
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceBiosynthetic selenoproteins with genetically-encoded photocaged selenocysteines†
Rasa
Rakauskaitė
a,
Giedrė
Urbanavičiūtė
a,
Audronė
Rukšėnaitė
a,
Zita
Liutkevičiūtė‡
a,
Robertas
Juškėnas
ab,
Viktoras
Masevičius
ab and
Saulius
Klimašauskas
*a
aInstitute of Biotechnology, Vilnius University, V. Graičiūno 8, Vilnius LT-02241, Lithuania. E-mail: saulius.klimasauskas@bti.vu.lt; Fax: +370 5260116
bFaculty of Chemistry, Vilnius University, Naugarduko 24, Vilnius LT-03225, Lithuania
First published on 5th February 2015
Abstract
Selenocysteine is a valuable component of both natural selenoproteins and designer biocatalysts; however the availability of such proteins is hampered by technical limitations. Here we report the first general strategy for the production of selenoproteins via genetically-encoded incorporation of a synthetic photocaged selenocysteine residue in yeast cells, and provide examples of light-controlled protein dimerization and targeted covalent labeling in vitro.
Engineering and in-cell production of recombinant proteins with desired catalytic capacity is widely exploited for structural and functional studies and for practical applications in medicine and industry.1L-Selenocysteine (Sec), the 21st amino acid, endows engineered proteins with new valuable properties due to its enhanced chemical reactivity (higher nucleophilicity, lower pKa, and a lower redox potential) as compared to cysteine. For example, targeted placement of selenocysteine permits expansion of the naturally evolved catalytic capacity of enzymes, and creates sites for selective protein conjugation, labeling, dimerization, or altered protein folding; it also includes valuable isotopes of Se for phase determination in X-ray crystallography, positron emission tomography PET (73Se), radiolabeling (75Se) and NMR studies (77Se) of proteins.2–4 Humans contain over 20 essential selenoproteins, and thus efficient methods for heterologous production of natural selenoproteins are highly desired.5,6
Despite its high technological potential, targeted incorporation of Sec into recombinant proteins is far from trivial. Naturally, biosynthesis of selenoproteins in prokaryotes requires the so called “selenocysteine insertion sequence” (SECIS) located in proximity to the target UGA codon to control its ribosomal suppression during translation.4,7 Therefore, Sec insertions/substitutions in recombinant selenoproteins synthesized in prokaryotic cells typically incur additional mutations. Alternative methods such as protein ligation,8 chemical serine conversion to Sec or protein expression in Cys auxotrophic E. coli strains9 are often cumbersome and/or do not achieve desired target flexibility and specificity. Some recently developed mammalian6 and E. coli10–13 systems no longer depend on the natural selenoprotein synthesis mechanism for site-directed incorporation of Sec but given the profound chemical reactivity of the selenol group under aerobic physiological conditions, the general utility of this strategy has yet to be proven for proteins carrying solvent accessible functionalities (note that most natural selenoproteins contain Se in the form of Se–S bridges).2–4 Targeted incorporation of genetically encoded unnatural amino acids (uAAs) is an alternative general approach that is based on the suppression of an engineered UAG nonsense codon in the presence of an additional orthogonal tRNA/tRNA aminoacyltransferase (tRNA/aaRS) pair that is specific for a particular uAA supplied in growth medium.14,15 A variety of chemical entities were incorporated in a chemically protected (caged) form, permitting the spatiotemporal control of the engineered proteins via subsequent chemical or photochemical uncaging of the incorporated residue.16–20 Using this technique, GFP and histone H3 proteins containing phenylselenocysteine have been produced,21,22 but no method for the deprotection of the incorporated residue to Sec has been proposed.23 Therefore a truly general and efficient method for targeted placement of selenocysteines in recombinant proteins is still lacking.
