Catalytic fluorination of unactivated C(sp3)–H bonds
Jun-An
Ma
* and
Shen
Li
Department of Chemistry, Key Laboratory of Systems Bioengineering (The Ministry of Education), Tianjin University, Collaborative Innovation Center of Chemical Science and Engineering (Tianjin), Tianjin 300072, P. R. China. E-mail: majun_an68@tju.edu.cn
First published on 4th April 2014
Abstract
Organofluorine compounds have found widespread applications in the pharmaceutical and agrochemical industries. Efficient construction of organofluorine molecules is highly desirable. Catalytic transformation of C(sp3)–H bonds into C(sp3)–F bonds provides the simplest and most straightforward way to organofluorine compounds. This Highlight discusses the most recent findings in the field.
Organofluorine compounds possess unique physical and chemical properties because of fluorine's small size and high electronegativity, thus efficient construction of C–F bonds has been the subject of intensive research in the fields of synthetic and medicinal chemistry.1 In this context, direct fluorination of sp3 hybridized carbon–hydrogen [C(sp3)–H] sites offers significant opportunities for the incorporation of fluorine into a broad range of hydrocarbon compounds. The traditional methods have focused on the use of the active C(sp3)–H bond-containing compounds such as carbonyl derivatives, phosphate esters, and sulfones (Fig. 1).2 However, the challenge entails achieving site selectivity in direct fluorination of structurally complex molecules that bear many unactivated C(sp3)–H bonds of comparable strengths and reactivity.
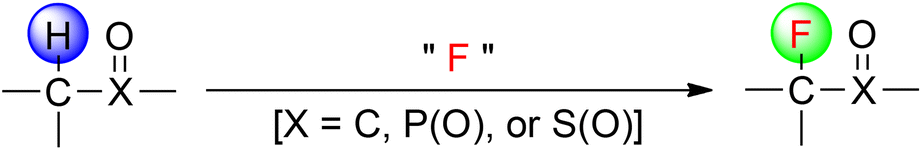 | ||
| Fig. 1 Fluorination of activated C(sp3)–H bonds. | ||
Early methods for the direct transformation of unactivated C(sp3)–H bonds into C(sp3)–F bonds with a fluorine source has been restricted to molecular F2, hypofluorites, and XeF2. Although these fluorinating reagents are commercially available, they are hazardous and exhibit uncontrollable reactivities. Recently, transition metal- and organic small molecule-catalyzed C(sp3)–H bond fluorination employing stoichiometric amounts of fluorinating agents has been reported. In 2006, Sanford and co-workers described a palladium-catalyzed fluorination of benzylic C(sp3)–H bonds (Scheme 1).3 By employing a quinoline directing group, the formation of benzylic C–F bonds was achieved using an electrophilic fluorinating reagent. Notably, this fluorinating reagent was believed to play two roles: serving as an oxidizing agent to generate high-valent palladium(IV) fluoride complexes from PdII catalysts and providing a source of fluorine that ends up in the final products. Later on, the same group demonstrated that the palladium-catalyzed quinoline-directed benzylic C–H fluorination of 8-methylquinolines was also achievable when using fluoride salts and hypervalent iodine oxidants ArI(OPiv)2 (Scheme 1).4 Although the primary active fluorinating species could not be established, a 19F NMR analysis of the crude reaction mixture provided preliminary evidence that an ArIF2 intermediate was formed under the reaction conditions.
