
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePhosphorus promotion and poisoning in zeolite-based materials: synthesis, characterisation and catalysis
Hendrik E.
van der Bij
and
Bert M.
Weckhuysen
*
Inorganic Chemistry and Catalysis, Debye Institute for Nanomaterials Science, Faculty of Science, Utrecht University, Universiteitsweg 99, 3584 CG, Utrecht, The Netherlands. E-mail: b.m.weckhuysen@uu.nl; Fax: +31-30-251-1027
First published on 5th June 2015
Abstract
Phosphorus and microporous aluminosilicates, better known as zeolites, have a unique but poorly understood relationship. For example, phosphatation of the industrially important zeolite H-ZSM-5 is a well-known, relatively inexpensive and seemingly straightforward post-synthetic modification applied by the chemical industry not only to alter its hydrothermal stability and acidity, but also to increase its selectivity towards light olefins in hydrocarbon catalysis. On the other hand, phosphorus poisoning of zeolite-based catalysts, which are used for removing nitrogen oxides from exhaust fuels, poses a problem for their use in diesel engine catalysts. Despite the wide impact of phosphorus–zeolite chemistry, the exact physicochemical processes that take place require a more profound understanding. This review article provides the reader with a comprehensive and state-of-the-art overview of the academic literature, from the first reports in the late 1970s until the most recent studies. In the first part an in-depth analysis is undertaken, which will reveal universal physicochemical and structural effects of phosphorus–zeolite chemistry on the framework structure, accessibility, and strength of acid sites. The second part discusses the hydrothermal stability of zeolites and clarifies the promotional role that phosphorus plays. The third part of the review paper links the structural and physicochemical effects of phosphorus on zeolite materials with their catalytic performance in a variety of catalytic processes, including alkylation of aromatics, catalytic cracking, methanol-to-hydrocarbon processing, dehydration of bioalcohol, and ammonia selective catalytic reduction (SCR) of NOx. Based on these insights, we discuss potential applications and important directions for further research.
 Hendrik E. van der Bij | Hendrik van der Bij received his PhD from the Inorganic Chemistry and Catalysis group of Utrecht University (The Netherlands) in 2014. His PhD research, under the supervision of Prof. Bert Weckhuysen, focused on the use of spectroscopy to study zeolite post-modifications. He currently works as a scientist at Albemarle Catalysts in Amsterdam, The Netherlands. |
 Bert M. Weckhuysen | Bert Weckhuysen received his master's degree in chemical and agricultural engineering from Leuven University (Belgium) in 1991. After obtaining his PhD from Leuven University in 1995 under the supervision of Prof. Robert Schoonheydt, he has worked as a postdoc with Prof. Israel Wachs at Lehigh University (USA) and with Prof. Jack Lunsford at Texas A&M University (USA). Weckhuysen has been since 2000 a full professor of inorganic chemistry and catalysis at Utrecht University (The Netherlands). His research interests are in the development of spectroscopy and microscopy techniques for elucidating the working and deactivation principles of catalytic solids, often applied under true reaction conditions. |
1. Introduction
Biomass conversion, cracking of crude oil, automotive catalysis and gas-to-liquid technology1–4 are just a few of the many chemical processes where zeolites are used or have the potential to be used as catalysts. Made from the abundant elements silicon, aluminium and oxygen, they are relatively cheap and environmentally friendly materials. In the expected transitional period where the global economy will move from fossil-based to truly sustainable resources, zeolites still have an important part to play. Therefore, it is essential to understand the chemistry of these fascinating shape-selective ‘micron-scale molecule making factories’ on a fundamental level, from the macroscale to the nanoscale. Insights into their morphology, accessibility, active sites, selectivity, stability and deactivation behaviour will allow us to further improve and extend their use as valuable catalyst materials.An influential example of a zeolite material – and one very relevant for this review – is zeolite H-ZSM-5. Fig. 1 shows H-ZSM-5 on different dimensional scales. Single H-ZSM-5 crystals are often coffin- or parallelepiped-shaped and can have sizes ranging from 100 nm to 100 μm.5–7 Inside the crystal we find a three-dimensional microporous channel system, as can be seen in Fig. 1b and c, which acts as transport channels for molecules. The channel system comprises straight and sinusoidal pores. Both pore types run parallel to the [010] and [100] crystallographic planes, respectively. As the two pore types are connected at regular intersections, molecules can diffuse in the [001] direction as well. The average pore size of these channels is around 5.5 × 5.5 Å.8 At the positions where the microporous channels intersect, cavities form, which are 6.36 Å in diameter.9 Consequently, molecules that exceed the dimensions of the channels are prevented from entering or exiting the zeolite. However, larger molecules can form in the cavities at the channel intersections. These shape-selective properties make H-ZSM-5 and zeolites in general effectively act as molecular sieves.
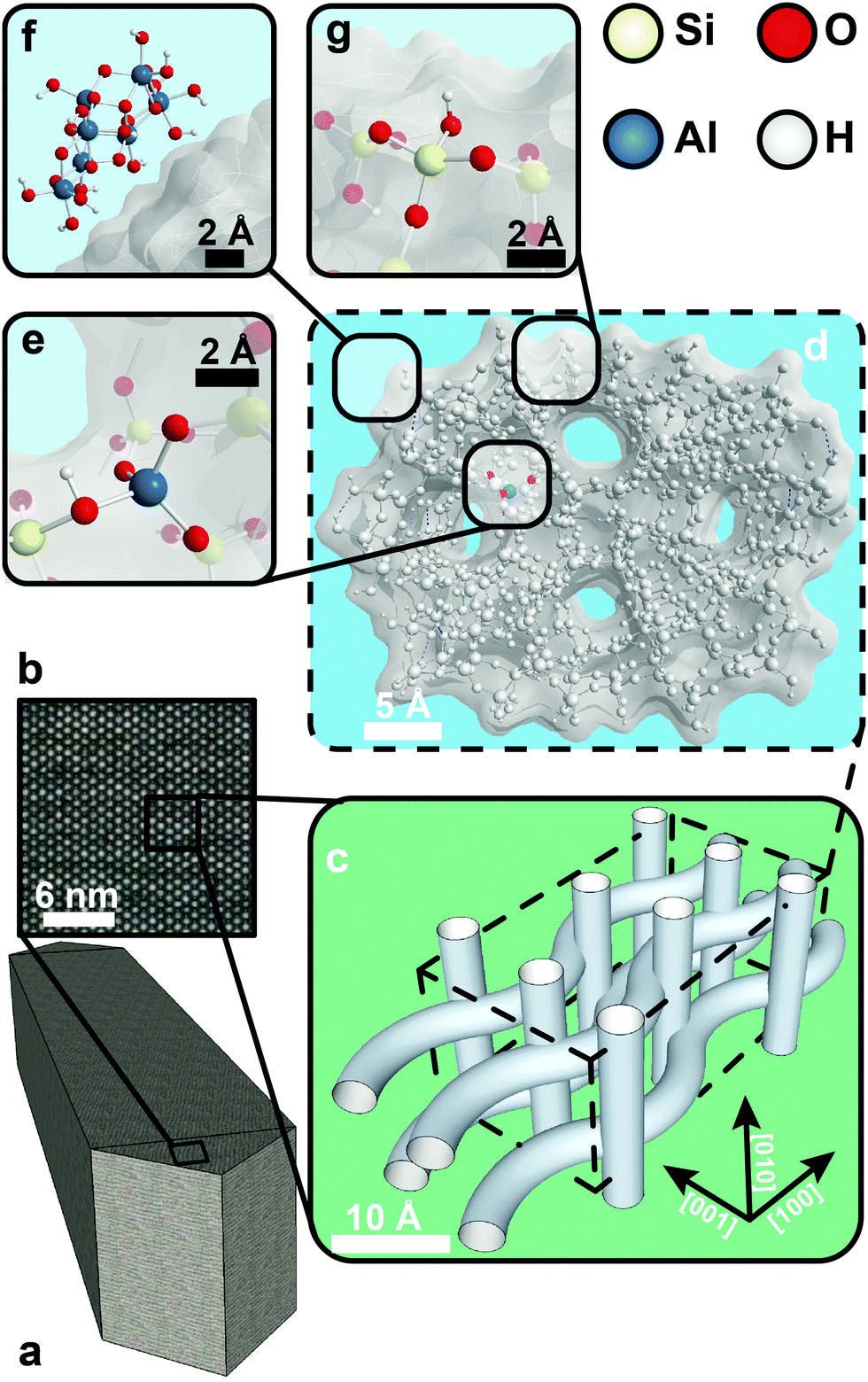 | ||
| Fig. 1 Overview of zeolite H-ZSM-5 on different dimensional scales. (a) A micron-sized coffin-shaped H-ZSM-5 crystal. (b) Closer inspection of the [001][100] plane, revealing straight pores. (c) The three-dimensional channel system of H-ZSM-5 consists of intersecting straight and sinusoidal pores. (d) The channel system is derived from the MFI framework, which comprises linked silica and alumina tetrahedra. (e) A tetrahedrally coordinated framework aluminium (TFAl) atom produces a net negative charge on the framework. In H-ZSM-5 this negative charge is compensated by a proton, which generates a Brønsted acid site. The OH group in SiOHAl is generally referred to as a bridging hydroxyl group. (f) Dealumination leads to the formation of extra-framework aluminium, here shown as boehmite. (g) Terminal silicon and aluminium atoms, which are generally found on the external surface of a zeolite, form silanol and aluminol groups. | ||
The channel system of H-ZSM-5 stems from a well-ordered MFI-type framework. All zeolite frameworks consist of silica and alumina tetrahedra, which are linked by shared oxygen atoms. The solid-acid character of zeolites arises from negatively charged AlO4− tetrahedra that are incorporated into the zeolite lattice. Each of these tetrahedra produces a net negative charge on the framework, which needs to be compensated by a counter-cation. If the counter-cations are protons, strong Brønsted acid sites are formed, as shown in Fig. 1e. If more aluminium is substituted in the framework, it leads to more acid sites. Therefore, the silicon to aluminium ratio in the framework is an important parameter for zeolites and is generally written as the Si/Al ratio. The combined shape selectivity and strong acidity of zeolites create the desired properties such as catalytic cracking of hydrocarbons.
Although the majority of zeolites are used as hydrocarbon cracking or isomerisation catalysts in oil-refining processes, zeolites are also used in many other catalytic processes, including alkylation of aromatics with oxygenates, conversion of (bio)alcohol to light olefins, dewaxing of hydrocarbons, aromatics and fuels, dehydrogenation of paraffins, and dehydration of (bio)alcohols, and in photocatalytic reactions.10–26 It is therefore the case that zeolites are not just catalysts for present-day oil-refining purposes, but are also important catalysts for future green/clean hydrocarbon production processes.
In the late 1970s, it was found that post-modification with a phosphorus precursor can change the acidic and shape-selective properties of zeolites.10,27–30 The most notable phosphorus-containing precursor is H3PO4, which is added to a zeolite by wet impregnation. After this step, the zeolite is thermally treated in air/oxygen, a process better known as calcination, which decomposes phosphorus precursors into (poly)phosphates and phosphorus pentoxide. It has been found that phosphorus can act towards zeolites either as a promoter, e.g., in the fluid catalytic cracking (FCC) process and the methanol-to-hydrocarbons (MTH) reaction, or as a poison, as in ammonia selective catalytic reduction (NH3-SCR) of exhaust gases, i.e., NOx.27,31,32
As a promoter, phosphorus is well known to improve the hydrothermal stability of zeolite H-ZSM-5.33,34 At the same time, phosphorus interacts with the acid sites of zeolites, leading to a reduction in the number and strength of acid sites.35 Furthermore, if phosphorus is located in the micropores of a zeolite, it can either favourably alter its shape-selective effects or block the micropores and reduce the overall accessibility of the zeolite material.36 Although the presence of phosphorus can be beneficial, for example in the MTH reaction where it boosts selectivity towards light olefins and especially towards propylene,31 it is deleterious for NH3-SCR nitrogen oxide-removing applications.37
The promotional effects of phosphorus are well known to the refining industry, which follows from the numerous patents involving all major refining companies.23,38–49 Not surprisingly, many academic groups have reported on the effects of phosphorus on zeolites. However, the exact nature of phosphorus–zeolite interaction has always been a point of discussion, which makes phosphorus–zeolite chemistry an important topic from a fundamental perspective as well. Many models of possible interactions have been proposed over the years and the main division of opinion regards the question of whether a permanent phosphorus–framework interaction exists or not.34,50–56
In the case of a permanent phosphorus–framework interaction, the most intuitive explanation would be the incorporation of phosphorus into the zeolitic framework. Examples of these suggestions are shown in Scheme 1a–d. These interactions can be subsequently categorized into (a–b) phosphorus bonded to the bridging oxygen group of Si–O–Al,52,57 (c) Si–O–P bonds,35 and (d) Al–O–P bonds.55,58 Other possible phosphorus–framework interactions have been suggested as being reversible, as shown in Scheme 1e and f.34,50,59 Examples of suggestions for reversible interactions are (e) intramolecular bonds59 or (f) protonation of phosphoric acid by bridging hydroxyl groups in the zeolite.34 It has also been proposed that (g) phosphorus does not interact at all with the zeolite framework and only interacts with extra-framework aluminium.56
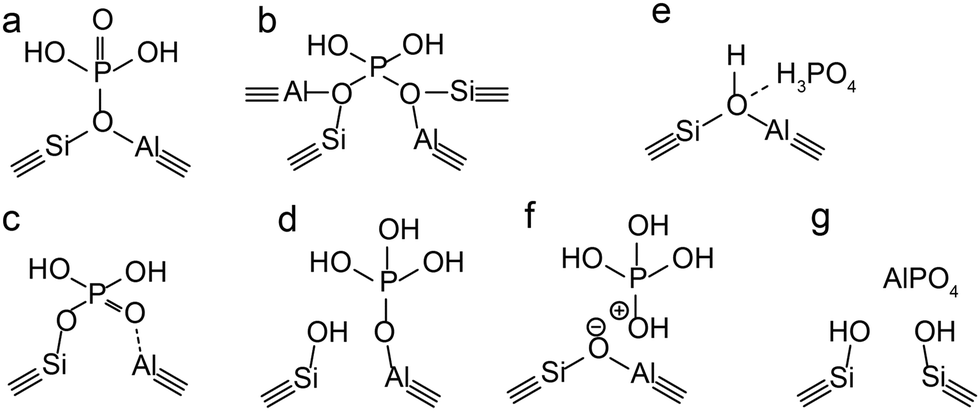 | ||
| Scheme 1 Suggestions from the literature for phosphorus–framework interactions. (a–c and f) Are most cited. (a) Kaeding et al.,52 (b) Xue et al.,57 (c) Lercher et al.,35 (d) Zhuang et al.,58 (e) Abubakar et al.,59 (f) Blasco et al.,34 and (g) Caro et al.56 | ||
We believe that the topic of phosphorus–zeolite chemistry will become increasingly important for three reasons: (i) hydrothermal stabilisation of zeolites is essential for the industrial application of zeolites as catalysts to produce hydrocarbons. Especially during regeneration after deactivation of coke, conversion of (bio)alcohol and dehydration, the presence and formation of water combined with high temperatures lead to permanent deactivation of zeolite catalysts.60–62 (ii) There is an ever-increasing demand for propylene and its selective production using phosphated zeolites is a serious candidate for selective large-scale production.31,63,64 (iii) The relatively high phosphorus content in biofuel poses a challenge for zeolites as NH3-SCR catalysts.65
Elucidation of the nature of phosphorus–zeolite chemistry is expected to reveal the reasons behind hydrothermal stabilisation, alteration of the acid site and changes in catalytic performance. This review article provides the reader with a thorough analysis of the academic literature on phosphorus–zeolite chemistry, which has, to the best of our knowledge, never been the subject of a review paper. The goal of this review article is to find structural and physicochemical trends in phosphorus-modified zeolites. Subsequently, we aim to link these structural properties to catalytic poisoning and promotional effects found in catalysis. The review article ends with an outlook to future research directions.
2. Methods of phosphorus introduction
When considering the vast literature on zeolites modified by phosphorus, it is clear that incipient wetness impregnation (IWI) or wet impregnation (WI) is the most practised method of phosphatation.31,33,34,50,51,53,57–60,63,64,66–85The precursor of choice is phosphoric acid.10,31,33,34,50,51,53,55,58,63,64,66–68,70,71,73,75,77,78,81–87 Less acidic precursors such as NH4H2PO434,36,88,89 and (NH4)2HPO457,60,69,74,76,79,80,86 are used as well. After impregnation, the materials obtained are first dried at temperatures ranging from 70 °C to 120 °C, sometimes at reduced pressures. Afterwards, samples are calcined at elevated temperatures ranging from 450 °C to 650 °C for durations ranging from 1 h to 6 h.
Another preparation method is quite similar to impregnation, with the difference that the sample is mixed under reflux conditions with an aqueous solution of H3PO4, where the concentration of H3PO4 corresponds to the desired phosphorus content. Water is removed by evaporation at reduced pressure. Subsequently, the samples are dried and calcined.35,54,56,86,87,90–92
Besides phosphoric acid and ammonium phosphates, different precursors are used as well. Especially popular in the 1980s was the use of trimethylphosphite (TMPT). This precursor was dissolved in n-octane and mixed with a zeolite slurry. The mixture was stirred under reflux conditions, then filtered, washed with, e.g., n-pentane and dried at 120 °C. Subsequently, the sample was calcined at elevated temperatures.29,52,54,90,93,94 Gas-phase deposition is also a technique used to transfer a phosphorus precursor into H-ZSM-5 zeolite. Precursors reported with this technique are trimethylphosphite (TMPT),78,94,95 triphenylphosphine (TPP),96,97 trimethylphosphine (TMP)54,59,90–92 and phosphorus pentachloride (PCl5).98 The zeolite was contacted with the vapour phase of these precursors at elevated temperatures ranging from 360 °C to 600 °C. In the case of TMP a cycle was repeated twice.
Different precursors used in combination with impregnation methods are diphenylphosphinous (DPP) acid, TPP and phosphorus trichloride (PCl3), dissolved in methylene chloride, carbon tetrachloride and benzene, respectively.17 However, it is important to understand that all these methods are followed by a post-calcination process, which converts the phosphorus precursors into phosphates.
Finally, attempts have been made to incorporate phosphorus into the MFI framework by synthesizing ZSM-5 with a phosphorus precursor in the reaction gel.99,100 Gao et al. used a phosphatation method which was named the hydrothermal dispersion method. Here, H-ZSM-5 is mixed with (NH4)2HPO4 and reacted at 140 °C and 0.3 MPa for 2 h.101,102
Although there are many different ways to introduce phosphorus, it can be seen in the following sections that the overall physicochemical effects on the zeolite structure are very similar. Nevertheless, significant differences that arise due to the method of phosphorus introduction used will be highlighted when applicable.
