Multi-stimuli responsive polymers – the all-in-one talents
Philipp
Schattling
ab,
Florian D.
Jochum
a and
Patrick
Theato
*b
aInstitute for Organic Chemistry, Johannes Gutenberg-University Mainz, Duesbergweg 10-14, 55099 Mainz, Germany
bInstitute for Technical and Macromolecular Chemistry, University Hamburg, Bundesstraβe 45, 20146 Hamburg, Germany. E-mail: theato@chemie.uni-hamburg.de; Fax: +49 40 42838 6008; Tel: +49 40 42838 6009
First published on 7th August 2013
Abstract
Stimuli-responsive polymers have gained increasing attention, which is attributed to the manifold applications they can be used for. Several years' intensive research was invested in stimuli-responsive polymers. Their stimuli-responsiveness led not only to novel responsive groups, which enabled the translation of an external physical impact into a change of a material property, but also to polymers that are equipped with more than one responsive group. The integration of several responsive moieties within one polymer yields smart polymers exhibiting complex responsive behaviour of the polymers. This review summarises recent developments in the area of multi-stimuli responsive polymers, laying the focus on the improved, multifaceted response of polymer materials depending on the impact of several external stimuli.
 Philipp Schattling | Philipp Schattling studied chemistry at the University of Mainz and Seoul National University, Republic of Korea, and received his Diploma in 2010 from the University of Mainz. He conducted his Ph.D. at the University of Mainz and the University of Hamburg, under the supervision of Prof. Theato. His research interests lie in the synthesis and nano-patterning of novel stimuli-responsive polymers and he received his Ph.D. in 2013. During his Ph.D. course he was awarded a scholarship from the International Research and Training Group 1404. During his graduate studies he spent more than 18 months at Seoul National University in the group of Prof. Kookheon Char and one month in the group of Prof. Nishide at Waseda University, Japan. |
 Florian D. Jochum | Florian D. Jochum studied chemistry at the University of Mainz (Germany). He obtained his Diploma in Chemistry in 2007 for his work on switchable amphiphilic materials in the research group of Prof. Zentel and Prof. Theato. Afterwards, he proceeded with his scientific career as a Ph.D. candidate under the supervision of Prof. Theato with the focus on stimuli-responsive polymers combining temperature- and light-responsive properties. In 2010, he received his Ph.D. in chemistry from the University of Mainz (Germany). One year later, he started a postdoctoral fellowship at the University of Louvain (Belgium) in the research group of Prof. Jean-Francois Gohy. During the last four years, Florian D. Jochum has authored and co-authored more than 20 peer-reviewed publications mainly focused on stimuli-responsive polymers. |
 Patrick Theato | Patrick Theato studied chemistry at the University of Mainz and the University of Massachusetts, Amherst, and obtained his Ph.D. degree from the University of Mainz in 2001. After postdoctoral studies at Seoul National University supported by Feodor Lynen Postdoctoral Research Fellowship and at Stanford University, he completed his Habilitation at the University of Mainz in 2007. From 2009 to 2012 he held a joint appointment at Seoul National University within the WCU program. In 2011, he accepted a prize senior lectureship at the University of Sheffield. Shortly after he moved to the University of Hamburg as an associate professor of polymer chemistry. |
Introduction
Today's polymers have become smart materials. They are able to shrink or expand, to change their optical properties or to change their electrical properties depending on environmental changes. The reason for this development is the change in our society's understanding of materials science: we expect the polymer to adopt or respond in real time to external changes and hence, adjust to our demands. Clearly, the necessity or desire for smart polymers is reflected in the rising number of publications – with a tenfold increase within the last decade – and manifests itself in the fact that smart polymers are indispensable in our life. Stimuli-responsive polymer materials are of different origin, are an important part in a range of applications and have been intensively reviewed recently.1–3 Within this review, we will focus only on stimuli-responsive polymers in aqueous solutions.Smart polymers are characterized by their stimuli-responsive behaviour, which is essentially dictated by the functional groups present within or on a polymer chain. In fact, a broad library of such functionalities has been studied and is covered appropriately in the literature. Different functionalities respond to different stimuli and the most commonly utilized stimuli are temperature, pH, light, ionic strength, electron transfer (redox) and host–guest interactions. In this context relatively new is the combination of several functional moieties within one polymer that can respond to different stimuli. As a result, dual- or multi-stimuli responsive polymers are obtained, which is the starting point to more sophisticated applications, due to the variability that is introduced to the responsiveness.
The combinatorial impact of two or more stimuli responsive groups can be parallel, serial or causal. Over the years, examples for all cases have been presented. Using these terms, parallel means that the response of one group does not affect the response of the other and vice versa. It can therefore be considered as an orthogonal stimuli-responsive system. One example is the dual responsive behaviour of poly(N-isopropylacrylamide) (PNIPAM), which has been statistically modified with pendent fulgimide moieties.4 PNIPAM is well known for its temperature sensitive behaviour, exhibiting a characteristic and distinct lower critical solution temperature (LCST) in water. On the other hand fulgimides feature light-induced photochromic properties. The copolymer shows the same LCST in water before and after irradiation and as such the fulgimide photochromic transition is independent of the LCST behaviour of PNIPAM. This behaviour corresponds to a parallel stimuli responsive system.
Examples of a serial interplay between two stimuli responsive groups are more common. In this context serial describes the amplified impact of the stimulated response of one group by the other. In other words, the impact of one further group exhibits an additive effect on the previous one. Just to mention one example: the group of Bergbreiter demonstrated that TEMPO moieties, which are classified as redox-active groups, can significantly raise the LCST of PNIPAM just by oxidizing and reducing the TEMPO groups within a statistical copolymer.5
Exceptionally fascinating is the causal interplay between two or more stimuli responsive groups. When an external stimulus is applied, the corresponding group will respond. The addressed functional group itself generates a new stimulus – whether by a structural change (occupation of a reactive side, conformational change) or by an environmental change through the whole polymer (e.g. hydrophilic to hydrophobic) – which then stimulates the second responsive group. As a result the second group responds subsequently, but in a different way compared to the first group. Although this interaction is very exciting, examples are rare. Iwai et al. synthesized a copolymer containing N,N-dimethylaminopropylacrylamide, N-t-butylacrylamide and a benzofurazan derivative, which was attached to the backbone via an ester linkage.6 The benzofurazan group is known for its variation in fluorescence wavelength when an environmental change is induced. For example, stimulation of the thermo-sensitive N-t-butylacrylamide moiety results in a polarity change of the polymer – it becomes more hydrophobic – and consequently the benzofurazan fluoresces with a different wavelength. This copolymer represents a fascinating example for a causal interplay.