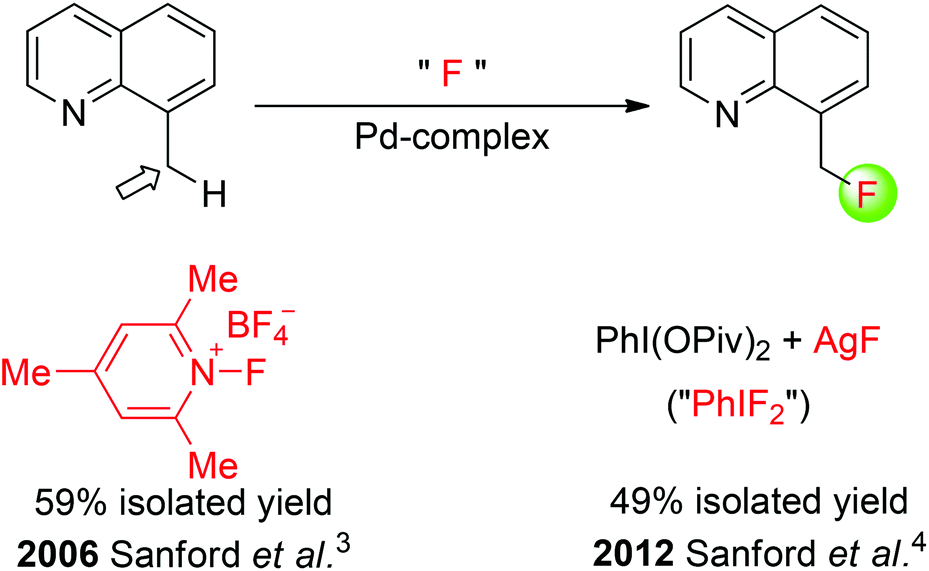 | ||
| Scheme 1 Pd-catalyzed benzylic C(sp3)–H fluorination. | ||
Lectka et al. developed a mild iron(II) complex-catalyzed fluorination of benzylic C(sp3)–H using commercially available Selectfluor [1-chloromethyl-4-fluorodiazoniabicyclo [2.2.2]-octane bis(tetrafluoroborate)] as the fluorine transfer reagent,5 whereas Groves and co-workers presented a (S,S)-(salen)MnCl-catalyzed oxidative benzylic C(sp3)–H fluorination reaction with Et3N·3HF or a combination of Et3N·3HF and AgF.6 These two systems could produce an array of benzylic fluorinated products in good yields and selectivity. As an example, a nonsteroidal anti-inflammatory drug (ibuprofen methyl ester) was selectively fluorinated at a single position (Scheme 2). Clearly, transition-metal complexes are crucial for reaction selectivity favoring the benzylic position over chemistry at the more acidic carbon.
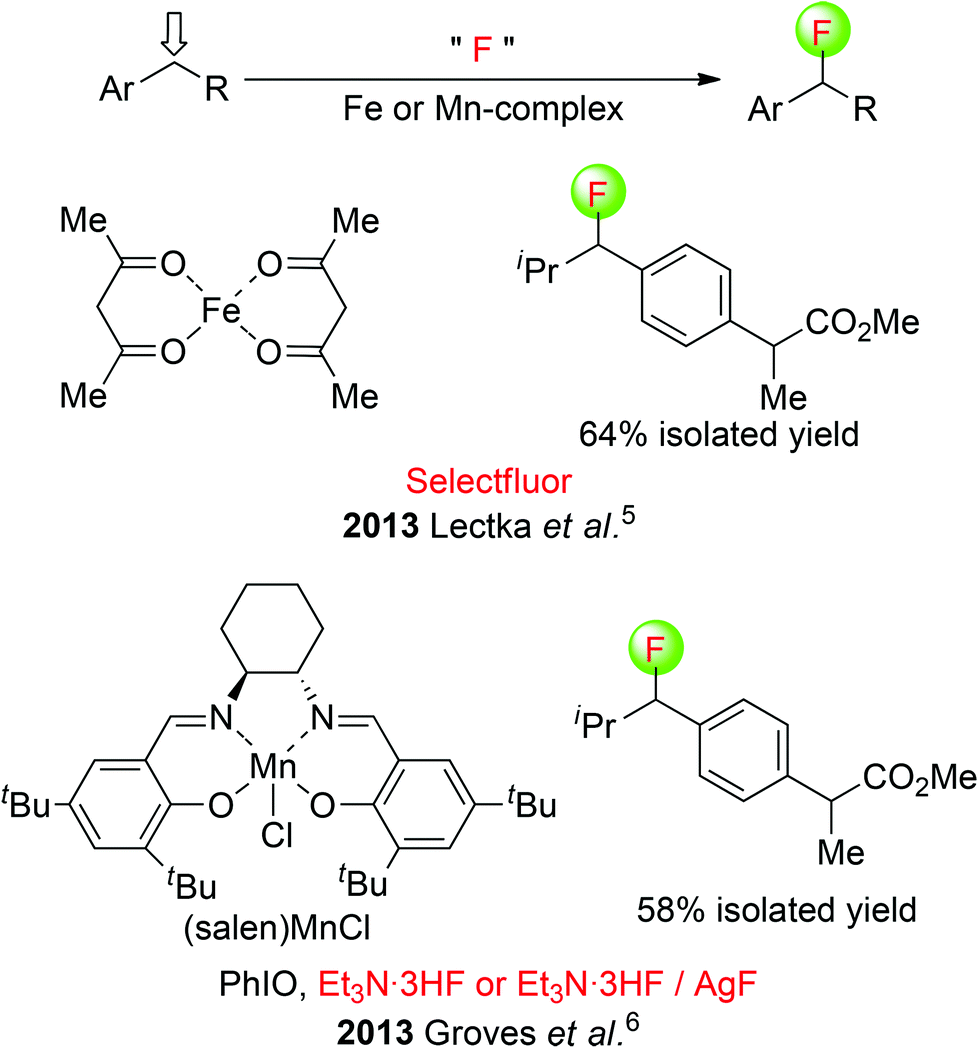 | ||
| Scheme 2 Metal-catalyzed fluorination of benzylic C(sp3)–H. | ||