In the present work we explored a novel strategy based on a photolabile (4,5-dimethoxy-2-nitrobenzyl, DMNB) group to protect Sec in producing cells and during protein isolation (Fig. 1a). We took advantage of a yeast expression system originally designed to incorporate DMNB-Ser residues at genetically-defined positions of a protein, and which showed a good acceptance of DMNB-Cys as well.19,24 Given their structural similarity, we reasoned that the orthogonal LeuRS may also use DMNB-Sec to aminoacylate the orthogonal tRNA. We first turned to the chemical synthesis of DMNB-Sec and DMNB-Cys amino acids such that incorporation of Sec and its sulphur analog could be directly compared. Although in general the sulphur and selenium chemistries have certain similarities, we found that a synthetic route established for production of DMNB-Cys from cystine25 required substantial changes to be efficient for the synthesis of DMNB-Sec from selenocystine (Fig. 1b and Scheme S1†). The major difference derived from a greater sensitivity of selenols to oxidation compared to that of thiols, which precluded isolation of N-Boc-L-selenocysteine in preparatively useful yields. We thus elaborated a one-pot synthesis of N-Boc-DMNB-Sec using sodium borohydride both as a reductant for bis-Boc-L-selenocystine (1) and as a base for generating a selenolate ion for the nucleophilic substitution reaction. Additionally, we found that DMNB-Sec (3) is less stable in acidic media (pH < 3) as compared to DMNB-Cys due to the ability of selenonucleophiles to attack protonated methoxy groups in the aromatic ring. This side reaction (appearance of a 2.06 ppm 1H-NMR resonance corresponding to the methylseleno group of Se-methyl-L-selenocysteine)26 was spotted under the conditions of prolonged (>1 hour) acidic removal of the protecting Boc group (not shown). Using the optimized procedures, both compounds were obtained in gram quantities required for the protein biosynthesis experiments.
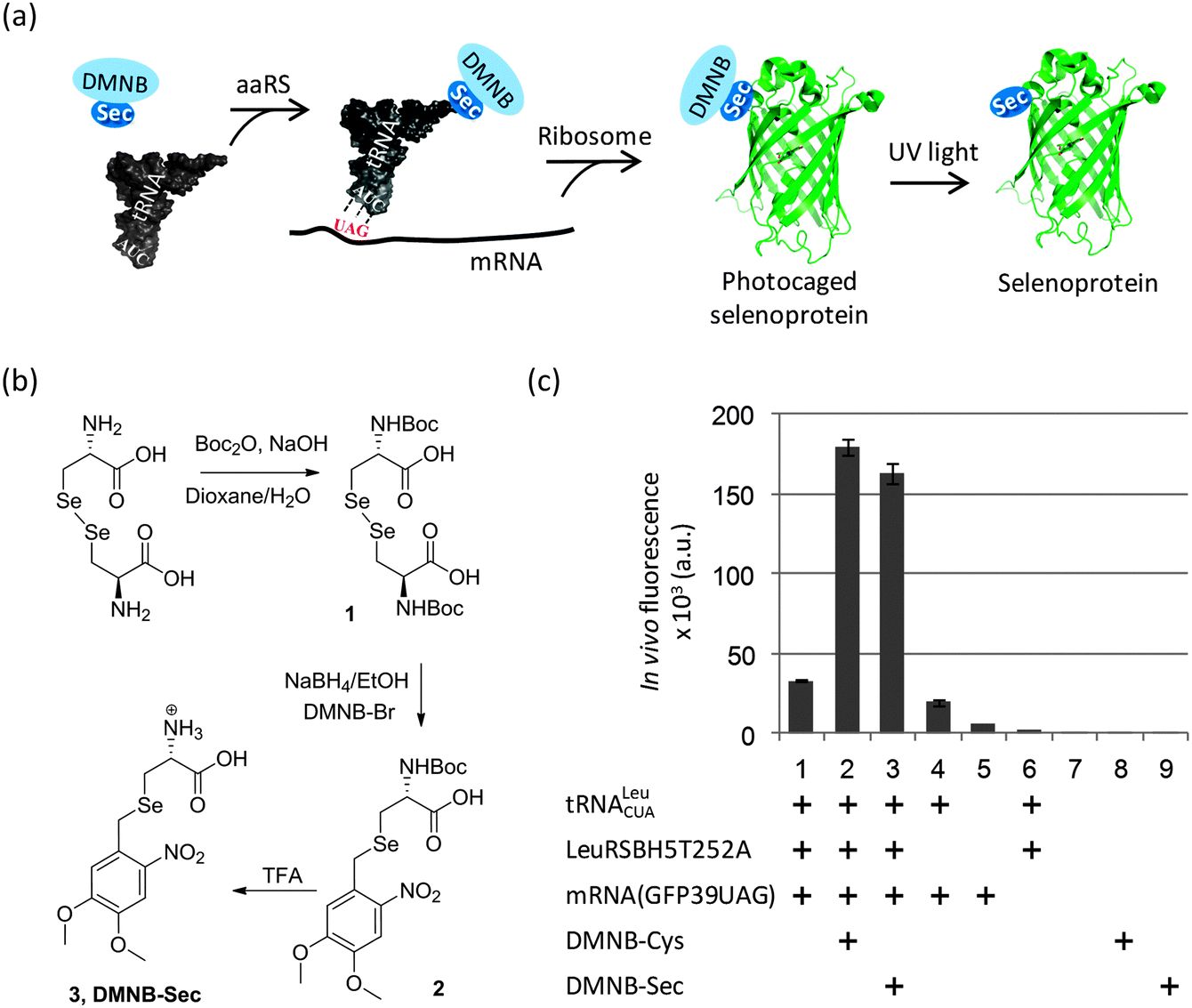 | ||
| Fig. 1 Targeted incorporation of Sec into recombinant proteins. (a) Strategy for in vivo incorporation of a photocaged L-selenocysteine (DMNB-Sec) into a genetically encoded position of a recombinant protein followed by its photochemical decaging. Shown are interactions between key components during incorporation of genetically encoded Sec in the form of a unnatural amino acid DMNB-Sec. (b) Chemical synthesis