Motivated by these examples, it is easy to imagine that the successive introduction of different stimuli responsive groups into one polymer can lead to a more pronounced or more detailed response of the polymer towards the impact of an external stimulus. Hence, the right combination of responsive groups will make it possible to design a multi-functional polymer, which exhibits multifaceted behaviour when applying one or more external stimuli. This review deals with multi-stimuli responsive polymers, bearing more than two responsive groups, combined in one polymer. The scope of this chapter is to illustrate the current possibilities and chances of multi-stimuli responsive polymers by highlighting selected fascinating examples, which have been published recently. For this purpose the relevant stimuli are organized at the beginning (Fig. 1) and the origin of the response after applying an external stimulus for each group is explained briefly. The main part describes a few examples sorted by parallel, serial or causal interaction of the responsive groups.
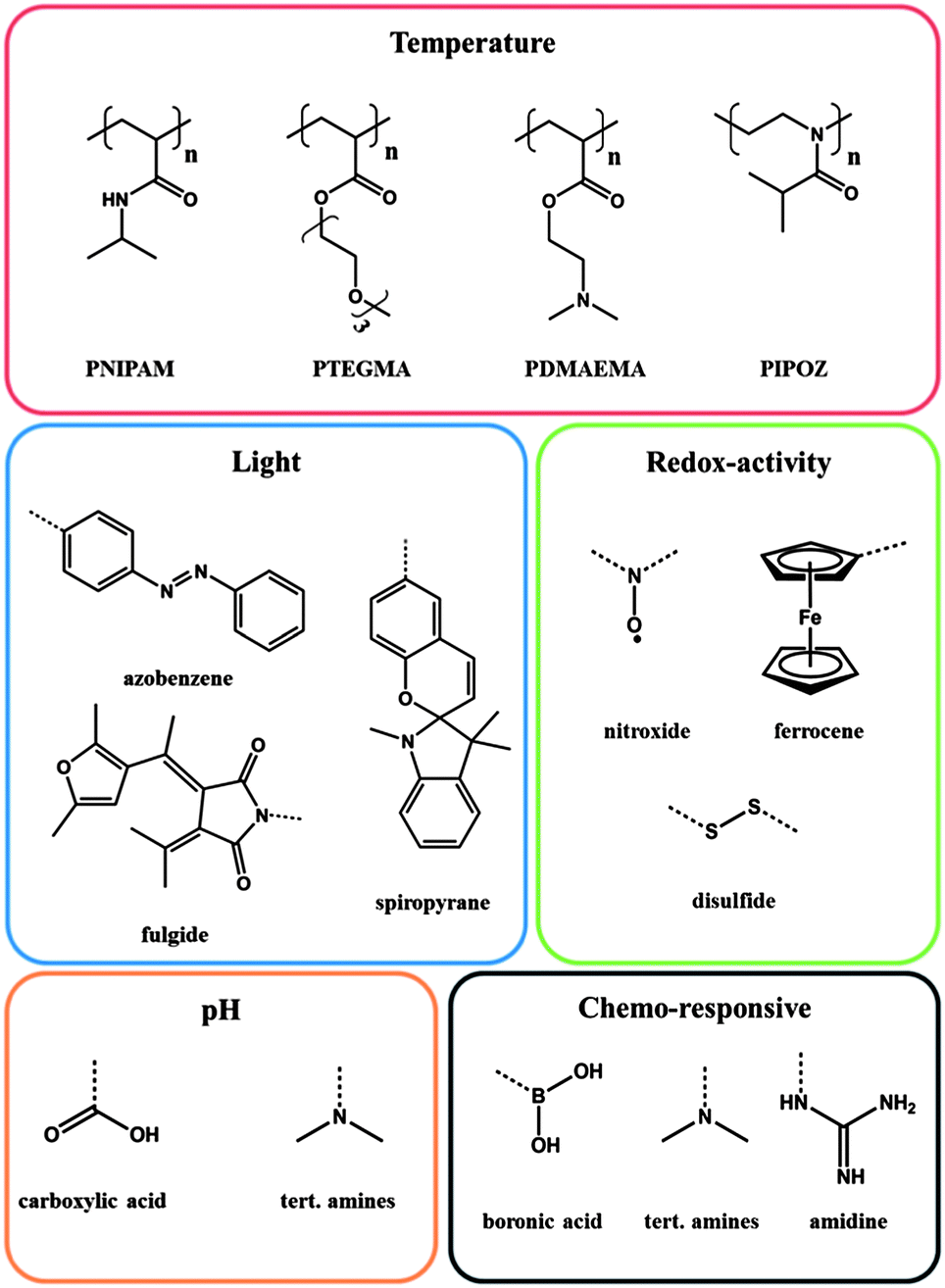 | ||
| Fig. 1 Selection of important polymers and functional groups, exhibiting stimuli responsive behaviour. | ||
Stimuli responsive groups
Stimulus: temperature
Among all available stimuli, changes in temperature play a unique role, because this stimulus can easily be applied from outside. In this context, in particular the lower critical solution temperature (LCST) should be mentioned, which is actually a general phenomenon, but becomes particularly exciting in water due to numerous proposed applications, such as tissue engineering,7 drug delivery,8 catalysis,9 surface engineering10 or information processing.4,11 Indeed, the LCST occurs in solution at a certain composition and it represents from the physico-chemical point of view the shared minima of the spinodal and coexistence curves in a binary mixture (polymer–solvent). At that point, a sharp transition from a stable one phase to an instable system, i.e. two phase, can be observed while increasing the temperature of the system. However, this phenomenon of a temperature-triggered hydrophobic-to-hydrophilic transition is fully reversible and cooling down the polymer solution can regenerate the initial state. The LCST is often observed in highly polar media such as water or alcohols, because of the hydrogen bonding in polymer–solvent interacting systems. Although poly(N-isopropylacrylamide) (PNIPAM)12 is indeed one of the most investigated LCST polymers in aqueous solution, many other polyacrylamides,13 polyvinylethers,14 polyoxazolines,15 as well as poly(oligoethylenenoxide)methacrylates16 should be mentioned here as other important classes of synthetic thermo-responsive polymers featuring a LCST in aqueous solution. At LCST, the polymer will start to phase separate from solution due to a molecular transition from a coiled, enthalpic favoured structure to a dense globular, entropically favoured structure. This process will minimize the free energy of the system considerably. Following this principle, the solution macroscopically starts to become turbid due to the phase separation of the polymer while heating. The LCST is further dependent on different parameters such as chain length,17 tacticity,18 and incorporation of co-monomers,19 pressure20 or even the chemical nature of the end group.21For the sake of completeness it should be mentioned that beside the LCST an upper critical solution temperature (UCST) exists as well. UCST exhibiting polymers are non-soluble below the UCST and become soluble as soon as the temperature is increased. Although there are very good examples in the literature, UCST behaving polymeric systems are beyond the scope of this chapter and the reader is referred to fascinating contributions in the literature.22
Stimulus: light
The irradiation with light can be easily realized by exposure of a sample to light of a specific wavelength. Next, to the control of the energy range, the utilization of light as a stimulus offers the additional feature of controlling the exposure time and intensity with the corresponding system. Virtually the whole light spectra, beginning in the hard UV right up to the deep infrared, can be applied that allow a diversity which cannot be offered by any other stimulus. Last but not least, light allows a localized stimulation within a defined area or volume. Within light responsive polymers, the impact on the corresponding light responsive moiety can be associated with photoinduced isomerization and/or photochromism,23 which renders light as a versatile stimulus. The most studied examples for materials exhibiting photo-responsive properties are azobenzene (trans–cis isomerization),24,25 spiropyran (spiro- to merocyanine form),26 spirooxazine (spiro- to merocyanine form)27 and fulgide (photochromic behaviour)4 derivatives. But also the formation of photocontrollable block copolymer micelles has found broad interest.28Stimulus: redox-activity
