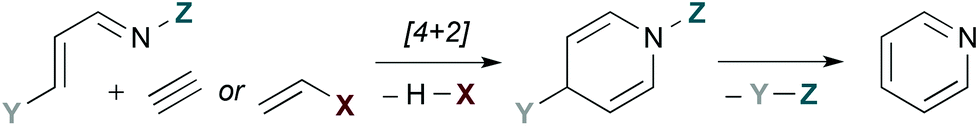DOI:
10.1039/C4QO00187G
(Highlight)
Org. Chem. Front., 2014,
1, 1010-1015
Pyridine synthesis by [4 + 2] cycloadditions of 1-azadienes: hetero-Diels Alder and transition metal-catalysed approaches†
Received
3rd July 2014
, Accepted 6th August 2014
First published on 13th August 2014
Abstract
Cycloaddition reactions are powerful tools in synthetic chemistry. Incorporation of a nitrogen-containing component via cycloaddition represents an attractive strategy for assembling aza-heterocycles. One approach takes advantage of [4 + 2] cycloaddition reactions of 1-azadiene derivatives and 2-carbon π-components to access pyridines, a particularly significant subset of aza-heterocycles. This highlight discusses two distinct modes of reactivity within this class: the thermal pericyclic or hetero-Diels Alder (hDA) reaction and the transition metal-catalysed formal [4 + 2] cycloaddition reaction.
 Jamie M. Neely, Tomislav Rovis | Jamie M. Neely received her B.Sc. in chemistry from the University of Missouri in Columbia, where she worked under the supervision of Professor Timothy Glass. She began her graduate studies in 2008 at Colorado State University under the direction of Professor Tomislav Rovis, defending her thesis in 2014. Her research interests are synthetic chemistry and transition metal catalysis. Tomislav Rovis earned his Ph.D. degree at the University of Toronto in 1998 under the direction of Professor Mark Lautens. From 1998–2000, he was an NSERC postdoctoral fellow at Harvard University with Professor David A. Evans. In 2000, he began his independent career at Colorado State University and was promoted in 2005 to Associate Professor and in 2008 to Professor. His group's accomplishments have been recognized by a number of awards including an NSF CAREER and a Roche Excellence in Chemistry award. He has been named a GlaxoSmithKline Scholar, Amgen Young Investigator, Eli Lilly Grantee, Alfred P. Sloan Fellow, a Monfort Professor at Colorado State University, a Fellow of the American Association for the Advancement of Science, a Katritzky Young Investigator in Heterocyclic Chemistry, and an Arthur C. Cope Scholar. He currently holds the John K. Stille Chair in Chemistry. |
Introduction
Substituted pyridines are important scaffolds in medicinal chemistry,1 as they represent the most common heterocyclic structure encountered in pharmaceutical drugs.2 Developing methods to access these frameworks is therefore a topic of continued interest for synthetic chemists.3 Multicomponent approaches that assemble the heterocyclic core are particularly attractive for pyridine synthesis. One such strategy couples a 1-azadiene to a 2-carbon π-component in a [4 + 2] cycloaddition reaction. This highlight focuses on recent applications of both thermal pericyclic (hDA)4 and transition metal-catalysed5 [4 + 2] cycloadditions of 1-azadienes and 2-carbon π-components to pyridine synthesis.6
Properties of 1-azadienes
1-Azadiene and butadiene differ only by the identity of one atom, yet substitution of carbon with nitrogen greatly differentiates 1-azadiene derivatives from their carbon analogues. Polarization toward the electronegative atom makes 1-azadienes susceptible to nucleophilic attack at the 2- and 4-positions as well as tautomerisation to the 1-aminobutadiene isomer.7 Therefore, 1-azadienes are typically less stable than their butadiene counterparts. From a thermodynamic perspective, exchange of a relatively strong C–N π-bond (79 kcal mol−1) for a relatively weak C–N σ-bond (69 kcal mol−1) makes thermal [4 + 2] cycloaddition of 1-azadienes8 and 6π-electrocyclization of 1-azatrienes9 (vide infra) less favourable than all-carbon versions. Finally, nitrogen's electronegativity serves to decrease molecular orbital energies compared to the butadiene system, most notably leading to a low-lying highest occupied molecular orbital (HOMO) and lowest unoccupied molecular orbital (LUMO).10
Hetero-Diels Alder approaches to pyridines
Fig. 1 provides a conceptual illustration of pyridine synthesis via hDA cycloaddition of a 1-azadiene and a 2-carbon π-component. Since a fully unsaturated ring is the objective, 2-carbon π-components are typically alkynes or alkenes substituted with a leaving group X. The latter act as alkyne surrogates, as elimination of HX after [4 + 2] cycloaddition provides an analogous dihydropyridine intermediate. The third π-bond of the aromatic ring is established by overall loss of Y–Z, a process that is generally accomplished by elimination.
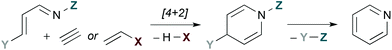 |
| | Fig. 1 Pyridine synthesis by hDA cycloadditions of 1-azadienes. | |
The low-lying 1-azadiene frontier molecular orbitals encourage one of two approaches: (a) Substitution of the 1-azadiene with electron donating groups. These raise the energy of the HOMO and allow normal electron demand hDA cycloadditions with electron deficient dienophiles to proceed. (b) Inverse electron demand hDA reactions with electron rich dienophiles. These take advantage of the low energy 1-azadiene LUMO.