3. Physicochemical changes
3.1 Physical and chemical phosphorus–framework interactions
In the following section we will describe three distinct phases that can be found in phosphated zeolites, which consist of (i) separate phosphate and zeolite phases (ii) a phosphate–zeolite interface phase and (iii) separate aluminium-phosphate and zeolite phases. The formation of these phases depends strongly on the pre- and post-treatment of the zeolite during phosphorus introduction. However, it is important to understand that these phases often exist simultaneously in phosphated zeolites.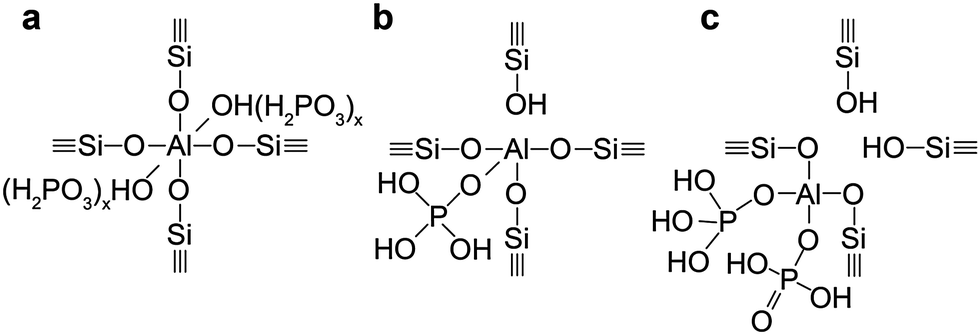 | ||
| Scheme 2 Suggestions for different phosphorus–zeolite interface phases found in phosphated and thermally treated H-ZSM-5. (a) TFAl forced into six fold coordination by physically bonded phosphate species (b) local SAPO interface (c) local SAPO interface with aluminium connected to two phosphates.55,58,71,86 | ||
Besides a change in the coordination of aluminium, the presence of phosphoric acid can distort the electronic environment of TFAl atoms, which has been suggested to arise from hydrogen bonding between hydrogen atoms in H3PO4 and framework oxygen and/or Brønsted acid sites and phosphate oxygen groups.59,86,103 Also, this type of interaction has been found to be fully reversible.86
Both types of interaction lead to an observable decrease in TFAl species and a reduction in Brønsted acid sites, as we will discuss in the following sections.30,50,67,86 Permanent damage to the framework before thermal treatment of phosphated zeolites has not been reported for the MFI topology. However, as we will see in Section 3.1.4, other topologies appear to suffer damage to the framework after phosphorus introduction.30
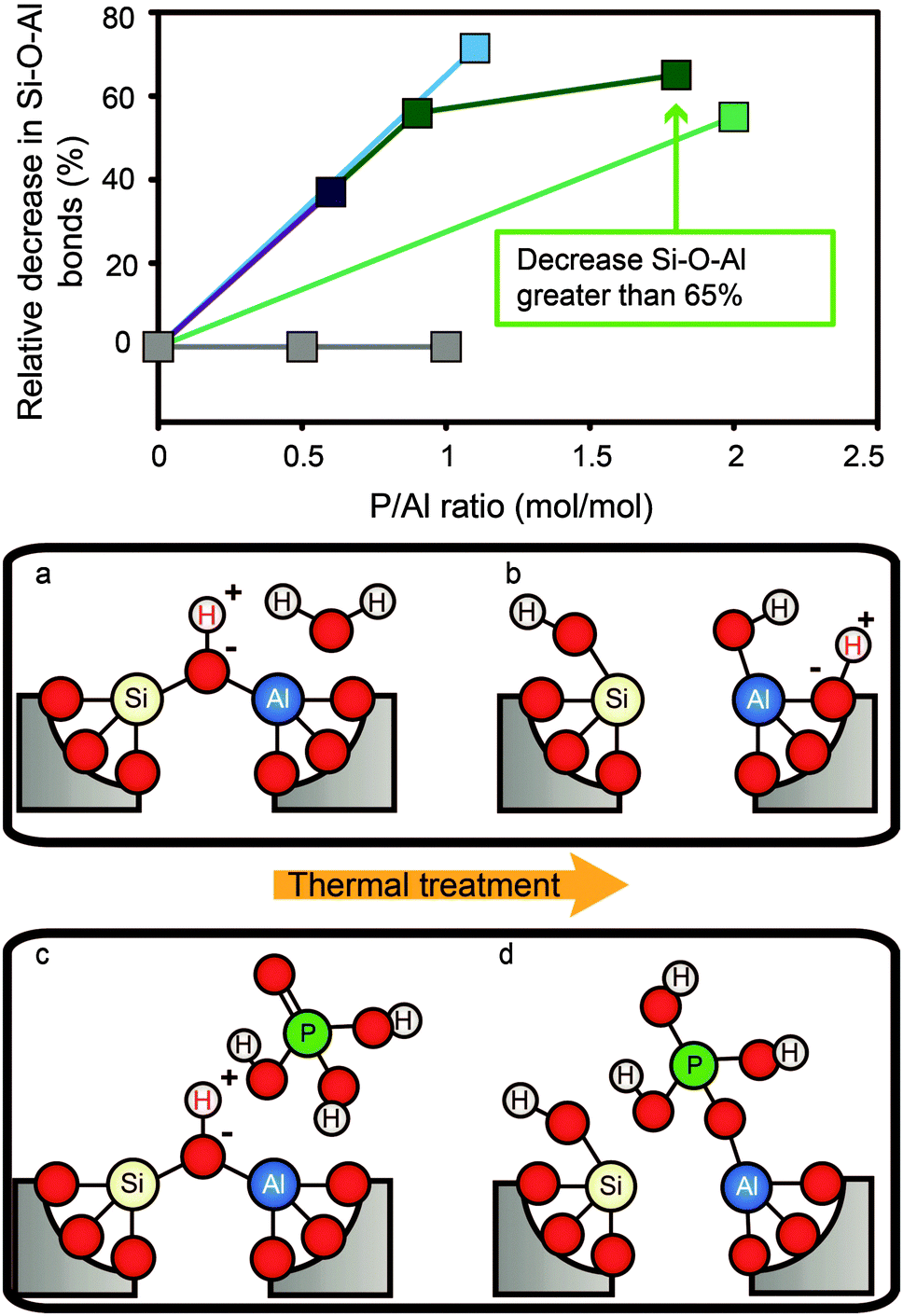 | ||
Fig. 2 Decrease in Si–O–Al bonds vs. P/Al ratio after phosphatation. Values are calculated from reported Si/Al ratios obtained by 29Si MAS NMR.  = Caro et al.56 = Caro et al.56 = Zhuang et al.58 = Zhuang et al.58 = Abubakar et al.59 = Abubakar et al.59 = Blasco et al.34 = Blasco et al.34 = Li et al.82 The decrease in TFAl for P/Al ratio 1.8 corresponds to a reported Si/Al ratio that is greater than 200, so the actual percentage is higher. (a–d) Postulated mechanism of “SAPO-fication” by hydrolysis of Si–O–Al bonds during thermal treatment in the presence of H2O and H3PO4.55,58,60,67,86,104,105 = Li et al.82 The decrease in TFAl for P/Al ratio 1.8 corresponds to a reported Si/Al ratio that is greater than 200, so the actual percentage is higher. (a–d) Postulated mechanism of “SAPO-fication” by hydrolysis of Si–O–Al bonds during thermal treatment in the presence of H2O and H3PO4.55,58,60,67,86,104,105 | ||
Due to the decrease in the percentage of Si–O–Al bonds, the crystallinity of zeolites may be reduced to a small extent after the introduction of phosphorus and subsequent thermal treatment, but the framework structure is maintained and no additional crystalline phases have been reported.30,53,58,63,64,71,73,76,77,81–84,101,110 A loss of crystallinity was found for samples treated with acidic and non-acidic precursors and with the use of impregnation solutions of different acidities, excluding acid leaching as the source of the loss of crystallinity.
After thermal treatment of phosphated H-ZSM-5, H-ITQ-13, and H-MCM-22, a new type of aluminium is formed at the expense of classic TFAl atoms.33,34,57,58,67,71,75,82,85–87,92,110,112 In other zeolite topologies these species are not observed.30,37,108 This new type of aluminium can mostly be attributed to four-coordinated but also to five- and six-coordinated aluminium species that are still present at lattice positions in a highly distorted environment due to chemical and spatial interactions with phosphate species.55,58,67,71,86 It is suggested that phosphate species bond with terminal Si–O–Al–OH groups of partially dislodged TFAl species that form during thermal treatment (Fig. 2b), which effectively leads to the formation of local silicoaluminophosphate (SAPO) interfaces (shown in Fig. 2c and d).58,71,86 It is expected that the positively charged [PO4]+ unit at the SAPO interface balances the negative charge on the zeolite framework. 27Al–31P INEPT-HETCOR NMR experiments have confirmed that these species contain Al–O–P bonds.55
During this “SAPO-fication” process the zeolite material undergoes a gradual increase in this new SAPO interface phase with an increase in phosphorus loading.34,53,67,71,82,92,107 However, a maximum amount of SAPO interfaces has been observed.67,71,107 Local SAPO interfaces do not disappear after washing with hot water.67,85,86 However, washing with NH4F did remove the species.34
Importantly, Damodaran et al. have shown that local SAPO interfaces are not a single defined species, but consist of a variety of framework aluminium–phosphorus interactions, e.g. phosphate mono- and bidentate ligands on four-, five-, and six-coordinated aluminium atoms, of which a few are shown in Scheme 2b and c.55,71 With higher phosphorus loadings the spectroscopic signatures of phosphorus in local SAPO interfaces become more like that of AlPO4.55,113 This indicates that more Si–O–Al bonds are broken and replaced by Al–O–P bonds with an increase in phosphorus content.55,58,113 The afore-mentioned increase in Si–OH groups with an increase in P content agrees with this model.53,64,94,107 Neither different pH values nor phosphorus precursors seem to have an influence on the appearance of local SAPO interfaces after thermal treatment.34,78,86 The SAPO-fication model has also been proposed in environmental and soil science studies, where mesoporous silicates impregnated with aluminium are effective absorbers of phosphates from the environment, as phosphates are suggested to form Si–O–Al–O–P(HO)3 monolayers.114
All the findings described in this section indicate an attraction between phosphorus and framework aluminium. Furthermore, there is a synergistic effect between the presence of phosphorus and subsequent heat treatment, as phosphorus actively promotes the dealumination of zeolites during thermal treatment, which indicates that the formation of Al–O–P bonds is energetically favoured.
Although in the last ten years more indirect evidence of framework-connected Si–O–Al–O–P(HO)3–R species has been reported, the existence of SAPO interfaces has not yet been directly proved.55,58,67,71,86,107 In order to clearly confirm bonding between phosphorus and framework aluminum we suggest the addition of J-coupling 27Al–29Si correlation NMR spectroscopy115 in combination with J-coupling 27Al–31P correlation NMR spectroscopy.55
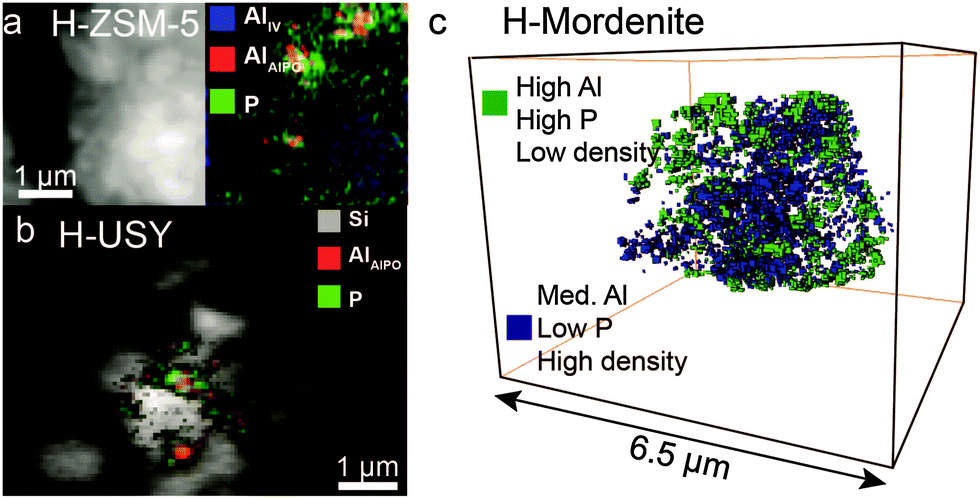 | ||
| Fig. 3 X-ray absorption microscopy results show AlPO4 ‘islands’ in phosphated (a) H-ZSM-5, (b) H-USY and (c) H-mordenite. Adapted from ref. 87 and 113. | ||
In the case of the zeolites mordenite, ZSM-11, MCM-22 and zeolite Y, treatment with a phosphate precursor does not only lead to the reaction of phosphoric acid with extra-framework aluminium. Framework aluminium is leached from the lattice by phosphorus and amorphous AlPO4 is formed.30,82,98,108,112,113,116 The tendency of framework aluminium to be leached from the lattice by acid treatment depends on the topology of the zeolite.117 In the case of USY, which has high aluminium content in its framework, extraction of framework aluminium leads to the collapse of the structure.113 In the case of mordenite, almost all TFAl atoms are leached out of the framework, while the overall framework remains intact.113 Although the effect of topology on the extraction of lattice aluminium by phosphorus would benefit from further systematic study, these findings exemplify the attraction between phosphorus and aluminium.
In a frequently cited study by Xue et al., it was proposed that phosphorus is incorporated into vacant framework positions in dealuminated H-ZSM-5, for which convincing spectroscopic evidence was given (see Scheme 1b).57 However, a detailed study on the formation of phosphosilicate glasses shows that the spectroscopic signatures Xue et al. attributed to Si–O–P species in fact stemmed from polyphosphate species that were trapped in a siloxane (or silica) framework and did not have any interaction with silicon species via a bond.124 In this work, it was convincingly argued that P–OH and Si–OH groups rather self-condense than form cross-links.124 The only Si–O–P bonds in phosphosilicate glasses that have been unequivocally assigned by means of J-coupling NMR spectroscopy have NMR resonances, which, to the best of our knowledge, have never been reported in the literature on phosphated zeolites.125
Therefore, due to the unstable character of Si–O–P bonds compared to the stable character of Al–O–P bonds and the lack of any experimental evidence,126 it seems highly implausible that Si–O–P bonds easily form in phosphated zeolites and complete framework incorporation of phosphorus is thus very unlikely.
3.2 Changes in porosity, accessibility and acidity induced by phosphorus
In the previous section we discussed what type of phases and interactions can be found in phosphated zeolites. The following section will discuss the effect of these phases and interactions on the porosity, accessibility and acidity of the zeolite.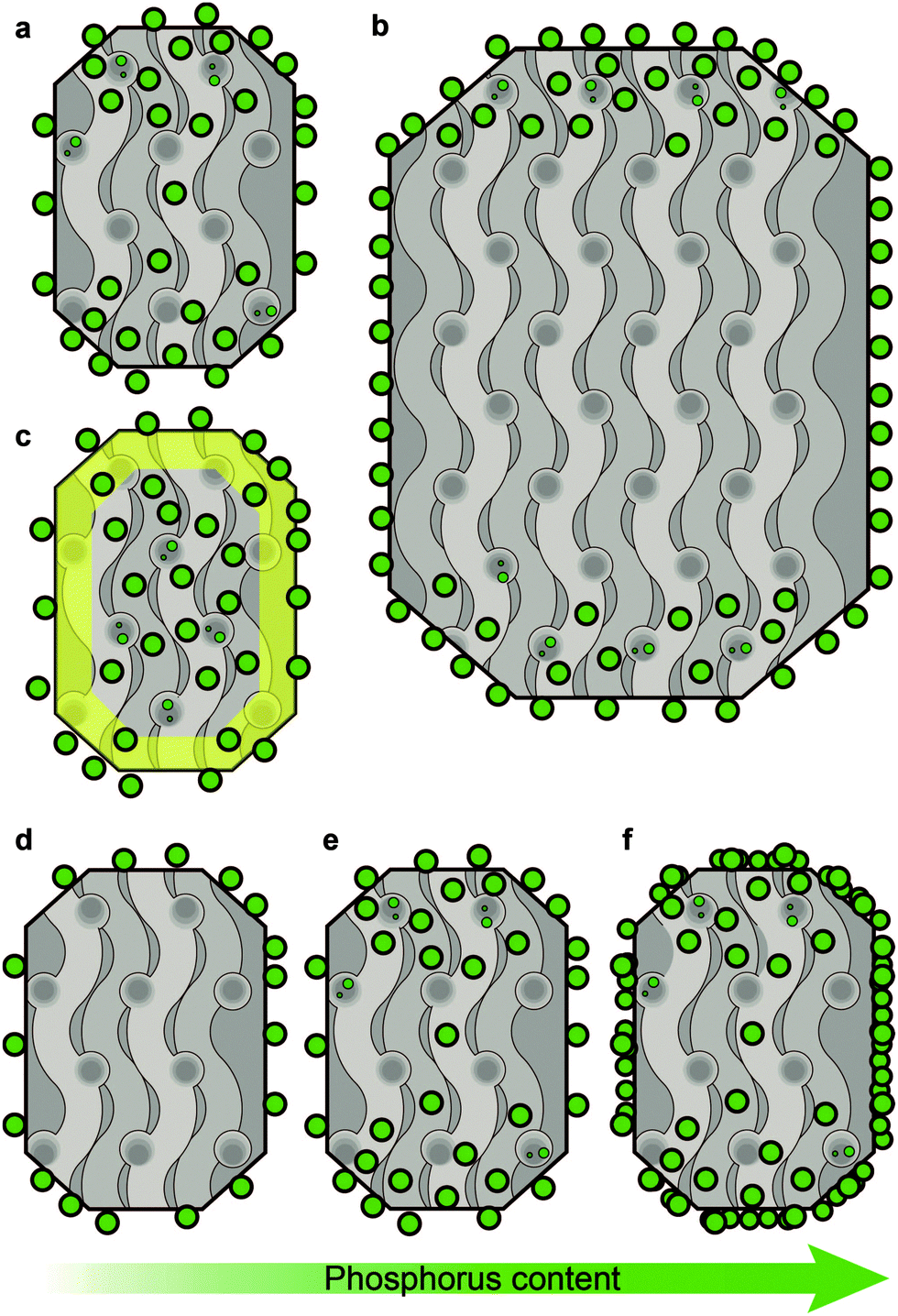 | ||
| Fig. 4 Proposed schematic model of the location of phosphorus species depending on crystal size, aluminium distribution and increasing weight loadings of phosphorus. The model is based on XPS data from ref. 29, 51 and 96, 129Xe NMR and 1H NMR tracer desorption from ref. 56 and 69 and STXM studies.86,87 (a) Typical distribution of phosphorus on H-ZSM-5. (b) Effect of crystal size. (c) Effect of aluminium distribution. Yellow indicates an aluminium-poor region. (d–f) Effect of increasing phosphorus loading. Due to diffusion limitations, the location of phosphorus is more dependent on the actual weight loading of P than on the P/Al ratio. Crystals are viewed along the [100][001] plane. | ||
Although most phosphate species present on the external surface have no interaction with the zeolite phase, it has been shown that the presence of phosphorus on the external surface of a zeolite is accompanied by a decrease or disappearance of surface hydroxyl groups after phosphatation.54,56,69,72–74,86,87,92,94 In agreement with the presence of a phosphorus distribution gradient is the fact that at low loadings, below 2 wt% P, the amount of surface hydroxyl groups decreases faster than that of those found in the bulk. Only when the surface is covered with surfactant do phosphorus species enter the zeolite bulk immediately at low loadings, as shown in Fig. 4d.54,56,69,72–74,86,87,92,94 The latter results would indicate that phosphate species first form a phosphate–zeolite interface phase with external silanol and aluminol groups.
With an increase in crystal size, the ratio of PSurface/PBulk species increased (Fig. 4b).51 This suggests that phosphorus species experience diffusion limitations. Interestingly, when the concentration of aluminium was higher in the zeolite bulk than on the external surface, the distribution of phosphorus was homogeneous (Fig. 4c).29 This latter finding is in accordance with the previously described attraction between framework aluminium and phosphorus.
