Synthesis and characterization of spin-labelled [2]rotaxanes containing tetrathiafulvalene and 1,5-dioxynaphthalene molecular stations†‡
Roberta
Manoni
,
Francesco
Romano
,
Costanza
Casati
,
Paola
Franchi
,
Elisabetta
Mezzina
* and
Marco
Lucarini
*
Department of Chemistry “G. Ciamician” – Alma Mater Studiorum-University of Bologna, Via San Giacomo 11, 40126, Bologna, Italy. E-mail: elisabetta.mezzina@unibo.it; marco.lucarini@unibo.it
First published on 27th March 2014
Abstract
The central component of one of the most studied programmable molecular switches reported by Stoddart and Heath is a [2]rotaxane, which consists of a cyclobis(paraquat-p-phenylene) shuttle (CBPQT4+)(PF6−)4 (the ring) encircling a finger and moving between two molecular stations, tetrathiafulvalene (TTF) and 1,5-dioxynaphthalene (DNP). In order to employ ESR spectroscopy for understanding the mechanism of this switch, we report here the synthesis and characterization of [2]rotaxanes having persistent nitroxide radicals as the stopper units and TTF or DNP as molecular stations. Isolation of the rotaxanes was successful when bulky nitroxide radicals containing cyclohexyl substituents at 2 and 6 positions of the piperidine-N-oxyl ring were used as terminal stoppers.
Introduction
Switchable, mechanically interlocked molecules (MIM) have attracted much interest in the last few years1 on account of their ability to change the relative positions of their dumbbell and ring components in response to external stimuli, such as variations in pH, absorption of light and removal or addition of electrons. This switching at a molecular level offers huge opportunities for the development of a series of applications ranging from molecular electronic devices2 to peptide synthesis by artificial molecular machines,3 including muscle-like molecular machines.4One of the most promising candidates for a programmable molecular switch is the “Stoddart–Heath type” bistable [2]rotaxane.5 The Stoddart–Heath molecular switch consists of the aromatic shuttle and station compounds, which are attached to other components such as linkers and bulky stoppers, and shows a reversible on/off switching which arises from moving the cyclobis-(paraquat-p-phenylene) (CBPQT4+; blue, Fig. 1) shuttle between the tetrathiafulvalene (TTF; green) station and the 1,5-dioxynaphthalene (DNP; red) one by oxidation and reduction of the TTF moiety.5
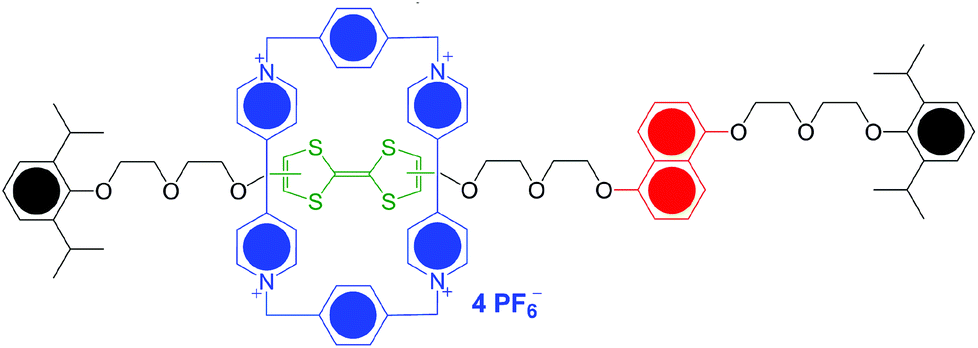 | ||
| Fig. 1 A Stoddart–Heath type bistable [2]rotaxane. | ||
Redox-responsive bistable [2]rotaxanes are of considerable interest because the redox switching process is usually rapid and precise and can be controlled within solid-state devices, as well as on surfaces.6
Still, many questions about the detailed structure and function of these structures need to be answered to maximize performance and to provide guidance in developing new devices. We have recently shown that it is possible to employ ESR spectroscopy7 for obtaining information on the rotaxane structure complementary to those obtained by traditional techniques by insertion of persistent cyclic nitroxide spin labels at the dumbbell and/or at the ring components. In particular we were able to determine the distances among the stopper-end units of a cucurbit[6]uril-based rotaxane,8 to unravel unidirectional threading in an α-cyclodextrin-based [2]rotaxane and to study quantitatively the dynamics of intramolecular collisions of the molecular fragments mechanically connected.9
Here we report the synthesis and ESR characterization of the first examples of [2]rotaxanes containing nitroxide spin labels as stoppers and TTF or DNP units as molecular stations. Bulky nitroxide radicals containing cyclohexyl substituents at 2 and 6 positions of the piperidine-N-oxyl ring were necessary to kinetically trap the CBPQT4+ wheel.
Results and discussion
To prepare the bis-labeled rotaxanes, we followed the approach reported by Stoddart and coworkers10 based on the formation of CBPQT4+ donor–acceptor pseudorotaxanes with 1,5-dioxynaphthalene (DNP) or tetrathiafulvalene (TTF) axial derivatives (1 and 14, respectively) carrying at both edges an azide group. The formation of the pseudorotaxane was followed by a Cu-(I)-catalyzed click reaction with alkyne terminated TEMPO stoppers (4-propargyloxy-2,2,6,6-tetramethylpiperidine-N-oxyl).11We have previously shown that the TEMPO unit is large enough to be used as the end-cap group in paramagnetic rotaxanes having cucurbit[6]urils8 or α-cyclodextrin12 as the wheel. Based on this, we verified the possibility of using TEMPO derivatives as suitable stoppers for CBPQT-based rotaxanes by monitoring the 1H-NMR behavior in d7-dimethylformamide of the diamagnetic dumbbell 2b in the absence and in the presence of equal amounts of the CBPQT4+ macrocycle. The diamagnetic dumbbell was used to improve the spectral resolution of the NMR spectrum. The absence of new spectral signals in the NMR spectrum recorded at different times after mixing the two components suggested to exclude threading processes of the bulky guest into the macrocyclic host and the use of TEMPO derivatives as paramagnetic stoppers.
Thus, we undertook the synthesis of the rotaxane 3 by mixing CBPQT·4PF6 in DMF at −10 °C, with the DNP derivative 1 carrying azide-terminated glycol chains following the Stoddart approach. Cycloaddition of the nitroxide alkyne 4-propargyloxy-TEMPO onto 1 was carried out by adding to the purple solution containing the pseudorotaxane a catalytic amount of the complex [Cu(MeCN)4]PF6 in the presence of (benzyltriazolylmethyl)amine (TBTA) as a stabilizer (Scheme 1). The reaction, however, did not yield the expected results. Although the formation of the rotaxane was detected by thin layer chromatography and ESI-MS analysis, we never obtained, in the purification step, fractions containing pure rotaxanes. Only mixtures of the interlocked complex together with the free host and dumbbell 2a were actually obtained (Scheme 1).