A redox stimulus is defined as an electrochemical addressing of the redox-sensitive group, which causes a change in its oxidation state. Indeed this kind of stimulus is more common in inorganic chemistry, especially in respect of transition metals. Nevertheless there are numerous examples of organic compounds that are able to respond towards an electric impulse, e.g. dithienylethenes,29 ferrocene30 or disulfides.31 Paramagnetic organic molecules, such as tetramethylpiperidine-1-oxyl (TEMPO) derivatives, also proved to be a reliable redox-responsive moiety with a long reversibility. TEMPO possesses an unpaired electron, which can be oxidized or reduced in a reversible fashion.32 This one electron process can be achieved either by anodic33 or by chemical reduction with reducing agents like ascorbic acid.5 During the reduction the TEMPO moiety becomes more hydrophilic due to the formed hydroxyl group and hence an improved interaction in aqueous media is the result.Another common redox-active group is ferrocene, because ferrocene captivates with a high chemical stability and a characteristic electrochemical response. The oxidation of the metallocene complex to cationic iron species is totally reversible and is associated with an increase of the polarity.34
Stimulus: pH
A polymer is considered as pH responsive when it features moieties that are capable of donating or accepting protons upon an environmental change in pH.3,35 The adjustment of the pH value triggers ionic interactions, which lead to extending or collapsing of the polymer chain in aqueous solution, induced by the electrostatic repulsion of the generated charges.36 In this context polyacids, such as poly(acrylic acid) with a pKa value around 5 (ref. 37), release their protons and swell under basic conditions, whereas polybases, such as poly(N,N-dimethyl aminoethyl methacrylate),38 accept protons under acidic conditions and hence the polymer chain tends to expand due to coulomb repulsion.Stimulus: chemo-responsive
Beside the different stimuli mentioned above, a more general group must be mentioned, summarizing responsive moieties, which are sensitive upon chemical changes in the environment. These moieties are not less important, even though they are gathered in just one category. They are just too specific and too numerous to be mentioned individually.Polymers featuring boronic acid functionalities are one example. They are sensitive to diols and found particular application in reaction with glucose.39 The diol sensitivity is based on the existence of an undissociated neutral form and an associated anionic form of boronic acid. The anionic species is able to bind diols in a reversible reaction and thus leads to an increase of the hydrophilicity, whereas the neutral form tends to be more hydrophobic.1
A second very fascinating class takes advantage of the formation of inclusion complexes induced by a host–guest interaction. Most known are host–guest interactions utilizing cyclodextrins (CDs). CD belongs to the family of cyclic-oligosaccharides and possesses a shallow truncated cone shape with a hydrophilic outside and comparatively more hydrophobic cavity.40 The inclusion complexes are driven by the geometrical compatibility of the guest molecule, van-der-Waals interactions and hydrophobic interactions. For that, cyclodextrins are used to increase the solubility of low polarity molecules in water as the guest molecule is shielded from the aqueous environment by the encapsulation process. The utilization of CDs in order to manipulate the LCST of a polymer was promoted by the group of Ritter during the last decade.41
Recently, functional groups that are responsive towards carbon dioxide have been the focus of research. These polymers usually feature amidine or tert-amine functionalities.42,43 Amidine groups can react with CO2 yielding a charged amidinium complex, which can be reversible and switched back during exposure to argon or nitrogen.44 By utilizing the amidine chemistry it is possible to render the polymeric system hydrophilic by introducing inexpensive CO2.