Normal electron demand hetero-Diels Alder cycloaddition
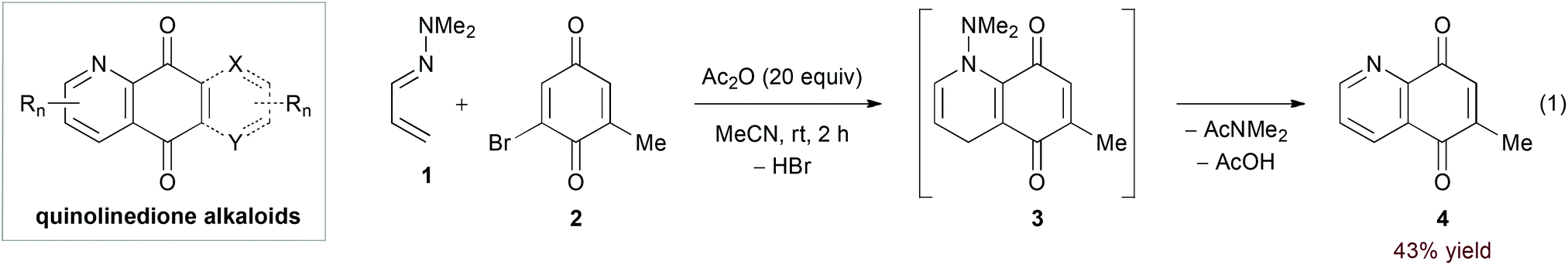
Strongly electron donating substituents can increase the energy of the 1-azadiene HOMO to allow for reactivity with electron deficient dienophiles. Ghosez's pioneering work11 identified α,β-unsaturated hydrazones as an important class of substrates in this manifold. In particular, the hDA reaction of α,β-unsaturated N,N-dialkylhydrazones and quinone derivatives has found frequent use in the synthesis of quinolinedione alkaloids (Scheme 1).12 Here, final aromatization occurs by 1,4-elimination of the dialkylamine N-substituent. In their recent report, Davioud-Charvet and coworkers added acetic anhydride as a scavenger for the amine byproduct in the reaction of hydrazone 1 and bromoquinone 2, observing a yield increase from 9% to 43% (eqn (1)).13| | 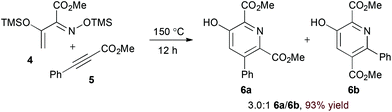 | (2) |
| |  | (3) |
| |  | (5) |
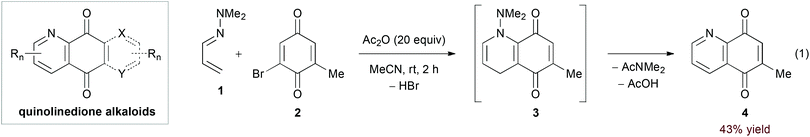 |
| | Scheme 1 Synthesis of quinolinedione alkaloids. | |
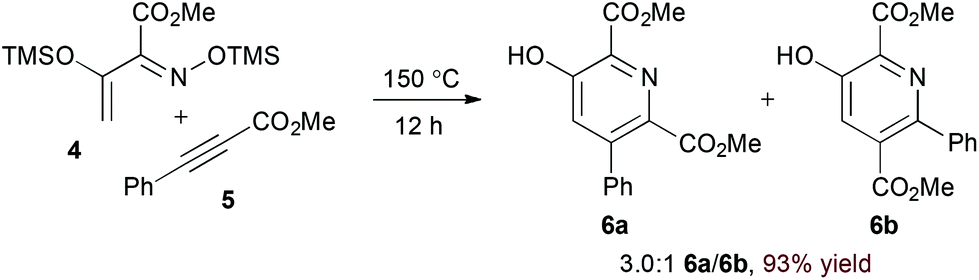
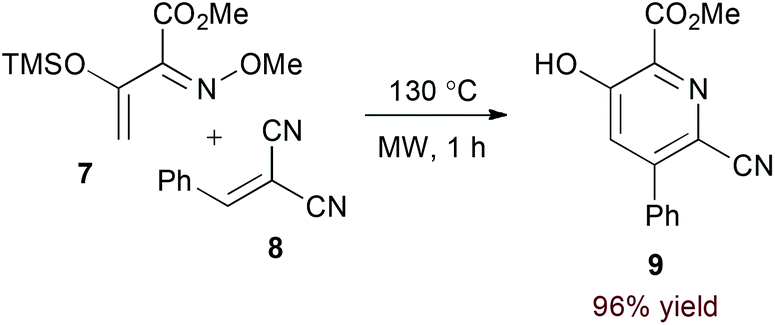
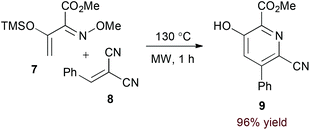
Appropriately activated α,β-unsaturated oxime derivatives may also serve as 1-azadiene components in hDA cycloadditions. Indeed, Arndt and coworkers demonstrated that α-siloxy α,β-unsaturated oxime ethers participate with both alkyne14 and electron deficient alkene15 partners to form pyridine products. Notably, the reaction of alkynes provides regioisomeric mixtures (6a and 6b, eqn (2)) while the reaction of alkylidene malononitriles (8) proceeds with complete regioselectivity (eqn (3)). DFT calculations support an asynchronous concerted pathway rather than a stepwise mechanism for cycloaddition in both cases.15