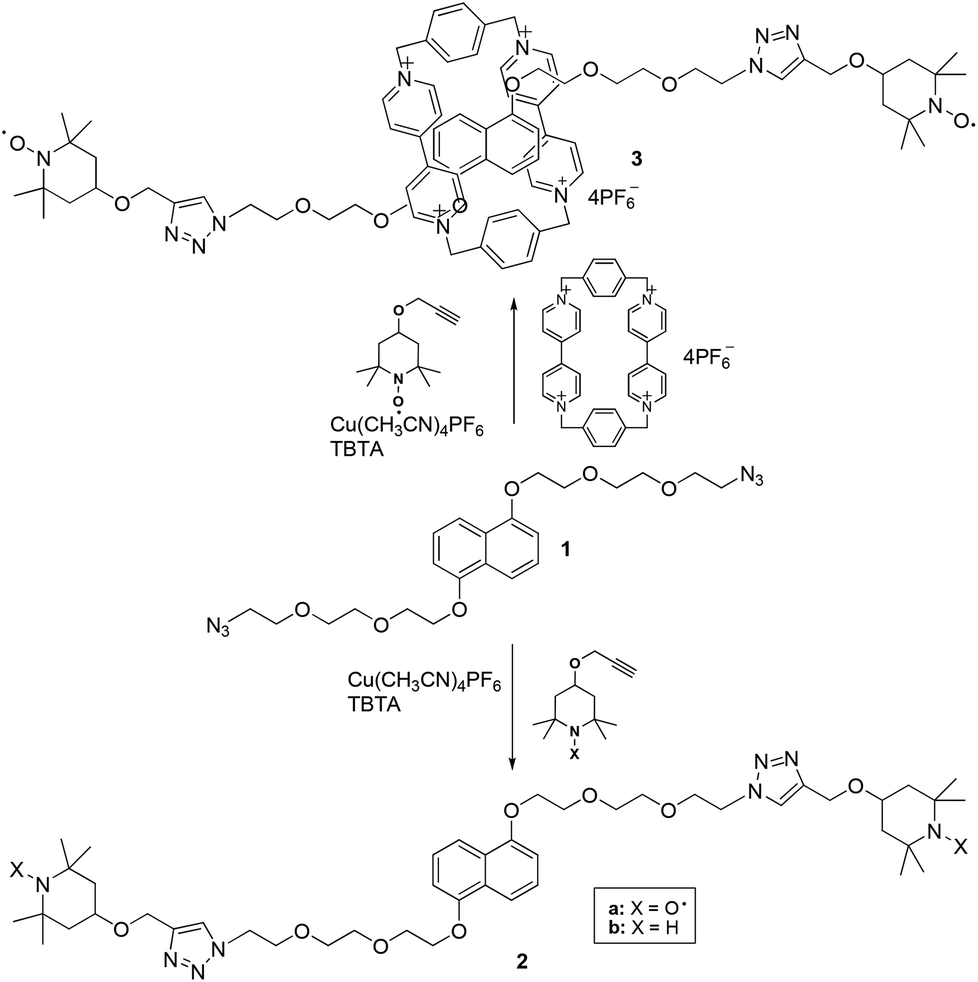 | ||
| Scheme 1 | ||
Although this result does contradict the previous NMR analysis, one can argue a partial ‘dethreading’ of the wheel by the dumbbell not sufficiently sterically shielded by four methyl groups at the 2,6-positions of the TEMPO fragment.
The difficulty in isolating the rotaxane from its components, also after further chromatographic separation of the fractions, induced us to reconsider the validity of the TEMPO unit as a stopper in the case of CBPQT-based rotaxanes. For this reason we decided to change the substitution at the 2,6-positions of the piperidine nitroxide with bulkier cyclohexyl fragments, and prepared the spirocyclic paramagnetic stopper 5a (Scheme 2).
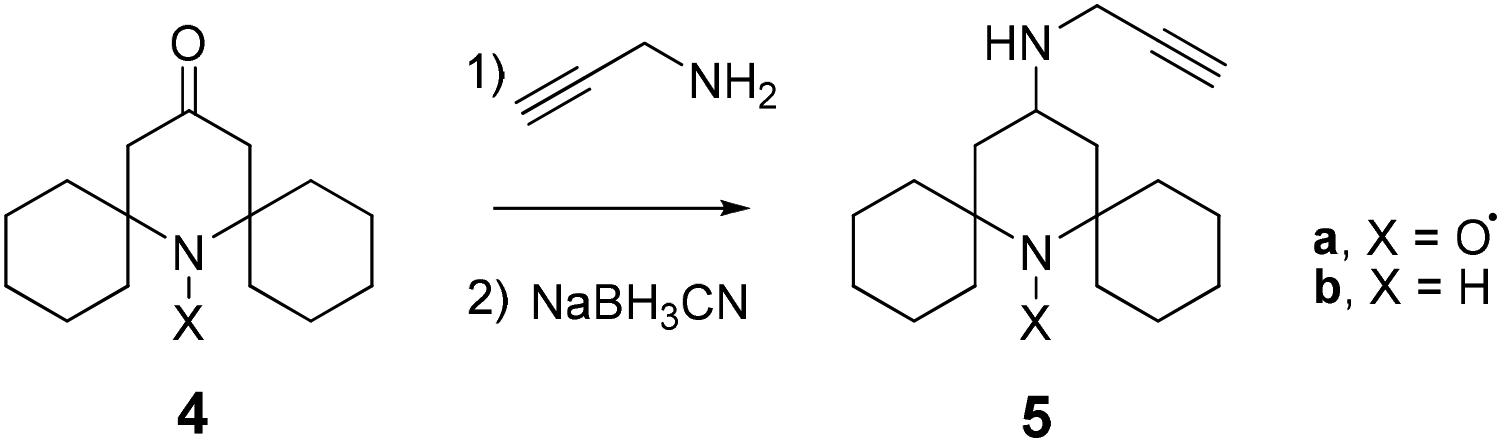 | ||
| Scheme 2 | ||
The new alkyne-terminated structure (5) was obtained in good yield by treating the piperidin-4-one derivative 413 with an excess of propargylamine without any solvent, and after in situ reduction of the corresponding Schiff base with sodium cyanoborohydride.
Then we repeated the click reaction between the pseudorotaxane CBPQT@1 and the new stopper alkyne 5a obtaining rotaxane 7a in 30% isolated yield (Scheme 3). The best results were reached using a CBPQT/1 molar ratio of 0.8.