Double-stimuli responsive polymers
Thermo- and light-responsive polymers
The majority of examples describing a serial impact of multiple stimuli deal with the combination of thermo-responsive polymers with light-responsive molecules, which are attached onto the polymer chain to photo-control the LCST.45 A prime example was reported by Kungwatchakun et al. in 1988.46 They have synthesized thermo-responsive copolymers by polymerization of N-isopropylacrylamide (NIPAM) with an N-(4-phenylazophenyl)acrylamide monomer. The intention of this work was to photo-control the phase separation temperature of aqueous solutions of PNIPAM by introducing photochromic azobenzene moieties. Consequently, a shift in the phase separation temperature from initial 21 °C to 27 °C after UV-light irradiation was observed and this reversible LCST-change was explained by the change of the dipole moment from 0 to 3 debye due to the trans-to-cis isomerisation of the chromophoric azobenzene moieties. The initial phase transition temperature of 21 °C was re-obtained after exposure to visible light, demonstrating the reversibility of the system. Building upon this seminal work, a variety of studies of thermo-responsive polymers that have been functionalized with azobenzene moieties have been conducted, exhibiting a serial interplay.24,48Thermo- and pH-responsive polymers
Next, changes of the pH have been established as an additional trigger within thermo-responsive polymers.49 These materials attracted great attention in the field of drug delivery because both variables are subject to changes within cancer tissue, which can be utilized to trigger an autonomous response. In such dual responsive systems, functional groups that can either dissociate into ionic groups or form ionic groups upon protonation are incorporated into the backbone of a LCST polymer. Typical examples are carboxylic acids or tertiary amines. It is noteworthy that some homopolymers show pH- and temperature responsive behaviour, namely poly(2-(dimethylamino)ethyl methacrylate) (PDMAEMA) has been found to have a cloud point in the range of 50 °C in neutral aqueous solution.50 However, increasing the pH can shift the cloud points to higher values due to the protonation of the amino-functionality and hence represents a dual-responsive system with serial interplay.Thermo- and redox-responsive polymers
Dual-responsive (thermo and redox) materials based on PNIPAM have been presented by Phillips and Gibson.31 These systems consist of PNIPAM macromonomers, which were linked via disulfide units. The redox sensitive disulfide moiety could be cleaved in the presence of glutathione, leading to PNIPAM fragments, exhibiting molecular-weight dependent cloud point behaviour and can be considered as a system with two stimuli having a causal impact.51One more example with serial interplay between both stimuli is based on a block copolymer composed of alkylacrylamide and vinylferrocene moieties. The polymer shows a characteristic LCST, which is reversibly increased by the ferrocene moiety once it is transferred into the hydrophilic cationic state.53
Multi-stimuli responsive polymers
The selected examples of polymers responsive to two stimuli demonstrate impressively the strength of combining several stimuli within one polymer. However, recently research activities to add one more stimulus, i.e. triple stimuli-responsive polymers, have gained momentum. The addition of one more stimulus is particularly advantageous because it can improve the degree of precision, enlarge the switching window or even change the switching conditions due to the higher level of complexity of the polymer.Light-, pH- and temperature responsive polymers
As already mentioned above, one way to obtain triple stimuli-responsive polymers with a serial interplay is the combination of building blocks that are both pH- and thermo sensitive, e.g. PDMAEMA, with another responsive moiety. For example, in 2010 Tang et al. synthesized an azobenzene terminated PDMAEMA polymer via ATRP (Fig. 2).47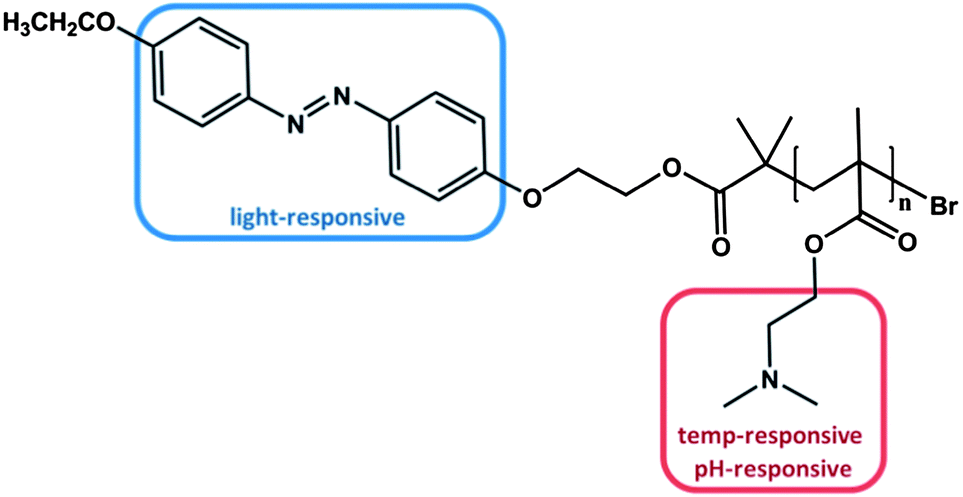 | ||
| Fig. 2 PDMAEMA polymer end-functionalized with azobenzene, which can be stimulated by light, temperature and change of the pH value.47 | ||
As designed, the polymer exhibited responsiveness towards pH, temperature and light and was considered to be triple stimuli-responsive. Indeed, changing the pH value could alter the LCST characteristic. At pH = 4 the dimethylamino functionality was fully protonated, resulting in an increased polarity and thus no LCST was observed. By decreasing the proton concentration, i.e. increasing the pH, the LCST was lowered to 68 °C at pH = 7 and to 30 °C at pH = 11 attributed to the consecutive deprotonation of the dimethyl amino group. The additional effect of the azobenzene end-group due to its photo-isomerization at different pH values led to a slightly higher LCST after irradiation with UV light in comparison to the LCST before irradiation at the corresponding pH value. The process was fully reversible. The less pronounced impact of the light-responsive moiety is not the consequence of the azobenzene group per se, but is rather attributed to the small fraction of azobenzene per polymer. Nevertheless, several studies focused on the effect of azobenzene moieties located at the chain end of stimuli-responsive polymers. All studies confirmed that the amount of azobenzene is proportional to the difference in LCST before and after irradiation.45,55 Please note that this relationship is only valid for azobenzene end group functionalized polymers, where the photo-isomerization is totally independent from any side group effects. In statistical copolymers containing azobenzene as side groups the phase transition behaviour is not necessarily affected in a linear relationship.24,56
Another example of serial interaction was investigated by Sumaru et al., who functionalized PNIPAM with spirobenzopyran. The obtained polymer acted as a logic gate due to the responsive behaviour towards temperature, light and pH.52 While the PNIPAM polymer provided the general thermo-responsive behaviour, the spiropyran acted in dual sense as a pH- and light-responsive unit (Fig. 3). Similarly, Achilleos and Vamvakaki described statistical copolymers comprising a DMAEMA unit and a spiropyran.57 In this case, not only the dimethylamino functionality acted as a pH-responsive unit, but also the spiropyran unit was able to respond to changes in pH, besides the photo-isomerization between the spiropyran and merocyanine isomers. Hence, both groups contributed a dual responsive property, again with a serial impact. PDMAEMA: temperature- and pH-responsive behaviour; spiropyran: light- and pH-responsive behaviour. Zhang et al. demonstrated a very interesting approach recently. They utilized a thermo-, pH-, and light-responsive hyperbranched polymer on the basis of hyperbranched polyethylenimine (HPEI).58 Termination of HPEI with isobutyramide groups, followed by supramolecular complexation of 4-(phenylazo)benzoic acid (PABA) led to a structure that was serial responsive to all three stimuli, similar to the linear copolymer.