Inoue and co-workers disclosed a metal-free fluorination of benzylic C(sp3)–H under thermal conditions, employing a simple reagent system composed of N,N-dihydroxypyromellitimide (NDHPI) and Selectfluor (Scheme 3: top).7 This radical-based reaction is initiated by hydrogen abstraction at the electron-rich position, and the carbon radical is intermolecularly trapped by Selectfluor, resulting in installation of a fluorine atom. Furthermore, this transformation is predictable toward the formation of benzylic C(sp3)–F bonds of aromatic compounds and tertiary C(sp3)–F bonds of aliphatic compounds. An alternative metal-free photolytic fluorination of benzylic C(sp3)–H bonds was developed by the Chen group (Scheme 3: bottom).8 It was found that visible light activated diarylketones to abstract a benzylic hydrogen atom selectively, and a fluorine radical donor delivered the benzylic fluoride. 9-Fluorenone catalyzes benzylic C(sp3)–H monofluorination, while xanthone catalyzes benzylic C(sp3)–H difluorination.
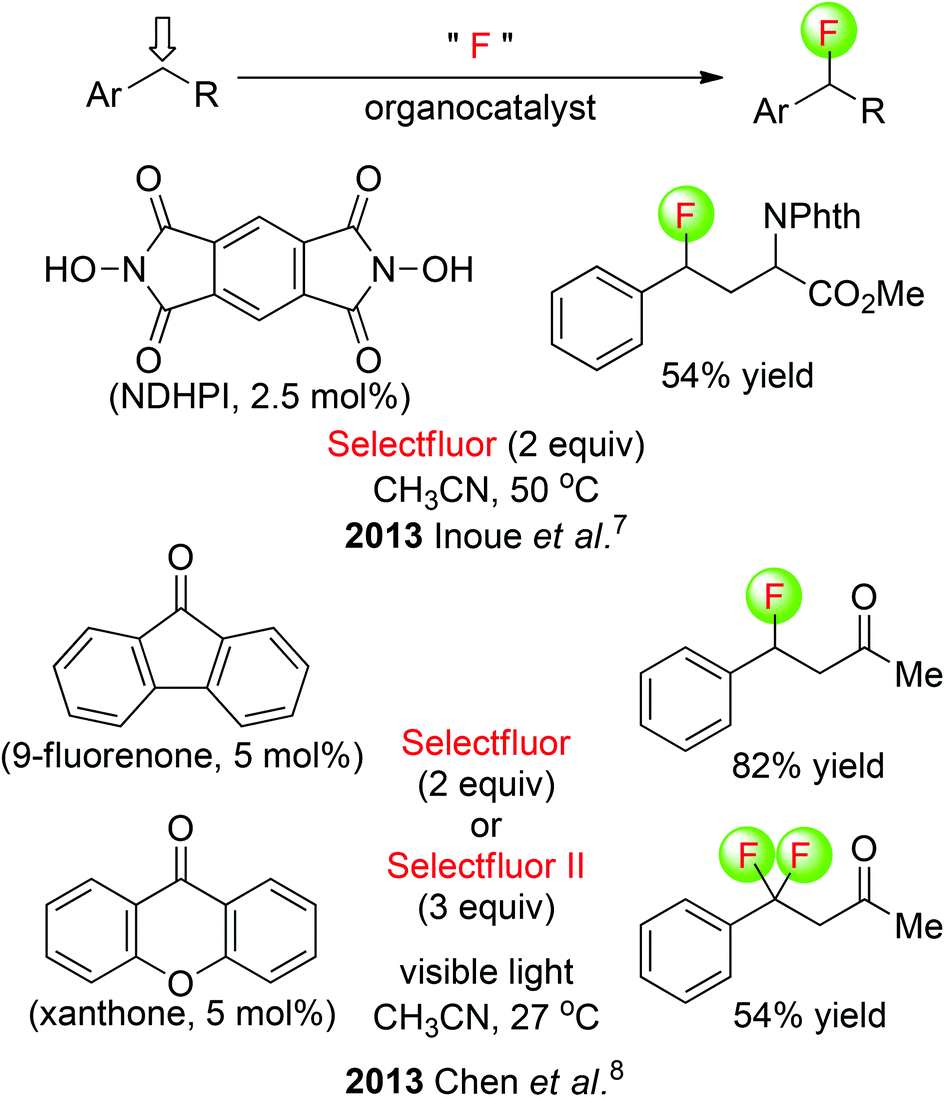 | ||
| Scheme 3 Organocatalytic fluorination of benzylic C(sp3)–H. Selectfluor II: 1-fluoro-4-methyl-1,4-diazoniabicyclo[2.2.2]octane bis(tetrafluoroborate). | ||
The chemistry of allylic fluorides has been intensively studied in recent years.9 Although many examples on transition-metal catalyzed allylic fluorination of pre-functionalized allyl precursors have emerged,10 yet only two reports by Lectka and Doyle groups have addressed the direct fluorination of allylic C(sp3)–H bonds. Lectka employed a multicomponent system of CuI-bis(imine) complex, N-hydroxyphthalimide (NHPI), and KB(C6F5)4 in the allylic C(sp3)–H fluorination. Two of α-fluoromethylstyrenes were obtained in moderate yields (Scheme 4: top).11 In Doyle's method, the combination of Pd(II)-sulfoxide, (R,R)-(salen)CrCl, and benzoquinone (BQ) was shown to enable the branched-selective synthesis of allylic fluorides from simple olefin substrates using Et3N·3HF.12 This allylic C(sp3)–H fluorination was further applied to the direct functionalization of a bioactive steroid, and the fluorinated product was isolated in 59% yield with a high branched/linear ratio (Scheme 4: bottom).