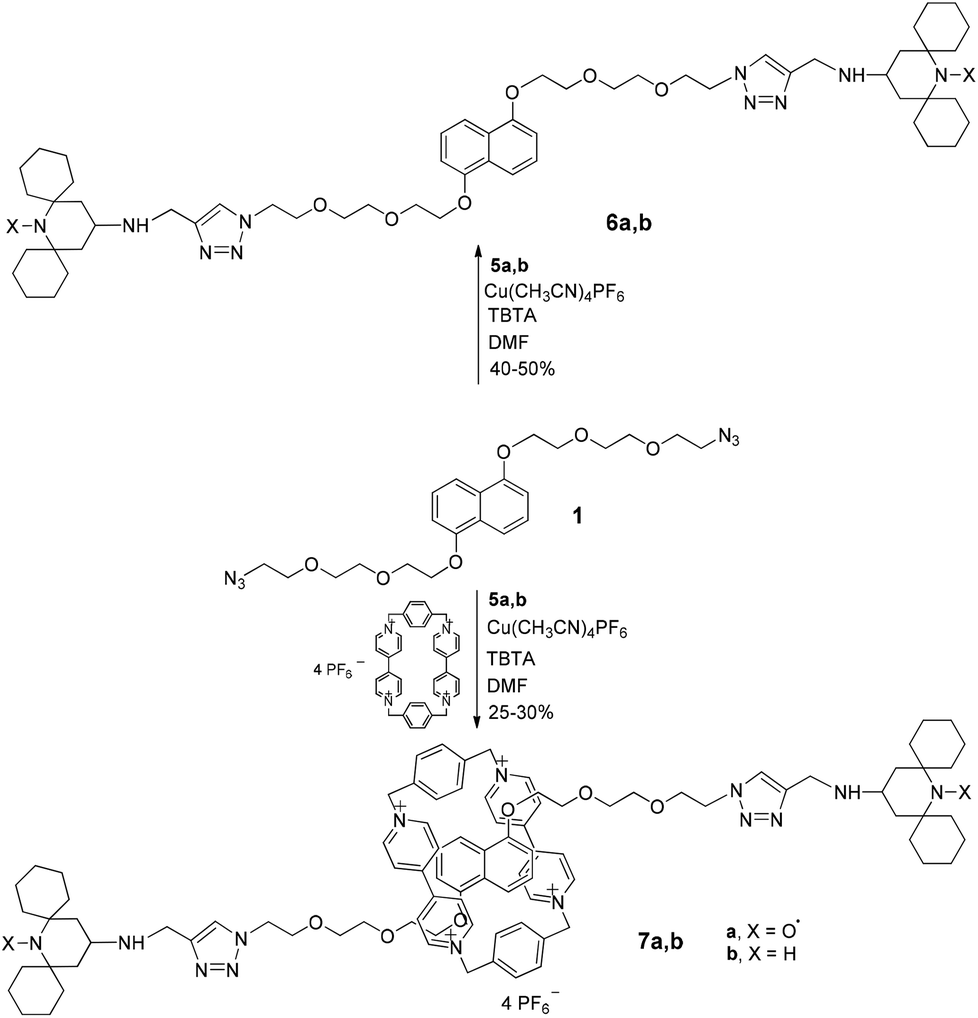 | ||
| Scheme 3 | ||
Both ESI-MS analysis and NMR spectroscopy were used to provide evidence of the formation of the interlocked structure. In all cases 1H NMR spectra of the paramagnetic compounds 6a and 7a were recorded also after in situ reduction of the samples by phenylhydrazine, to obtain the corresponding diamagnetic N-hydroxy forms.
In order to better understand the ‘threading’ process by NMR analysis we performed the ‘rotaxanation’ also with the diamagnetic stopper 5b affording the corresponding MIM 7b (see ESI‡).
Partial spectra of the dumbbell 6a, the rotaxane 7a, and the free host in CD3CN are reported in Fig. 2. In trace b the resonances for the DNP protons gave clear evidence of the interlocked structure, undergoing large upfield shifts at 6.27, 5.98, and 2.41 ppm when the aromatic DNP ring is mechanically trapped inside the macrocycle. Shift values are typical of rotaxanes containing the CBPQT4+ host and the DNP recognition unit.10 Also labeled signals (evidenced by red dashed lines) of the CBPQT4+ are diagnostic of the complexation process showing both a general shielding and broadening of all peaks, the latter being an indication of slow rotation of the CBPQT4+ rings on the 1H NMR time scale at 298 K. In addition, splittings recorded for bipyridinium and p-phenylene resonances point out that the exchange processes between the relevant protons slow down yet further, allowing to distinguish nonequivalent proton signals.
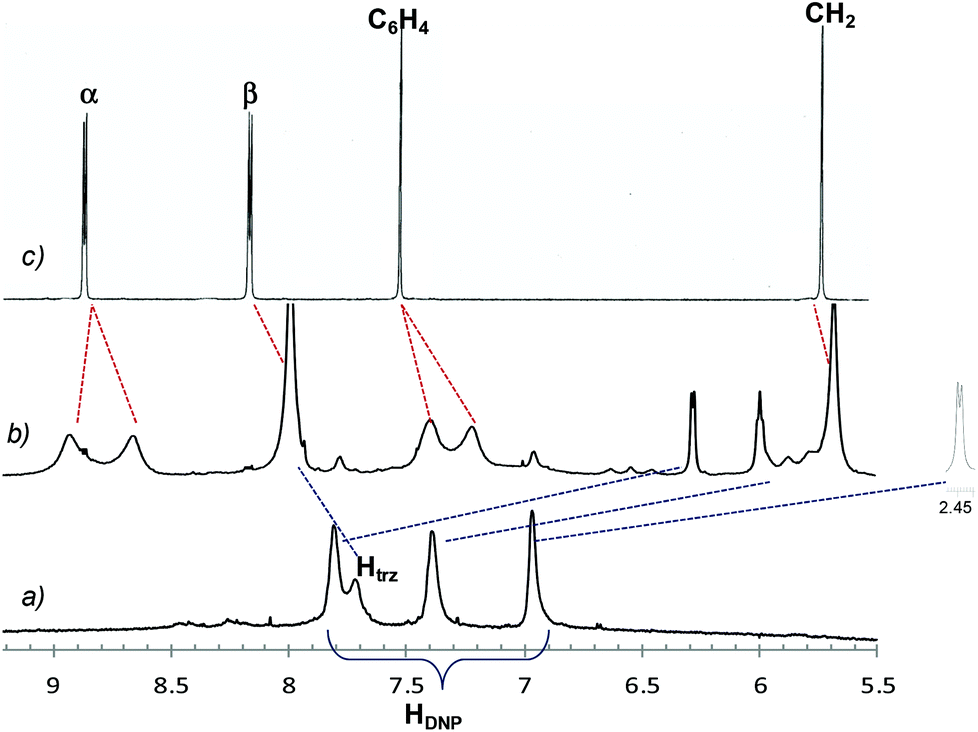 | ||
| Fig. 2 Partial 1H NMR spectra (600 MHz, CD3CN, 298 K) of (a) dumbbell 6a; (b) rotaxane 7a; (c) CBPQT4+ host. Blue dashed lines evidence the marked shielding of DNP protons caused by the presence of CBPQT4+ around the electron donor heterocycle and red dashed lines show the broadening and splitting of the macrocyclic signals due to reduced rotation of CBPQT4+ rings in the rotaxane complex. Htrz = triazole proton, HDNP = DNP aromatic protons, α,β = CBPQT4+ bipyridinium α and β protons, respectively; C6H4 = host p-phenylene protons; CH2 = host benzyl protons. | ||
After verifying the feasibility of the threading-followed-by-stoppering approach to the synthesis of DNP paramagnetic CBPQT-based rotaxane using the spirocyclic stopper 5a we decided to extend our investigation to the electron donor guest with a TTF backbone.