When phosphorus loadings are increased, phosphorus species tend to move further into the zeolite, as it was found that all aluminium species and corresponding acid sites are affected by the presence of phosphorus (Fig. 4e).33,34,53,56,58,71,77,81,84,86,87 However, with progressively increasing phosphorus loadings the concentration of phosphorus surface species increases faster than that of those in the bulk and after phosphorus loadings exceed 5 wt% excess phosphorus is mostly deposited on the external surface.35,37,53,55,56,69,78,81 Platelets of excess polyphosphates have been found to be present on the outer surface.35 A schematic drawing of this effect is presented in Fig. 4f. Evidence for the aggregation of zeolite particles by phosphorus modification can be found in the literature as well and it has been suggested that phosphorus promotes the self-annealing of external Si–OH groups.56,67,87
The distribution gradient of phosphorus has recently been illustrated by our group in a spatially resolved manner using X-ray absorption microscopy and representative images are shown in Fig. 5. From the figure, it can also be observed that by introduction of phosphorus using wet impregnation heterogeneities in the phosphorus distribution between particles can be found.86,87
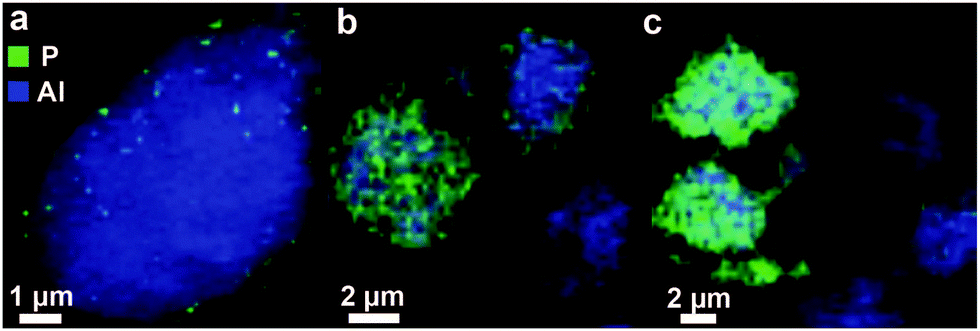 | ||
Fig. 5 X-ray absorption microscopy results, modified from ref. 86 and 87. Chemical maps of clusters of zeolite H-ZSM-5 (Si/Al = 11.5), modified by wet impregnation with a phosphate precursor followed by calcination (P/Al = 0.5), are constructed from aluminium K-edge spectra and phosphorus K-edge spectra stacks. Precursors used are (a and b) H3PO4 and (c) (NH4)2HPO4.  = Al = Al  = P, resolution is 60 × 60 nm. = P, resolution is 60 × 60 nm. | ||
In summary, the weight loading of phosphorus, P/Al ratio, surface OH groups, aluminium distribution, crystal size and phosphorus introduction technique all play a role in the eventual distribution of phosphorus species.
When a phosphate precursor is used to introduce phosphorus, most of the phosphorus is present as ortho-, pyro-, and polyphosphates before thermal treatment.67,86 As a proportion, up to the majority, of these soluble species can be removed by washing with hot water before thermal treatment, the external surface area and micropore volume of phosphated zeolites can be restored to 95% of those of the parent material.53 However, washing a phosphated zeolite after thermal treatment leads only to a partial restoration of surface area and micropore volume.36,53,85,89 One of the reasons for this situation is that after thermal treatment phosphorus species become condensed and occluded and have decreased solubility in water, whereas the other is the formation of a permanent phosphorus–zeolite interface phase, as discussed in Section 3.1.67,86
Nevertheless, it can be observed that most of the loss in micropore volume and surface area that is detectable by physisorption techniques is caused by the presence of water-soluble (poly)phosphate species. Seo et al. suggested that changes in micropore volume caused by (permanent) phosphorus–framework interactions were too small to detect by conventional physisorption methods, but could be distinguished by 129Xe NMR spectroscopy.69
With the breaking of Si–O–Al bonds, new acid sites can form. These are silanol groups that form at defect sites and extra-framework aluminium species.111 Furthermore, framework aluminium species that are only partially connected to the framework can form Lewis or Brønsted acid sites.86,130,131
After modification of zeolites with phosphorus followed by thermal treatment there is a gradual decrease in bridging hydroxyl groups and silanol groups, with an increase in phosphorus content.29,31,34–36,50,53,54,56–58,60,64,66–70,72,73,76–79,81–87,89,90,92,94–96,110,112,116,127–129 Within a single study, as can be seen in Fig. 6, the relative decrease in Brønsted acid sites correlates quite well with the P/Al ratio.34 In the same study, it was established that all acid sites were accessible to molecules of probes and that the decrease in the number of acid sites can therefore most likely not be attributed to physical blocking of acid sites.
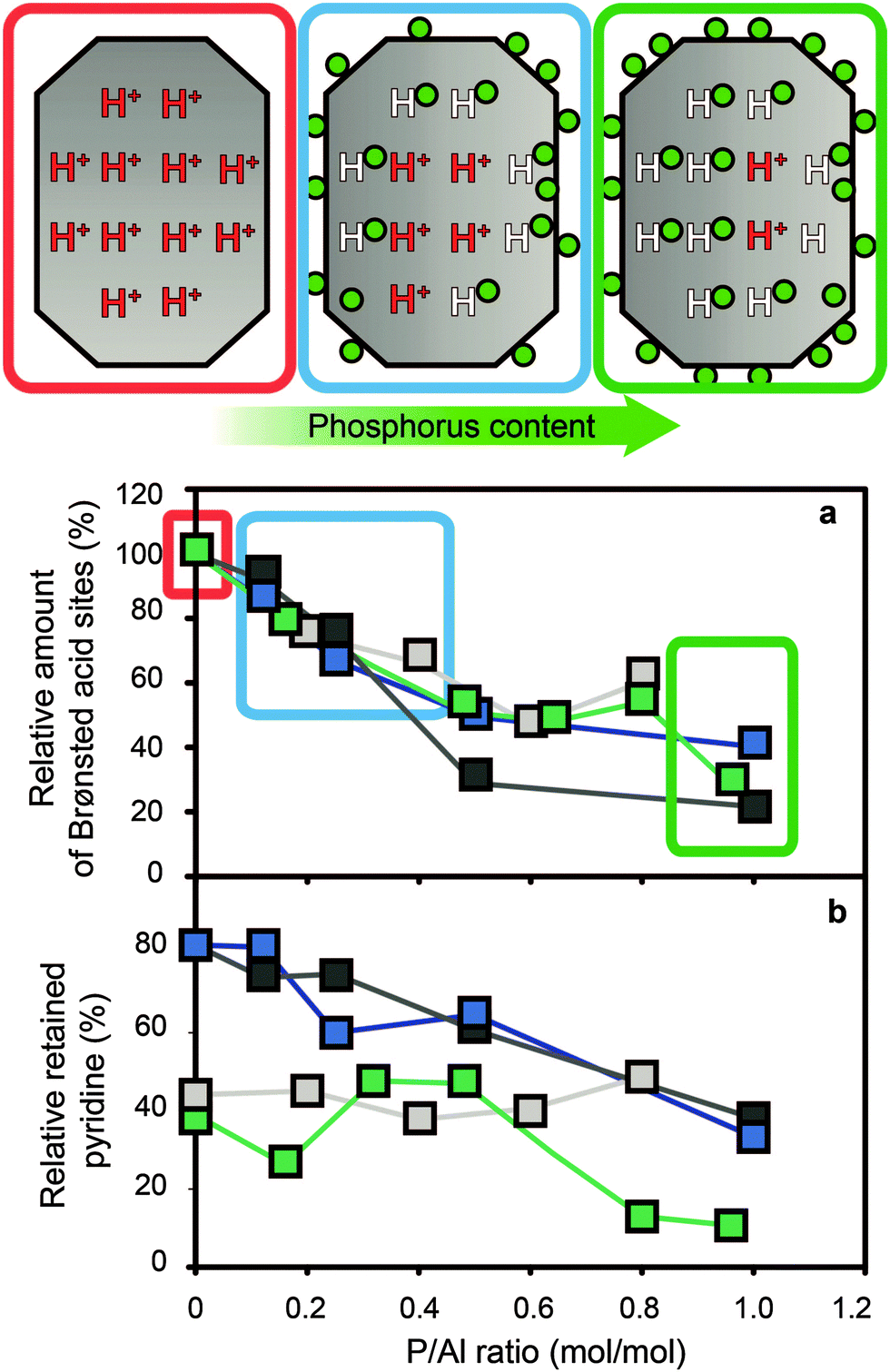 | ||
Fig. 6 (a) Relative amount of Brønsted acid sites vs. P/Al ratio, as determined by adsorption of pyridine at 150 °C. The concentration is relative to the parent material. (b) Relative acid site strength of Brønsted acid sites vs. P/Al ratio, as determined by adsorption of pyridine at 150 °C and 350 °C. The percentage is the concentration of protonated pyridinium ions of a sample at 350 °C, divided by the concentration of protonated pyridinium ions of the same sample at 150 °C.  = H-ZSM-5 + NH4H2PO4 (Si/Al = 15) = H-ZSM-5 + NH4H2PO4 (Si/Al = 15)  = H-ZSM-5 + NH4H2PO4 (Si/Al = 25) = H-ZSM-5 + NH4H2PO4 (Si/Al = 25)  = H-ZSM-5 + H3PO4 (Si/Al = 25) = H-ZSM-5 + H3PO4 (Si/Al = 25)  = H-ZSM-5 + NH4H2PO4 (Si/Al = 40). Data obtained from the work of Blasco et al.34 = H-ZSM-5 + NH4H2PO4 (Si/Al = 40). Data obtained from the work of Blasco et al.34 | ||
As all reported examples have been thermally treated after phosphatation, one obvious cause of the loss of Brønsted acid sites is dealumination induced by thermal treatment. The breaking of Si–O–Al bonds and formation of Al–OH groups have been observed after phosphatation and subsequent calcination.56,58,59,82,84,86,94,132 Thermal treatment can contribute to at least 45% of the loss in strong acid sites.86 Furthermore, as phosphorus promotes the hydrolysis of Si–O–Al bonds, a gradual decrease in acid sites with an increase in phosphorus loading is understandable.56,58,59,82,87 Also, the expected formation of neutral local SAPO interfaces will decrease the overall negative charge on the zeolitic framework, which reduces the amount of protons that can act as counter-cations.
In the literature it is reported that with weight loadings of phosphorus above 5 wt% strong acid sites have almost completely disappeared.35,68,77,81 The Brønsted/Lewis acid site ratio is found to decrease in samples modified by phosphorus, which would indicate that Lewis acid sites are not as much affected by phosphorus as Brønsted acid sites.35,64,76,86,91,96,100,102
However, introduction of phosphorus without any thermal treatment also leads to a decrease in strong acid sites.50,67,86 This decrease is completely reversible, as washing the sample leads to full recovery of all strong acid sites without any signs of permanent dealumination.50,86 Furthermore, it was calculated that after thermal treatment the number of Brønsted acid sites does not correspond to the amount of tetrahedrally coordinated framework aluminium (TFAl) species in phosphated samples.67,92 In other words, 20% more TFAl species were present than Brønsted acid sites. When the sample was mixed with alumina or washed with hot water, these phosphorus species reacted and 20% of Brønsted acid sites were recovered.67,92 The recovery of acid sites after thermal treatment by washing with hot water was confirmed in other studies.36,67,85,86
It is important to understand from the former results that phosphorus is not only a dealuminating agent, but reduces the number of Brønsted acid sites in a reversible way. Therefore, although breaking of Si–O–Al bonds and the formation of local SAPO interfaces during SAPO-fication are reasons for permanent loss of acid sites, reversible interactions of phosphorus with framework aluminium as described in Section 3.1.2 lead to a reversible loss of acid sites as well. At this moment the exact nature of the reversible loss of acid sites is not well understood, but a recent computational study suggested that hydrogen bonding between orthophosphoric acid and framework oxygen could lead to the formation of a H3PO4H+ group, similar to a suggestion of Blasco et al.34,103 We are hopeful that future computational studies will elucidate the origin of reversible acid loss in phosphated zeolites.
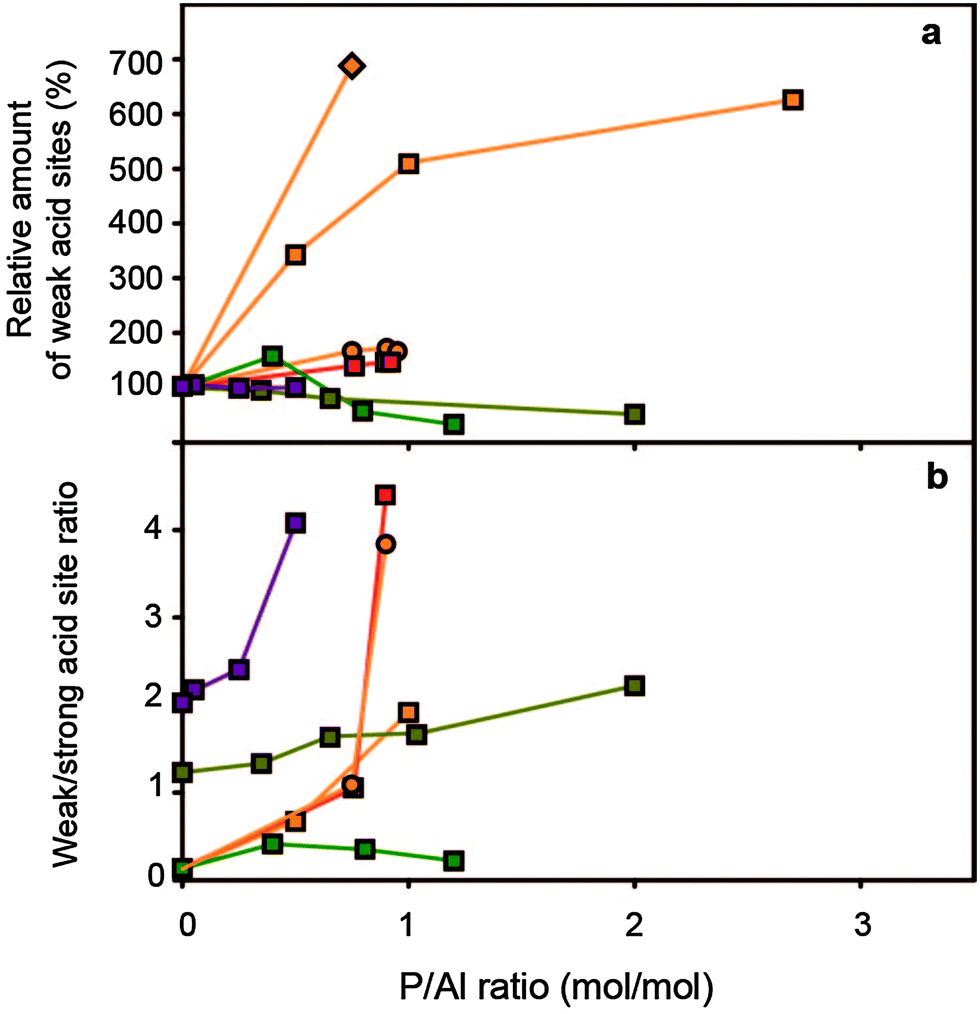 | ||
Fig. 7 (a) Relative concentration of weak acid sites (%) vs. P/Al ratio (b) weak/strong acid site ratio vs. P/Al ratio. Concentrations are relative to the respective parent H-ZSM-5 material.  = Rumplmayr et al.91 precursor: TMP, probe: pyridine, = Rumplmayr et al.91 precursor: TMP, probe: pyridine,  = Vinek et al.90 precursor: □ = H3PO4, ○ = TMP, ◇ = TMPT, probe: pyridine = Vinek et al.90 precursor: □ = H3PO4, ○ = TMP, ◇ = TMPT, probe: pyridine  = Jiang et al.76 precursor: (NH4)H2PO4, probe: NH3 = Jiang et al.76 precursor: (NH4)H2PO4, probe: NH3 = Lee et al.78 precursor: H3PO4, probe: NH3, = Lee et al.78 precursor: H3PO4, probe: NH3,  = Caeiro et al.,33 precursor: H3PO4, probe: n-propylamine. = Caeiro et al.,33 precursor: H3PO4, probe: n-propylamine. | ||
First of all, the maximum temperature at which molecules of probes are desorbed from bridging hydroxyl groups shifts to lower temperatures with an increase in phosphorus content, which indicates that these sites exhibit a decrease in acid site strength.29,31,34,35,50,53,54,57,63–65,74–77,85–87,90,91,94,102,110 Theoretical calculations have indicated that intermolecular bonds between phosphoric acid and the zeolitic framework lead to a decrease in acid site strength.103 Conversely, rates of H/D exchange for propene and adsorption of acetonitrile indicated that the remaining acid sites in H-ZSM-5 modified by phosphorus were not altered in their acid site strength.59,89
Secondly, relatively more strong acid sites are affected by phosphatation than weak acid sites. The corresponding increase in the ratio of weak/strong acid sites leads to an overall decrease in the average acid site strength.31,50,53,63,64,68,69,75–78,82,83,85,91,94,116,128,129,133
Thirdly, there have been reports where the number of weak acid sites actually increases.35,50,54,57,81,84,90 The formation of these new weak acid sites has been attributed to the formation of P–OH groups, hydrogen-bonded Si–OH groups in silanol nests and framework-incorporated phosphate species.35,50,54,57,94 With weight loadings of phosphorus above 2 wt%, P–OH groups and internal Si–OH groups have been observed.34,53,54,56,64,82,86,89,92,94,96,107,134 It has been reported that these P–OH groups are able to protonate pyridine.34,54 In contrast, Rahman et al. reported that P–OH groups have no acidic character and attributed the sites to excess phosphates.96,97 Other attributions have been P–OH groups in framework-incorporated phosphate species and orthophosphoric acid.56,58,87,92,94 Nevertheless, at this moment the nature of the weak acid sites that form in phosphated zeolites remains elusive. Systematic studies on these new acid types should be performed in order to attribute them to specific acid sites.
4. Hydrothermal stability
As mentioned in the introduction, hydrothermal stabilisation of zeolites is a topic of extreme importance for their use in future clean/green chemical processes. Therefore, we dedicate a separate section of this review paper to the promotional role that phosphorus plays in hydrothermal stabilisation of zeolites.4.1 Hydrothermal stability of zeolites
Zeolites are metastable, as they form under hydrothermal conditions as a transitional phase of quartz.135 Therefore, the continued application of hydrothermal conditions eventually leads to the collapse of their framework structure. In the presence of steam and at temperatures above 400 °C framework Si–O–Al bonds will be gradually hydrolysed and aluminium will be expelled from the framework, forming extra-framework aluminium (EFAl) species (Scheme 3).104,105,136–138 During this process, silicon can also be extracted from the framework, leading to the formation of extra-framework silica-alumina.139 When Si and Al atoms are extracted from the zeolite lattice, mesopores form and amorphisation of the crystal lattice can take place.5,137 Due to the removal of negatively charged [AlO4]− units from the zeolite lattice, this so-called dealumination will reduce the amount of counter-cations that the zeolitic framework can retain. In the case of H-zeolites, this means that the active sites for catalysis are lost.140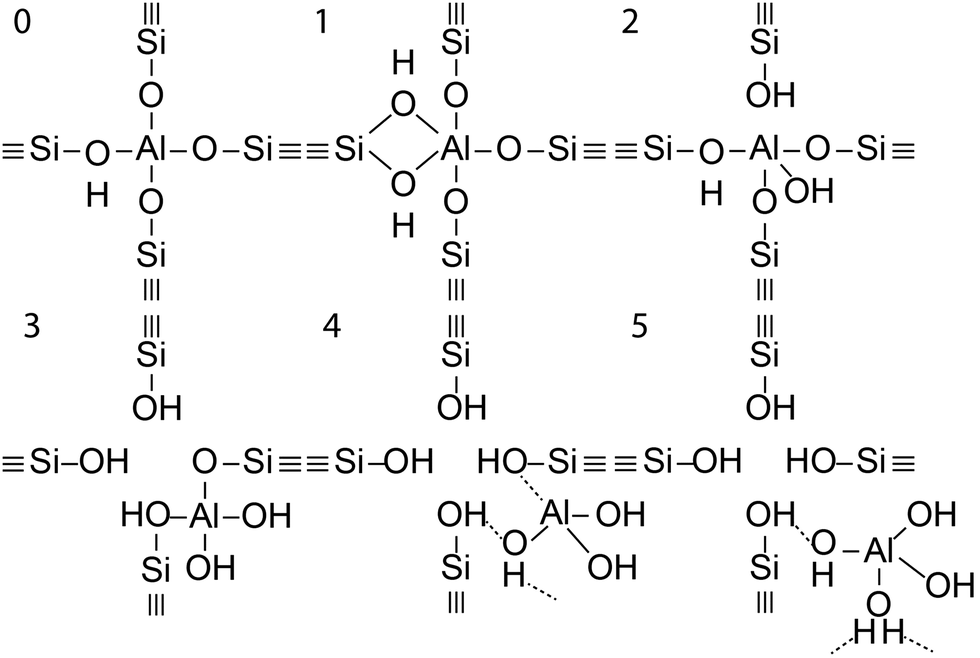 | ||
| Scheme 3 Reaction steps with intermediate configurations found for dealumination. A solid line is a covalent bond. A dashed line is a hydrogen bond. Based on Malola et al.105 | ||
The exact nature of extra-framework aluminium is not clear, but it has been shown to consist of four-, five-, and six-coordinated aluminium species.137,141 Furthermore, it has been suggested that extra-framework aluminium can be cationic and can replace protons in bridging hydroxyl groups.142,143 The hydrothermal stability of a zeolite indicates how prone the framework is to dealumination under hydrothermal conditions. Therefore, a zeolite that does not undergo dealumination and retains acid sites during treatment by steam exhibits high hydrothermal stability. The hydrothermal stability of a zeolite depends on the number of aluminium atoms in the lattice, the framework type, and the type of counter-cations.137,144 Increases in temperature, the volume of steam, or the duration of hydrothermal treatment all lead to more severe dealumination.145,146
As briefly mentioned in the introduction, dealumination poses a challenge to the industrial application of zeolites as acid catalysts. For example, in the FCC process a regeneration step of zeolites is performed at high temperatures and in the presence of steam.25 Although this step is effective in the removal of coke deposits, dealumination induced by steam leads to gradual permanent deactivation of the catalyst.25,140 This catalytic deactivation of zeolites will also take place in high-temperature reactions in which H2O will form as a reaction by-product, examples being the methanol-to-hydrocarbons (MTH) process and the dehydration of alcohols.61,62 Therefore, hydrothermal stabilization is essential for the industrial application of zeolites.