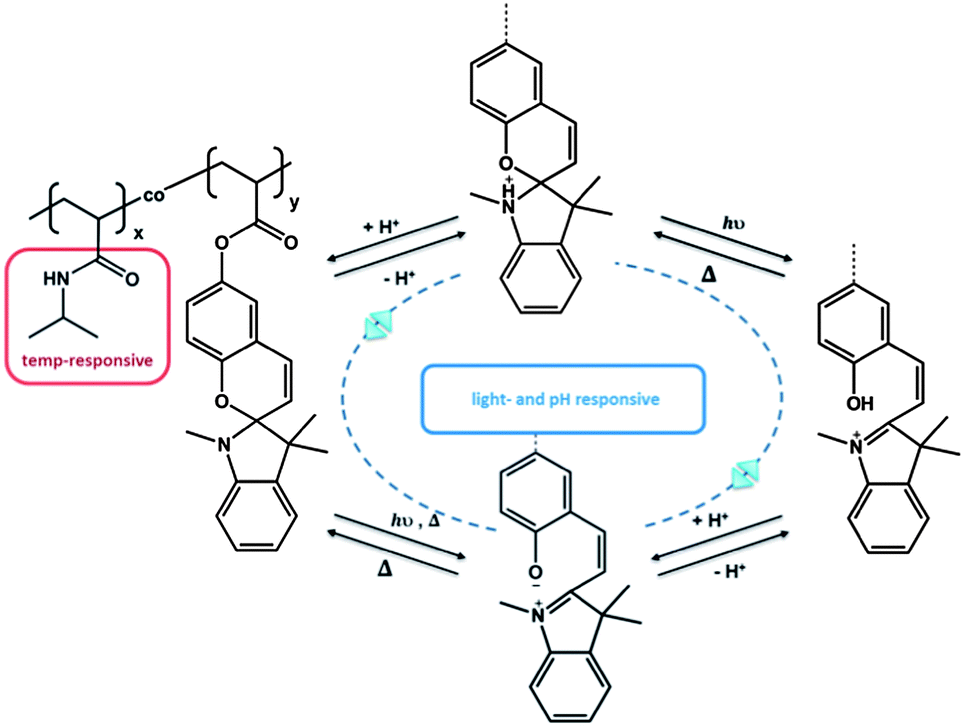 | ||
| Fig. 3 Triple responsive system, based on the dual responsive behavior of spiropyran.52 | ||
In another system, with a serial impact as well, a statistical copolymer was prepared from N-hydroxymethylacrylamide (NHMA), NIPAM and 2-diazo-1,2-naphthoquinone-5-sulfonyl-methylacrylamide (DNQ), which exhibits irreversible triple responsive behavior (Fig. 4).54 The NHMA moiety was considered to be pH responsive, whereas the DNQ was classified as a light-sensitive moiety, due to the photo-induced Wolff rearrangement, even though it is an irreversible process. After UV irradiation, the photoproduct, a 3-indenecarboxylic acid derivative, led to an increase of the polarity and thus to a change of the hydrophilic/hydrophobic balance of the entire polymer, resulting in a change in LCST. Compared to the previous example each moiety is responsive towards only one stimulus, i.e. each group can be addressed independently. It is noteworthy that, as a single functional group, PDMAEMA is responsive to two stimuli, namely pH and temperature. This might be advantageous, because the density of responsive groups within one polymer can be increased. However, it has to be mentioned that the two stimuli cannot be applied independently, which might be unfavourable in some applications. It therefore can be that the subsequently applied second stimulus is suppressed by the state that was generated by the impact of the first applied stimulus. In the case of the poly(NIPAM-co-NHMA-co-DNQ) polymer each responsive moiety can be stimulated independently, even after the application of one orthogonal stimulus. In other words, the LCST can still be shifted by increasing or decreasing the pH value, even after UV irradiation. Clearly, one drawback in this system is the irreversibility of the photo-induced rearrangement, which led to an increase of the LCST, but this change has permanently no chance to recover the initial LCST values.