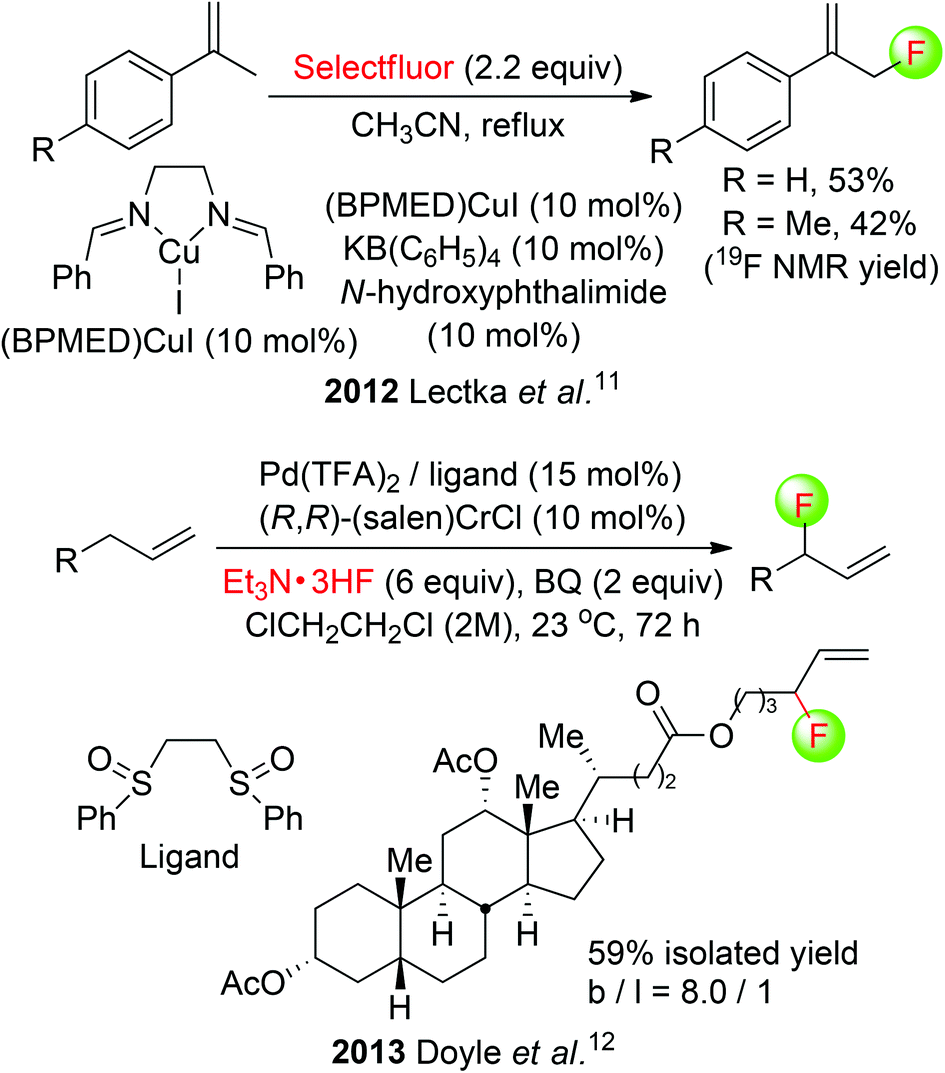 | ||
| Scheme 4 Metal-catalyzed allylic C(sp3)–H fluorination. | ||
Although direct fluorination of aliphatic unactivated C(sp3)–H bonds with elemental fluorine and electrophilic fluorinating agents, as developed by Rozen, Sandford, and Chambers,13 is a viable and very useful method, recent advancement has also been made in the development of catalytic procedures. Lectka et al. demonstrated their multicomponent system to be effective for aliphatic C(sp3)–H fluorination as well.11 The optimized reaction conditions include CuI-bis(imine), KB(C6F5)4, KI, and NHPI (Scheme 5: top). In this procedure, a radical mechanism was proposed with NHPI, known to form the phthalimide N-oxygen radical in situ in the presence of redox active metals, serving as a radical initiator. More recently, the same group disclosed another approach for direct fluorination of aliphatic unactivated C(sp3)–H bonds using a photocatalytic system including ultraviolet light and a photosensitizer (1,2,4,5-tetracyanobenzene: TCB).14 A series of substrates, from simple hydrocarbons to complex natural products, could be readily fluorinated by employing a simple UV lamp, a water bath, and a culture tube containing the reagents under an inert atmosphere of N2.