of 4,5-dimethoxy-2-nitrobenzyl-L-selenocysteine (DMNB-Sec) from seleno-L-cystine. Boc, tert-butyloxycarbonyl; DMNB-Br, 4,5-dimethoxy-2-nitrobenzyl bromide. (c) DMNB-Sec-dependent synthesis of a reporter protein (EGFP) in engineered yeast cells with an expanded genetic code. In vivo EGFP fluorescence assay shows effective production of EGFP only in the presence of the orthogonal tRNA, orthogonal aaRS (LeuRSBH5T252A), a recombinant GFP39TAG gene and externally supplied DMNB-Sec (or DMNB-Cys). Error bars denote standard deviations derived from 3 replicates. | ||
To monitor the production of the target proteins in yeast cells, we used a reporter EGFP expression system that contained the TAG codon replacing a non-essential surface position (Tyr39) of the EGFP gene.27 The DMNB-Ser-specific LeuRSBH5T252A19 gene was integrated in the original construct pSNR-LeuRS27 to create a plasmid bearing the orthogonal pair tRNA/LeuRSBH5T252A. The new construct was co-transformed with the reporter plasmid pGFP39TAG into the LWUPF1Δ yeast cells (see ESI†). Growing these cells with either DMNB-Cys or DMNB-Sec produced a 5-fold increase in EGFP expression levels compared to cells grown with neither uAA (Fig. 1c, columns 1–3), indirectly suggesting that production of the full length protein was strongly dependent on the incorporation of a DMNB-protected uAA. Expression of EGFP in the absence of the uAAs occurred at a substantially lower level (Fig. 1c, columns 1, 4) most likely due to residual charging of the orthogonal tRNA with Leu or Ile by the cellular LeuRS, and/or the orthogonal LeuRSBH5T252A.19 The UAG nonsense suppression by cellular translation machinery, as well as yeast autofluorescence and background fluorescence of the uAAs was negligible (Fig. 1c, columns 5–9). The corresponding DMNB-Sec-EGFP and DMNB-Cys-EGFP proteins were isolated from the yeast cells and purified using Ni-IMAC to greater than 80% purity (Fig. S1†). HPLC/ESI-MS analysis (Fig. 2b (upper) and Fig. S1 and S2†) confirmed the replacement of a Tyr with a single DMNB-Sec or DMNB-Cys residue in the EGFP protein. The incorporation of DMNB-Sec into EGFP was highly efficient: the amount of the EGFPTyr39Leu/Ile in protein preparations was below 2.5% at 2 mM DMNB-Sec, and was undetectable by HPLC/ESI-MS in the presence of 3 mM DMNB-Sec in cell medium. The yield of the caged selenoproteins was typically in the range of 1–2 mg per L of culture.