| |  | (6) |
| |  | (7) |
Inverse electron demand hetero-Diels Alder cycloaddition
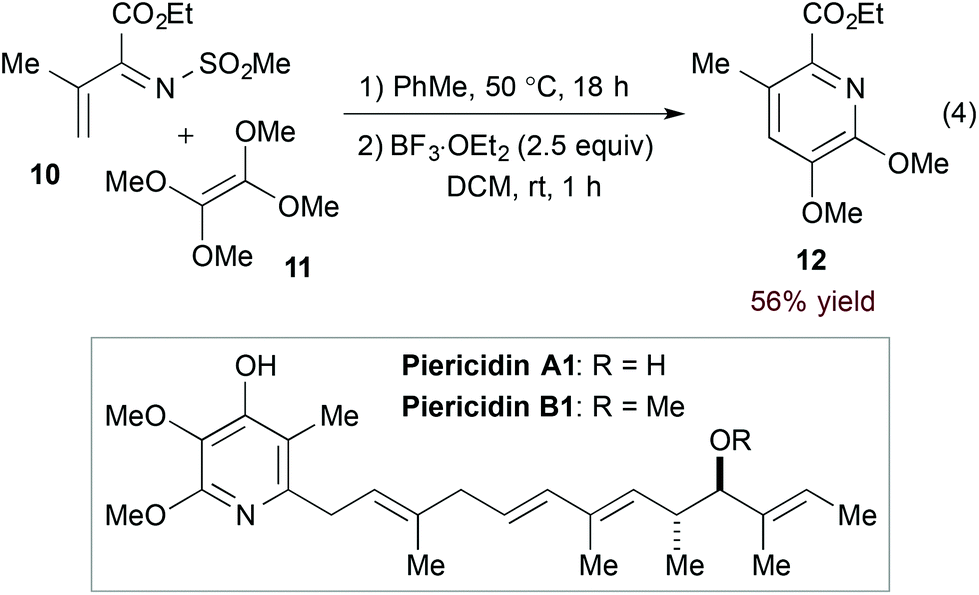
Inverse electron demand hDA cycloadditions of 1-azadienes and electron rich dienophiles take advantage of the low-lying 1-azadiene LUMO. Additional activation by the electron withdrawing N-substituent of α,β-unsaturated N-sulfonyl imines makes them particularly suited for this type of transformation. Complementing their long-standing contribution to hDA reactions of azadienes16 including α,β-unsaturated N-sulfonyl imines,17 Boger and coworkers exploited a hDA approach in their syntheses of piericidins A1 and B1 (Scheme 2).18 Namely, [4 + 2] cycloaddition of imine 10 and tetramethoxyethylene (11) followed by Lewis acid-mediated aromatization accesses the functionalized pyridine core (12, eqn (4)).
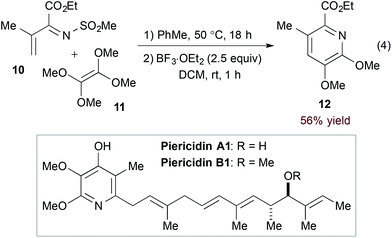 |
| | Scheme 2 Key hDA step in the synthesis of piericidins A1 and B1. | |
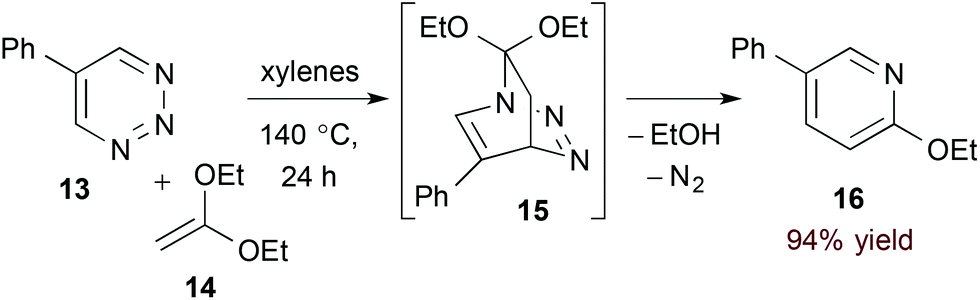
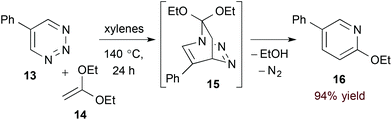
Azadienes embedded within certain 6-membered heteroaromatic rings are particularly useful substrates in the hDA approach to pyridines. In such cases, the initial bicyclic adduct undergoes subsequent retro-[4 + 2] cycloaddition, extruding a 2-atom leaving group and affording the pyridine product directly.19 Boger and coworkers have explored this strategy with a variety of heteroaromatic substrates20 including 1,2,3-triazines (13, eqn (5)).21 Here, after formation of transient bicyclic intermediate 15, retro-[4 + 2] cycloaddition occurs, releasing dinitrogen and the pyridine product 16. 5-Substituted 1,2,3-triazines couple to a variety of electron rich dienophiles with exquisite regioselectivity by this method.