To obtain the TTF diazide derivative 14 we followed the route described in Scheme 4 by using the previously reported14 reaction of TBA2[Zn(DMIT)2] [8, bis(tetrabutylammonium)bis(1,3-dithiole-2-thione-4,5-dithiolato)zinc complex)] with 3-bromopropionitrile which affords the key intermediate compound 9. Conversion of 9 to 10 by conventional methods14,15 and coupling of 10 in the presence of triethylphosphite led to the TTF derivative 11.15 After removal of 2-cyanoethyl groups followed by reaction with a large excess of the diiodoelectrophile 12,16 compound 13 was obtained in satisfactory yield. Exchange reaction of 13 with NaN3 and subsequently click reaction with the spirocyclic stopper 5 converted the diazide 14 into the corresponding TTF dumbbell 15 which was purified and characterized by NMR and ESI-MS analyses.
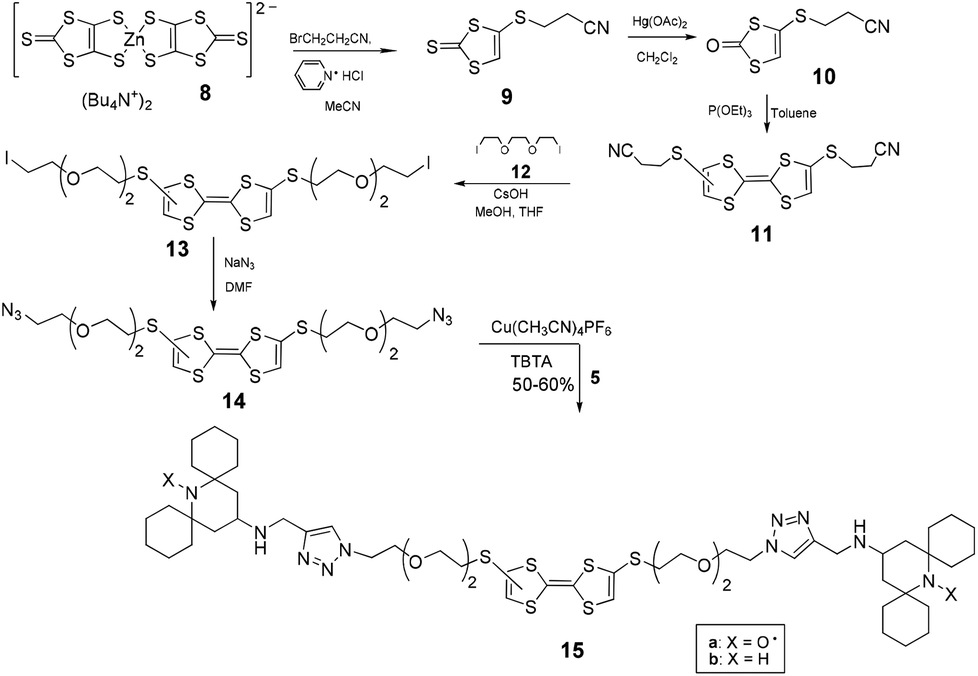 | ||
| Scheme 4 | ||
We then applied the experimental procedure described above for the synthesis of the rotaxane containing a DMP unit, obtaining the desired paramagnetic complex 16a in satisfactory yield (Scheme 5). Again the NMR spectra of the diamagnetic analogue 16b helped us to confirm the interlocked structures (see ESI‡).
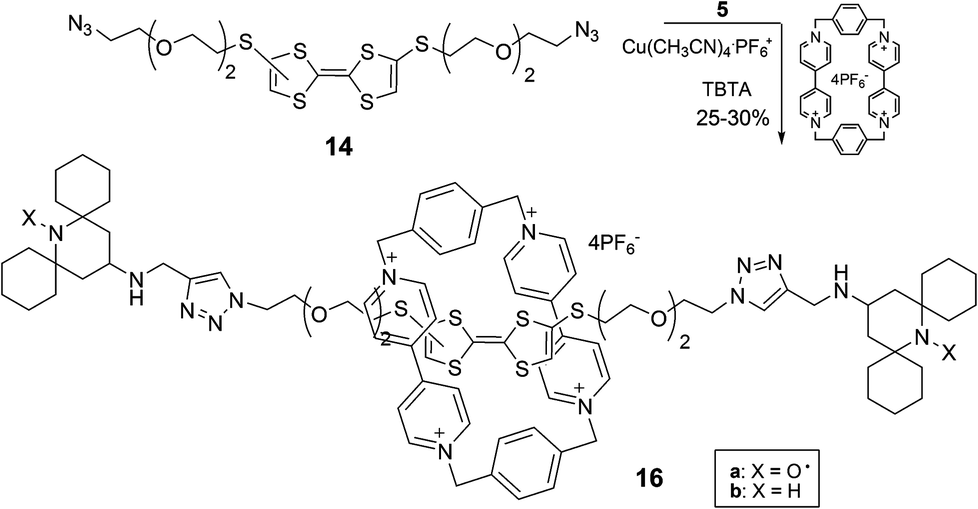 | ||
| Scheme 5 | ||
The partial spectrum of the dumbbell 15a and that of the paramagnetic rotaxane 16a are reported in Fig. 3 (traces a and b, respectively). The dashed lines point out (a) the upfield shift of the TTF proton signal and (b) the downfield shifts of the signals belonging to the triazole and to some carbon chain protons of the guest when trapped by the tetracationic host. This behaviour, typical of the complexation process, indicates that the TTF unit is encircled by CBPQT4+, while the glycol pendants and the triazole rings are not directly involved being placed outside the host.