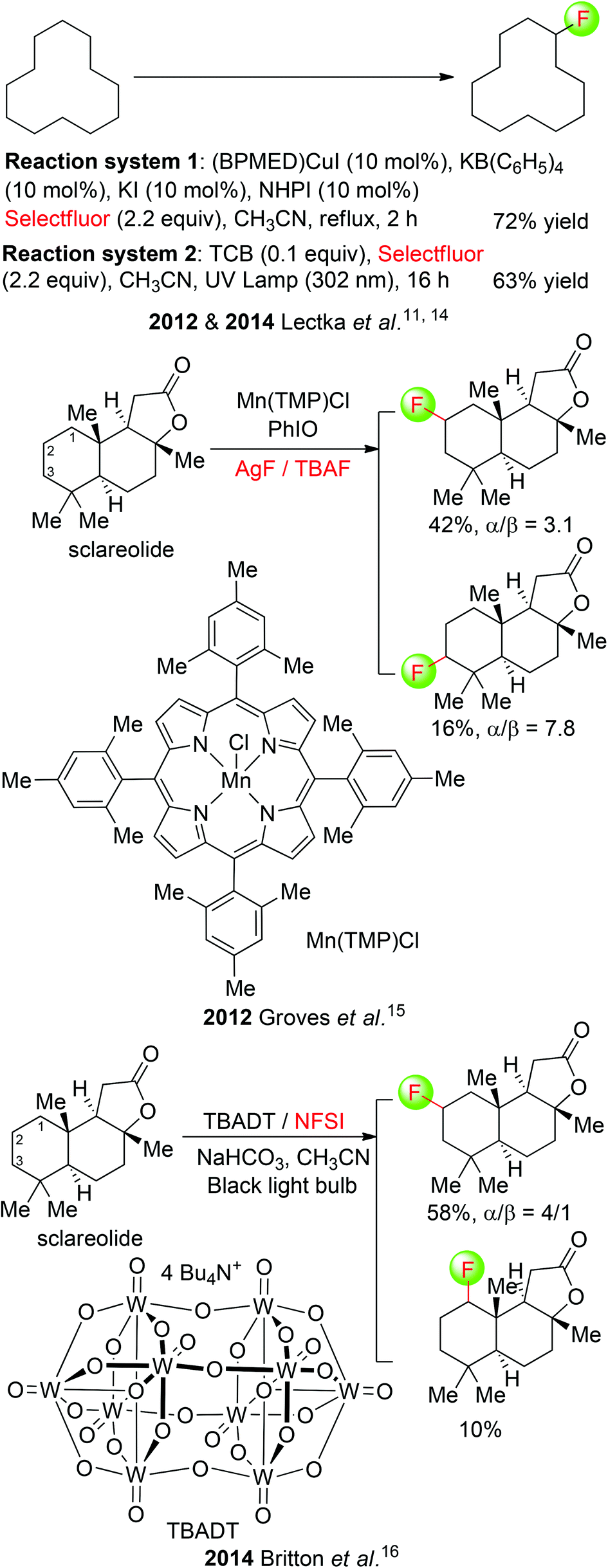 | ||
| Scheme 5 Metal- or/and photocatalyzed fluorination of unactivated aliphatic C(sp3)–H bonds. | ||
Simultaneously, Groves and co-workers described the direct fluorination of aliphatic unreactive C(sp3)–H bonds with a combination of manganese porphyrin complex [Mn(TMP)Cl], AgF, PhIO and a catalytic amount of TBAF.15 The unique reactivity of this multicomponent system was employed in the fluorination of a number of polycyclic natural/unnatural compounds. It is noteworthy that among the 26 aliphatic C(sp3)–H bonds of sclareolide, only those at C2 and C3 methylene reacted to give methylene-fluorinated products in 58% yield (Scheme 5: middle). The reaction mechanism could involve H-abstraction by the in situ formed oxomanganese intermediate [Mnv(O) (TMP)] and the radical-trapping with fluorine by a trans-MnIV(TMP)F2 species.
During our revision of this manuscript, Britton and co-workers presented an alternative approach to direct photocatalytic fluorination of unactivated C(sp3)–H bonds.16 Under the irradiation of two 15 watt black light bulbs, the reaction was carried out using a readily prepared decatungstate TBADT as the photocatalyst and NFSI as the fluorine transfer agent, producing a variety of fluorinated organics, including natural products, acyl fluorides, and fluorinated amino acid derivatives in good yields. The TBADT/NFSI system was also highly efficient for fluorination of sclareolides, similarly delivering C2-fluorinated sclareolides as the major product (Scheme 5: bottom).