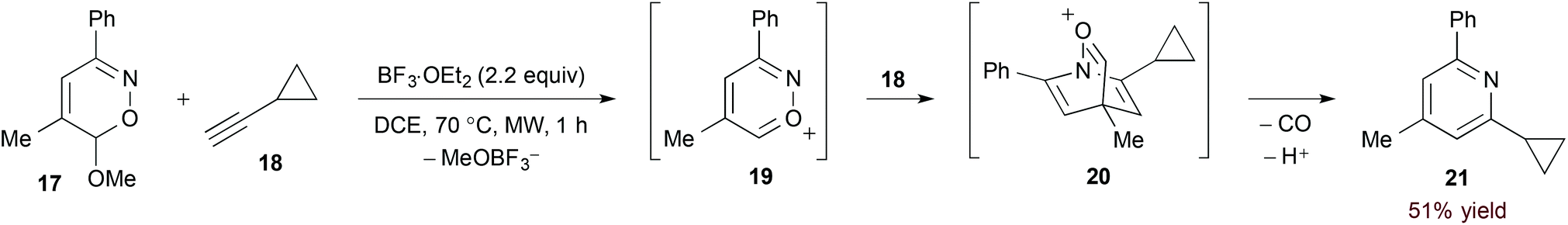
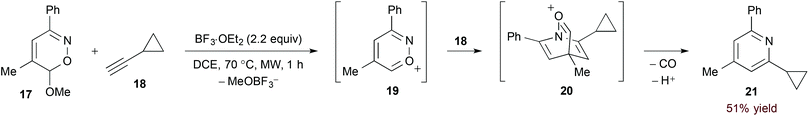
The considerable electron deficiency of a cationic heteroaromatic system presumably imparts a very low energy to the 1-azadiene LUMO. [4 + 2]-Cycloaddition of less activated dienophiles should therefore be possible with such substrates. Reissig and Zimmer investigated this concept in their study of 6H-1,2-oxazine derivatives (17, eqn (6)).22 In this case, Lewis acid-mediated ionization accesses azapyrylium intermediate 19. Cycloaddition proceeds with both electron neutral and rich dienophiles, and extrusion of carbon monoxide affords pyridine products with complete regioselectivity.
Transition metal-catalysed approaches to pyridines
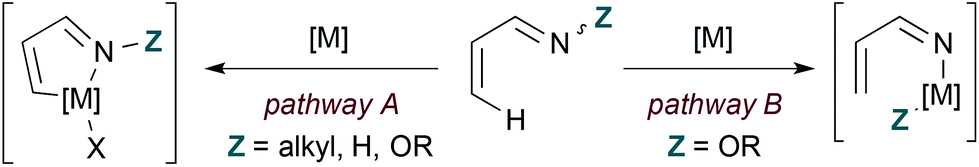
Transition metal catalysis offers distinct pathways for coupling 1-azadienes to 2-carbon π-components by formal [4 + 2] cycloaddition (Fig. 2). First, the nitrogen lone pair can function as a ligand to direct C–H bond activation at the 4-position (pathway A). Alternatively, insertion to the weak N–O bond may occur with N-oxy-substituted 1-azadiene substrates (pathway B). These α,β-unsaturated oxime derivatives may engage in either C–H or N–O bond activation, and the pathway taken depends on the identity of the metal. In general, rhodium catalysts appear to favour C–H activation mechanisms while first row transition metals are thought to operate by N–O insertion pathways.| | 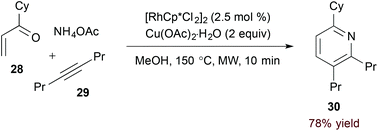 | (8) |
| |  | (9) |
| |  | (10) |
| |  | (11) |
| |  | (12) |
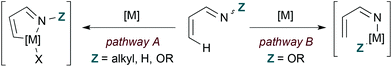 |
| | Fig. 2 Transition metal-catalysed C–H (pathway A) and N–O (pathway B) bond activation of 1-azadienes. | |
Reaction mechanisms and intermediates differ in the following examples, but some general considerations for obtaining pyridine products by transition metal catalysis still apply as illustrated for hDA cycloaddition in Fig. 1. 2-Carbon π-components are typically alkynes, and access to the fully unsaturated ring requires overall loss of Y–Z. The process by which Y–Z (Y = H in these cases) is lost varies by example and sometimes is accomplished by use of an oxidant.
Nitrogen-directed C–H bond activation
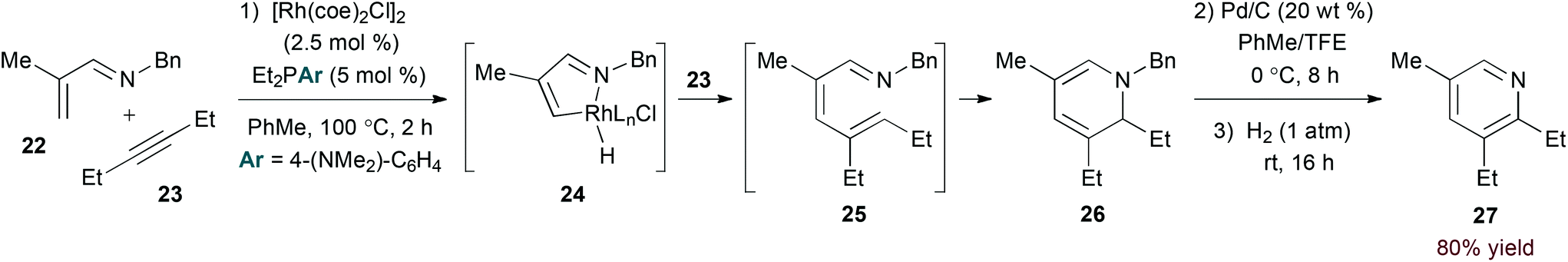
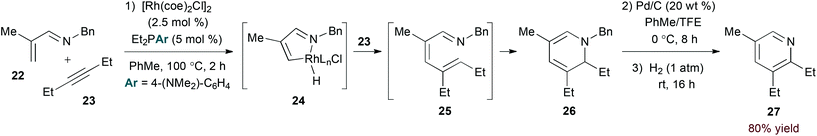
The imine functionality of N-alkyl-substituted 1-azadienes provides a suitable directing group for rhodium-catalysed C–H bond activation.23 Bergman and Ellman provided evidence for C–H activation by an oxidative addition mechanism in the rhodium(I)-catalysed coupling of α,β-unsaturated N-benzyl imines and alkynes (eqn (7)).24 Specifically, stoichiometric reaction of imine 22 afforded a rhodium hydride complex analogous to 24 that was characterized by X-ray analysis. From 24, the authors propose migratory insertion of the alkyne substrate and subsequent reductive elimination to give 1-azatriene intermediate 25. 6π-Electrocyclization affords isolable dihydropyridine 26. Oxidative debenzylation may be conducted in the same flask to provide pyridine products in a three step, one pot sequence.