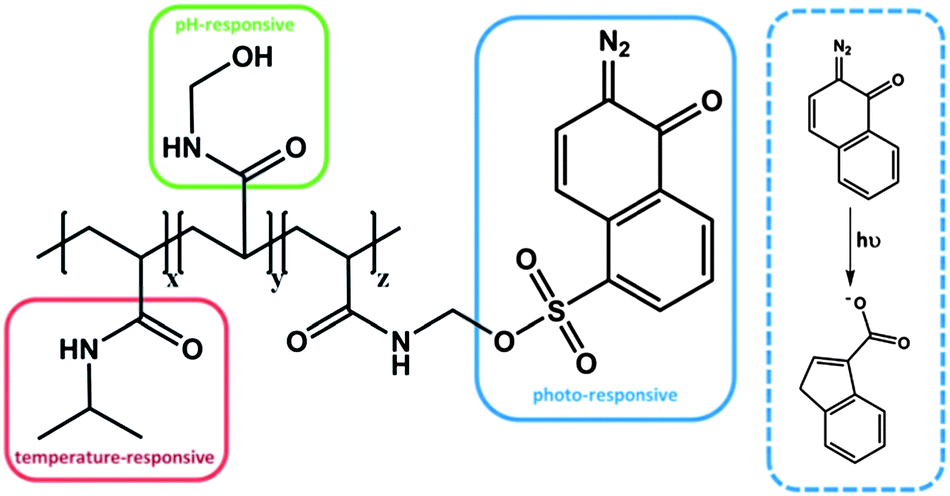 | ||
| Fig. 4 Triple responsive system, bearing thermo-, pH- and photo-sensitive moieties.54 | ||
Light-, redox- and temperature-responsive polymers
Depending on the redox-active moiety employed, different classes of triple light-, redox- and temperature-responsive polymers can be defined. At first, the redox-active group 2,2,6,6-tetramethylpiperidine-1-oxyl (TEMPO) is discussed. Theato et al. prepared PNIPAM copolymers, containing 4-amino-2,2,6,6-tetramethylpiperidine-1-oxyl (amino-TEMPO) and amino functionalized azobenzene moieties linked via an amide bond of the polymer backbone, and it can be considered as a system with serial interplay.11 The TEMPO moiety is redox-sensitive and can be reduced and re-oxidized in a reversible fashion, either chemically using ascorbic acid as a mild reducing agent and red prussiate as an oxidising agent or electrochemically.The reduction of the TEMPO moiety to the corresponding hydroxylamine resulted in a shift of the hydrophilic/hydrophobic balance to an increased hydrophilicity and thus led to an increase in LCST. It is noteworthy that the LCST could be fine-tuned simply by the degree of reduction of the TEMPO moieties. Additional irradiation with UV light in order to stimulate the azobenzene chromophore led to a further increase of LCST. The independent addressing of the two responsive groups was investigated by the sequence of the applied stimuli. Although there was no difference in terms of the final LCST, a huge difference in the “intermediate” LCST was observed depending on the stimulation sequence (Fig. 5). When the azobenzene was stimulated first, the impact of the azobenzene moiety was 60% stronger in comparison to an already reduced copolymer, which is believed due to the increasing polarity influence of the azobenzene isomerized in a hydrophobic copolymer compared to the influence of the isomerization within an already hydrophilic copolymer. Due to the reversibility in combination with the stability of each state, this terpolymer was proposed as a logic gate.
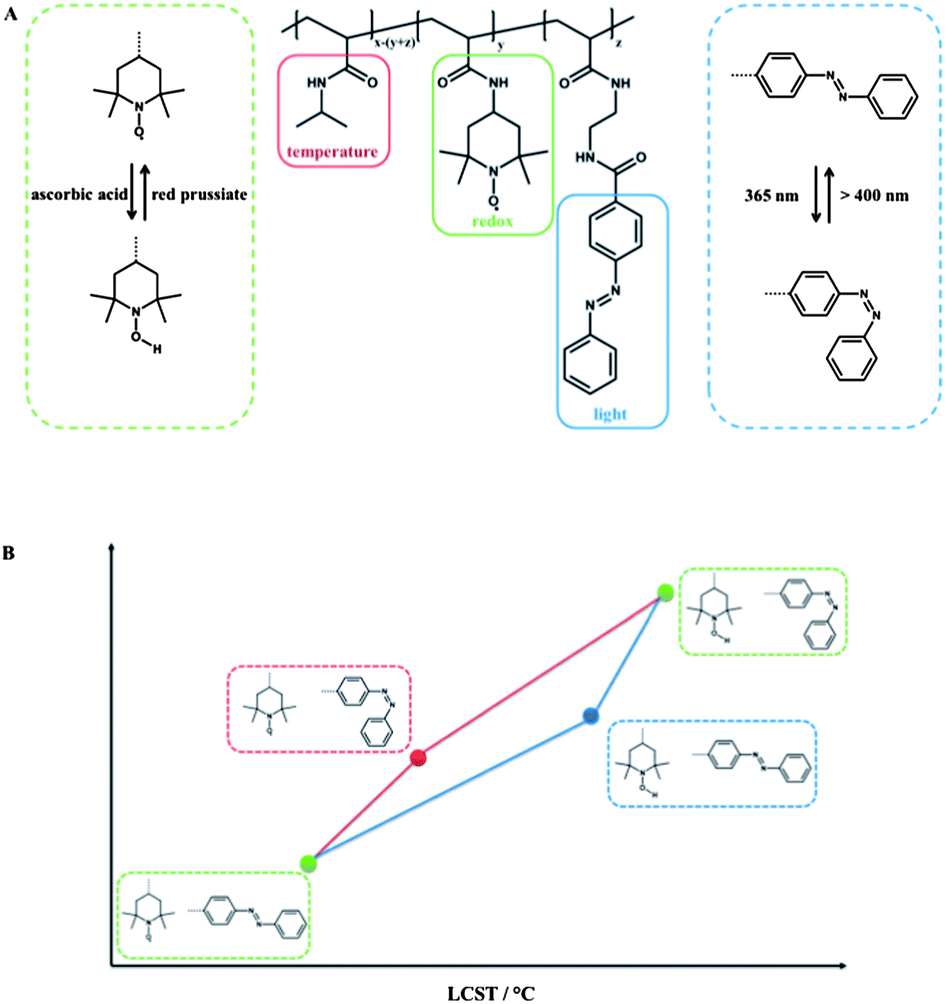 | ||
| Fig. 5 Triple-responsive polymer, equipped with the redox-sensitive moiety TEMPO, the light-responsive azobenzene and NIPAM, which is sensitive towards temperature (A) LCST depending on the stimulation sequence (B). | ||
Environmental-, pH- and temperature-responsive polymers
The group of Uchiyama prepared a random copolymer composed of PNIPAM, poly(dimethylaminopropyl acrylamide) (PDMAPAM) and N-{2-[(7-N,N-dimethylaminosulfonyl)-2,1,3-benzoxadiazol-4-yl-](methyl)amino}ethyl-N-methylacrylamide (DBD-AA) (Fig. 6).6 Besides the already well-known PNIPAM, the PDMAPAM moiety belongs to the class of pH-responsive groups, while the benzofurazan derivative is considered as a polarity sensitive fluorophore. Keeping the temperature fixed at 50 °C (i.e. above LCST of PNIPAM), the copolymer existed in the hydrated open form under acidic conditions, due to the high hydrophilicity induced by the protonated dimethylamino group. As soon as the pH was increased the copolymer chains collapsed. The improved hydrophobic interaction between PNIPAM and DMAPAM units led to an increase of the DBD-AA fluorescence, because the water molecules were repelled from the collapsed polymer chain. For the hydrophobic collapsed polymer chain the fluorescence intensity was 6.6-fold stronger compared to the hydrophilic regime and exhibited a very sharp and distinct transition (within almost one unit), which suggests these materials as advanced pH sensors. They found that the pH range can be fine-tuned by variation of the ionic moiety: incorporation of a weaker proton receptor resulted in a transition under more acidic conditions, while a stronger proton receptor led to a shift of the fluorescence response at more basic pH values.6 In another study Uchiyama et al. reported about a similar polymer and suggested its potential application for information processing.59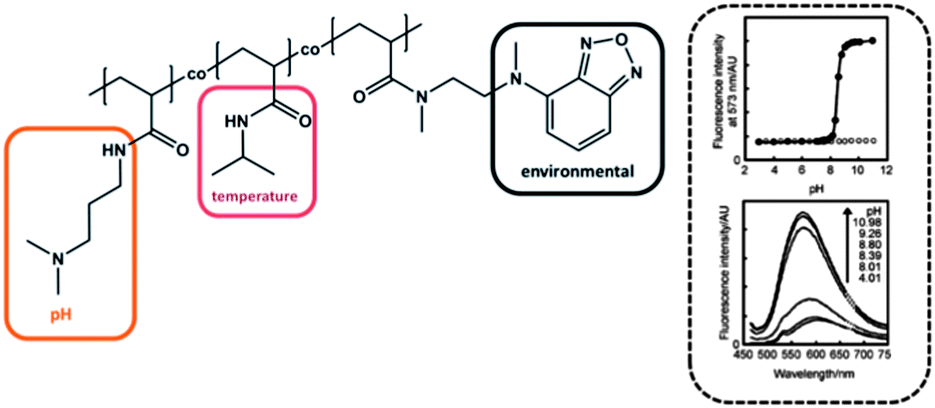 | ||
| Fig. 6 Stimuli responsive polymer system with causal interaction (left). The fluorescence intensity depending on the pH value (right). Graphs reprinted from ref. 6. | ||
The causal interaction of multi-stimuli responsive groups in copolymers is rather fascinating, because these kinds of materials offer the possibility of converting signals from one dimension into another. In the above-mentioned example the impact of ionic strength or temperature was converted into light response, i.e. emission of the fluorescent dye.
Responsive block copolymer structures