For zeolite Y, the most important cracking catalyst, rare-earth cations and ultra-stabilization by hydrothermal treatment are used to stabilize the material.147,148 In MCM-41, hydrothermal stabilization can be achieved by the addition of salts during synthesis.149,150 In the case of zeolite H-ZSM-5, hydrothermal stabilization can be achieved by the addition of phosphorus.27,33,34,50,58 Therefore, most of the studies on stabilization induced by phosphorus have been performed on H-ZSM-5, although it has been found that zeolite IM-5 can be stabilized by phosphorus as well.127
4.2 Promotion of hydrothermal stability by phosphorus
If steam treatment is more severe, a higher absolute number of acid sites remain for phosphated H-ZSM-5.33,34,50,51,57,58,78 The phosphorus content plays a role as increasingly higher loadings lead to an increasingly higher number of acid sites being retained after steaming.34,51 However, optimal loadings of phosphorus have been reported, which lead to a maximum number of strong acid sites being retained as can be seen in Fig. 8.33,34,78 The corresponding P/Al ratios that were reported are different, i.e., 0.5, 0.65 and 1. However, the weight loadings are somewhat more similar, i.e., 1.3 wt%,78 1.4 wt%,33 and 2 wt%.34 The reason for the optimum number of retained strong acid sites most likely stems from the two effects of phosphorus on zeolite H-ZSM-5, i.e., the progressive decrease in acid sites with an increase in phosphorus loading on the one hand and the increase in hydrothermal stability on the other.33 This effect is schematically drawn in Fig. 8.
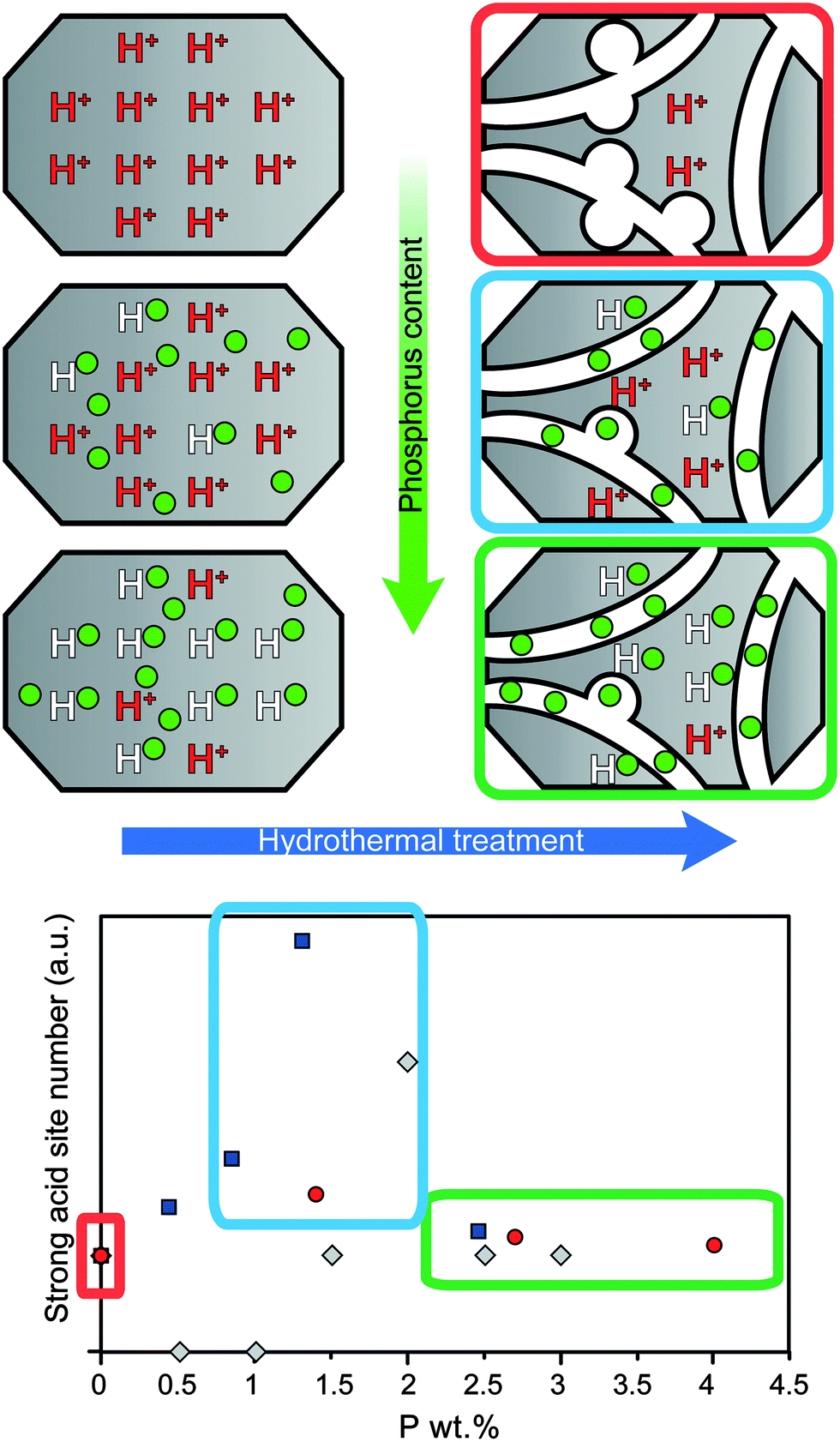 | ||
Fig. 8 Schematic representation of the promotional effect on the number of acid sites during hydrothermal treatment with increasing weight loading of phosphorus. The number of strong acid sites is normalized to the amount of acid sites in the parent material after hydrothermal treatment.  = Caeiro et al.33 = Caeiro et al.33 = Lee et al.78 = Lee et al.78 = Blasco et al.34 = Blasco et al.34 | ||
Generally, the number of weak acid sites is not as much affected by steam treatment as the number of strong acid sites.33,50,51,57,64,75,78,107 This phenomenon leads to an even stronger shift towards a lower average strength of acid sites after steam treatment. Furthermore, Lischke et al. demonstrated the formation of a new type of weak acid site by following the decomposition of ammonium for steam-treated NH4+-exchanged P/NH4-ZSM-5 samples. Washing with hot water did not remove this new weak type of Brønsted acid site, but washing with HNO3 could. The authors attributed the newly formed type of weak acid site to surface-bonded phosphoric acid species, which cannot be removed by washing with hot water.50 This type of weak acid site was confirmed in another work and also attributed to (pyro)phosphoric acid.33
After post-steam treatment, the amount of smaller phosphate species decreases and the amount of more condensed polyphosphate species increases.33,34,50,55,57,58,64,78,85 The amount of condensed species can be reduced after a washing step, which would indicate that the condensed species are either washed out or hydrolysed into smaller species.50,59,85 Furthermore, spectroscopic results indicate that most phosphorus appears to be present at local SAPO interfaces,33,34,50,55,57,58,64,78,85 especially for samples with P/Al ratios above 0.6.33,34,50,55,57,58,64,78,85
A recent 3-D X-ray tomography study shown in Fig. 9 indicated that local SAPO interfaces are located throughout zeolite H-ZSM-5 and hold aluminium atoms fixed in the framework, which was previously suggested by Zhuang et al. and Damodaran et al.55,58,71 This explains why the pore structure of the zeolite remains intact during steam treatment. The study further showed that the location of phosphorus plays an important role in the phenomenon of hydrothermal stabilization. As previously discussed, phosphorus establishes a gradient and does not penetrate completely into the interior of the zeolite. Therefore, aluminium atoms that are located far from the external surface will not be in the presence of phosphorus.55,71,86,87 Consequently, these classic TFAl atoms are expelled from the framework during hydrothermal treatment.33,34,55,71,85–87,107 High steaming temperatures and H2O lead to migration of this extra-framework aluminium to the external surface of the zeolite, where these species react with excess phosphorus to form AlPO4.33,34,51,55,71,107,151,152 This means that phosphate species also effectively act as a sink for EFAl species, which is another possible promotional effect of phosphorus.92 As EFAl species have been suggested to be present as debris in the pores and form cationic Al species that replace protons on bridging oxygen groups, removal of these species could explain the higher number of strong acid sites that were retained.136,153
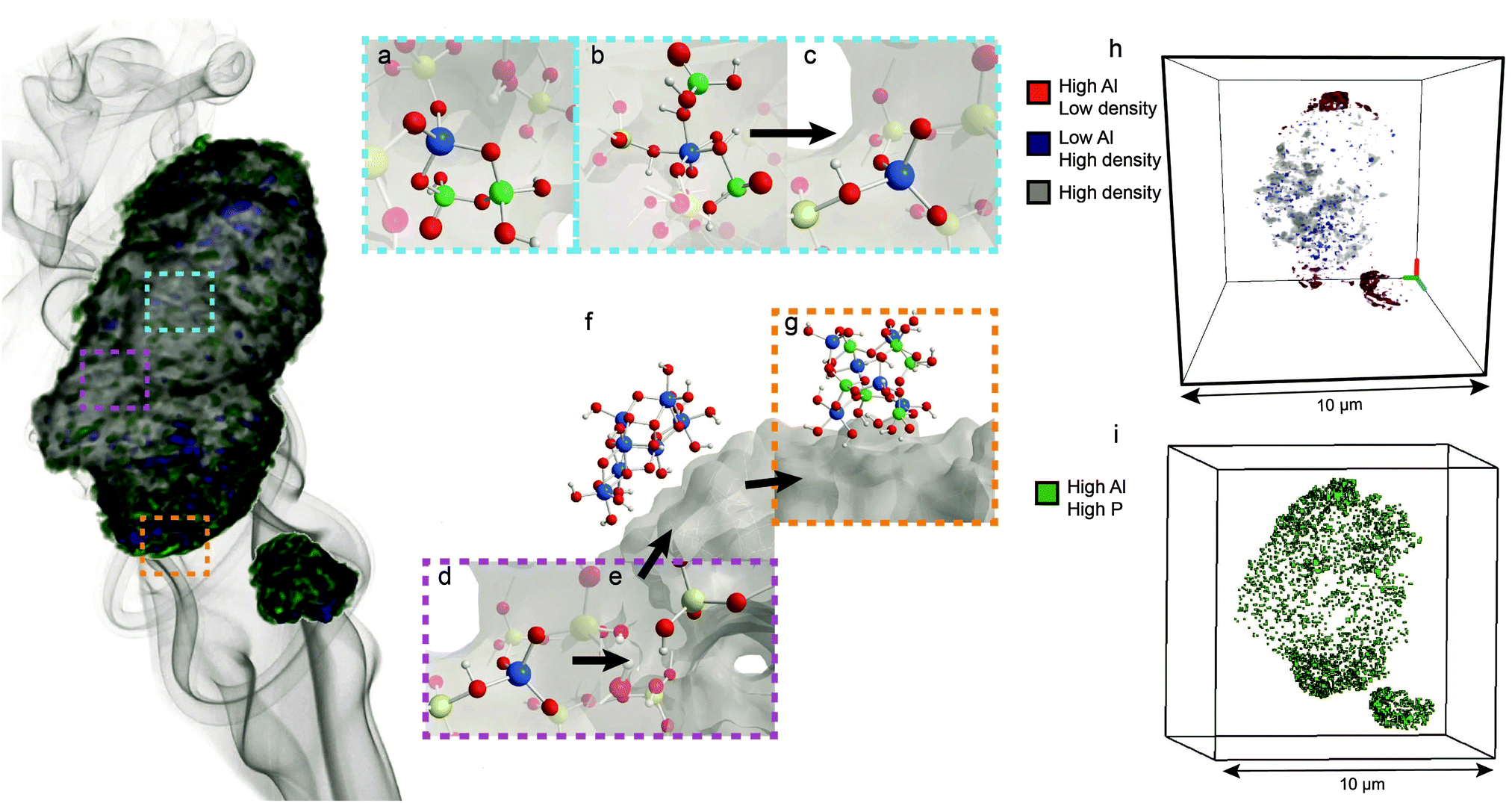 | ||
| Fig. 9 Phosphated and steam-treated H-ZSM-5 sample, adapted from ref. 107 and the main effects that take place during steam treatment. (a) Local framework SAPO interfaces are not affected by steam treatment and remain in the framework. (b) Physically coordinated TFAl species are also resistant to steam, whereas (c) removal of physically coordinated phosphates by washing with hot water leads to recovery of acid sites. (d) TFAl atoms that are not interacting with phosphorus are (e) expelled from the framework and (f) migrate as AlO(OH) species to the surface, where they (g) react with phosphates to form aluminium phosphates. (h) Statistical analysis of the particle showing the presence of zones enriched in Al on the external surface and regions depleted in Al in the interior of the crystal. (i) Statistical analysis of the particle showing high concentrations of phosphorus and aluminium on the external surface, indicating the presence of extra-framework AlPO4 islands that form on the surface. | ||
The improved hydrothermal stability of H-ZSM-5 after phosphatation (or SAPO-fication) can be rationalized by the presence of local SAPO interfaces. It is well known that SAPO materials are extremely hydrothermally stable and have been reported to withstand temperatures up to 1000 °C in steam without significant loss of crystal structure.154–156 Therefore, the formation of patches of SAPO throughout the zeolite framework reinforces its structure, keeping the framework intact during prolonged exposure to hydrothermal conditions.
Nevertheless, although phosphorus has a promotional effect on the hydrothermal stability of TFAl species, increasing the time and temperature of hydrothermal treatment eventually leads to the complete removal of aluminium species from the framework. As hydrothermal treatment gradually breaks all Si–O–Al bonds at one aluminium site, more Al–O–P bonds are formed in the presence of phosphorus.58,71,104 For steam treatment at elevated temperatures and prolonged times the formation of an extra-framework crystalline AlPO4 phase has been observed.58,85 Crystalline AlPO4 is only formed under severe steaming conditions, as AlPO4 is not observed after steaming P/H-ZSM-5 at temperatures lower than 760 °C for less than 2 h.55,58,71,87,107 However, in the case of Caeiro et al., an AlPO4 phase was not observed even after steaming phosphated H-ZSM-5 at 800 °C for 20 h.33
It is important to realise that local SAPO interfaces do not prevent dealumination during hydrothermal treatment altogether, but most likely retard the process, as shown in Scheme 3. The formation of AlPO4 is gradual, as illustrated in Fig. 10.
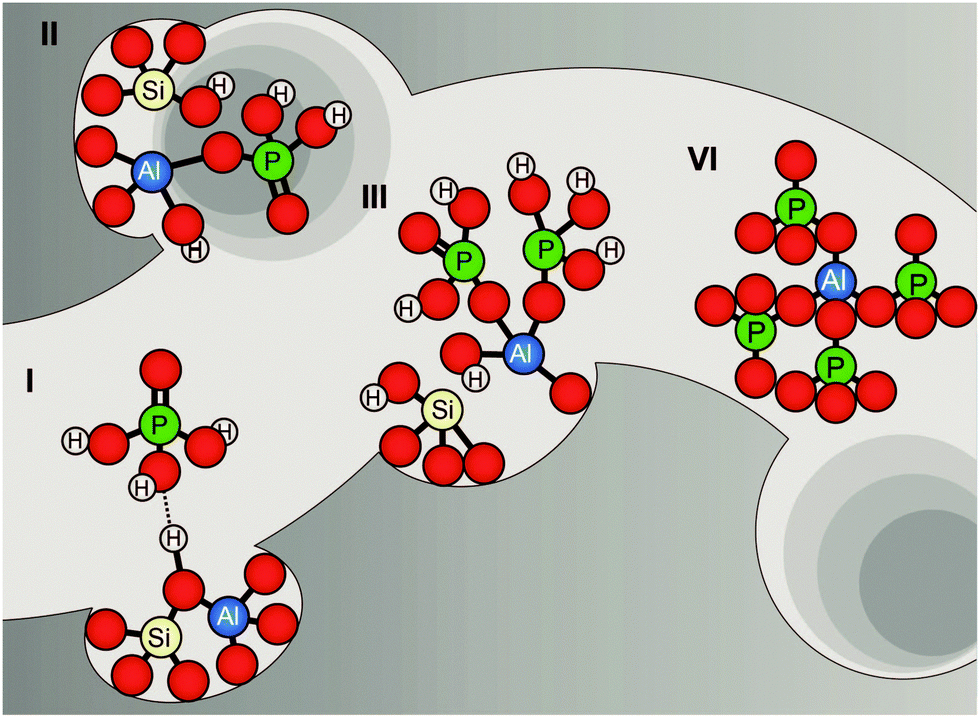 | ||
| Fig. 10 Proposed model of different stages of dealumination in the presence of phosphorus. Model is based on suggestions by Abubakar et al., Zhuang et al., and Cabral de Menezes et al.58,59,71 | ||
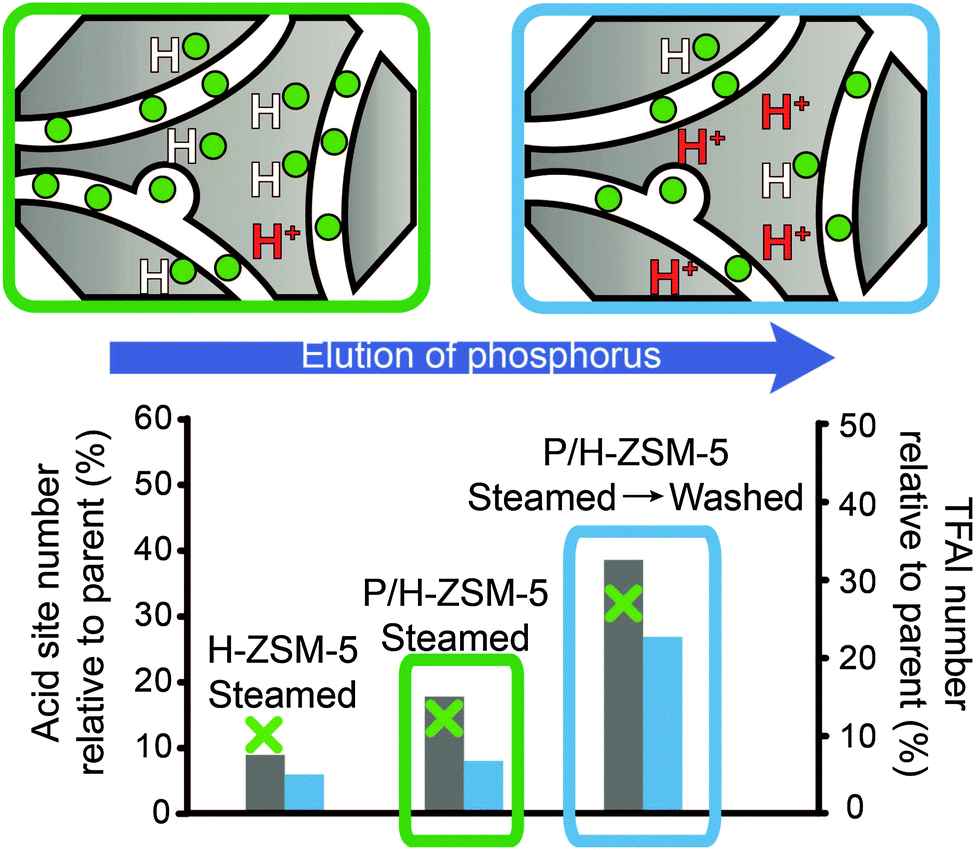 | ||
Fig. 11 Schematic representation of the recovery of the number of acid sites and TFAl species by washing after hydrothermal treatment. The acid site number is relative to the respective parent materials. Left axis: acid site number was determined using adsorption of pyridine.  = Lischke et al.50 (steamed at 700 °C for 30 min. Washed using hot water) = Lischke et al.50 (steamed at 700 °C for 30 min. Washed using hot water)  = Blasco et al.34 (steamed at 750 °C for 5 h. Washed using NH4F) right axis: TFAl number was determined using 27Al MAS NMR. = Blasco et al.34 (steamed at 750 °C for 5 h. Washed using NH4F) right axis: TFAl number was determined using 27Al MAS NMR.  = Lischke et al. (steamed at 700 °C for 30 min. Washed using hot water).50 = Lischke et al. (steamed at 700 °C for 30 min. Washed using hot water).50 | ||
In addition, Liu et al. found a small quantity of a new type of strong Brønsted acid site that was formed after a steaming and washing step.85 They reported that subsequent steam treatment removed the acid site, whereas a further washing step caused it to reappear. Similarly to the recovery of acid sites after hydrothermal treatment, the amount of TFAl species that can be recovered by elution of phosphorus is much lower than before steaming.50 Still, a considerable amount of aluminium species can be retrieved as TFAl species. That is, of the 30% of the original amount of TFAl species that remained after steaming P/H-ZSM-5 under mild conditions, 70% of the original amount of TFAl species could be obtained after washing. With increasingly severe hydrothermal treatment conditions, this amount decreased, i.e., from 15% of the original amount of TFAl species after steaming to 30% of the original amount of TFAl species after washing, as shown in Fig. 11.50 Similar results have been reported in other works.85,107
Although these findings are not quite understood at the moment, an important consequence of the recovery of the number of strong acid sites after washing is that the hydrothermal stabilization effect of phosphorus does not seem to require irreversible phosphorus–aluminium interactions. Recently, a study performed by our group has shown that the recovery of strong acid sites and TFAl species is accompanied by the decrease in lattice aluminium that was attributed to six-coordination by phosphates.85,107 These findings indicate that TFAl atoms, which adopt six-fold coordination in the presence of phosphate species, are hydrothermally stabilized as well.
