M. Green and C. A. Russell, Chem. Commun., 2010, 46, 2313 RSC.
- (a) A. S. K. Hashmi, T. Lauterbach, P. Nösel, M. H. Vilhelmsen, M. Rudolph and F. Rominger, Chem. – Eur. J., 2013, 19, 1058 CrossRef CAS PubMed; (b) M. Wieteck, Y. Tokimizu, M. Rudolph, F. Rominger, H. Ohno, N. Fujii and A. S. K. Hashmi, Chem. – Eur. J., 2014, 20, 16331 CrossRef CAS PubMed.
- D. Zuccaccia, L. Belpassi, L. Rocchigiani, F. Tarantelli and A. Macchioni, Inorg. Chem., 2010, 49, 3080 CrossRef CAS PubMed.
- L. Biasiolo, M. Trinchillo, P. Belanzoni, L. Belpassi, V. Busico, G. Ciancaleoni, A. D'Amora, A. Macchioni, F. Tarantelli and D. Zuccaccia, Chem. – Eur. J., 2014, 20, 14594 CrossRef CAS PubMed.
- (a) A. Das, C. Dash, M. Yousufuddin, M. A. Celik, G. Frenking and H. V. R. Dias, Angew. Chem., Int. Ed., 2012, 51, 3940 CrossRef CAS PubMed; (b) A. Das, C. Dash, M. A. Celik, M. Yousufuddin, G. Frenking and H. V. R. Dias, Organometallics, 2013, 32, 3135 CrossRef CAS.
- R. Casado, M. Contel, M. Laguna, P. Romero and S. Sanz, J. Am. Chem. Soc., 2003, 125, 11925 CrossRef CAS PubMed.
- R. Usón, A. Laguna and M. L. Arese, Synth. React. Inorg. Met.-Org. Chem., 1984, 14, 557 CrossRef.
- N. Savjani, D.-A. Roşca, M. Schormann and M. Bochmann, Angew. Chem., Int. Ed., 2013, 52, 874 CrossRef CAS PubMed.
- (a) D. B. Powell and N. Sheppard, J. Chem. Soc., 1960, 2519 RSC; (b) R. Cramer, Inorg. Chem., 1965, 4, 445 CrossRef CAS.
- M. H. Chisholm, H. C. Clark, L. E. Manzer and J. B. Stothers, J. Am. Chem. Soc., 1972, 94, 5087 CrossRef CAS.
- M. H. Chisholm and H. C. Clark, Inorg. Chem., 1973, 12, 991 CrossRef CAS.
- W. Partenheimer, J. Am. Chem. Soc., 1976, 98, 2779 CrossRef CAS.
- J. A. Wright, personal communication. For details of computational methods see ref. 52.
- E. Langseth, M. L. Scheuermann, D. Balcells, W. Kaminsky, K. I. Goldberg, O. Eisenstein, R. H. Heyn and M. Tilset, Angew. Chem., Int. Ed., 2013, 52, 1660 CrossRef CAS PubMed.
- E. Langseth, A. Nova, E. A. Tråseth, F. Rise, S. Øien, R. H. Heyn and M. Tilset, J. Am. Chem. Soc., 2014, 136, 10104 CrossRef CAS PubMed.
- F. Rekhroukh, R. Brousses, A. Amgoune and D. Bourissou, Angew. Chem., Int. Ed., 2015, 54, 1266 CrossRef CAS PubMed.
- By contrast, the polymerization of styrene and vinyl ethers in the presence of AuCl/AgSbF6 mixtures proceeds most probably via a carbocationic mechanism: A. S. K. Hashmi, S. Schäfer, V. Göker, C. D. Eisenbach, K. Dirnberger, Z. Zhao-Karger and P. Crewdson, Aust. J. Chem., 2014, 67, 500 CrossRef CAS.
- W. Manchot and H. Gall, Ber. Dtsch. Chem. Ges., 1925, 58, 2175 CrossRef.
- (a) M. S. Kharash and H. S. Isbell, J. Am. Chem. Soc., 1930, 52, 2919 CrossRef; (b) D. Belli Dell'Amico and F. Calderazzo, Gazz. Chim. Ital., 1973, 103, 1099 Search PubMed; (c) D. Belli Dell'Amico and F. Calderazzo, Inorg. Synth., 1986, 24, 236 CrossRef.
- (a) F. Calderazzo, J. Organomet. Chem., 1990, 400, 303 CrossRef CAS; (b) D. Belli Dell'Amico, F. Calderazzo, P. Robino and A. Segre, J. Chem. Soc., Dalton Trans., 1991, 3017 RSC.
- H. Willner and F. Aubke, Inorg. Chem., 1990, 29, 2195 CrossRef CAS.
- H. Willner, J. Schaebs, G. Hwang, F. Mistry, R. Jones, J. Trotter and F. Aubke, J. Am. Chem. Soc., 1992, 114, 8972 CrossRef CAS.
- H. V. R. Dias and W. Jin, Inorg. Chem., 1996, 35, 3687 CrossRef CAS.