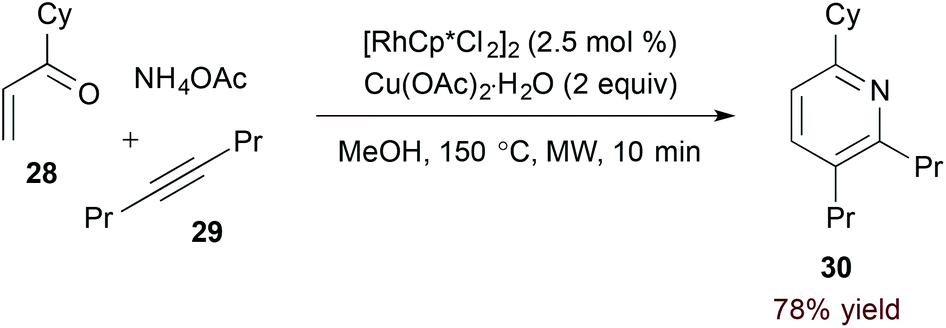
Use of an unsubstituted (N-H) 1-azadiene simplifies the oxidative process required for pyridine formation. Jun and coworkers demonstrated that formal [4 + 2] cycloaddition of α,β-unsaturated N-H imine derivatives and alkynes accesses pyridines directly in the presence of a copper(II) oxidant (eqn (8)).25 Given their instability, imine substrates are generated in situ by condensation of the corresponding ketone (28).
| |  | (13) |
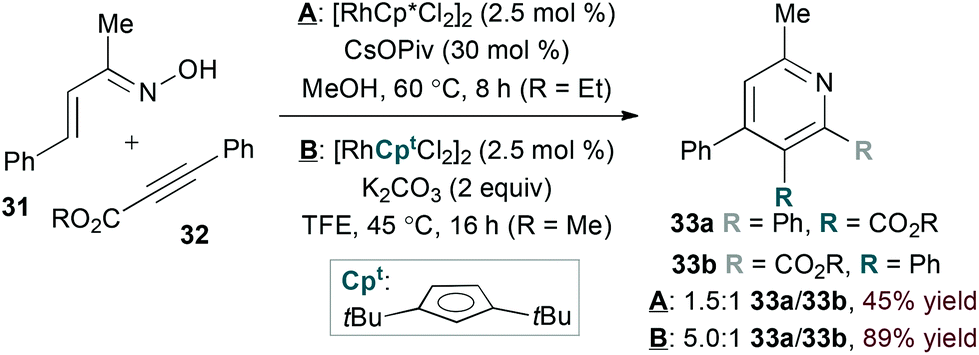
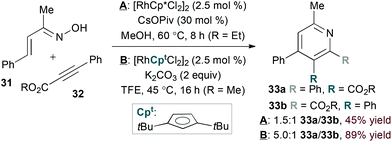
As in hDA approaches, the N-oxy-substituent of an α,β-unsaturated oxime derivative can act as a leaving group to obviate the need for an external oxidant. Cheng and coworkers exploited this strategy in the rhodium(I)-catalysed coupling of α,β-unsaturated oximes26 and alkynes.27 Li and Chiba found that rhodium(III) catalysis28 also achieves the desired transformation (conditions A, eqn (9)).29 In both rhodium(I)- and rhodium(III)-catalysed reactions, unsymmetrical alkyne substrates lead to regioisomeric mixtures.
Modifying the steric environment around a metal centre can influence selectivity in transition metal-catalysed reactions. Indeed, Rovis and Hyster showed that use of a bulky di-tert-butylcyclopentadienyl ligand (Cpt, conditions B, eqn (9)) provides substantial improvement in regioselectivity for the reaction of unsymmetrical alkyne substrates.30 Mechanistic studies support a concerted metalation deprotonation pathway for C–H activation by the rhodium(III) catalyst, differentiating the rhodium(I)- and rhodium(III)-catalysed pathways.