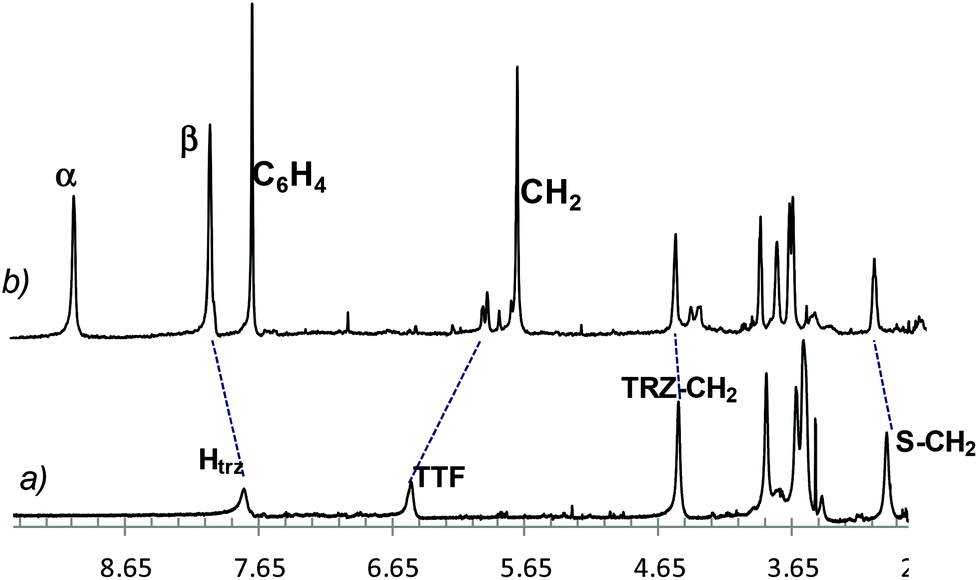 | ||
| Fig. 3 Partial 1H NMR spectra (600 MHz, CD3CN, 298 K) of (a) dumbbell 15a; (b) rotaxane 16a. Blue dashed lines evidence the proton signal deshielding of TTF glycol chain and the marked shielding of TTF protons caused by the presence of CBPQT4+ around the electron donor heterocycle. Htrz = triazole proton, TTF = TTF protons, TRZ-CH2 = N-methylene protons of the triazole, S-CH2 = TTF-S-methylene protons, α,β = CBPQT4+ bipyridinium α and β protons, respectively; C6H4 = host p-phenylene protons; CH2 = host benzyl protons. | ||
The ESR spectrum of the dumbbell containing a DNP unit, 6a, recorded in ACN at 338 K (aN = 15.22 G, g = 2.0058) is reported in Fig. 4a.17 The spectrum is characterized by five lines, due to the exchange interaction between the paramagnetic fragments linked by the polyether chain, as the exchange coupling constant between unpaired electrons, J, is greater than the hyperfine splitting, aN.18 This exchange interaction, which leads to the appearance of extra lines in the ESR spectra, is operating through space and depends on the frequency of collisions between the paramagnetic moieties.
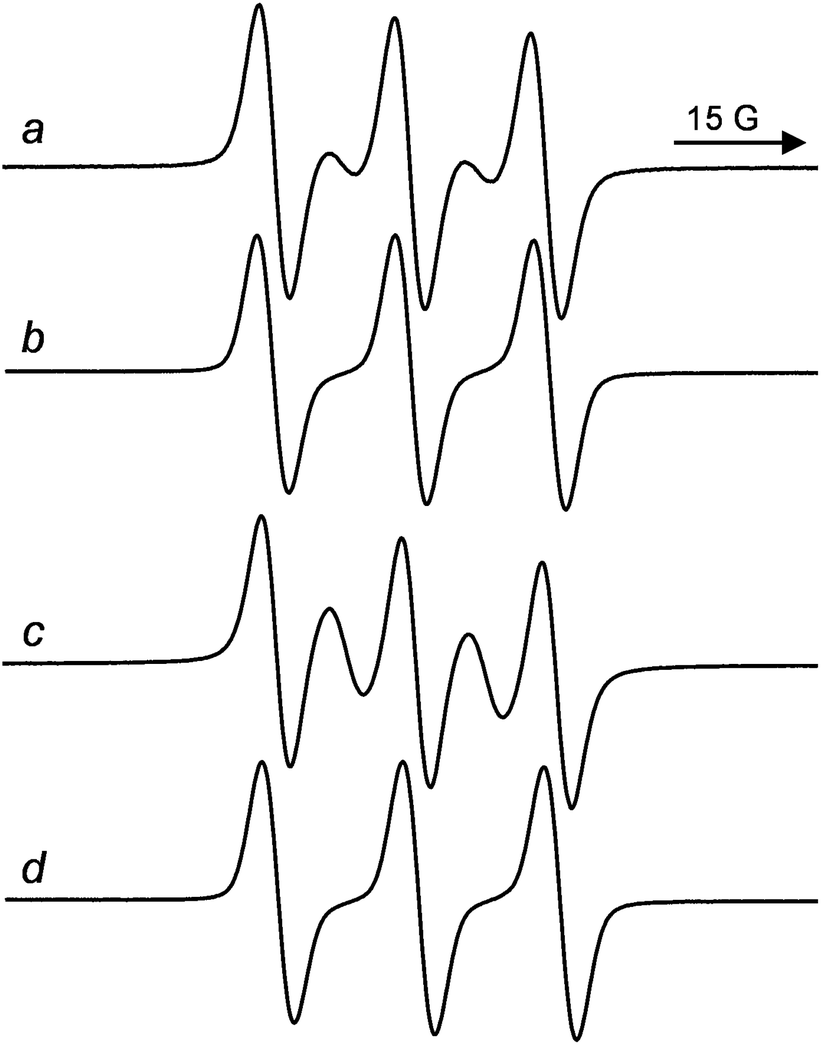 | ||
| Fig. 4 ESR spectra of dumbbells 6a (a), 15a (c) and rotaxanes 7a (b), and 16a (d) in ACN at 338 K. | ||
The ratio of line intensities, however, differs essentially from 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2:3:2:1, which is expected in case all the conformations of the biradical were characterized by strong exchange between nitroxide stopper units. Thus, the spectrum of 6a is the superposition of the ESR signals due to biradicals where the two labels are too far and the radical centers have no opportunity to interact (three line spectrum) and to biradicals where the TEMPO radical centers get some possibilities to give exchange coupling (five lines spectrum). Following Parmon's model9,18 and by measuring the relative ESR line intensities it was possible to estimate the presence of ca. 40% of conformations at 338 K in ACN where the two nitroxide units are relatively “close” in the space and a strong electron exchange (J ≫ aN) is observable.
:2:3:2:1, which is expected in case all the conformations of the biradical were characterized by strong exchange between nitroxide stopper units. Thus, the spectrum of 6a is the superposition of the ESR signals due to biradicals where the two labels are too far and the radical centers have no opportunity to interact (three line spectrum) and to biradicals where the TEMPO radical centers get some possibilities to give exchange coupling (five lines spectrum). Following Parmon's model9,18 and by measuring the relative ESR line intensities it was possible to estimate the presence of ca. 40% of conformations at 338 K in ACN where the two nitroxide units are relatively “close” in the space and a strong electron exchange (J ≫ aN) is observable.
It is interesting to note that these exchanging conformations are not present in the corresponding rotaxane. Actually, the ESR spectrum of rotaxane 7a in ACN at 338 K (aN = 15.30 G, g = 2.0058, see Fig. 4b) consists of only three lines as expected for a nitroxide biradical in which single radical units do not interact with each other. This implies that rotaxanation by the aromatic wheel CBPQT4+ significantly reduce the probability of collisions of the nitroxide termini in the biradical dumbbell.