- S. Martinez-Salvador, J. Forniés, A. Martín and B. Menjon, Angew. Chem., Int. Ed., 2011, 50, 6571 CrossRef CAS PubMed.
- M. Adelhelm, W. Bacher, E. G. Höhn and E. Jacob, Chem. Ber., 1991, 124, 1559 CrossRef CAS.
- P. K. Hurlburt, J. J. Rack, J. S. Luck, S. F. Dec, J. D. Webb, O. P. Anderson and S. H. Strauss, J. Am. Chem. Soc., 1994, 116, 10003 CrossRef CAS.
- R. Küster and K. Seppelt, Z. Anorg. Allg. Chem., 2000, 626, 236 CrossRef.
- (a) C. Dash, P. Kroll, M. Yousufuddin and H. V. R. Dias, Chem. Commun., 2011, 47, 4478 RSC; (b) H. V. R. Dias, C. Dash, M. Yousufuddin, M. A. Celik and G. Frenking, Inorg. Chem., 2011, 50, 4253 CrossRef CAS PubMed; (c) M. A. Celik, C. Dash, V. A. K. Adiraju, A. Dias, M. Yousufuddin, G. Frenking and H. V. R. Dias, Inorg. Chem., 2013, 52, 729 CrossRef CAS PubMed.
- G. Ciancaleoni, N. Scafuri, G. Bistoni, A. Macchioni, F. Tarantelli, D. Zuccaccia and L. Belpassi, Inorg. Chem., 2014, 53, 9907 CrossRef CAS PubMed.
- Reviews: (a) S. H. Strauss, J. Chem. Soc., Dalton Trans., 2000, 1 RSC; (b) A. J. Lupinetti, S. H. Strauss and G. Frenking, Prog. Inorg. Chem., 2001, 49, 1 CrossRef CAS.
- Review: H. Willner and F. Aubke, Organometallics, 2003, 22, 3612 CrossRef CAS.
- M. Joost, L. Estévez, S. Mallet-Ladeira, K. Miqueu, A. Amgoune and D. Bourissou, Angew. Chem., Int. Ed., 2014, 53, 14512 CrossRef CAS PubMed.
- The reverse reaction, i.e. the extrusion of CO from an Au(I) acyl intermediate, has recently been postulated: J. Bucher, T. Stößer, M. Rudolph, F. Rominger and A. S. K. Hashmi, Angew. Chem., Int. Ed., 2015, 54, 1666 CrossRef CAS PubMed.
- W. Grünert, D. Großmann, H. Noei, M.-M. Pohl, I. Sinev, A. De Toni, Y. Wang and M. Muhler, Angew. Chem., Int. Ed., 2014, 53, 3245 CrossRef PubMed.
- J. Donau, Monatsh. Chem., 1905, 26, 525 CrossRef CAS.
- L. A. Pretzer, Q. X. Nguyen and M. S. Wong, J. Phys. Chem. C, 2010, 114, 21226 CAS.
- (a) P. Schützenberger, Ann. Chim. Phys., Ser. 4, 1868, 15, 100 Search PubMed; (b) P. Schützenberger, Ann. Chim. Phys. Ser. 4, 1870, 21, 350 Search PubMed.
- D.-A. Roşca, J. Fernandez-Cestau, J. Morris, J. A. Wright and M. Bochmann, Sci. Adv., 2015, 1, e1500761 Search PubMed.
- T. L. LeValley, A. R. Richard and M. H. Fan, Int. J. Hydrogen Energy, 2014, 30, 16983 CrossRef.
- (a) M. Gonzalez-Castaño, T. R. Reina, S. Ivanova, M. A. Centeno and J. A. Odriozola, J. Catal., 2014, 314, 1 CrossRef; (b) J. A. Rodriguez, S. D. Senanayake, D. Stacchiola, P. Liu and J. Hrbek, Acc. Chem. Res., 2014, 47, 773 CrossRef CAS PubMed; (c) A. Hussain, J. Gracia, B. E. Nieuwenhuys and J. W. Niemantsverdriet, ChemCatChem, 2013, 5, 2479 CrossRef CAS.
- Q. Fu, H. Saltsburg and M. Flytzani-Stephanopoulos, Science, 2003, 301, 935 CrossRef CAS PubMed.
- M. Shekhar, J. Wang, W.-S. Lee, W. D. Williams, S. M. Kim, E. A. Stach, J. T. Miller, W. N. Delgass and F. H. Ribeiro, J. Am. Chem. Soc., 2012, 134, 4700 CrossRef CAS PubMed.
- G. Bond, Gold Bull., 2009, 42, 337 CrossRef CAS.
- W. Song and E. J. M. Hensen, ACS Catal., 2014, 4, 1885 CrossRef CAS.
- (a) M. Yang, S. Li, Y. Wang, J. A. Herron, Y. Xu, L. F. Allard, S. Lee, J. Huang, M. Mavrikakis and M. Flytzani-Stephanopoulos, Science, 2014, 346, 1498 CrossRef CAS PubMed; (b) M. Flytzani-Stephanopoulos, Acc. Chem. Res., 2014, 47, 783 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2015 |