5. Phosphorus and zeolite-based catalysis
In the previous sections we have shown that phosphorus affects the acidity, porosity and hydrothermal stability of zeolites. As expected, this has a profound effect on the catalytic properties of zeolites in a variety of catalytic processes, as can be seen in the following sections. However, since catalytic conditions and materials differ in most of the literature, direct comparisons concerning performance are almost impossible to make. Therefore, all comparisons and figures in the following section are only meant to show rough trends and the reader should keep it in mind that differences between, e.g., reaction temperature, conversion, Si/Al ratio, crystal size and space velocity have not been taken into account.5.1 Automotive catalysis
Transition metal-exchanged zeolites are used in automotive catalysis to remove NOx formed during the combustion of transportation fuels and are especially suited for diesel engines.4,37,159 The process is known as ammonia selective catalytic reduction (NH3-SCR) and examples of catalysts are Fe-ZSM-5, Fe-beta and Cu-SSZ-13, although small-pore zeolites such as SSZ-13 were found to be most active.4,37,65 In short, NO is oxidised to NO2 by transition metal ions (TMI), e.g., Fe3+ and Cu2+, which act as redox centres.159,160 When NO or NO2 reacts with NH3 chemisorbed on Brønsted acid sites, N2 and H2O are formed.159,160Deactivation of NH3-SCR zeolite catalysts during operation occurs mainly due to thermal conditions and chemical poisoning.4 Although many chemicals present in the feed, e.g., Pt, Zn, and Ca, poison the catalyst, poisoning by phosphorus is especially damaging.37,161 Phosphorus compounds are present in lubricant oils and biofuels and come into contact with the zeolite catalyst during operation.65 NH3-SCR zeolites that are treated with phosphorus show a strong decrease in nitrogen oxide-removing activity.32,37,65,161 It was found that there is a linear relationship between phosphorus content and decrease in catalytic activity.32
Effective poisoning with phosphorus is caused by several deactivation mechanisms, which are related to the previously discussed physicochemical changes induced by phosphorus.37 First of all, there is a decreased amount of NH3 that can be chemisorbed.32,37,65,161 This can be attributed to a loss of acid sites and to blocking of active sites by phosphate species. However, it was found that the decrease in NH3 uptake was only minor compared to the decrease in chemisorbed NO and NO2 species and is therefore only a partial contributor to deactivation of the catalyst.65,161
The decrease in chemisorbed NOx species indicates that there is a decrease in TMI centres, which was confirmed by an increase in Fe2+ and CuO species after the introduction of phosphorus.37,65 Furthermore, it was found that after modification by phosphorus Fe2+ could not be reduced to Fe0 at low temperatures.161 In the case of Fe, it was suggested that Fe3+ reacts with phosphorus.65,161 However, another explanation of the observed effects was suggested to arise from partial dealumination caused by phosphorus.37 In this manner the negative charge on the framework decreases and Cu2+ counter-cations are forced into CuO agglomerates. It was further observed that these CuO agglomerates moved out of the zeolite and were deposited on the external surface, as can be seen in Fig. 12.37 Their presence was suggested to decrease the accessibility of the material even more. While further studies should be performed to determine the exact effect of phosphorus–TMI interactions, the decrease in acid sites, dealumination and pore blocking all follow from previously discussed phosphorus–zeolite interactions. The attraction between phosphorus and framework aluminium and the formation of neutral SAPO interfaces are good explanations why phosphorus is a much more effective poison than Zn, Pt, and Ca.
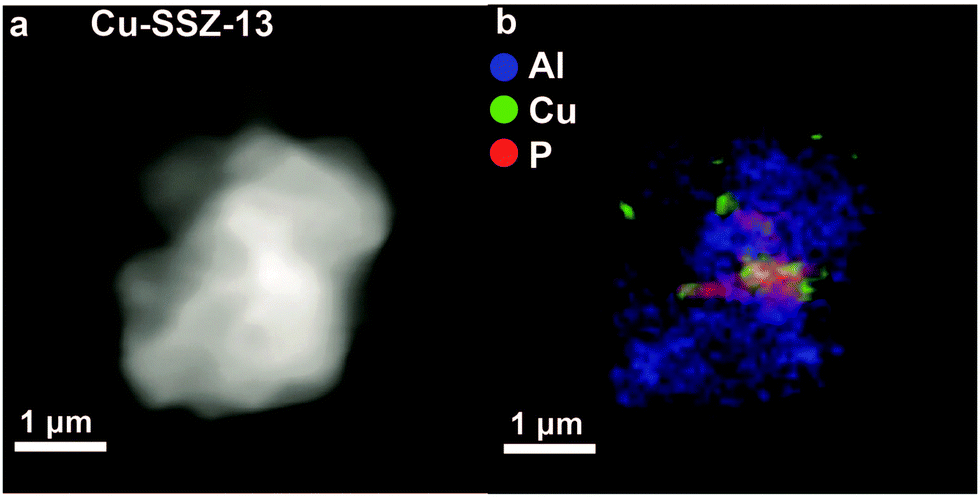 | ||
| Fig. 12 X-ray absorption microscopy results for a Cu-SSZ-13 crystal agglomerate poisoned by phosphorus (0.34 mmol g−1 zeolite). (a) Density map. (b) Elemental map. It can be seen that phosphorus and copper are located on the external surface of the agglomerated squares. Adapted from ref. 37. | ||
5.2 Hydrocarbon catalysis
Protonation by Brønsted acid sites of C–C bonds, C–H bonds, or –OH groups in hydrocarbons and alcohols leads to the formation of carbonium, carbenium, or oxonium groups.162–164 Subsequently, these groups can be cracked, dehydrogenated, dehydrated, isomerised, or oligomerised. As H-zeolites have Brønsted acid sites and shape-selective properties, they are widely used in industry in these types of reactions to produce hydrocarbons. Changes induced by phosphorus in the number and strength of acid sites and shape selectivity lead to different catalytic properties. Furthermore, the catalytic lifetime is increased as the hydrothermal stability increases for phosphated materials.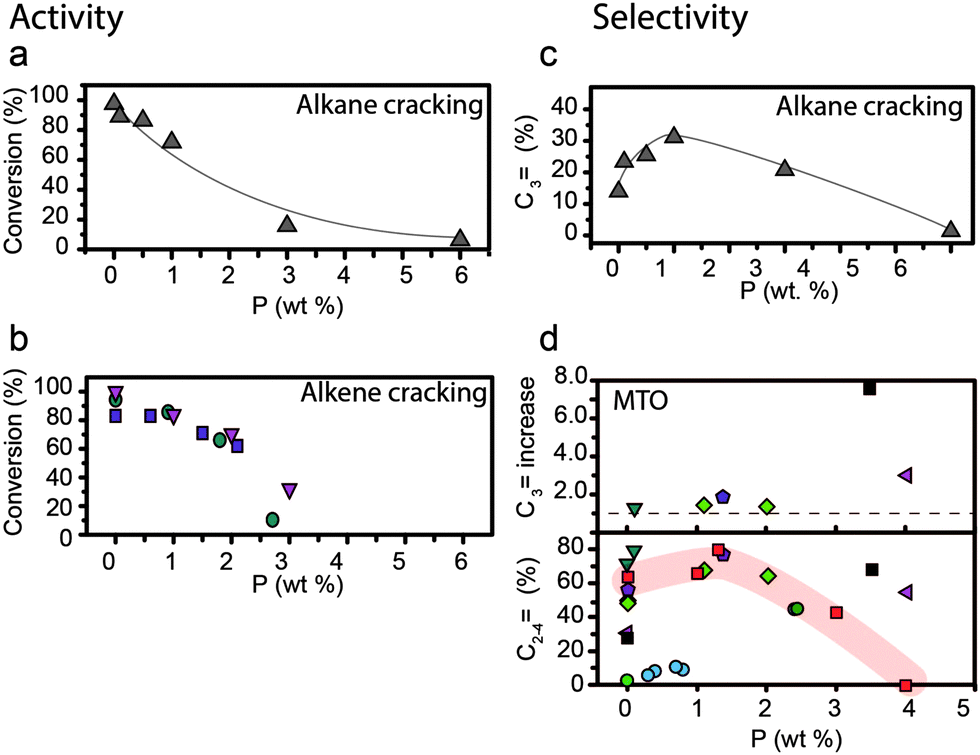 | ||
Fig. 13 Effect of weight loading of phosphorus on (a) alkane cracking activity, conversion of reactant.76 (b) Alkene cracking, conversion of reactant.  = Zhao et al.64 = Zhao et al.64 = Xue et al.57 = Xue et al.57 = Xue et al.79 Differences between reaction temperature, Si/Al ratio, crystal size and space velocity have not been taken into account. (c) Alkane cracking selectivity, selectivity for propylene (%)76 (d) (top) methanol-to-olefins (MTO) selectivity, increase in selectivity for propylene. The increase in selectivity for propylene is relative to that of the respective parent material. (d) (bottom) Actual C2–C4 selectivity vs. weight loading of phosphorus in MTO. ■ = Kaeding et al.52 = Xue et al.79 Differences between reaction temperature, Si/Al ratio, crystal size and space velocity have not been taken into account. (c) Alkane cracking selectivity, selectivity for propylene (%)76 (d) (top) methanol-to-olefins (MTO) selectivity, increase in selectivity for propylene. The increase in selectivity for propylene is relative to that of the respective parent material. (d) (bottom) Actual C2–C4 selectivity vs. weight loading of phosphorus in MTO. ■ = Kaeding et al.52 = Rahman et al.96 = Rahman et al.96 = Rahman et al.97 = Rahman et al.97 = Védrine et al.29 = Védrine et al.29 = Abubakar et al.59 = Abubakar et al.59 = Liu et al.31 = Liu et al.31 = Li et al.82 = Li et al.82 = Dyballa et al.89 The red highlight focuses on a trend in optimum loading of phosphorus reported in ref. 89. Reaction temperatures are between 400 °C and 460 °C. Differences between Si/Al ratio, crystal size and space velocity have not been taken into account. = Dyballa et al.89 The red highlight focuses on a trend in optimum loading of phosphorus reported in ref. 89. Reaction temperatures are between 400 °C and 460 °C. Differences between Si/Al ratio, crystal size and space velocity have not been taken into account. | ||
For the cracking of alkanes, it was established that there is a linear correlation between a high number of Brønsted acid sites and cracking activity in phosphated zeolites.33,90 In the cases of the methanol-to-hydrocarbons reaction and dehydration of alcohol, 100% conversion can be obtained with an increase in temperature.29,31,59,67,77,81–84,96,97,112 For the MTH reaction these results are not surprising as Hunger et al. showed that zeolite materials with a very low number of Brønsted acid sites could achieve 100% conversion of methanol at 400 °C.165
Besides activity, a decrease in the number of acid sites can change the selectivity in hydrocarbon catalysis reactions as well, provided that it reduces the chance of secondary reactions. This is shown in Fig. 13c and d. For the MTH reaction, selectivity towards light olefins and especially propylene increases for unmodified zeolite materials with a low number of acid sites.31,59,112,165–167 It has been suggested that a low density of acid sites reduces the chance of the primary MTH products (light olefins) undergoing hydride transfer and cyclisation reactions during diffusion, thus decreasing the formation of larger products.31,167,168 In agreement with this, the observed decrease in strong acid sites in phosphated zeolites leads to improved selectivity toward light olefins in the MTH reaction.29,31,59,67,82,96,97,112 In Fig. 13d it can be observed that the selectivity towards light olefins increases with an increase in phosphorus loading, i.e., a decrease in the number of acid sites.
It is expected that the decrease in the number of acid sites also influences the product selectivity in catalytic cracking of alkanes. Catalytic cracking over zeolites with a low number of acid sites generally follows the protolytic cracking route (monomolecular mechanism).169 This is because a low density of acid sites reduces the chance of alkanes reacting with carbenium ions (bimolecular mechanism).170 The bimolecular mechanism is in essence a secondary reaction of the monomolecular pathway, as carbenium ions are formed in the latter.162,163,171,172 For more information on monomolecular and bimolecular cracking mechanisms we refer the reader to some of the excellent reviews that have been written on the subject.162,163,171,172
Nevertheless, typical products that are formed by the monomolecular cracking mechanism are methane, ethane, propylene, hydrogen and high olefin/paraffin (C=/C) product ratios,163,169 whereas the bimolecular cracking mechanism produces more paraffins, less olefins and more branched C4+ products. Therefore, phosphated materials are expected to yield a product distribution that corresponds to the monomolecular cracking mechanism. Indeed, as can be seen in Fig. 13c, phosphated zeolites applied in alkane cracking display increased selectivity towards light olefins.33,76,85,97,101,107 However, when high alkane cracking temperatures are applied, i.e., above 650 °C, product distributions are very similar for phosphated and unmodified H-ZSM-5, as at these high temperatures carbenium ions become unstable and the monomolecular mechanism is dominant.171,173
However, the formation of weak acid sites does have a pronounced effect on catalytic performance in the cracking of alkenes and dehydration of alcohols. For the cracking of alkenes, modification of zeolites with phosphorus leads to an increase in propylene product yield.57,64,66,79,102,110 Lin et al. showed in their study that the weak acid sites that form during phosphatation prefer a different reaction pathway to strong acid sites, which leads to higher yields of propylene.66 Higher loadings of phosphorus, i.e., a lower average acid site strength, lead to higher selectivity toward propylene.66 However, as can be seen in Fig. 14b, the selectivity towards propylene decreases when weight loadings of phosphorus reach above 2 wt%.57,64,79,102
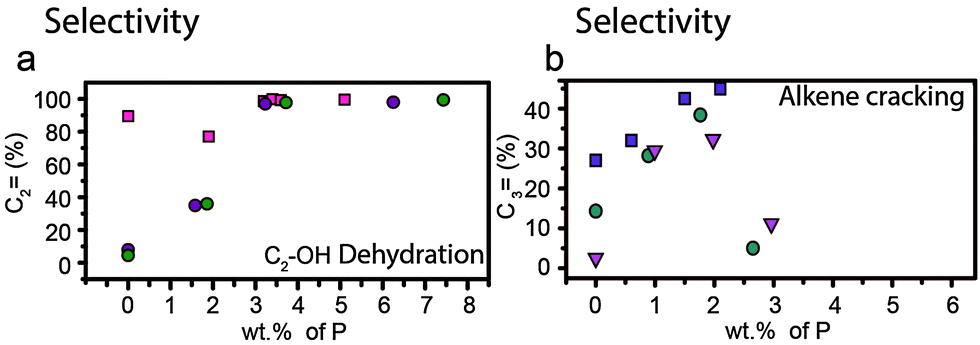 | ||
Fig. 14 Effect of weight loading of phosphorus on (a) dehydration of ethanol and selectivity for ethylene at temperatures >400 °C  = Zhang et al.77 = Zhang et al.77 = Ramesh et al.81 = Ramesh et al.81 = Ramesh et al.84 Differences between reaction temperature, Si/Al ratio, crystal size and space velocity have not been taken into account. (b) Alkene cracking and selectivity for propylene (%). = Ramesh et al.84 Differences between reaction temperature, Si/Al ratio, crystal size and space velocity have not been taken into account. (b) Alkene cracking and selectivity for propylene (%).  = Zhao et al.64 = Zhao et al.64 = Xue et al.57 = Xue et al.57 = Xue et al.79 Differences between reaction temperature, Si/Al ratio, crystal size and space velocity have not been taken into account. = Xue et al.79 Differences between reaction temperature, Si/Al ratio, crystal size and space velocity have not been taken into account. | ||
For the dehydration of alcohols, the selectivity towards ethylene increases with increases in phosphorus content and reaction temperature, as shown in Fig. 14a.63,77,81,83,84,175 If all strong acid sites are removed, at weight loadings of phosphorus above 3 wt% the selectivity towards ethylene reaches 99.4% as only dehydration takes place.63,77,81,83,84 If the catalyst still contains a small amount of strong Brønsted acid sites, higher hydrocarbons are also detected. At weight loadings of 1.3–1.9 wt% P the selectivity towards propylene has been reported to reach a maximum and can be as high as 33% at temperatures between 400 °C and 500 °C.63,81,84,175 Selectivity for propylene is maintained with time on-stream, whereas the unmodified material gradually loses selectivity.63
In a summary of the effect of changes in acidity on catalysis, a decrease in the number of acid sites has a promotional effect on the selectivity of the methanol-to-hydrocarbons reaction toward light olefins, especially propylene. A decrease in the strength of acid sites improves the pure dehydration of alcohols to olefins. For the cracking of hydrocarbons, decreases in both the strength of acid sites (alkenes) and number of acid sites (alkanes) lead to increased selectivity towards light olefins, especially propylene.