Similar ESR results were also observed with the dumbbell 15a (aN = 15.32 G, g = 2.0058) and the rotaxane 16a (aN = 15.26 G, g = 2.0058) containing the TTF unit (see Fig. 4). In this case, however, the intensity of the exchange lines (the second and the fourth lines) in the dumbbell 15a is much higher than in 6a (the presence of conformations where J ≫ aN were estimated to be >80%). This makes the difference between the spectra of 15a and 16a more evident and thus the possibility of distinguishing them easier. It should be remarked that this favourable ESR feature can be extremely useful to unambiguously distinguish the [2]rotaxane biradical from the corresponding free dumbbell during the purification procedure of spin labelled rotaxanes.
Conclusions
In conclusion, we demonstrated that it is possible to isolate spin-labelled rotaxanes containing cyclobis-(paraquat-p-phenylene) as the wheel and tetrathiafulvalene or 1,5-dioxynaphthalene as the molecular station in the thread by using bulky nitroxide radicals containing cyclohexyl substituents at 2 and 6 positions of piperidine-N-oxyl ring as stoppers. This paved the way for the use of ESR in the measurement of spin-to-spin distance and in the extraction of information on intrinsic flexibility of the redox-responsive “Stoddart–Heath type” molecular switch. Work in this direction is actually under investigation.Experimental sections
2b: To a solution of diazide 1 (0.049 g, 0.1 mmol) and 4-propargyloxy-TMP (0.042 g, 0.215 mmol) in DMF (5 mL), tris(benzyltriazolylmethyl)amine (TBTA) (0.011 mg, 0.02 mmol) and Cu(CH3CN)4PF6 (0.007 g, 0.02 mmol) were added, and the resulting mixture was stirred at room temperature for 24 h, at which time the solvent was evaporated. Purification of the crude mixture by a silica gel column using CH3CN–H2O–30% NH3 90:10:2 affords 2b (0.052 g, 0.06 mmol) in 60% yield. 1H NMR (600 MHz, d6-DMSO): δ 0.92 (t, J = 11.0 Hz, 4H), 1.02 (s, 12H), 1.07 (m, 12H), 1.82 (dd, J = 11.0 and 3.0 Hz, 4H), 3.56–3.60 (m, 4H), 3.62–3.66 (m, 4H), 3.74–3.78 (m, 2H), 3.81–3.86 (m, 8H), 4.23 (br s, 4H), 4.54 (s, 4H), 4.50 (t, J = 5.2 Hz, 4H), 6.98 (d, J = 8.0 Hz, 2H), 7.37 (t, J = 8.0 Hz), 7.71 (d, J = 8.0 Hz, 2H), 8.00 (s, 2H). ESI-MS: m/z 433.0 (M + 2H)2+. Elemental analysis: calculated for C46H72N8O8 C, 63.86; H, 8.39; N, 12.95. Found: C, 63.64; H, 8.45; N, 12.81.
2a: The above procedure was followed using diazide 1 (0.012 g, 0.025 mmol), 4-propargyloxy-TEMPO (0.013 g, 0.063 mmol) in DMF (1 mL), TBTA (0.003 mg, 0.0054 mmol) and Cu(CH3CN)4PF6 (0.0054 mmol, 0.002 g). Purification of the crude mixture by a silica gel column using CH2Cl2–MeOH 90:10 affords the biradical 2a (0.0123 g, 0.014 mmol) in 55% yield. 1H NMR (600 MHz, d6-DMSO): 3.59 (br s), 3.64 (s, 4H), 3.84 (br s), 4.23 (br s), 4.39 (br s), 4.52 (br s), 7.00 (br s), 7.39 (br s), 7.73 (br s), 8.01 (br s). ESI-MS: m/z 895.5 (M + H)+. Elemental analysis: calculated for C46H70N8O10 C, 61.72; H, 7.88; N, 12.52. Found: C, 61.64; H, 7.97; N, 12.45.
6a: To a solution of diazide 1 (0.016 g, 0.034 mmol) and alkyne functionalized nitroxide 5a (0.024 g, 0.084 mmol) in DMF (1 mL), Cu(CH3CN)4PF6 (0.002 g, 0.006 mmol) and TBTA (0.003 g, 0.006 mmol) were added, and the resulting mixture was stirred at room temperature for 24 h, at which time the solvent was evaporated. Purification of the crude mixture by a silica gel column using CH2Cl2–MeOH 90:10 affords biradical 6a (0.018 g, 50% yield). 1H NMR (600 MHz, CD3CN):19 3.61 (br s, 4H), 3.68 (br s, 4H), 3.80–3.90 (m, 8H), 4.26 (br s, 4H), 4.46 (br s, 4H), 6.95 (br s, 2H), 7.38 (br s, 2H), 7.70 (br s, 1H), 7.79 (br s, 2H). ESI-MS: m/z 1054.4 (M + H)+. Elemental analysis: calculated for C58H88N10O8 C, 66.13; H, 8.42; N, 13.30. Found: C, 65.96; H, 8.35; N, 13.38.
6b: The above procedure was followed using diazide 1 (0.016 g, 0.034 mmol) and alkyne functionalized amine 5b (0.024 g, 0.084 mmol) in DMF (1 mL), Cu(CH3CN)4PF6 (0.002 g, 0.006 mmol) and TBTA (0.003 g, 0.006 mmol). Purification of the crude mixture by a silica gel column using CH2Cl2–MeOH 90:10 and then CH2Cl2–MeOH–30% NH3 90:10:1 affords 6b (0.0135 g, 39% yield).
1H NMR (600 MHz, d6-DMSO): δ 0.62 (t, J = 11.7 Hz, 4H), 1.10–1.70 (m, 40H), 1.90 (d, J = 11.7 Hz, 4H), 2.68–2.78 (m, 2H), 3.54–3.60 (m, 4H), 3.60–3.68 (m, 4H), 3.74 (s, 4H), 3.81 (t, J = 5.0 Hz, 4H), 3.82–3.84 (m, 4H), 4.20–4.26 (m, 4H), 4.48 (t, J = 5.0 Hz, 4H), 6.98 (d, J = 8.0 Hz, 2H), 7.37 (t, J = 8.0 Hz, 2H), 7.71 (d, J = 8.0 Hz, 2H), 7.87 (s, 2H). ESI-MS: m/z 1024.2 (M + H)+. Elemental analysis: calculated for C58H90N10O6 C, 68.07; H, 8.86; N, 13.69. Found: C, 68.15; H, 8.80; N, 13.59.