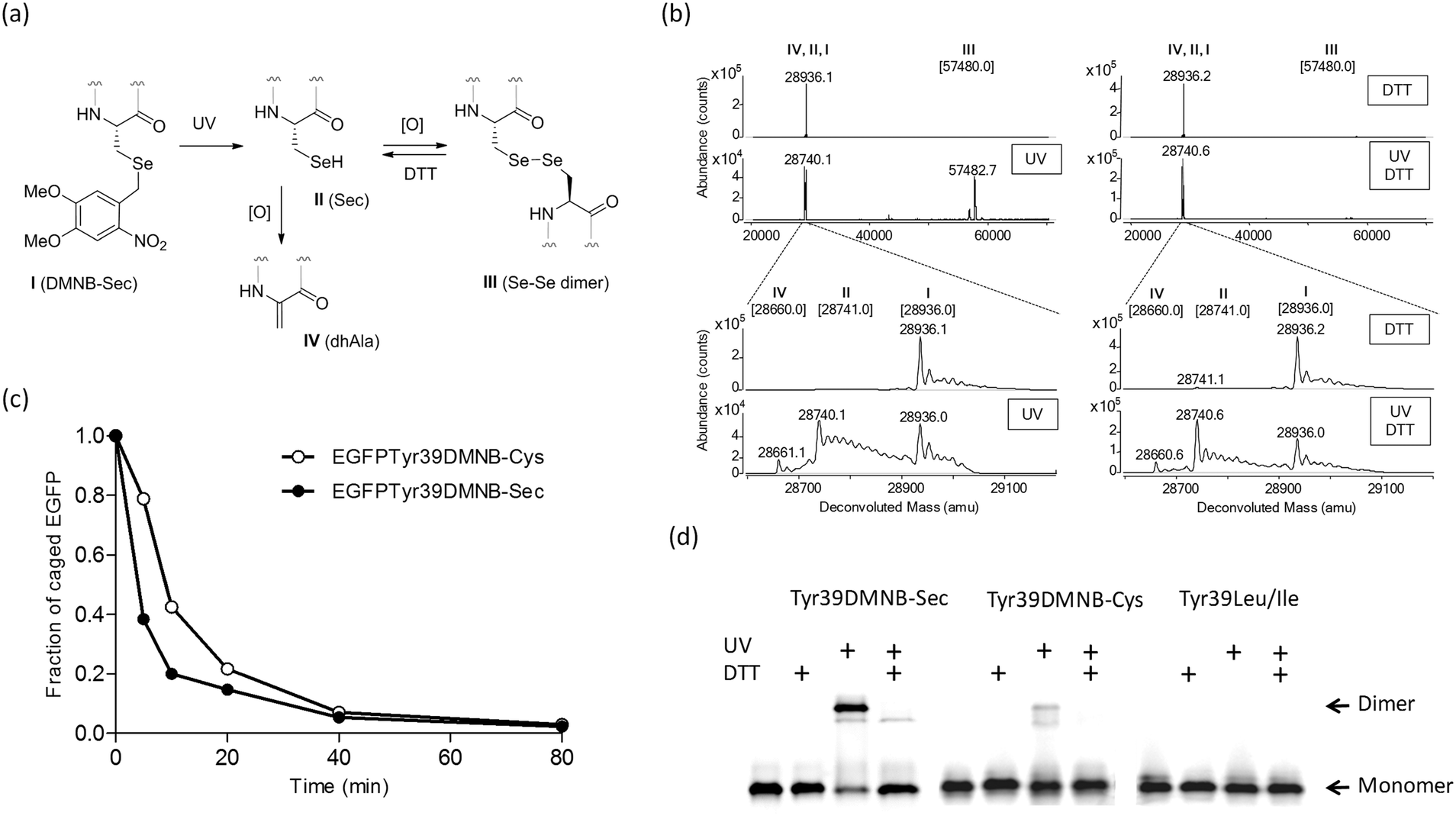 | ||
| Fig. 2 Photochemical decaging of the EGFPTyr39DMNB-Sec protein. (a) Transformations of the incorporated DMNB-Sec residue (I) into selenocysteine (II), diselenide dimer (III) or dehydroalanine (IV). (b) ESI-MS analysis of photolysis products before (upper) and after 20 min exposure (lower) of isolated EGFPTyr39DMNB-Sec protein to UV light. Photodecaging reactions were performed under non-reducing (left) or reducing (DTT, right) conditions. Samples were resolved on an Agilent 1290 Infinity HPLC system equipped with a Poroshell 300SB-C8 column via elution with 1% formic acid in a gradient of water/acetonitrile at a flow rate of 0.4 mL min−1. High-resolution mass spectra were acquired on an in-line Agilent Q-TOF 6520 mass analyzer (100–3200 m/z range, positive ion mode) and analyzed using MassHunter software. Theoretical masses of EGFP variants I–IV are shown in brackets.§ (c) Representative time courses of photochemical decaging under reducing conditions analyzed by HPLC/ESI-MS (Fig. S2†). (d) Semi-native SDS-PAGE gel analysis of fluorescent EGFP variants (EGFPTyr39DMNB-Sec, EGFPTyr39DMNB-Cys, and EGFPTyr39Leu/Ile) before and after UV illumination (UV) under non-reducing and reducing (DTT) conditions. | ||
The DMNB caging group was removed by exposing a solution of the isolated protein to UV light (330–385 nm) (Fig. 2a). HPLC/ESI-MS analysis of reaction products showed an efficient and fast formation of the expected EGFPTyr39Sec protein along with a protein product of a double mass (Fig. 2b and c) and a lower SDS-PAGE mobility (Fig. 2d). Given the fact that a decaged Sec residue is highly reactive and is exposed on the protein surface, we assumed that EGFPTyr39Sec dimerized under aerobic conditions making a diselenide bridge. The dimers were readily converted back to monomers in the presence of DTT (Fig. 2b and d). As expected, the dimerization was much less pronounced in the decaged EGFPTyr39Cys protein, and was completely absent in EGFPTyr39Leu (Fig. 2d). The time course of the photolysis reaction revealed that >95% of EGFPTyr39Sec and EGFPTyr39Cys molecules were decaged (Fig. 2c and Fig. S2†). The decaging process was similar in both tested proteins except that a small fraction of EGFPTyr39Sec was transformed into EGFPTyr39Dha (Fig. 2a, b and Fig. S2a†) which is a common product of selenoprotein oxidation.28