Conclusions and outlook
In conclusion, catalytic fluorination of benzylic, allylic, and aliphatic unactivated C(sp3)–H bonds has emerged as a particularly promising approach for accessing organofluorines. Organic chemists have succeeded in designing and developing several novel catalytic systems for direct transformation of unactivated C(sp3)–H bonds into C(sp3)–F bonds. Of course, there are still important gaps to be filled by future studies, such as high-yielding, site-selective fluorination of C(sp3)–H bonds, and industrial application.Acknowledgements
This work was supported by the National Natural Science Foundation of China (no. 21172170, 21225208 and 21302137) and the National Basic Research Program of China (973 program, 2014CB745100).Notes and references
- For selected reviews: (a) T. Liang, C. N. Neumann and T. Ritter, Angew. Chem., Int. Ed., 2013, 52, 8214 CrossRef CAS PubMed; (b) M. Tredwell and V. Gouverneur, Angew. Chem., Int. Ed., 2012, 51, 11426 CrossRef CAS PubMed; (c) C.-P. Zhang, Q.-Y. Chen, Y. Guo, J.-C. Xiao and Y.-C. Gu, Chem. Soc. Rev., 2012, 41, 4536 RSC; (d) T. Besset, C. Schneider and D. Cahard, Angew. Chem., Int. Ed., 2012, 51, 5048 CrossRef CAS PubMed; (e) T. Furuya, A. S. Kamlet and T. Ritter, Nature, 2011, 473, 470 CrossRef CAS PubMed; (f) S. Lectard, Y. Hamashima and M. Sodeoka, Adv. Synth. Catal., 2010, 352, 2708 CrossRef CAS; (g) I. Ojima, Fluorine in Medicinal Chemistry and Chemical Biology, Wiley-Blackwell, Chichester, 2009 Search PubMed; (h) J. Hu, W. Zhang and F. Wang, Chem. Commun., 2009, 7465 RSC; (i) K. L. Kirk, Org. Process Res. Dev., 2008, 12, 305 CrossRef CAS; (j) S. Purser, P. R. Moore, S. Swallowb and V. Gouverneur, Chem. Soc. Rev., 2008, 37, 320 RSC; (k) D. O'Hagan, Chem. Soc. Rev., 2008, 37, 308 RSC; (l) W.-D. Meng and F.-L. Qing, Curr. Top. Med. Chem., 2006, 6, 1499 CrossRef CAS; (m) M. Shimizu and T. Hiyama, Angew. Chem., Int. Ed., 2005, 44, 214 CrossRef CAS PubMed; (n) P. Kirsch, Modern Fluoroorganic Chemistry: Synthesis Reactivity, Applications, Wiley-VCH, Weinheim, 2004 Search PubMed.
- For selected reviews: (a) M. P. Sibi and Y. Landais, Angew. Chem., Int. Ed., 2013, 52, 3570 CrossRef CAS PubMed; (b) D. Cahard, X. Xu, S. Couve-Bonnaire and X. Pannecoucke, Chem. Soc. Rev., 2010, 39, 558 RSC; (c) J.-A. Ma and D. Cahard, Chem. Rev., 2008, 108, PR1 CrossRef CAS; (d) N. Shibata, S. Mizuta and H. Kawai, Tetrahedron: Asymmetry, 2008, 19, 2633 CrossRef CAS PubMed; (e) P. M. Pihko, Angew. Chem., Int. Ed., 2006, 45, 544 CrossRef CAS PubMed; (f) J.-A. Ma and D. Cahard, Chem. Rev., 2004, 104, 6119 CrossRef CAS PubMed; (g) K. Mikami, Y. Itoh and M. Yamanaka, Chem. Rev., 2004, 104, 1 CrossRef CAS PubMed.
- K. L. Hull, W. Q. Anani and M. S. Sanford, J. Am. Chem. Soc., 2006, 128, 7134 CrossRef CAS PubMed.