[2]Rotaxane 7a
DNP diazide derivative 1 (0.012 g, 0.025 mmol), CBPQT·4PF6 (0.022 g, 0.02 mmol) and the alkyne functionalized stopper 5a (0.018 g, 0.062 mmol) were dissolved in DMF (0.200 mL) at −10 °C under an N2 atmosphere, forming a deep purple solution. TBTA (0.002 g, 0.004 mmol) and Cu(CH3CN)4PF6 (0.0014 g, 0.004 mmol) were added to the solution and the resulting mixture was stirred at room temperature for 48 h, at which time the solvent was evaporated. The crude reddish solid was purified by column chromatography (SiO2: 1% w/v NH4PF6 solution in Me2CO, i.d. 10 mm, h 19.5 cm). The purple fractions in Me2CO were collected, concentrated to a minimum volume, and the product was precipitated from this solution through the addition of an excess of cold water. The resulting solid was collected by filtration, and washed with water to remove the excess of NH4PF6 to afford the [2]rotaxane 7a·4PF6 as a purple powder (0.0162 g, 30% yield). 1H NMR (600 MHz, CD3CN):19δ 2.42 (d, J = 7.2 Hz, 2H), 3.90 (br s, 8H), 4.01 (br s, 8H), 4.21 (br s, 4H), 4.30 (br s, 4H), 4.57 (br s, 4H), 5.67 (br s, 8H), 5.94–6.02 (m, 2H), 6.27 (d, J = 7.2 Hz, 2H), 7.21 (br s, 4H), 7.38 (br s, 4H). 7.98 (br s, 10H), 8.65 (br s. 4H), 8.92 (br s, 4H). ESI-MS: m/z 2154.8 (M + H)+. Elemental analysis: calculated for C94H120F24N14O8P4 C, 52.42; H, 5.62; N, 9.10. Found: C, 52.39; H, 5.70; N, 8.97. UV-Vis: λmax = 504 nm, ε = 4184 L mol−1 cm−1 in acetonitrile (ACN) at 298 K.[2]Rotaxane 7b
The above procedure was followed using the DNP derivative 1 (0.016 g, 0.034 mmol), CBPQT·4PF6 (0.026 g, 0.027 mmol), the alkyne functionalized stopper 5b (0.024 g, 0.085 mmol), DMF (0.200 mL), TBTA (0.0031 g, 0.006 mmol) and Cu(CH3CN)4PF6 (0.0021 g, 0.006 mmol).The resulting [2]rotaxane 7b·4PF6 was obtained as a purple powder (0.0186 g, 25% yield). 1H NMR (600 MHz, d6-DMSO): δ 1.12–1.20 (m, 4H), 1.34–1.48 (m, 8H), 1.50–1.70 (m, 28H), 1.80–1.90 (m, 4H), 2.08–2.18 (m, 4H), 2.31 (d, J = 7.8 Hz, 2H), 2.70–2.76 (m, 2H), 3.86–3.90 (m, 8H), 3.96–4.06 (m, 8H), 4.21 (m, 4H), 4.30 (m, 4H), 4.55–4.70 (m, 4H), 5.78 (br s, 8H), 5.84 (t, J = 7.8 Hz, 2H), 6.15 (d, J = 7.8 Hz, 2H), 7.63–7.78 (m, 8H), 8.09 (s, 8H), 8.24 (s, 2H), 9.11 (d, J = 6.5 Hz, 8H). ESI-MS: m/z 2124.8 (M + H)+. Elemental analysis: calculated for C94H122F24N14O6P4 C, 53.16; H, 5.79; N, 9.23. Found: C, 52.99; H, 5.89; N, 9.15.
15a: To a solution of the diazide TTF derivative 14 (0.021 g, 0.036 mmol) and alkyne functionalized nitroxide 5a (0.026 g, 0.09 mmol) in DMF (1 mL), Cu(CH3CN)4PF6 (0.0022 g, 0.006 mmol) and TBTA (0.0031 g, 0.006 mmol) were added, and the resulting mixture was stirred at room temperature for 24 h, at which time the solvent was evaporated. Purification of the crude mixture by a silica gel column using CH2Cl2–MeOH 90:10 affords biradical 15a (0.021 g, 50% yield). 1H NMR (600 MHz, CD3CN):19δ 2.93 (br s, 4H) 3.56 (br s, 8H), 3.61 (br s, 4H), 3.69 (br s, 4H), 3.83 (br s, 4H), 4.48 (br s, 4H), 6.50 (br s, 2H), 7.71 (br s, 2H). ESI-MS: m/z 1161.6 (M + H)+. Elemental analysis: calculated for C54H84N10O6S6 C, 55.83; H, 7.29; N, 12.06; S, 16.56. Found: C, 55.72; H, 7.33; N, 11.98; S, 16.44.
15b: The above procedure was followed using the diazide TTF derivative 14 (0.021 g, 0.036 mmol), alkyne 5b (0.026 g, 0.09 mmol) in DMF (1 mL), Cu(CH3CN)4PF6 (0.0022 g, 0.006 mmol) and TBTA (0.0031 g, 0.006 mmol). Purification of the crude mixture by a silica gel column using CH2Cl2–MeOH–30% NH3 70:30:1 affords biradical 15b (0.015 g, 36%). 1H NMR (600 MHz, CD3CN):19δ 0.70 (t, J = 12 Hz, 4H), 1.26–1.44 (m, 20H), 1.50–1.66 (m, 20H), 2.01 (dd, J = 12.0 and 2.4 Hz, 4H), 2.84 (tt, J = 12.0 and 2.4 Hz, 2H), 2.94 (t, J = 6.0 Hz, 4H), 3.51–3.56 (m, 8H), 3.59 (t, J = 6.1 Hz, 4H), 3.81 (t, J = 5.0 Hz, 4H), 3.86 (s, 4H), 4.47 (t, J = 5.0 Hz, 4H), 6.54 (s, 2H), 7.68 (s, 2H). ESI-MS: m/z 1131.5 (M + H)+. Elemental analysis: calculated for C54H86N10O4S6 C, 57.31; H, 7.66; N, 12.38; S, 17.00. Found: C, 57.72; H, 7.33; N, 12.26; S, 17.11.