This effect is especially obvious in the alkylation and disproportionation of aromatics, as shown in Fig. 15a. The addition of phosphorus to H-ZSM-5 boosts the selectivity towards p-xylene to 95%.10,17,36,70,90,93,95 In general, the increase in selectivity can be attributed to the effective reduction in channel pore sizes, channel intersections and pore openings.10,17,95 Bulkier o- and m-xylenes will diffuse more slowly out of the pores and the rapid decrease in the concentration of p-xylenes will promote the isomerization of the former two isomers into the latter. In a recent work by Janardhan et al. it was found that washing out excess phosphorus from the pores improved the activity in alkylation and disproportionation of aromatics, as pore blocking was reduced. However, the selectivity remained 95% for p-isomers. This indicates that phosphates that are irreversibly connected to the framework are responsible for a decrease in pore volume, which restricts the formation and diffusion of the bulkier ortho and meta isomers.36
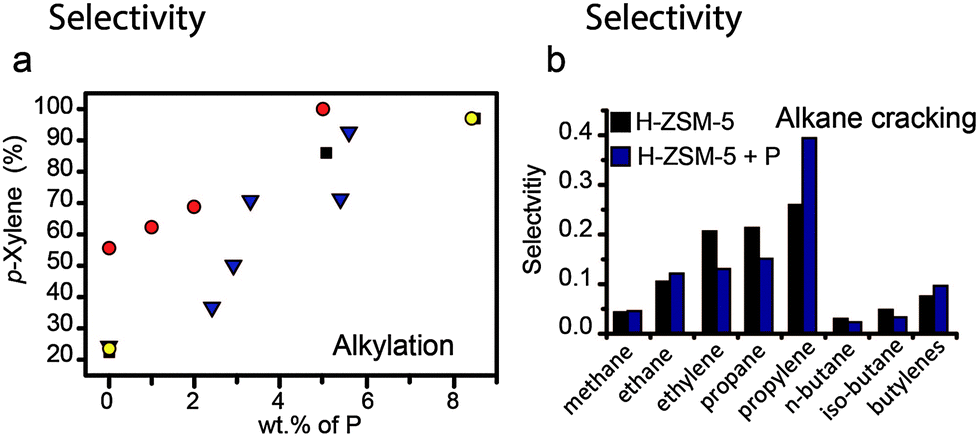 | ||
Fig. 15 Effect of weight loading of phosphorus on (a) alkylation of toluene, selectivity for p-xylene (%).  = Chen et al.10 = Chen et al.10 = Young et al.16 ■ = Kaeding et al.17 = Young et al.16 ■ = Kaeding et al.17 = Vinek et al.90 Differences between reaction temperature, Si/Al ratio, crystal size and space velocity have not been taken into account. (b) Alkane cracking, selectivity in moles for the main products formed during the catalytic cracking of n-hexane over a full temperature program ■ = H-ZSM-5 = Vinek et al.90 Differences between reaction temperature, Si/Al ratio, crystal size and space velocity have not been taken into account. (b) Alkane cracking, selectivity in moles for the main products formed during the catalytic cracking of n-hexane over a full temperature program ■ = H-ZSM-5  = phosphated steam-treated and eluted H-ZSM-5. Adapted from ref. 107. = phosphated steam-treated and eluted H-ZSM-5. Adapted from ref. 107. | ||
The presence of phosphates in the zeolite channels also has an effect on the product distribution in alkane cracking and the methanol-to-hydrocarbon reaction. If one compares the product distributions using phosphated multidimensional 10-MR zeolites in the methanol-to-hydrocarbon reaction at 400 °C it can be observed that there is an increase in selectivity for alkenes, a decrease in selectivity for aromatics and an increase in C3+ species.29,52,59,82,112 The strong decrease in aromatic species is not observed for zeolites with a low number of acid sites and can therefore be attributed to the presence of phosphorus species.59 Interestingly, similar product distributions were reported for the methanol-to-olefins reaction over zeolite H-ZSM-22, which has a 1-dimensional pore system and no channel intersections.176 In the latter topology a decrease in selectivity for ethylene was also observed, which can also be found in the studies of Kaeding et al.52 and Abubakar et al.,59 but not in other works. Nevertheless, an important suggestion to make is that although modification by phosphorus alters the acidity of zeolites, it appears that a change in topology plays a major role in their selective properties as well, especially reducing the presence of aromatics in the product stream.
In a recent study performed by our group we found that the product distribution for the cracking of n-hexane over phosphated H-ZSM-5 resembles that for zeolites that are 1-dimensional along 10-membered ring (MR) channels, such as H-ZSM-22 and H-ferrierite.107,177,178 Similarly to these materials, an increase in selectivity for methane, ethane, propylene, and butylene and a decrease for ethylene, butane and pentane was observed, as can be seen in Fig. 15b.
As mentioned in the previous section, this shift in the product distribution for alkane cracking is typical when the monomolecular cracking mechanism becomes dominant.163 The change in the product distribution in the MTH reaction stems from a larger contribution from the alkene cracking cycle.1,176 This change in mechanism in both catalytic processes stems from the same origin. More specifically, the change in selectivity arises from the ability of a zeolitic framework to stabilize carbenium ions, which are the intermediate species in the bimolecular cracking mechanism and the aromatic-based cycle in MTH.1,163,171,176 In unmodified multidimensional medium-pore zeolites, the channel intersections can more successfully stabilize these voluminous carbenium ions.169,171,179 However, phosphates – presumably connected to local SAPO interfaces – that are present in the channel intersections will hinder the formation of voluminous carbenium ions. This can be observed in one study on H-MCM-22, where, after modification by phosphorus, a decrease in the formation of enyl carbenium ions is observed during the MTH reaction.112
In summary, it is plausible that the presence of phosphate species suppresses the aromatic-based hydrocarbon pool mechanism during the MTH process, whereas in the catalytic cracking of alkanes the bimolecular cracking mechanism is inhibited. Whether this is considered a promotional or poisonous effect of phosphorus depends on the desired product distributions.
However, the major reason for the increased catalytic lifetime is the improved hydrothermal stability induced by phosphorus. After hydrothermal treatment, samples modified by phosphorus have higher catalytic activities than their non-phosphated counterparts.33,34,57,60,64,75,78,79,85,88,107,173 If reactions are performed under hydrothermal conditions, phosphated catalysts maintain activity for a longer time on-stream.60,63,64,88,127,175 Samples from which phosphorus was eluted after steaming regained strong acid sites and consequently their cracking activity increased.34,85,107
As we have seen in Section 4.2, phosphated zeolites are expected to perform better in catalysis after steam treatment than their non-phosphated counterparts, because they contain more strong acid sites. However, the direct link between the number of strong acid sites and cracking activity is not as straightforward after hydrothermal treatment. Samples with high weight loadings of phosphorus and a higher number of strong acid sites after hydrothermal treatment had lower cracking activity than samples with lower weight loadings of phosphorus and a lower or equal number of acid sites.34 Furthermore, it was found that steam-treated parent H-ZSM-5 had a higher number of Brønsted acid sites, whereas steam-treated phosphated H-ZSM-5 had a lower amount. Interestingly, the cracking activity for the latter sample was higher.33 Therefore, other factors such as the distribution of acid sites, accessibility and limitations to the formation of coke will probably play a role.33 In the cracking of alkenes, it was reported that hydrothermal treatment increased the activity for phosphated samples, which would indicate that the accessibility of phosphated zeolites improves after steam treatment.57,79
As mentioned in Section 4, adding more phosphorus is not the key to retaining more active sites after steaming, as there are several (varying) optimum loadings of phosphorus (as shown in Fig. 16a and b).33,34,57,60,75,78,79,88 In accordance with what was discussed in Sections 3 and 4, phosphatation leads to pore blockage, reduced accessibility and a decrease in the number of acid sites, which is amplified by increasing the loading of phosphorus. Therefore, a decrease in cracking activity is expected with an increase in phosphorus content. However, after steaming the promotion of hydrothermal stability by phosphorus comes into effect. As such, when comparing a steam-treated phosphated sample with a steam-treated non-phosphated sample, the former will perform better in cracking, especially after more severe steaming conditions have been applied. The effect will decline when the phosphorus loading becomes very high, when the poisonous effects of loss of acid sites and reduced accessibility start to dominate again.33
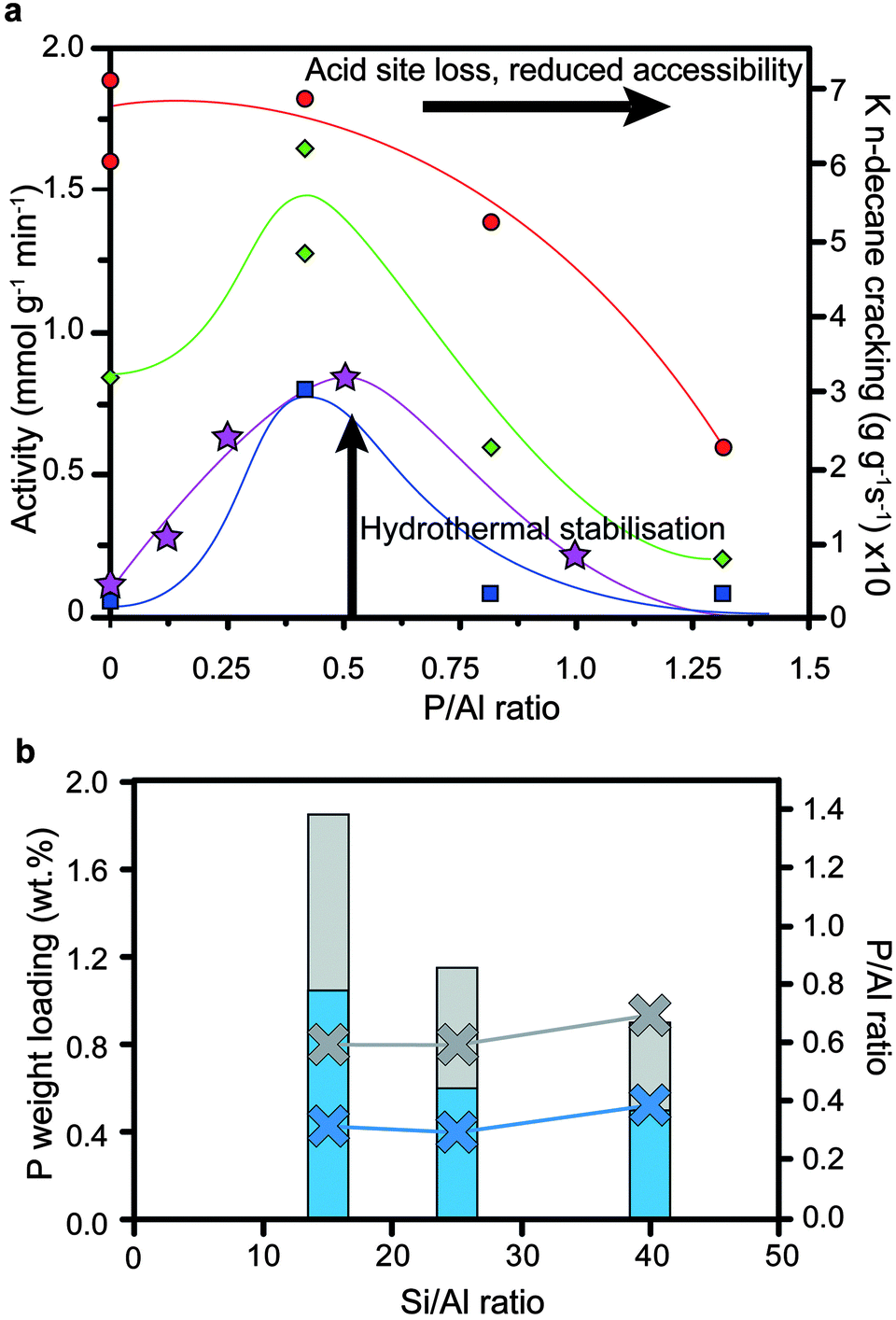 | ||
Fig. 16 (a) Effect of phosphorus and post-steam treatment on the cracking of n-hexane33 (left axis) and n-decane34 (right axis) by H-ZSM-5. Adapted from ref. 33. Activity is plotted against weight loading of phosphorus (H3PO4 precursor). Left axis:33 = H-ZSM-5 (Si/Al = 13). = H-ZSM-5 (Si/Al = 13).  =H-ZSM-5 steamed at 800 °C for 5 h. =H-ZSM-5 steamed at 800 °C for 5 h.  = H-ZSM-5 steamed at 800 °C for 20 h. Reaction temperature is 500 °C. Right axis:34 = H-ZSM-5 steamed at 800 °C for 20 h. Reaction temperature is 500 °C. Right axis:34 = H-ZSM-5 (Si/Al = 25) steamed at 750 °C for 5 h. Reaction temperature is 500 °C. Lines are not fits and are meant to guide the eye. (b) = H-ZSM-5 (Si/Al = 25) steamed at 750 °C for 5 h. Reaction temperature is 500 °C. Lines are not fits and are meant to guide the eye. (b)  = optimal average cumulative catalytic conversion during cracking of naphtha with additional H2O in the feed at 650 °C after 1 h for H-ZSM-5 with different Si/Al ratios vs. P weight loadings and P/Al ratios (NH4H2PO4 precursor).88 = optimal average cumulative catalytic conversion during cracking of naphtha with additional H2O in the feed at 650 °C after 1 h for H-ZSM-5 with different Si/Al ratios vs. P weight loadings and P/Al ratios (NH4H2PO4 precursor).88 = optimal first-order kinetic rate constants in cracking of n-decane with H-ZSM-5 steamed at 750 °C for 5 h.34 Left axis in bars: phosphorus weight loading. Right axis as crosses with lines: P/Al ratio. = optimal first-order kinetic rate constants in cracking of n-decane with H-ZSM-5 steamed at 750 °C for 5 h.34 Left axis in bars: phosphorus weight loading. Right axis as crosses with lines: P/Al ratio. | ||
5.3 Future applications
Although hydrocarbon cracking and MTH reactions are the most widely studied reactions for phosphated zeolites, there are also new promising directions where phosphated zeolites can be utilized, an example being the catalytic dehydration of bioalcohols, such as dehydration of ethanol to ethylene. As discussed in the previous sections, carrying out a phosphatation step decreases the number and strength of acid sites in a zeolite, which makes phosphated zeolites very suitable for the selective dehydration of bioalcohols to alkenes.63,77,81,83,84 However, if these reactions are performed in the liquid phase the reversible interactions of phosphorus should be considered (Scheme 4a). The formation of H2O during dehydration removes the reversible phosphorus–aluminium interactions and strong acid sites will reappear, leading to undesired cracking reactions (Scheme 4b). A possible solution to overcome this problem is to perform a pre-leaching step followed by post-steam treatment. In this manner, the physically interacting phosphorus is removed and the strong acid sites that form as a consequence can be removed by hydrothermal treatment (Scheme 4a–d). It is expected that this material has exclusively weak acid sites, which remain weak in aqueous environments.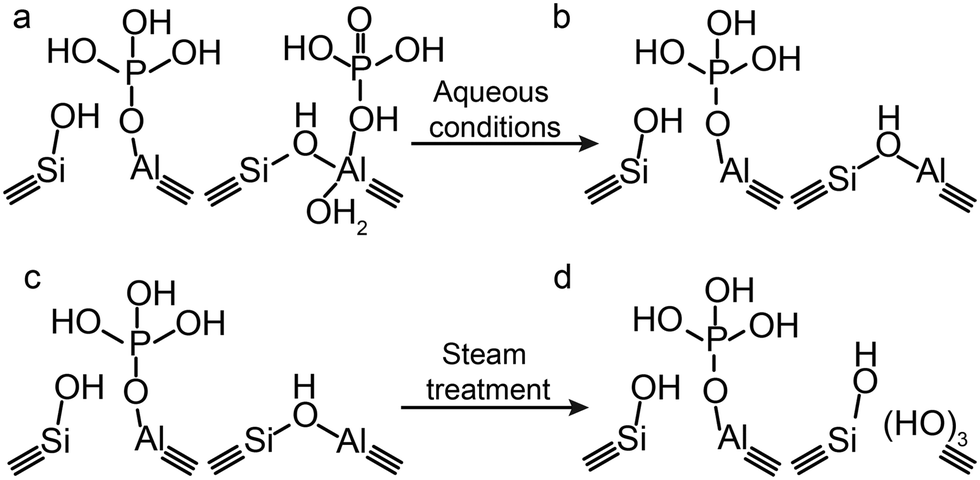 | ||
| Scheme 4 Schematic representation of the effect of aqueous conditions on permanent and reversible phosphorus–aluminium interactions. | ||
Another key potential application for phosphated H-ZSM-5 is in catalytic fast pyrolysis of biomass.2,12 In this process Ga-ZSM-5 is used to produce large amounts of aromatics from lignocellulosic biomass.12 However, a major obstacle is the formation of coke during the process, which is overcome by thermal regeneration. Stabilization by phosphorus could be a successful method for providing additional stability during thermal regeneration. A study by Furomoto et al. has shown that the addition of phosphorus to Ga-ZSM-5 improves stability and suppresses carbonaceous deposits and the release of gallium from the framework during ethanol-to-propylene reactions.182 Therefore, we hope to see future studies on the use of phosphorus as a promoter in zeolite-catalysed pyrolysis of biomass.
The concepts presented in this review article help the aim to modify zeolites by phosphatation more rationally. For example, by exclusive synthesis of local SAPO interfaces at channel intersections of a multidimensional 10-MR framework, one obtains a material that has a multidimensional framework structure, but not the internal cavities that promote the formation of carbenium ions. In these materials, transport of molecules is still fast, while the formation of coke and reaction mechanisms that use carbenium ions are reduced.180,181 As the dislodged Si–O–Al–OH species that form during thermal treatment were found to act as anchoring points for phosphoric acid, thermal treatment before phosphatation is a first step in the exclusive formation of local SAPO interfaces.86
However, more precise synthesis methods should be explored than the impregnation methods that are often applied to introduce phosphorus.31,33–35,50,51,53,54,56–60,63,64,66–87,90–92,107 As we have seen in the previous sections, introduction of phosphoric acid under these conditions leads to inter- and intra-particle heterogeneities. Gao et al. have shown that introduction of phosphorus by means of hydrothermal dispersion leads to a more homogeneous distribution of phosphorus species than by impregnation.101,102 Janardhan et al. reported on the formation of phosphate monolayer islands by performing phosphatation followed by an elution step.36 The authors argued that the removal of excess phosphorus species is essential for the creation of accessible materials. Although we agree, the elution of phosphorus by washing with hot water also removes reversible phosphorus–aluminium interactions. This can be undesirable, as is the case in the dehydration of bioalcohols. Gas-vapour deposition as a means of introducing phosphorus has been reported to lead to more phosphorus–aluminium interactions than wet impregnation.78 Furthermore, pre-steam treatment followed by extraction of extra-framework aluminium to form mesopores could be a method of obtaining more efficient transport routes for phosphorus precursors. Extraction of extra-framework aluminium is essential in this step if the formation of extra-framework AlPO4 is not desired.
Furthermore, if one were to leave the extra-framework aluminium, it is possible to synthesise an aluminium phosphate binder from the zeolite's own supply of aluminium. Especially in the field of catalytic hydrocarbon cracking, the addition of AlPO4 to zeolites leads to improved selectivity for light olefins, hydrothermal stabilization, improved mechanical strength and resistance to attrition.23,46–48,183 Furthermore, there are several arguments in favour of creating a binder phase from a zeolite's own supply of aluminium.113 First of all, it allows the use of zeolites with low Si/Al ratios, which are cheaper and more environmentally friendly to produce, as the use of organic templates is not required.184,185 Secondly, the formation of mesopores caused by the dealumination step creates a hierarchical material, which facilitates the access and transport of reactant and product molecules during catalysis.186,187 Finally, careful synthesis of AlPO4 from extra-framework aluminium (EFAl) allows one to form AlPO4 species inside the zeolite channel/cage system, altering its shape-selective properties. Although not many studies have yet been performed in this direction, we believe these promising avenues of phosphorus–zeolite chemistry deserve to be investigated.