To demonstrate the functional utility of the engineered selenocysteine proteins, we performed a Sec-specific labeling reaction with maleimide-PEG2-biotin. Sec residues can be labeled with the same reagents used for Cys modification, but the former are selectively reactive in acidic milieu.29,30 Indeed, using HPLC/ESI-MS analysis we for the first time directly observed that, at pH 5.0, the decaged EGFPTyr39Sec quantitatively reacted with one equivalent of the maleimide reagent, whereas the protected EGFPTyr39DMNB-Sec protein, which contains two Cys residues, remained unaltered under these conditions (Fig. S3a†). Since the reactivity of Cys residues may vary significantly depending on their location, and the incorporated Sec residue is exposed on the surface of the protein, we performed similar control reactions with the EGFPTyr39Cys and EGFPTyr39Leu proteins (Fig. S3b and c†). We observed no protein modification products corresponding to maleimide-PEG2-biotin at pH 5.0; however efficient biotinylation (presumably at Cys residues) occurred in reactions performed at neutral pH (Fig. S3d†). The Sec protein was also nearly quantitatively labeled in a mixture of decaged EGFPTyr39Sec and EGFPTyr39Cys proteins at pH 5.0 and in the presence of 10 mM DTT, with Cys-biotinylation product detectable by ESI-MS at the level of ∼5% of the Sec-product (Fig. 3). The observed pH-programmable selectivity of the incorporated Sec residue towards maleimide probes well supersedes those reported in previous studies.30
 | ||
| Fig. 3 Selective labeling of a Sec-containing EGFP using a maleimide probe. HPLC/ESI-MS analysis of an equimolar mixture of DMNB-caged EGFPTyr39Sec and EGFPTyr39Cys proteins (upper) and following its UV-illumination and treatment with maleimide-PEG2-biotin (lower) under reducing and slightly acidic conditions (pH 5.0). Analysis was performed as described in Fig. 2b. Deconvoluted spectrograms are scaled to a signal intensity of the most abundant peak (8.5 × 105 counts). Theoretical masses are shown in brackets. | ||
We present the first general strategy for efficient biosynthesis of selenoproteins containing photocaged selenocysteine residues at genetically predetermined positions. The caging group protects highly reactive Sec from undesired side reactions inside producing cells and during subsequent manipulations, which is especially important for Sec residues incorporated at solvent-exposed positions. We also demonstrate a photolytic removal of a protecting group from a Se atom, which has not been previously described for any protein or a synthetic peptide.31 Examples of light-controlled dimerization and site-specific labeling of such recombinant proteins further illustrate robustness and practical utility of the new technique. The generality of this approach is attested by our recent successful production of a HpaII DNA cytosine-5 methyltransferase32,33 fusion protein (see Fig. S4†), in which an essential catalytic Cys is replaced with Sec. This paves the way to a direct comparison of S- and Se-nucleophiles in the natural34 and atypical33,35–37 reactions potentially leading to the design of improved molecular tools for genome studies.