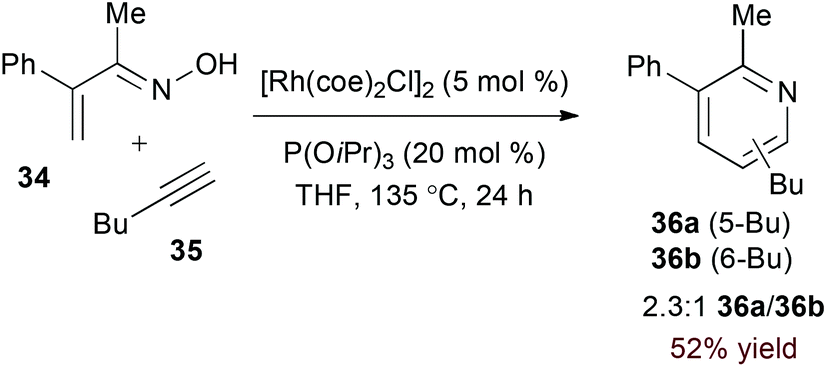
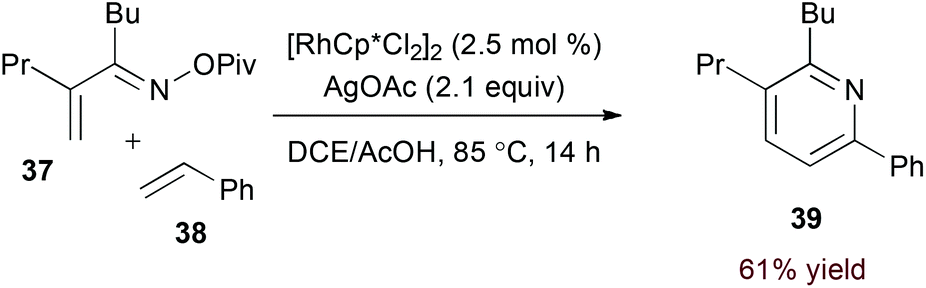
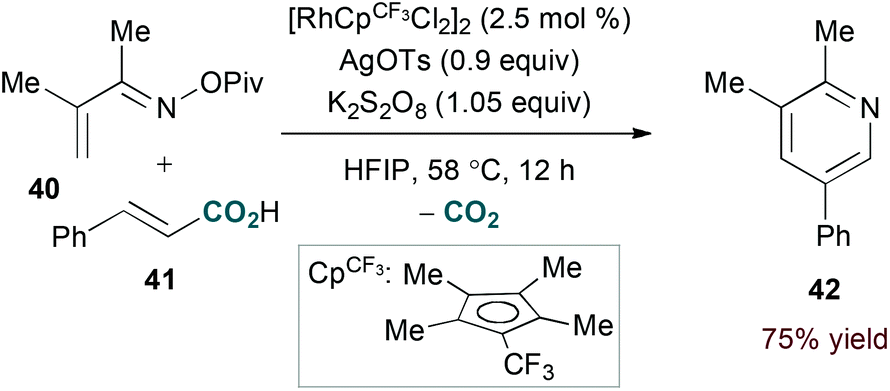
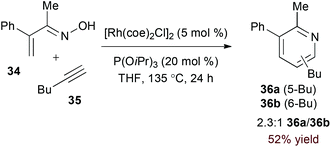
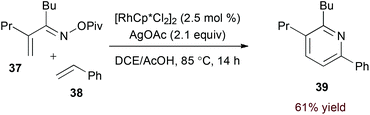
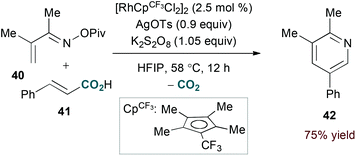
Terminal alkyne substrates often prove to be problematic in the above-mentioned transition metal-catalysed reactions, since they are susceptible to a variety of side reactions.31 Bergman and Ellman identified a rhodium(I)/phosphite catalyst system that favours pyridine formation in the reaction of α,β-unsaturated oximes and terminal alkynes, providing mixtures of 5- and 6-substituted products (36a and 36b, eqn (10)).32 Use of a terminal alkene substrate in combination with a stoichiometric oxidant represents an alternative route to products with these substitution patterns. Rovis and Neely demonstrated that 6-substituted pyridines (39, eqn (11)) are accessed with complete regioselectivity by rhodium(III)-catalysed coupling of O-pivaloyl α,β-unsaturated oxime esters and activated alkenes in the presence of a silver(I) oxidant.33 In a complementary method, the carboxylic acid of acrylic acid derivatives (41, eqn (12)) serves as a traceless activating group for selective formation of 5-substituted pyridines by decarboxylative cycloaddition.34
| |  | (14) |
N–O bond activation of α,β-unsaturated oxime derivatives
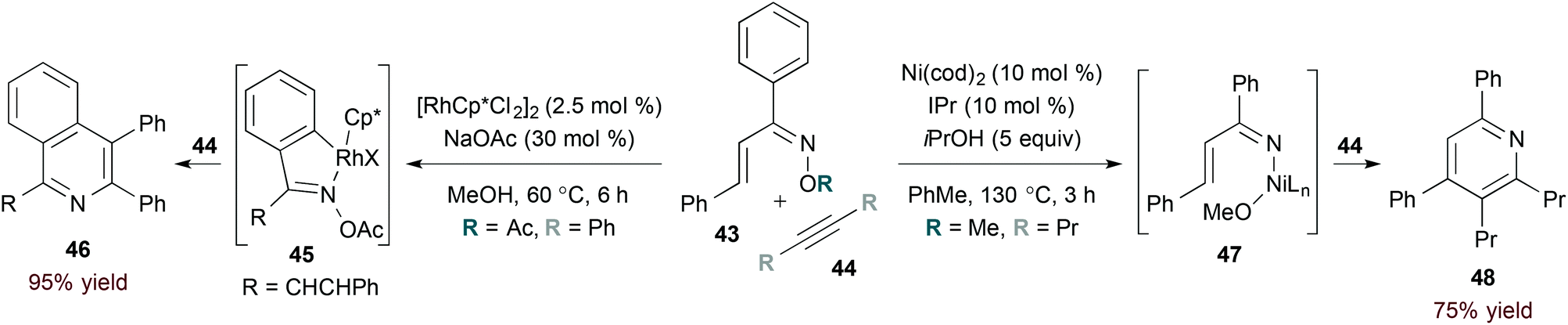
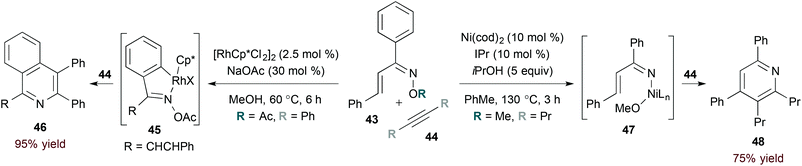
In contrast to rhodium-catalysed methods, catalysis by first row transition metals likely proceeds by activation of the weak N–O bond in α,β-unsaturated oxime substrates. The rhodium- and nickel-catalysed reactions of oxime 43 highlight this complementarity (eqn (13)). Specifically, rhodium(III) catalysis leads to formation of isoquinoline derivative 46 (eqn (13), left),35 while a nickel(0) catalyst provides pyridine 48 selectively (eqn (13), right).36 The latter conditions are useful for accessing pyridine derivatives from α,β-unsaturated oxime ethers, as shown by Kurahashi and Matsubara.36
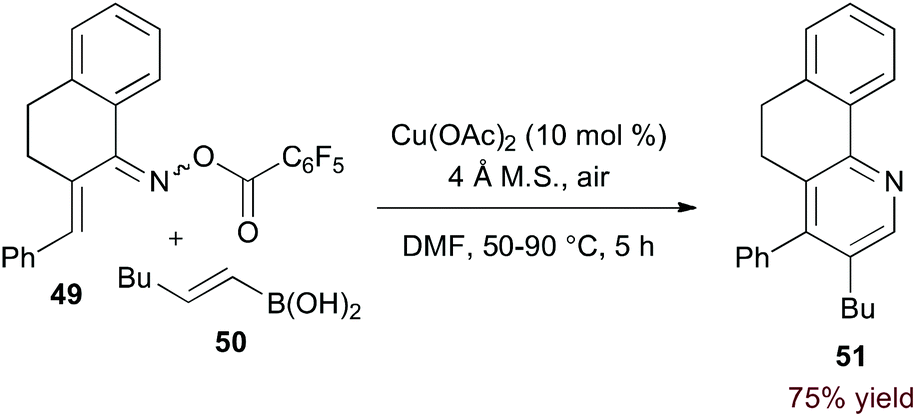
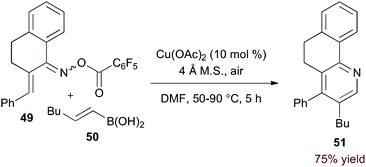