On a final note, the interaction of phosphorus with zeolites could act as a model system for understanding the chemistry between zeolites and other group VA elements such as arsenic and antimony. Zeolites are very effective materials for removing arsenic from wastewater.188–190 The suggested condensation reaction of arsenate with the Al–OH groups of surface framework aluminium has certain analogies to the formation of local SAPO interfaces.190 Similarly to phosphorus, modification of H-ZSM-5 with antimony oxide exhibited increased para-selectivity in the disproportionation of toluene and a decrease in strong acid sites in H-ZSM-5.191,192 As this type of inorganic chemistry has an effect on fields such as conversion of biomass, hydrocarbon catalysis, automotive catalysis and wastewater treatment, we hope to see future studies that explore and ultimately obtain a unified view of group VA chemistry with zeolite materials.
6. Summary
The study and comparison of the existing literature on phosphorus–zeolite chemistry has revealed many universal physicochemical effects that take place. One of the most important considerations should be that the attraction between phosphorus and aluminium is the driving force behind the physicochemical changes in phosphated zeolites, such as the decrease in the number of acid sites, decrease in framework negative charge, improved hydrothermal stability and changes in catalytic shape selectivity. Some of these changes are permanent and caused by SAPO-fication, i.e., the formation of local silicoaluminophosphate (SAPO) interfaces in the zeolite framework. However, a significant other part of these changes can simply be reversed by the elution of physically bonded phosphate species. In catalysis, the physicochemical changes caused by modification by phosphorus can be either promotional or poisonous.During the modification with phosphorus of zeolites by wet impregnation, phosphorus species are deposited on the external surface before entering the zeolite channels. Phosphorus enters the zeolite channels with increasing loadings, but limitations on diffusion lead to a distribution gradient with a higher concentration of phosphorus at the outer surface. With weight loadings of phosphorus above 5 wt%, the formation of large polyphosphates on or near the external surface is observed. Phosphorus species do not cause severe pore blockage at average loadings of phosphorus (above 2 wt%). However, treatment with phosphorus always leads to a decrease in surface area and micropore volume. This decrease is amplified with an increase in phosphorus content.
Excess phosphorus is present as ortho-, pyro-, and polyphosphates. With an increase in phosphorus content the amount of excess phosphorus increases. Excess phosphorus is dehydrated at high temperatures forming condensed polyphosphates. This effect is reversible. Rehydration leads to the formation of smaller phosphate species. Phosphorus species that do not interact with the zeolite framework can easily be eluted by washing with hot water.
Depending on the P/Al ratio and the location of phosphorus, only a certain amount of phosphorus can chemically interact with aluminium in a zeolite. Phosphorus can reversibly interact with tetrahedrally coordinated framework aluminium (TFAl) species, forcing TFAl species into octahedral coordination. Elution of phosphorus by washing with hot water reverses this effect. Phosphorus preferentially reacts with extra-framework aluminium (EFAl) or partially dislodged tetrahedrally coordinated framework aluminium (TFAldis) species. This leads to the formation of (amorphous) extra-framework AlPO4 species and local framework SAPO interfaces. Washing with hot water cannot remove these species. Due to the lack of experimental evidence and the unstable character of Si–O–P bonds it seems unlikely that [PO4] units are fully substituted in the framework to replace [AlO4] and [SiO4] tetrahedra.
Introduction of phosphorus leads to a reduction in the number of strong acid sites. This reduction increases with an increase in the amount of phosphorus. Phosphorus actively promotes dealumination during thermal treatment. The loss of Si–O–Al bonds and the expected reaction of phosphorus with Si–O–Al–OH species to form neutral local SAPO interfaces leads to permanent loss of Brønsted acid sites. Furthermore, the reversible interactions of phosphorus with framework aluminium before thermal treatment have been shown to lead to a loss of acid sites as well. At this moment, the exact nature of this reversible loss of acid sites is not understood. The choice of phosphorus precursor does not have a strong influence on the final effects of modification after thermal treatment, as phosphorus precursors decompose into (poly)phosphate species.
Besides a reduction in the number of acid sites, phosphatation also reduces the average strength of acid sites. An increase in phosphorus content leads to a further decrease in the average strength of acid sites. There are three causes for the observed decrease in average site strength. (i) A decrease in the acid site strength of bridging hydroxyl groups. (ii) Relatively more strong acid sites are removed by phosphatation than weak acid sites. (iii) An actual increase in the number of weak acid sites. Possible candidates for these new weak acid sites are silanol nests, extra-framework aluminium species and P–OH groups. The question of how the average acid site strength is reduced and what the source of new weak acid sites is demands more detailed and systematic characterisation studies.
Phosphorus has been shown to have a promotional effect on the hydrothermal stability of zeolites H-ZSM-5 and H-IM-5. Relatively more strong acid sites are retained during hydrothermal treatment and the amount of tetrahedrally coordinated framework aluminium (TFAl) species that are expelled from the framework is either relatively or absolutely less than in unmodified samples. Like silicoaluminophosphates, local SAPO interfaces have been found to remain unaffected during steam treatment and, as aluminium remains fixed in the framework, the pore structure is better retained. With prolonged steam treatment all Si–O–Al bonds at the SAPO interfaces are hydrolysed, leading to the formation of extra-framework AlPO4. Therefore, we suggest that SAPO interfaces form metastable intermediates during dealumination in the zeolite H-ZSM-5 and H-IM-5 channel systems. It is not at all certain if phosphorus has a similar promotional effect on other framework topologies, which should receive more emphasis in further research.
All these physicochemical effects change the activity and selectivity of phosphated zeolites in a range of catalytic applications. In the selective catalytic reduction of NOx with ammonia over transition metal-exchanged zeolites, the presence of phosphorus is extremely damaging. The breaking of Si–O–Al bonds, leading to a reduction in framework negative charge and acid sites, leads to a decrease in chemisorbed NH3 and NOx species. In addition, the subsequent formation of metal oxides and phosphate species reduces the accessibility of the zeolites even more, leading to severe deactivation.
In catalytic processes to produce hydrocarbons, zeolites modified by phosphorus have generally higher selectivity towards light olefins, especially propylene, and retain higher activities with time on-stream compared to their non-phosphated counterparts, due to reduced formation of coke and improved hydrothermal stability. The decrease in the number of strong Brønsted acid sites and the increase in weak Brønsted acid sites or the weak/strong acid site ratio have most often been attributed as the origin of most changes in performance in catalytic reactions such as hydrocarbon cracking, methanol-to-olefins, dehydration of ethanol and alkylation of aromatics. Reactions that demand strong acid sites achieve reduced conversion with an increase in the phosphorus content. Often, applying higher reaction temperatures increases the overall catalytic conversion. The decrease in acid site number and average acid site strength leads to a reduction in secondary reactions, such as hydride transfer, cracking and cyclisation reactions. This prevents oligomerization of light olefins and formation of coke, as well as the cracking of products.
Besides chemical alterations, the introduction of phosphorus into the zeolite channel leads to decreased pore dimensions and reduced accessibility, which in turn lead to longer diffusion pathways for reactants and products. Therefore, products that diffuse rapidly out of the zeolite will be formed with improved selectivity. Phosphate species that are present in the pores act as steric impediments and hinder the formation of bulky (intermediate) products such as carbenium ions. Therefore, it is suggested that modification by phosphorus of H-ZSM-5 inhibits the bimolecular cracking mechanism in alkane cracking and the aromatic-based cycle in methanol-to-hydrocarbon reactions.
As post-modification of zeolites with phosphorus is a relatively inexpensive way to boost catalyst lifetime and selectivity towards light olefins, and especially as zeolites are important potential agents in future sustainable hydrocarbon production processes, it is of great importance to obtain a more fundamental understanding of the underlying physicochemical effects of phosphorus–zeolite chemistry. The universal concepts on phosphorus–zeolite chemistry that are presented in this review article help the aim to modify zeolites by phosphorus more rationally and can act as a frame of reference in future studies to fundamentally understand the interaction of zeolites with group VA elements in general.
Notes and references
- U. Olsbye, S. Svelle, M. Bjørgen, P. Beato, T. V. W. Janssens, F. Joensen, S. Bordiga and K. P. Lillerud, Angew. Chem., Int. Ed., 2012, 51, 5810–5831 CrossRef CAS PubMed.
- G. W. Huber, S. Iborra and A. Corma, Chem. Rev., 2006, 106, 4044–4098 CrossRef CAS PubMed.
- M. Guisnet and J.-P. Gilson, Zeolites for cleaner technologies, Imperial College Press, London, 2002 Search PubMed.
- S. Brandenberger, O. Kröcher, A. Tissler and R. Althoff, Catal. Rev., 2008, 50, 492–531 CAS.
- L. Karwacki, M. H. F. Kox, D. A. Matthijs de Winter, M. R. Drury, J. D. Meeldijk, E. Stavitski, W. Schmidt, M. Mertens, P. Cubillas, N. John, A. Chan, N. Kahn, S. R. Bare, M. Anderson, J. Kornatowski and B. M. Weckhuysen, Nat. Mater., 2009, 8, 959–965 CrossRef CAS PubMed.
- G. Reding, T. Mäurer and B. Kraushaar-Czarnetzki, Microporous Mesoporous Mater., 2003, 57, 83–92 CrossRef CAS.
- Y. Yan, M. E. Davis and G. R. Gavalas, Ind. Eng. Chem. Res., 1995, 34, 1652–1661 CrossRef CAS.
- D. H. Olson, G. T. Kokotailo, S. L. Lawton and W. M. Meier, J. Phys. Chem., 1981, 85, 2238–2243 CrossRef CAS.
- http://www.iza-structure.org .
- N. Y. Chen, W. W. Kaeding and F. G. Dwyer, J. Am. Chem. Soc., 1979, 101, 6783–6784 CrossRef CAS.
- Z. Y. Zakaria, N. A. S. Amin and J. Linnekoski, Biomass Bioenergy, 2013, 55, 370–385 CrossRef CAS PubMed.
- J. Jae, R. Coolman, T. J. Mountziaris and G. W. Huber, Chem. Eng. Sci., 2014, 108, 33–46 CrossRef CAS PubMed.
- M. Takeuchi, T. Kimura, M. Hidaka, D. Rakhmawaty and M. Anpo, J. Catal., 2007, 246, 235–240 CrossRef CAS PubMed.
- R. K. Grasselli, D. L. Stern and J. G. Tsikoyiannis, Appl. Catal., A, 1999, 189, 1–8 CrossRef CAS.
- W. O. Haag, R. M. Lago and P. G. Rodewald, J. Mol. Catal., 1982, 17, 161–169 CrossRef CAS.
- L. B. Young, S. A. Butter and W. W. Kaeding, J. Catal., 1982, 76, 418–432 CrossRef CAS.
- W. W. Kaeding, C. Chu, L. B. Young, B. Weinstein and S. A. Butter, J. Catal., 1981, 67, 159–174 CrossRef CAS.
- C. D. Chang and A. J. Silvestri, J. Catal., 1977, 47, 249–259 CrossRef CAS.
- W. R. Moser, R. W. Thompson, C.-C. Chiang and H. Tong, J. Catal., 1989, 117, 19–32 CrossRef CAS.
- T. F. Degnan, C. M. Smith and C. R. Venkat, Appl. Catal., A, 2001, 221, 283–294 CrossRef CAS.
- K. Tanabe and W. F. Hölderich, Appl. Catal., A, 1999, 181, 399–434 CrossRef CAS.
- H. Mooiweer, K. De Jong, B. Kraushaar-Czarnetzki, W. Stork and B. Krutzen, Stud. Surf. Sci. Catal., 1994, 84, 2327–2334 CAS.
- G. Cao, L. R. M. Martens, J. L. White, T. J. Chen and M. J. Shah, US6080303 A, 2000.
- C. Plank, E. Rosinski and W. Hawthorne, Ind. Eng. Chem. Prod. Res. Dev., 1964, 3, 165–169 CrossRef CAS.
- J. E. Otterstedt, S. B. Gevert, S. G. Jäås and P. G. Menon, Appl. Catal., 1986, 22, 159–179 CrossRef CAS.
- J. W. Ward, Fuel Process. Technol., 1993, 35, 55–85 CrossRef CAS.
- T. F. Degnan, G. K. Chitnis and P. H. Schipper, Microporous Mesoporous Mater., 2000, 35–36, 245–252 CrossRef CAS.
- A. Corma, V. Fornes, W. Kolodziejski and L. J. Martinez-Triguero, J. Catal., 1994, 145, 27–36 CrossRef CAS.
- J. C. Védrine, A. Auroux, P. Dejaifve, V. Ducarme, H. Hoser and S. Zhou, J. Catal., 1982, 73, 147–160 CrossRef.
- A. F. Costa, H. S. Cerqueira, J. M. M. Ferreira, N. M. S. Ruiz and S. M. C. Menezes, Appl. Catal., A, 2007, 319, 137–143 CrossRef CAS PubMed.
- J. Liu, C. Zhang, Z. Shen, W. Hua, Y. Tang, W. Shen, Y. Yue and H. Xu, Catal. Commun., 2009, 10, 1506–1509 CrossRef CAS PubMed.
- R. Silver, M. Stefanick and B. Todd, Catal. Today, 2008, 136, 28–33 CrossRef CAS PubMed.
- G. Caeiro, P. Magnoux, J. M. Lopes, F. R. Ribeiro, S. M. C. Menezes, A. F. Costa and H. S. Cerqueira, Appl. Catal., A, 2006, 314, 160–171 CrossRef CAS PubMed.
- T. Blasco, A. Corma and J. Martínez-Triguero, J. Catal., 2006, 237, 267–277 CrossRef CAS PubMed.
- J. A. Lercher and G. Rumplmayr, Appl. Catal., 1986, 25, 215–222 CrossRef CAS.
- H. L. Janardhan, G. V. Shanbhag and A. B. Halgeri, Appl. Catal., A, 2014, 471, 12–18 CrossRef CAS PubMed.
- I. Lezcano-Gonzalez, U. Deka, H. E. van der Bij, P. Paalanen, B. Arstad, B. M. Weckhuysen and A. M. Beale, Appl. Catal., B, 2014, 154, 339–349 CrossRef PubMed.
- A. K. Ghosh, N. Kulkarni and P. L. Harvey, WO2014025371 A1, 2014.
- D. H. Harris, WO2014042641 A1, 2014.
- S. Mignard, A. Corma, J. Martinez-Triguero, E. Benazzi, S. Lacombe and G. Mabilon, US6306286 B1, 2001.
- W. F. Lai, M. A. Hamilton and S. J. Mccarthy, WO2013059176 A1, 2013.
- M. S. Al-Ghrami and C. Ercan, WO2013177388 A1, 2013.
- G. Burghels, E. Corresa Mateu, S. Klingelhofer, J. Martinez-Triguero, M. Frauenrath and A. Corma, WO2012123558 A1, 2012.
- L. A. Chewter, W. J. Van and F. Winter, WO2010133643 A2, 2010.
- D. L. Johnson, K. E. Nariman and R. A. Ware, WO2001004237 A2, 2001.
- A. Corma, WO1999002260 A1, 1999.
- T. G. Roberie and F. T. I. I. John, US5286369 A, 1994.
- G. W. Kirker, M. E. Landis and J. H. Yen, US4724066 A, 1988.
- A. J. Tissler and E. T. C. Vogt, WO2001037994 A3, 2001.
- G. Lischke, R. Eckelt, H. G. Jerschkewitz, B. Parlitz, E. Schreier, W. Storek, B. Zibrowius and G. Öhlmann, J. Catal., 1991, 132, 229–243 CrossRef CAS.
- M. J. B. Cardoso, D. D. O. Rosas and L. Y. Lau, Adsorption, 2005, 11, 577–580 CrossRef CAS PubMed.
- W. W. Kaeding and S. A. Butter, J. Catal., 1980, 61, 155–164 CrossRef CAS.
- M. Göhlich, W. Reschetilowski and S. Paasch, Microporous Mesoporous Mater., 2011, 142, 178–183 CrossRef PubMed.
- A. Jentys, G. Rumplmayr and J. A. Lercher, Appl. Catal., 1989, 53, 299–312 CrossRef CAS.
- K. Damodaran, J. W. Wiench, S. M. Cabral de Menezes, Y. L. Lam, J. Trebosc, J. P. Amoureux and M. Pruski, Microporous Mesoporous Mater., 2006, 95, 296–305 CrossRef CAS PubMed.
- J. Caro, M. Bülow, M. Derewinski, J. Haber, M. Hunger, J. Kärger, H. Pfeifer, W. Storek and B. Zibrowius, J. Catal., 1990, 124, 367–375 CrossRef CAS.
- N. Xue, X. Chen, L. Nie, X. Guo, W. Ding, Y. Chen, M. Gu and Z. Xie, J. Catal., 2007, 248, 20–28 CrossRef CAS PubMed.
- J. Zhuang, D. Ma, G. Yang, Z. Yan, X. Liu, X. Liu, X. Han, X. Bao, P. Xie and Z. Liu, J. Catal., 2004, 228, 234–242 CrossRef CAS PubMed.
- S. M. Abubakar, D. M. Marcus, J. C. Lee, J. O. Ehresmann, C. Y. Chen, P. W. Kletnieks, D. R. Guenther, M. J. Hayman, M. Pavlova, J. B. Nicholas and J. F. Haw, Langmuir, 2006, 22, 4846–4852 CrossRef CAS PubMed.
- A. Yamaguchi, D. Jin, T. Ikeda, K. Sato, N. Hiyoshi, T. Hanaoka, F. Mizukami and M. Shirai, Catal. Lett., 2014, 144, 44–49 CrossRef CAS PubMed.
- S. M. Campbell, D. M. Bibby, J. M. Coddington and R. F. Howe, J. Catal., 1996, 161, 350–358 CrossRef CAS.
- A. T. Aguayo, A. G. Gayubo, A. M. Tarrío, A. Atutxa and J. Bilbao, J. Chem. Technol. Biotechnol., 2002, 77, 211–216 CrossRef CAS PubMed.
- Z. Song, A. Takahashi, I. Nakamura and T. Fujitani, Appl. Catal., A, 2010, 384, 201–205 CrossRef CAS PubMed.
- G. Zhao, J. Teng, Z. Xie, W. Jin, W. Yang, Q. Chen and Y. Tang, J. Catal., 2007, 248, 29–37 CrossRef CAS PubMed.
- S. Shwan, J. Jansson, L. Olsson and M. Skoglundh, Appl. Catal., B, 2014, 147, 111–123 CrossRef CAS PubMed.
- L. Lin, C. Qiu, Z. Zhuo, D. Zhang, S. Zhao, H. Wu, Y. Liu and M. He, J. Catal., 2014, 309, 136–145 CrossRef CAS PubMed.
- M. Derewinski, P. Sarv, X. Sun, S. Müller, A. C. Van Veen and J. A. Lercher, J. Phys. Chem. C, 2014, 118, 6122–6131 CAS.
- K. H. Chandawar, S. B. Kulkarni and P. Ratnasamy, Appl. Catal., 1982, 4, 287–295 CrossRef CAS.
- G. Seo and R. Ryoo, J. Catal., 1990, 124, 224–230 CrossRef CAS.
- V. N. Romannikov, A. J. Tissler and R. Thome, React. Kinet. Catal. Lett., 1993, 51, 125–134 CrossRef CAS.
- S. M. Cabral de Menezes, Y. L. Lam, K. Damodaran and M. Pruski, Microporous Mesoporous Mater., 2006, 95, 286–295 CrossRef CAS PubMed.
- M. Ghiaci, A. Abbaspur, M. Arshadi and B. Aghabarari, Appl. Catal., A, 2007, 316, 32–46 CrossRef CAS PubMed.
- M. Ghiaci, A. Abbaspur and R. J. Kalbasi, Appl. Catal., A, 2006, 298, 32–39 CrossRef CAS PubMed.