We are grateful to Peter G. Schultz and Lei Wang for their kind gifts of the LWUPF1Δ strain and associated plasmids. This work was supported by the Research Council of Lithuania (grant MIP-115/2012) and the European Social Fund under National Integrated Programme Biotechnology & Biopharmacy (grant VP1-3.1-ŠMM-08-K01-005).
Notes and references
- E. M. Brustad and F. H. Arnold, Curr. Opin. Chem. Biol., 2011, 15, 201–210 CrossRef CAS PubMed.
- N. Metanis, J. Beld and D. Hilvert, PATAI’S Chemistry of Functional Groups, John Wiley & Sons, 2009 Search PubMed.
- E. S. Arner, Exp. Cell Res., 2010, 316, 1296–1303 CrossRef CAS PubMed.
- L. Johansson, G. Gafvelin and E. S. Arner, Biochim. Biophys. Acta, 2005, 1726, 1–13 CrossRef CAS PubMed.
- E. S. Arner, Methods Enzymol., 2002, 347, 226–235 CAS.
- H. Liu, L. Yin, P. G. Board, X. Han, Z. Fan, J. Fang, Z. Lu, Y. Zhang and J. Wei, Protein Expression Purif., 2012, 84, 59–63 CrossRef CAS PubMed.
- S. Yoshizawa and A. Bock, Biochim. Biophys. Acta, 2009, 1790, 1404–1414 CrossRef CAS PubMed.
- T. W. Muir, Annu. Rev. Biochem., 2003, 72, 249–289 CrossRef CAS PubMed.
- S. Muller, H. Senn, B. Gsell, W. Vetter, C. Baron and A. Bock, Biochemistry, 1994, 33, 3404–3412 CrossRef CAS.
- M. J. Bröcker, J. M. L. Ho, G. M. Church, D. Söll and P. O'Donoghue, Angew. Chem., Int. Ed., 2014, 53, 319–323 CrossRef PubMed.
- C. Aldag, M. J. Bröcker, M. J. Hohn, L. Prat, G. Hammond, A. Plummer and D. Söll, Angew. Chem., Int. Ed., 2013, 52, 1441–1445 CrossRef CAS PubMed.
- R. Thyer, A. Filipovska and O. Rackham, J. Am. Chem. Soc., 2013, 135, 2–5 CrossRef CAS PubMed.
- K.-i. Haruna, M. H. Alkazemi, Y. Liu, D. Söll and M. Englert, Nucleic Acids Res., 2014, 42, 9976–9983 CrossRef CAS PubMed.
- C. C. Liu and P. G. Schultz, Annu. Rev. Biochem., 2010, 79, 413–444 CrossRef CAS PubMed.
- A. Dumas, L. Lercher, C. D. Spicer and B. G. Davis, Chem. Sci., 2015, 6, 50–69 RSC.
- J. Li, J. Yu, J. Zhao, J. Wang, S. Zheng, S. Lin, L. Chen, M. Yang, S. Jia, X. Zhang and P. R. Chen, Nat. Chem., 2014, 6, 352–361 CrossRef CAS PubMed.
- C. W. Riggsbee and A. Deiters, Trends Biotechnol., 2010, 28, 468–475 CrossRef CAS PubMed.
- A. Deiters, D. Groff, Y. Ryu, J. Xie and P. G. Schultz, Angew. Chem., Int. Ed. Engl., 2006, 45, 2728–2731 CrossRef CAS PubMed.
- E. A. Lemke, D. Summerer, B. H. Geierstanger, S. M. Brittain and P. G. Schultz, Nat. Chem. Biol., 2007, 3, 769–772 CrossRef CAS PubMed.
- J.-Y. Kang, D. Kawaguchi, I. Coin, Z. Xiang, D. D. M. O'Leary, P. A. Slesinger and L. Wang, Neuron, 2013, 80, 358–370 CrossRef CAS PubMed.