Incorporation of a 2-carbon π-component may also be achieved by transmetalation. Indeed, Liebeskind and Liu demonstrated that O-pentafluorobenzoyl α,β-unsaturated oxime esters (49, eqn (14)) are coupled to vinyl boronic acids by copper catalysis.37 Presumably, after N–O activation, transmetalation and C–N bond formation provide a 3-azatriene intermediate. 6π-Electrocyclization and aerobic oxidation afford the pyridine product. Importantly, this method of C–N bond formation precludes formation of regioisomeric products.
Conclusion
[4 + 2] Cycloaddition reactions of 1-azadienes and 2-carbon π-components represent a powerful approach to substituted pyridines. The hetero-Diels Alder reaction and transition metal-catalysed formal cycloaddition offer distinct pathways for accomplishing the transformation. Both approaches provide efficient methods for synthesizing pyridine derivatives, and will likely find continued use in the area.
Acknowledgements
We acknowledge NIGMS (GM80442) for support of our work in this area and Johnson Matthey for a generous loan of Rh salts.
Notes and references
- S. D. Roughley and A. M. Jordan, J. Med. Chem., 2011, 54, 3451 CrossRef CAS PubMed
 ; J. S. Carey, D. Laffan, C. Thomson and M. T. Williams, Org. Biomol. Chem., 2006, 4, 2337 Search PubMed .
; J. S. Carey, D. Laffan, C. Thomson and M. T. Williams, Org. Biomol. Chem., 2006, 4, 2337 Search PubMed .
- R. D. Taylor, M. MacCoss and A. D. G. Lawson, J. Med. Chem., 2014, 57, 5845 Search PubMed .
- For reviews of pyridine synthesis, see: M. D. Hill, Chem. – Eur. J., 2010, 16, 12052 CrossRef CAS PubMed ; G. D. Henry, Tetrahedron, 2004, 60, 6043 Search PubMed .
- M. Behforouz and M. Ahmadian, Tetrahedron, 2000, 56, 5259 CrossRef CAS .
- A. V. Gulevich, A. S. Dudnik, N. Chernyak and V. Gevorgyan, Chem. Rev., 2013, 113, 3084 CrossRef CAS PubMed .
- For a review that covers earlier [4 + 2] cycloadditions of 1-azadienes, see: B. Groenendaal, E. Ruijter and R. V. A. Orru, Chem. Commun., 2008, 5474 RSC .
-
D. L. Boger and S. M. Weinreb, Hetero Diels–Alder Methodology in Organic Synthesis, Academic Press, San Diego, CA, 1987, pp. 240–255 Search PubMed .