Block copolymers are able to self-organize in solution or bulk.60 A copolymer consisting of at least two segments, which are thermodynamically incompatible, can phase separate and nanoscale morphologies can be achieved. The obtained morphology is dictated by the molecular architecture, i.e. the choice of the monomer, the composition ratio and the degree of polymerization.61The most commonly used method for realizing a block copolymer self-assembly in solution is the utilization of amphiphilic diblock copolymers, for example in water. In general one block can be solubilized in a surrounding solvent and the second block tends to separate from the main phase, leading to self-assembly in solution. In many cases spherical micelles are obtained.62 The micelles are not necessarily a stable construct, but are rather fragile and hence they can be disrupted by small perturbations, e.g. by adjusting the pH value when the self-organization is driven by electrostatic interactions,63 by changing the solvent composition,64 by addition of CO2 (ref. 43, 44 and 65) or by changing the temperature.66 Or in other words, such micelles can be rendered stimuli responsive.
Double-hydrophilic block copolymers
Despite the requirement of incompatible segments in order to form micelles, Laschewsky and coworkers demonstrated that even double-hydrophilic block copolymers, i.e. polymers containing two hydrophilic blocks, are able to self-assemble into micelles.67 They synthesized a diblock copolymer consisting of poly(N-isopropylacrylamide) (PNIPAM), exhibiting a LCST characteristic, and poly(3-[N-(3-methylacrylamidopropyl)-N,N-diemthyl]ammoniumpropane sulfonate) (PSPP), which is known for its upper critical solution temperature (UCST) behaviour in aqueous solution. At low temperatures the polarity of PSPP decreases, the block collapses, which is accompanied by the exclusion of water, and accordingly a micelle with a PNIPAM corona is formed. When the temperature is increased above UCST, but still below LCST, the micelle disrupts and a clear homogeneous solution is obtained. But when the temperature is further increased above the LCST of PNIPAM, the PNIPAM block collapses and micelles are formed again. However, in this case the core consists of the “hydrophobic” PNIPAM and the shell of PSPP. The combination of both LCST and UCST in one polymer enables not only a controlled micelle organization at two specified temperatures, but also the thermally triggered exchange of molecular fragments, in order to control the composition of the inner core as well as the shell of the micelle.Schubert and coworkers reported a similar double hydrophilic block copolymer, poly[2-(dimethylamino)ethyl methacrylate]-block-poly[di(ethyleneglycol) methyl ether methacrylate] poly(DMAEMA)-b-poly(DEGMA), which undergoes a two-step thermo-induced self-assembly. First, multilamellar vesicles are formed at 33 °C. Heating to 50 °C resulted in the rearrangement into unilamellar vesicles.69 The self-assembly of double hydrophilic block copolymers into micellar structures has also been utilized in biological context. For example, double thermo-responsive diblock copolymers, poly(oligo(ethylene glycol)monomethyl ether methacrylate)-block-poly(N-isopropyl methacrylamide) (POEGMA-b-PNIPMAM), have been prepared in order to selectively screen a biotin end-group in the hydrophobic PNIPMAM micelle core, which is obtained when heated above 45 °C.70
Further examples of polymers exhibiting similar behaviour could be achieved with POEGMA-b-PDEAM (poly(diethylacrylamide)),71 poly(N-isopropyl acrylamide)-block-poly(dendronized methacrylates),72 poly(ethylene oxide)-block-poly(glycerol monoacrylate)73 or poly(ethylene glycol)-block-poly(N-isopropyl acrylamide) polymers functionalized with cyclodextrin moiety induced self-assembly to rod-shaped nanostructures below LCST and reconstruction into micellar structures above LCST.74
Schizophrenic block copolymers
The inversion of micelle core and corona, also called “schizophrenic” behaviour,75 inspired other groups to develop even more advanced diblock copolymers. In the examples mentioned above each block responses differently to one stimulus. The next step was the investigation of block copolymers in which each block can be addressed independently by applying a different stimulus. The Sumerlin group reported on a diblock copolymer based on poly(3-acrylamidophenylboronic acid) (PABA) and PNIPAM.76 The block copolymer combines both the pH- and diol-sensitive solubilities of boronic acid with the thermo-responsive nature of PNIPAM leading to triple responsive block copolymers, which are capable of self-assembling into micelles or inverse micelles depending on the environmental conditions. Below LCST of PNIPAM the polymer solution contains molecularly dissolved unimers, which self-assemble into micelles with a PNIPAM core and a PABA shell once the temperature is raised above LCST. However, changing the pH at temperatures below LCST results in micelles with a PABA core as soon as pH falls