- J. Wang, S. M. Schiller and P. G. Schultz, Angew. Chem., Int. Ed., 2007, 46, 6849–6851 CrossRef CAS PubMed.
- J. Guo, J. Wang, J. S. Lee and P. G. Schultz, Angew. Chem., Int. Ed. Engl., 2008, 47, 6399–6401 CrossRef CAS PubMed.
- K. M. Clark, W. A. van der Donk and Y. Lu, Methods Enzymol., 2009, 462, 97–115 CAS.
- T. S. Young, I. Ahmad, A. Brock and P. G. Schultz, Biochemistry, 2009, 48, 2643–2653 CrossRef CAS PubMed.
- O. Busnel, F. Carreaux, B. Carboni, S. Pethe, S. V. Goff, D. Mansuy and J. L. Boucher, Bioorg. Med. Chem., 2005, 13, 2373–2379 CrossRef CAS PubMed.
- I. Andreadou, W. M. Menge, J. N. Commandeur, E. A. Worthington and N. P. Vermeulen, J. Med. Chem., 1996, 39, 2040–2046 CrossRef CAS PubMed.
- Q. Wang and L. Wang, J. Am. Chem. Soc., 2008, 130, 6066–6067 CrossRef CAS PubMed.
- S. Ma, R. M. Caprioli, K. E. Hill and R. F. Burk, J. Am. Soc. Mass Spectrom., 2003, 14, 593–600 CrossRef CAS.
- B. Q. Shen, K. Xu, L. Liu, H. Raab, S. Bhakta, M. Kenrick, K. L. Parsons-Reponte, J. Tien, S. F. Yu, E. Mai, D. Li, J. Tibbitts, J. Baudys, O. M. Saad, S. J. Scales, P. J. McDonald, P. E. Hass, C. Eigenbrot, T. Nguyen, W. A. Solis, R. N. Fuji, K. M. Flagella, D. Patel, S. D. Spencer, L. A. Khawli, A. Ebens, W. L. Wong, R. Vandlen, S. Kaur, M. X. Sliwkowski, R. H. Scheller, P. Polakis and J. R. Junutula, Nat. Biotechnol., 2012, 30, 184–189 CrossRef CAS PubMed.
- Q. Cheng, L. Johansson, J. O. Thorell, A. Fredriksson, E. Samen, S. Stone-Elander and E. S. Arner, ChemBioChem, 2006, 7, 1976–1981 CrossRef CAS PubMed.
- S. Flemer, Jr., Molecules, 2011, 16, 3232–3251 CrossRef PubMed.
- C. O. Card, G. G. Wilson, K. Weule, J. Hasapes, A. Kiss and R. J. Roberts, Nucleic Acids Res., 1990, 18, 1377–1383 CrossRef CAS PubMed.
- J.-C. Shen, W. M. Rideout III and P. A. Jones, Cell, 1992, 71, 1073–1080 CrossRef CAS.
- R. Gerasimaitė, E. Merkienė and S. Klimašauskas, Nucleic Acids Res., 2011, 39, 3771–3780 CrossRef PubMed.
- R. K. Neely, P. Dedecker, J. I. Hotta, G. Urbanavičiūtė, S. Klimašauskas and J. Hofkens, Chem. Sci., 2010, 1, 453–460 RSC.
- Z. Liutkevičiūtė, E. Kriukienė, I. Grigaitytė, V. Masevičius and S. Klimašauskas, Angew. Chem., Int. Ed. Engl., 2011, 50, 2090–2093 CrossRef PubMed.
- Z. Liutkevičiūtė, E. Kriukienė, J. Ličytė, M. Rudytė, G. Urbanavičiūtė and S. Klimašauskas, J. Am. Chem. Soc., 2014, 136, 5884–5887 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental details, Scheme S1 and Fig. S1–S4. See DOI: 10.1039/c4cc07910h |
| ‡ Present address: Center for Physiology and Pharmacology, Medical University of Vienna, A-1090 Vienna, Austria. |
| § Minor peaks to the right from the major peaks manifest both inherent and apparent heterogeneity of the EGFP proteins in solution, which comprises inadvertent oxidation (increments of +16 amu) and/or bound counterions (Na+, K+). Oxidation of side chains (Met, His, Cys) in proteins commonly occurs to different degrees during biosynthesis and handling or due to ESI-MS artifacts under aerobic conditions. |
| This journal is © The Royal Society of Chemistry 2015 |