- K. B. McMurtrey, J. M. Racowski and M. S. Sanford, Org. Lett., 2012, 14, 4094 CrossRef CAS PubMed.
- S. Bloom, C. R. Pitts, R. Woltornist, A. Griswold, M. G. Holl and T. Lectka, Org. Lett., 2013, 15, 1722 CrossRef CAS PubMed.
- W. Liu and J. T. Groves, Angew. Chem., Int. Ed., 2013, 52, 6024 CrossRef CAS PubMed.
- Y. Amaoka, M. Nagatomo and M. Inoue, Org. Lett., 2013, 15, 2160 CrossRef CAS PubMed.
- J.-B. Xia, C. Zhu and C. Chen, J. Am. Chem. Soc., 2013, 135, 17494 CrossRef CAS PubMed.
- (a) M. C. Pacheco, S. Purser and V. Gouverneur, Chem. Rev., 2008, 108, 1943 CrossRef CAS PubMed; (b) T. Ishimaru, N. Shibata, T. Horikawa, N. Yasuda, S. Nakamura, T. Toru and M. Shiro, Angew. Chem., Int. Ed., 2008, 47, 4157 CrossRef CAS PubMed.
- (a) M. H. Katcher and A. G. Doyle, J. Am. Chem. Soc., 2010, 132, 17402 CrossRef CAS PubMed; (b) C. Hollingworth, A. Hazari, M. N. Hopkinson, M. Tredwell, E. Benedetto, M. Huiban, A. D. Gee, J. M. Brown and V. Gouverneur, Angew. Chem., Int. Ed., 2011, 50, 2613 CrossRef CAS PubMed; (c) M. H. Katcher, A. Sha and A. G. Doyle, J. Am. Chem. Soc., 2011, 133, 15902 CrossRef CAS PubMed; (d) J. J. Topczewski, T. J. Tewson and H. M. Nguyen, J. Am. Chem. Soc., 2011, 133, 19318 CrossRef CAS PubMed; (e) A. M. Lauer and J. Wu, Org. Lett., 2012, 14, 5138 CrossRef CAS PubMed; (f) E. Benedetto, M. Tredwell, C. Hollingworth, T. Khotavivattana, J. M. Brown and V. Gouverneur, Chem. Sci., 2013, 4, 89 RSC; (g) Z. Zhang, F. Wang, X. Mu, P. Chen and G. Liu, Angew. Chem., Int. Ed., 2013, 52, 7549 CrossRef CAS PubMed.
- S. Bloom, C. R. Pitts, D. C. Miller, N. Haselton, M. G. Holl, E. Urheim and T. Lectka, Angew. Chem., Int. Ed., 2012, 51, 10580 CrossRef CAS PubMed.
- M.-G. Braun and A. G. Doyle, J. Am. Chem. Soc., 2013, 135, 12990 CrossRef CAS PubMed.
- (a) S. Rozen, Acc. Chem. Res., 1988, 21, 307 CrossRef CAS; (b) R. D. Chambers, M. Parsons, G. Sandford and R. Bowden, Chem. Commun., 2000, 959 RSC; (c) R. D. Chambers, A. M. Kenwright, M. Parsons, G. Sandford and J. S. Moilliet, J. Chem. Soc., Perkin Trans. 1, 2002, 2190 RSC; (d) S. Rozen, Eur. J. Org. Chem., 2005, 2433 CrossRef CAS; (e) G. Sandford, J. Fluorine Chem., 2007, 128, 90 CrossRef CAS PubMed.
- S. Bloom, J. L. Knippel and T. Lectka, Chem. Sci., 2014, 5, 1175 RSC.
- W. Liu, X. Huang, M.-J. Cheng, R. J. Nielsen, W. A. Goddard and J. T. Groves, Science, 2012, 337, 1322 CrossRef CAS PubMed.
- S. D. Halperin, H. Fan, S. Chang, R. E. Martin and R. Britton, Angew. Chem., Int. Ed., 2014, 53 DOI:10.1002/anie.201400420 , ASAP.
| This journal is © the Partner Organisations 2014 |