[2]Rotaxane 16a
TTF diazide derivative 14 (0.012 g, 0.02 mmol), CBPQT·4PF6 (0.017 g, 0.015 mmol) and the alkyne functionalized stopper 5a (0.014 g, 0.051 mmol) were dissolved in DMF (0.200 mL) at room temperature under an N2 atmosphere, forming a green solution. TBTA (0.0018 g, 0.003 mmol) and Cu(CH3CN)4PF6 (0.0013 g, 0.003 mmol) were added to the solution and the resulting mixture was stirred at room temperature for 48 h, at which time the solvent was evaporated. The crude solid was purified by column chromatography (SiO2: 1% w/v NH4PF6 solution in Me2CO, i.d. 10 mm, h 19.5 cm). The green fractions in Me2CO were collected, concentrated to a minimum volume, and the product was precipitated from this solution through the addition of an excess of cold water. The resulting solid was collected by filtration, and washed with water to remove the excess of NH4PF6 to afford the [2]rotaxane 16a·4PF6 as a green powder (0.0114 g, 25%). 1H NMR (600 MHz, CD3CN): δ 3.07–3.13 (m, 4H), 3.66–3.76 (m, 12H), 3.83 (bs, 4H), 3.92–3.97 (m, 4H), 4.58 (br s, 4H), 5.74 (s, 8H), 5.96 (s, 1H), 6.00 (s, 1H), 7.71 (br s, 8H), 8.02 (br s, 10H), 9.02 (br s, 8H). ESI-MS: m/z 2263.4 (M + H)+. Elemental analysis: calculated for C90H116F24N14O6P4S6 C, 47.78; H, 5.17; N, 8.67; S, 8.50. Found: C, 47.65; H, 5.25; N, 8.37; S, 8.40. UV-Vis: λmax = 796 nm, ε = 4780 L mol−1 cm−1 in ACN at 298 K.[2]Rotaxane 16b
The above procedure was followed using the TTF derivative 14 (0.01 g, 0.017 mmol), CBPQT·4PF6 (0.015 g, 0.013 mmol), the alkyne functionalized stopper 5b (0.011 g, 0.042 mmol), DMF (0.200 mL), TBTA (0.0013 g, 0.002 mmol) and Cu(CH3CN)4PF6 (0.0009 g, 0.002 mmol).The resulting [2]rotaxane 16b·4PF6 was obtained as a green powder (0.0129 g, 30%). 1H NMR (600 MHz, d6-DMSO): δ 1.10–1.20 (m, 4H), 1.30–1.45 (m, 12H), 1.52–1.72 (m, 28H), 1.82–1.89 (m, 4H), 2.10–2.16 (m, 4H), 2.70–2.76 (m, 2H), 3.13 (br s, 4H), 3.65–3.70 (m, 8H), 3.80–3.86 (m, 4H), 3.92 (br s, 4H), 4.46 (br s, 4H), 4.66 (br s, 4H), 5.82 (br s, 8H), 6.23 (s, 2H), 7.78 (s, 8H), 8.23 (s, 2H), 8.40–8.48 (m, 8H), 9.40–9.48 (m, 8H). ESI-MS: m/z 2233.4 (M + H)+. Elemental analysis: calculated for C90H118F24N14O4P4S6 C, 48.43; H, 5.33; N, 8.78; S, 8.62. Found: C, 48.20; H, 5.25; N, 8.67; S, 8.50.
Acknowledgements
This work was supported by MIUR (Italian network for the development of multivalent nanosystems, MULTINANOITA) and University of Bologna (FARB, Programme for Basic Research, SLaMM Project).Notes and references
- For a review see: C. O. Dietrich-Buchecker and J. P. Sauvage, Chem. Rev., 1987, 87, 795 CrossRef CAS; V. Balzani, M. Gomez-Lopez and J. F. Stoddart, Acc. Chem. Res., 1998, 31, 405 CrossRef; A. Coskun, M. Banaszak, R. D. Astumian, J. F. Stoddart and B. A. Grzybowski, Chem. Soc. Rev., 2012, 41, 19 RSC; S. F. M. van Dongen, S. Cantekin, J. A. A. W. Elemans, A. E. Rowan and R. J. M. Nolte, Chem. Soc. Rev., 2014, 43, 99 RSC.
- For a recent review see: A. Coskun, J. M. Spruell, G. Barin, W. R. Dichtel, A. H. Flood, Y. Y. Botrosghi and J. F. Stoddart, Chem. Soc. Rev., 2012, 41, 4827 RSC.
- For a recent example see: B. Lewandowski, G. De Bo, J. W. Ward, M. Papmeyer, S. Kuschel, M. J. Aldegunde, P. M. E. Gramlich, D. Heckmann, S. M. Goldup, D. M. D. Souza, A. E. Fernandes and D. A. Leigh, Science, 2013, 339, 189 CrossRef CAS PubMed.
- For a recent example see: C. J. Bruns, J. Li, M. Frasconi, S. T. Schneebeli, J. Iehl, H.-P. Jacquot de Rouville, S. I. Stupp, G. A. Voth and J. F. Stoddart, Angew. Chem., Int. Ed., 2014, 53, 1953 CrossRef CAS PubMed.
- R. A. Bissell, E. Cordova, A. E. Kaifer and J. F. Stoddart, Nature, 1994, 369, 133 CrossRef CAS.
- A. C. Fahrenbach, C. J. Bruns, D. Cao and J. F. Stoddart, Acc. Chem. Res., 2012, 45, 1581 CrossRef CAS PubMed.
- P. Franchi, M. Lucarini and G. F. Pedulli, Curr. Org. Chem., 2004, 8, 1831 CrossRef CAS.
- R. Pievo, C. Casati, P. Franchi, E. Mezzina, M. Bennati and M. Lucarini, ChemPhysChem, 2012, 13, 2659 CrossRef CAS PubMed; E. Mileo, C. Casati, P. Franchi, E. Mezzina and M. Lucarini, Org. Biomol. Chem., 2011, 9, 2920 Search PubMed.
- C. Casati, P. Franchi, R. Pievo, E. Mezzina and M. Lucarini, J. Am. Chem. Soc., 2012, 134, 19108 CrossRef CAS PubMed.
- R. W. Dichtel, Š. M. Ognjen, J. M. Spruell, J. R. Heath and J. F. Stoddart, J. Am. Chem. Soc., 2006, 128, 10388 CrossRef PubMed.
- J. Luo, C. Pardin, W. D. Lubell and X. X. Zhu, Chem. Commun., 2007, 2136 RSC.
- E. Mezzina, M. Fanì, F. Ferroni, P. Franchi, M. Menna and M. Lucarini, J. Org. Chem., 2006, 71, 3773 CrossRef CAS PubMed.
- A. Rajca, V. Kathirvelu, S. K. Roy, M. Pink, S. Rajca, S. Sarkar, S. S. Eaton and G. R. Eaton, Chem. – Eur. J., 2010, 16, 5778 CrossRef CAS PubMed; K. Sakai, K. Yamada, T. Yamasaki, Y. Kinoshita, F. Mito and H. Utsumi, Tetrahedron, 2010, 66, 2311 CrossRef PubMed.
- C. Jia, D. Zhang, W. Xu and D. Zhu, Org. Lett., 2001, 3, 1941 CrossRef CAS PubMed.
- X. Guo, D. Zhang, H. Zhang, Q. Fan, W. Xu, X. Ai, L. Fan and D. Zhu, Tetrahedron, 2003, 4843 CrossRef CAS and references cited therein; N. Svenstrup, K. M. Rasmussen, T. K. Hansen and J. Becher, Synthesis, 1994, 809 CrossRef PubMed.
- G. Trippé, F. Le Derf, M. Mazari, N. Mercier, D. Guilet, P. Richomme, A. Gorgues, J. Becher and M. Sallé, C. R. Chim., 2003, 6, 573 CrossRef.
- In order to decrease the line width of the exchanging lines, ESR spectra have been recorded at 338 K.
- V. N. Parmon, A. I. Kokorin, G. M. Zhidomirov and K. I. Zamarev, Mol. Phys., 1975, 30, 695 CrossRef CAS.
- For detailed spectral data of the bis-nitroxide in CD3CN and d6-DMSO, and the corresponding N-hydroxy amines see ESI.‡.
Footnotes |
| † Dedicated to Professor Max Malacria on the occasion of his 65th birthday. |
| ‡ Electronic supplementary information (ESI) available: General information on synthetic procedures and characterization, synthetic details and additional NMR data. See DOI: 10.1039/c4qo00065j |
| This journal is © the Partner Organisations 2014 |