- S. M. Bachrach and M. Liu, J. Org. Chem., 1992, 57, 6736 CrossRef CAS .
- K. Tanaka, H. Mori, M. Yamamoto and S. Katsumura, J. Org. Chem., 2001, 66, 3099 Search PubMed .
-
I. Fleming, Frontier Orbitals and Organic Chemical Reactions, Wiley, Chichester, U. K., 1976 Search PubMed .
- B. Serckx-Poncin, A.-M. Hesbain-Frisque and L. Ghosez, Tetrahedron Lett., 1982, 23, 3261 Search PubMed .
- R. Lazny and A. Nodzewska, Chem. Rev., 2010, 110, 1386 CrossRef CAS PubMed and references therein.
- D. A. Lanfranchi, E. Cesar-Rodo, B. Bertrand, H.-H. Huang, L. Day, L. Johann, M. Elhabiri, K. Becker, D. L. Williams and E. Davioud-Charvet, Org. Biomol. Chem., 2012, 10, 6375 CAS .
- J.-Y. Lu and H.-D. Arndt, J. Org. Chem., 2007, 72, 4205 CrossRef CAS PubMed .
- J.-Y. Lu, J. A. Keith, W.-Z. Shen, M. Schürmann, H. Preut, T. Jacob and H.-D. Arndt, J. Am. Chem. Soc., 2008, 130, 13219 Search PubMed .
- D. L. Boger, Bull. Soc. Chim. Belg., 1990, 99, 599 Search PubMed .
- R. C. Clark, S. S. Pfeiffer and D. L. Boger, J. Am. Chem. Soc., 2006, 128, 2587 CrossRef CAS PubMed and references therein.
- M. J. Schnermann, F. A. Romero, I. Hwang, E. Nakamaru-Ogiso, T. Yagi and D. L. Boger, J. Am. Chem. Soc., 2006, 128, 11799 CrossRef CAS PubMed .
- Aza-heterocycle synthesis by the combination of hDA cycloaddition and subsequent retro-[4 + 2] cycloaddition was reviewed recently. See: R. A. A. Roster and M. C. Willis, Chem. Soc. Rev., 2013, 42, 63 RSC .
- D. L. Boger, Chem. Rev., 1986, 86, 781 CrossRef CAS .
- E. D. Anderson and D. L. Boger, J. Am. Chem. Soc., 2011, 133, 12285 CrossRef CAS PubMed .
- I. Linder, M. Gerhard, L. Schefzig, M. Andrä, C. Bentz, H.-U. Reissig and R. Zimmer, Eur. J. Org. Chem., 2011, 6070 CrossRef CAS PubMed .
- D. A. Colby, A. S. Tsai, R. G. Bergman and J. A. Ellman, Acc. Chem. Res., 2012, 45, 814 CrossRef CAS PubMed and references therein.
- D. A. Colby, R. G. Bergman and J. A. Ellman, J. Am. Chem. Soc., 2008, 130, 3645 CrossRef CAS PubMed .
- H. Lee, Y.-K. Sim, J.-W. Park and C.-H. Jun, Chem. – Eur. J., 2014, 20, 323 CrossRef CAS PubMed ; Y.-K. Sim, H. Lee, J.-W. Park, D.-S. Kim and C.-H. Jun, Chem. Commun., 2012, 48, 11787 Search PubMed .
- For the Rh(I)-catalyzed intramolecular reaction of ε-alkynyl α,β-unsaturated oxime ethers, see: A. Saito, M. Hironaga, S. Oda and Y. Hanzawa, Tetrahedron Lett., 2007, 48, 6852 CAS .
- K. Parthasarathy and C.-H. Cheng, Synthesis, 2009, 1400 CAS ; K. Parthasarathy, M. Jeganmohan and C.-H. Cheng, Org. Lett., 2008, 10, 325 Search PubMed .
- Dong and coworkers reported the analogous coupling of α,β-unsaturated N-sulfonyl imines and alkynes. See: Q.-R. Zhang, J.-R. Huang, W. Zhang and L. Dong, Org. Lett., 2014, 16, 1684 Search PubMed .
- P. C. Too, T. Noji, Y. J. Lim, X. Li and S. Chiba, Synlett, 2011, 2789 CAS .
- T. K. Hyster and T. Rovis, Chem. Commun., 2011, 47, 11846 RSC .
- V. Ritleng, C. Sirlin and M. Pfeffer, Chem. Rev., 2002, 102, 1731 CrossRef CAS PubMed , also see ref. 24.
- R. M. Martin, R. G. Bergman and J. A. Ellman, J. Org. Chem., 2012, 77, 2501 Search PubMed .
- J. M. Neely and T. Rovis, J. Am. Chem. Soc., 2013, 135, 66 CrossRef CAS PubMed .
- J. M. Neely and T. Rovis, J. Am. Chem. Soc., 2014, 136, 2735 Search PubMed .
- P. C. Too, Y.-F. Wang and S. Chiba, Org. Lett., 2010, 12, 5688 CrossRef CAS PubMed .
- Y. Yoshida, T. Kurahashi and S. Matsubara, Chem. Lett., 2012, 41, 1498 CrossRef CAS .
- S. Liu and L. S. Liebeskind, J. Am. Chem. Soc., 2008, 130, 6918 CrossRef CAS PubMed .
Footnote |
| † Dedicated to Professor Max Malacria on the occasion of his 65th birthday. |
|
| This journal is © the Partner Organisations 2014 |
Click here to see how this site uses Cookies. View our privacy policy here.