- R. J. Kalbasi, M. Ghiaci and A. R. Massah, Appl. Catal., A, 2009, 353, 1–8 CrossRef CAS PubMed.
- R. Lü, Z. Cao and X. Liu, J. Nat. Gas Chem., 2008, 17, 142–148 CrossRef.
- G. Jiang, L. Zhang, Z. Zhao, X. Zhou, A. Duan, C. Xu and J. Gao, Appl. Catal., A, 2008, 340, 176–182 CrossRef CAS PubMed.
- D. Zhang, R. Wang and X. Yang, Catal. Lett., 2008, 124, 384–391 CrossRef CAS.
- Y.-J. Lee, J. M. Kim, J. W. Bae, C.-H. Shin and K.-W. Jun, Fuel, 2009, 88, 1915–1921 CrossRef CAS PubMed.
- N. Xue, L. Nie, D. Fang, X. Guo, J. Shen, W. Ding and Y. Chen, Appl. Catal., A, 2009, 352, 87–94 CrossRef CAS PubMed.
- Z. Wang, G. Jiang, Z. Zhao, X. Feng, A. Duan, J. Liu, C. Xu and J. Gao, Energy Fuels, 2009, 24, 758–763 CrossRef.
- K. Ramesh, L. M. Hui, Y.-F. Han and A. Borgna, Catal. Commun., 2009, 10, 567–571 CrossRef CAS PubMed.
- P. Li, W. Zhang, X. Han and X. Bao, Catal. Lett., 2010, 134, 124–130 CrossRef CAS PubMed.
- N. Zhan, Y. Hu, H. Li, D. Yu, Y. Han and H. Huang, Catal. Commun., 2010, 11, 633–637 CrossRef CAS PubMed.
- K. Ramesh, C. Jie, Y.-F. Han and A. Borgna, Ind. Eng. Chem. Res., 2011, 49, 4080–4090 CrossRef.
- D. Liu, W. C. Choi, C. W. Lee, N. Y. Kang, Y. J. Lee, C.-H. Shin and Y. K. Park, Catal. Today, 2011, 164, 154–157 CrossRef CAS PubMed.
- H. E. van der Bij and B. M. Weckhuysen, Phys. Chem. Chem. Phys., 2014, 16, 9892–9903 RSC.
- H. E. van der Bij, L. R. Aramburo, B. Arstad, J. J. Dynes, J. Wang and B. M. Weckhuysen, ChemPhysChem, 2014, 15, 283–292 CrossRef CAS PubMed.
- A. Corma, J. Mengual and P. J. Miguel, Appl. Catal., A, 2012, 421, 121–134 CrossRef PubMed.
- M. Dyballa, E. Klemm, J. Weitkamp and M. Hunger, Chem. Eng. Technol., 2013, 85, 1719–1725 CAS.
- H. Vinek, G. Rumplmayr and J. A. Lercher, J. Catal., 1989, 115, 291–300 CrossRef CAS.
- G. Rumplmayr and J. A. Lercher, Zeolites, 1990, 10, 283–287 CrossRef CAS.
- N. Xue, R. Olindo and J. A. Lercher, J. Phys. Chem. C, 2010, 114, 15763–15770 CAS.
- W. W. Kaeding, C. Chu, L. B. Young and S. A. Butter, J. Catal., 1981, 69, 392–398 CrossRef CAS.
- P. Tynjälä, T. T. Pakkanen and S. Mustamaki, J. Phys. Chem. B, 1998, 102, 5280–5286 CrossRef.
- J. Nunan, J. Cronin and J. Cunningham, J. Catal., 1984, 87, 77–85 CrossRef CAS.
- A. Rahman, G. Lemay, A. Adnot and S. Kaliaguine, J. Catal., 1988, 112, 453–463 CrossRef CAS.
- A. Rahman, A. Adnot, G. Lemay, S. Kaliaguine and G. Jean, Appl. Catal., 1989, 50, 131–147 CrossRef CAS.
- M. Kojima, F. Lefebvre and Y. Ben Taârit, Zeolites, 1992, 12, 724–727 CrossRef CAS.
- W. Reschetilowski, B. Meier, M. Hunger, B. Unger and K. P. Wendlandt, Angew. Chem., Int. Ed., 1991, 30, 686–687 CrossRef PubMed.
- B. Viswanathan and A. C. Pulikottil, Catal. Lett., 1993, 22, 373–379 CrossRef CAS.
- X. Gao, Z. Tang, D. Ji and H. Zhang, Catal. Commun., 2009, 10, 1787–1790 CrossRef CAS PubMed.
- X. Gao, Z. Tang, H. Zhang, C. Liu, Z. Zhang, G. Lu and D. Ji, Korean J. Chem. Eng., 2010, 27, 812–815 CrossRef CAS PubMed.
- Y. Huang, X. Dong, M. Li, M. Zhang and Y. Yu, RSC Adv., 2014, 4, 14573–14581 RSC.
- T. Fjermestad, S. Svelle and O. Swang, J. Phys. Chem. C, 2013, 117, 13442–13451 CAS.
- S. Malola, S. Svelle, F. L. Bleken and O. Swang, Angew. Chem., Int. Ed., 2012, 51, 652–655 CrossRef CAS PubMed.
- S. M. Campbell, D. M. Bibby, J. M. Coddington, R. F. Howe and R. H. Meinhold, J. Catal., 1996, 161, 338–349 CrossRef CAS.
- H. E. van der Bij, F. Meirer, S. Kalirai, J. Wang and B. M. Weckhuysen, Chem. – Eur. J., 2014, 20, 16922–16932 CrossRef CAS PubMed.
- S. M. Bradley and R. F. Howe, Microporous Mater., 1997, 12, 13–19 CrossRef CAS.
- Z. Liu, Z.-X. Chen, W. Ding, G.-J. Kang and Z. Li, THEOCHEM, 2010, 948, 99–101 CrossRef CAS PubMed.
- P. H. Zeng, Y. Liang, S. F. Ji, B. J. Shen, H. H. Liu, B. J. Wang, H. J. Zhao and M. F. Li, J. Energy Chem., 2014, 23, 193–200 CrossRef CAS.
- M. Hunger, Zeolites and Catalysis: Synthesis, Reactions and Applications, Wiley-VCH, Weinheim, 2010, vol. 2, pp. 493–546 Search PubMed.
- X. Wang, W. Dai, G. Wu, L. Li, N. Guan and M. Hunger, Microporous Mesoporous Mater., 2012, 151, 99–106 CrossRef CAS PubMed.
- H. E. Van der Bij, D. Cicmil, J. Wang, F. Meirer, F. M. F. de Groot and B. M. Weckhuysen, J. Am. Chem. Soc., 2014, 136, 17774–17787 CrossRef CAS PubMed.
- E. W. Shin, J. S. Han, M. Jang, S.-H. Min, J. K. Park and R. M. Rowell, Environ. Sci. Technol., 2003, 38, 912–917 CrossRef.
- J. W. Wiench, G. Tricot, L. Delevoye, J. Trebosc, J. Frye, L. Montagne, J.-P. Amoureux and M. Pruski, Phys. Chem. Chem. Phys., 2006, 8, 144–150 RSC.
- Y. Wang, B. Shen, L. Wang, B. Feng, J. Li and Q. Guo, Fuel Process. Technol., 2013, 106, 141–148 CrossRef CAS PubMed.
- M. Müller, G. Harvey and R. Prins, Microporous Mesoporous Mater., 2000, 34, 135–147 CrossRef.
- G. Sastre, D. W. Lewis and C. R. A. Catlow, J. Phys. Chem., 1996, 100, 6722–6730 CrossRef CAS.
- D. Barthomeuf, Zeolites, 1994, 14, 394–401 CrossRef CAS.
- J. Tan, Z. Liu, X. Bao, X. Liu, X. Han, C. He and R. Zhai, Microporous Mesoporous Mater., 2002, 53, 97–108 CrossRef CAS.
- S. L. Suib, A. M. Winiecki and A. Kostapapas, Langmuir, 1987, 3, 483–488 CrossRef CAS.
- R. Lü, Z. Cao and S. Wang, THEOCHEM, 2008, 865, 1–7 CrossRef PubMed.
- R. M. Barrer and M. Liquornik, J. Chem. Soc., Dalton Trans., 1974, 2126–2128 RSC.
- N. J. Clayden, S. Esposito, P. Pernice and A. Aronne, J. Mater. Chem., 2001, 11, 936–943 RSC.
- C. Coelho, T. Azaïs, L. Bonhomme-Coury, G. Laurent and C. Bonhomme, Inorg. Chem., 2007, 46, 1379–1387 CrossRef CAS PubMed.
- G. H. Kühl and K. D. Schmitt, Zeolites, 1990, 10, 2–7 CrossRef.
- A. Corma, J. Mengual and P. J. Miguel, Appl. Catal., A, 2013, 460–461, 106–115 CrossRef CAS PubMed.
- Q. Cui, Y. Zhou, Q. Wei, G. Yu and L. Zhu, Fuel Process. Technol., 2013, 106, 439–446 CrossRef CAS PubMed.
- Y. Wang, B. Shen, K. Hao, J. Li, L. Wang, B. Feng and Q. Guo, Catal. Commun., 2012, 25, 59–63 CrossRef CAS PubMed.
- E. Loeffler, U. Lohse, C. Peuker, G. Oehlmann, L. M. Kustov, V. L. Zholobenko and V. B. Kazansky, Zeolites, 1990, 10, 266–271 CrossRef CAS.
- J. A. van Bokhoven, A. M. J. van der Eerden and D. C. Koningsberger, J. Am. Chem. Soc., 2003, 125, 7435–7442 CrossRef CAS PubMed.
- B. Zhao, H. Pan and J. H. Lunsford, Langmuir, 1999, 15, 2761–2765 CrossRef CAS.
- C. Liu, X. Gao, Z. Zhang, H. Zhang, S. Sun and Y. Deng, Appl. Catal., A, 2004, 264, 225–228 CrossRef CAS PubMed.
- P. Tynjälä and T. T. Pakkanen, Microporous Mesoporous Mater., 1998, 20, 363–369 CrossRef.
- J. Gopalakrishnan, Chem. Mater., 1995, 7, 1265–1275 CrossRef CAS.
- S. van Donk, A. H. Janssen, J. H. Bitter and K. P. de Jong, Catal. Rev., 2003, 45, 297–319 CAS.
- M.-C. Silaghi, C. Chizallet and P. Raybaud, Microporous Mesoporous Mater., 2014, 191, 82–96 CrossRef CAS PubMed.
- O. Lisboa, M. Sánchez and F. Ruette, J. Mol. Catal. A: Chem., 2008, 294, 93–101 CrossRef CAS PubMed.
- T. Chevreau, A. Chambellan, J. Lavalley, E. Catherine, M. Marzin, A. Janin, J. Hemidy and S. Khabtou, Zeolites, 1990, 10, 226–234 CrossRef CAS.
- C. S. Triantafillidis, A. G. Vlessidis, L. Nalbandian and N. P. Evmiridis, Microporous Mesoporous Mater., 2001, 47, 369–388 CrossRef CAS.
- T.-H. Chen, K. Houthoofd and P. J. Grobet, Microporous Mesoporous Mater., 2005, 86, 31–37 CrossRef CAS PubMed.
- J. Jiao, J. Kanellopoulos, W. Wang, S. S. Ray, H. Foerster, D. Freude and M. Hunger, Phys. Chem. Chem. Phys., 2005, 7, 3221–3226 RSC.
- Z. Yu, S. Li, Q. Wang, A. Zheng, X. Jun, L. Chen and F. Deng, J. Phys. Chem. C, 2011, 115, 22320–22327 CAS.
- I. Wang, T.-J. Chen, K.-J. Chao and T.-C. Tsai, J. Catal., 1979, 60, 140–147 CrossRef CAS.
- L. R. Aramburo, E. de Smit, B. Arstad, M. M. van Schooneveld, L. Sommer, A. Juhin, T. Yokosawa, H. W. Zandbergen, U. Olsbye, F. M. F. de Groot and B. M. Weckhuysen, Angew. Chem., Int. Ed., 2012, 51, 3616–3619 CrossRef CAS PubMed.
- S. M. T. Almutairi, B. Mezari, E. A. Pidko, P. C. M. M. Magusin and E. J. M. Hensen, J. Catal., 2013, 307, 194–203 CrossRef CAS PubMed.
- A. Corma, M. Faraldos and A. Mifsud, Appl. Catal., 1989, 47, 125–133 CrossRef CAS.
- G. T. Kerr, J. Phys. Chem., 1967, 71, 4155–4156 CrossRef CAS.
- C. S. Cundy and P. A. Cox, Chem. Rev., 2003, 103, 663–702 CrossRef CAS PubMed.
- R. Ryoo and S. Jun, J. Phys. Chem. B, 1997, 101, 317–320 CrossRef CAS.
- L. R. Aramburo, Y. Liu, T. Tyliszczak, F. M. F. de Groot, J. C. Andrews and B. M. Weckhuysen, ChemPhysChem, 2013, 14, 496–499 CrossRef CAS PubMed.
- L. R. Aramburo, L. Karwacki, P. Cubillas, S. Asahina, D. A. M. de Winter, M. R. Drury, I. L. C. Buurmans, E. Stavitski, D. Mores, M. Daturi, P. Bazin, P. Dumas, F. Thibault-Starzyk, J. A. Post, M. W. Anderson, O. Terasaki and B. M. Weckhuysen, Chem. – Eur. J., 2011, 17, 13773–13781 CrossRef CAS PubMed.
- J. Jiao, S. Altwasser, W. Wang, J. Weitkamp and M. Hunger, J. Phys. Chem. B, 2004, 108, 14305–14310 CrossRef CAS.
- M. Briend, A. Shikholeslami, M.-J. Peltre, D. Delafosse and D. Barthomeuf, J. Chem. Soc., Dalton Trans., 1989, 1361–1362 RSC.
- S. Wilson and P. Barger, Microporous Mesoporous Mater., 1999, 29, 117–126 CrossRef CAS.
- J. P. Loureno, M. F. Ribeiro, F. R. Ribeiro, J. Rocha, Z. Gabelica and E. G. Derouane, Microporous Mater., 1995, 4, 445–453 CrossRef.
- L. Huang and Q. Li, Chem. Lett., 1999, 829–830 CrossRef CAS.
- M. Bjørgen, F. Joensen, M. Spangsberg Holm, U. Olsbye, K.-P. Lillerud and S. Svelle, Appl. Catal., A, 2008, 345, 43–50 CrossRef PubMed.
- U. Deka, I. Lezcano-Gonzalez, B. M. Weckhuysen and A. M. Beale, ACS Catal., 2013, 3, 413–427 CrossRef CAS.
- M. Devadas, O. Kröcher, M. Elsener, A. Wokaun, G. Mitrikas, N. Söger, M. Pfeifer, Y. Demel and L. Mussmann, Catal. Today, 2007, 119, 137–144 CrossRef CAS PubMed.
- P. Kern, M. Klimczak, T. Heinzelmann, M. Lucas and P. Claus, Appl. Catal., B, 2010, 95, 48–56 CrossRef CAS PubMed.
- S. Kotrel, H. Knözinger and B. C. Gates, Microporous Mesoporous Mater., 2000, 35–36, 11–20 CrossRef CAS.
- A. Corma and A. V. Orchillés, Microporous Mesoporous Mater., 2000, 35–36, 21–30 CrossRef CAS.
- C. B. Phillips and R. Datta, Ind. Eng. Chem. Res., 1997, 36, 4466–4475 CrossRef CAS.
- W. Dai, X. Wang, G. Wu, L. Li, N. Guan and M. Hunger, ChemCatChem, 2012, 4, 1428–1435 CrossRef CAS PubMed.
- C. D. Chang, C. T. W. Chu and R. F. Socha, J. Catal., 1984, 86, 289–296 CrossRef CAS.
- C. Mei, P. Wen, Z. Liu, H. Liu, Y. Wang, W. Yang, Z. Xie, W. Hua and Z. Gao, J. Catal., 2008, 258, 243–249 CrossRef CAS PubMed.
- F. L. Bleken, S. Chavan, U. Olsbye, M. Boltz, F. Ocampo and B. Louis, Appl. Catal., A, 2012, 447–448, 178–185 CrossRef CAS PubMed.
- A. F. H. Wielers, M. Vaarkamp and M. F. M. Post, J. Catal., 1991, 127, 51–66 CrossRef CAS.
- B. S. Greensfelder, H. H. Voge and G. M. Good, Ind. Eng. Chem., 1949, 41, 2573–2584 CrossRef CAS.
- N. Rahimi and R. Karimzadeh, Appl. Catal., A, 2011, 398, 1–17 CrossRef CAS PubMed.
- F. C. Jentoft and B. C. Gates, Top. Catal., 1997, 4, 1–13 CrossRef CAS.
- K. Kubo, H. Iida, S. Namba and A. Igarashi, Microporous Mesoporous Mater., 2014, 188, 23–29 CrossRef CAS PubMed.
- F. Bleken, M. Bjørgen, L. Palumbo, S. Bordiga, S. Svelle, K.-P. Lillerud and U. Olsbye, Top. Catal., 2009, 52, 218–228 CrossRef CAS PubMed.
- A. Takahashi, W. Xia, I. Nakamura, H. Shimada and T. Fujitani, Appl. Catal., A, 2012, 423, 162–167 CrossRef PubMed.
- S. Teketel, S. Svelle, K.-P. Lillerud and U. Olsbye, ChemCatChem, 2009, 1, 78–81 CrossRef CAS PubMed.
- F. Bager, N. L. Salas and S. Ernst, Oil Gas Eur. Mag., 2012, 38, 107–111 CAS.
- R. Bastiani, Y. L. Lam, C. A. Henriques and V. Teixeira da Silva, Fuel, 2013, 107, 680–687 CrossRef CAS PubMed.
- O. Bortnovsky, P. Sazama and B. Wichterlova, Appl. Catal., A, 2005, 287, 203–213 CrossRef CAS PubMed.
- D. Mores, E. Stavitski, M. H. F. Kox, J. Kornatowski, U. Olsbye and B. M. Weckhuysen, Chem. – Eur. J., 2008, 14, 11320–11327 CrossRef CAS PubMed.
- M. J. Wulfers and F. C. Jentoft, J. Catal., 2013, 307, 204–213 CrossRef CAS PubMed.
- Y. Furumoto, Y. Harada, N. Tsunoji, A. Takahashi, T. Fujitani, Y. Ide, M. Sadakane and T. Sano, Appl. Catal., A, 2011, 399, 262–267 CrossRef CAS PubMed.
- Y.-J. Lee, Y.-W. Kim, N. Viswanadham, K.-W. Jun and J. W. Bae, Appl. Catal., A, 2010, 374, 18–25 CrossRef CAS PubMed.
- G. Majano, L. Delmotte, V. Valtchev and S. Mintova, Chem. Mater., 2009, 21, 4184–4191 CrossRef CAS.
- F. J. Machado, C. M. López, M. a. A. Centeno and C. Urbina, Appl. Catal., A, 1999, 181, 29–38 CrossRef CAS.
- M. Hartmann, Angew. Chem., Int. Ed., 2004, 43, 5880–5882 CrossRef CAS PubMed.
- J. Perez-Ramirez, C. H. Christensen, K. Egeblad, C. H. Christensen and J. C. Groen, Chem. Soc. Rev., 2008, 37, 2530–2542 RSC.
- Y. Xu, T. Nakajima and A. Ohki, J. Hazard. Mater., 2002, 92, 275–287 CrossRef CAS.
- S. Shevade and R. G. Ford, Water Res., 2004, 38, 3197–3204 CrossRef CAS PubMed.
- P. Chutia, S. Kato, T. Kojima and S. Satokawa, J. Hazard. Mater., 2009, 162, 440–447 CrossRef CAS PubMed.
- D. Mao, J. Xia, B. Zhang and G. Lu, Energy Convers. Manage., 2010, 51, 1134–1139 CrossRef CAS PubMed.
- S. Zheng, A. Jentys and J. A. Lercher, J. Catal., 2003, 219, 310–319 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2015 |