below the pKa value of boronic acid. These aggregates dissociate as soon as they are exposed to either an increase of the pH value or the addition of small molecular weight diols. This example demonstrated impressively the possibility of constructing micelles with different molecular design for the corona as well as for the core triggered by the impact of orthogonal stimuli. It is noteworthy that, the molecular fragment that forms the corona was determined by the “quantity” of one stimulus, i.e. the amount of diol that was added. In comparison with the previous single-stimulus responsive examples this approach may prove to be unfavourable when the switching window is very narrow and the response will not be precise or when systemic temperature fluctuations are quite large. Decoupling the different stimuli from each other not only improves the degree of precision and complexity of the system but also promotes the simplicity of the application likewise.The fragile character coupled with the responsiveness allows a specific assembly or rupture of the micelle structure. This is extremely attractive in modern biotechnology applications for the delivery of pharmaceutical drugs, which are mostly of hydrophobic nature. Using this concept, micelles can be utilized for the non-covalent drug delivery by encapsulation of drug molecules in the hydrophobic micelle core and the triggered rupture of the micelle can be achieved with spatial control. During the last few decades, numerous examples on micelles have been published, whose assembly and disassembly can be triggered by applying an external stimulus.60,77 For example, in 1999 Okano et al. synthesized a diblock copolymer comprised of thermo-responsive poly(N-isopropylacrylamide) (PNIPAM) and poly(n-butylmethacrylate) (PnBMA), by coupling hydroxyl functionalized PNIPAM (molecular weight 6.100 g mol−1) and carboxylic acid terminated PBMA (molecular weight 8.900 g mol−1) (Fig. 7).68
 | ||
| Fig. 7 Diblock copolymer based on PNIPAM and PnBMA investigated in the context of stimuli responsive drug delivery. Reprinted from ref. 68. | ||
The block copolymer self-assembled into micelles in water, which is attributed to the hydrophobicity of the PnBMA block. Once the temperature was increased above LCST, the local polarity of the PNIPAM block abruptly decreased, larger aggregates formed and the entire solution became turbid due to aggregation of the collapsed PNIPAM shells. When the micelle was loaded with the anticancer drug adriamycin (ADR), the drug was fully incorporated into the hydrophobic PBMA core below LCST, but was released when the temperature exceeded this critical temperature. The reversible character of the micelle formation facilitates on/off switching behaviour of the drug release and hence this system can guide in the direction of an advanced controlled drug delivery, where the drug release is not dependent on the diffusion of the load from the core into the solution or any micelle degradation processes. Because of the introduction of the stimuli responsive moiety, the load and the efficiency of the release can be triggered simply by temperature. This concept has been the focus of several other studies, which utilized acid-sensitive amphiphilic block copolymers to deliver anticancer drugs upon changes in pH,79 temperature and pH-responsive block copolymers,80 pH- and light-responsive microgels,81 pH-responsive hyperbranched double hydrophilic block copolymer micelles,82 or temperature- and CO2-responsive block copolymers.83
Multi-stimuli responsive micellation
Most presented examples just respond towards a single external stimulus. However, in nature the change of macromolecular behaviour usually is not induced by a single stimulus, but a consequence of the influence on several environmental changes. Therefore there is a need to design materials that are able to mimic these features. Thayumanavan and coworkers published one very fascinating example in 2009. They synthesized an amphiphilic diblock copolymer comprised of a PNIPAM block and a tetrahydropyran protected poly(hydroxyethyl methacrylate) (PHEMA) block (Fig. 8).78 The hydrophilic PNIPAM block was linked with the hydrophobic HEMA block by a disulfide group. Due to the combination of the three functional elements, the prepared polymer was responsive towards (a) temperature, due to the PNIPAM moieties, (b) pH, due to the acetal protected HEMA moieties and (c) reducing agents, due to the disulfide linkage, which can be cleaved resulting in the release of the homopolymers. This triple-responsive polymer was further investigated in respect of its micelle formation and micelle disruption depending on the influence of an external stimulus.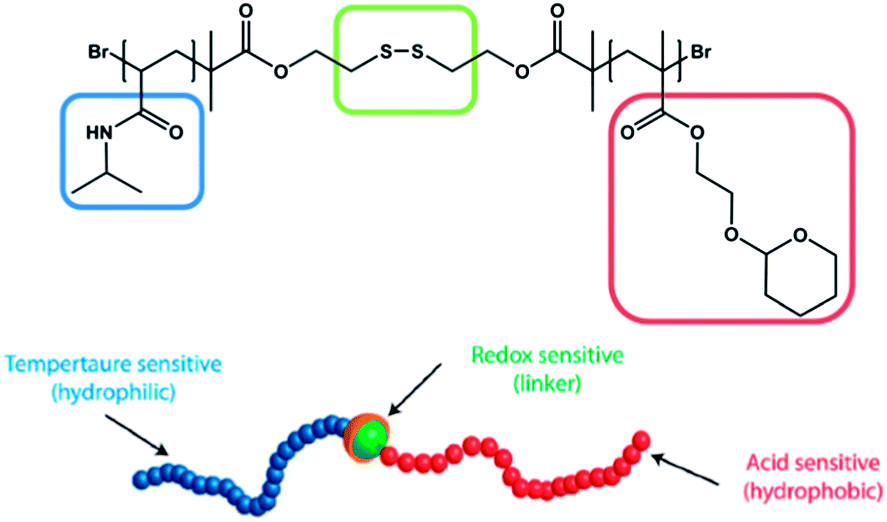 | ||
| Fig. 8 Triple-responsive disulfide linked diblock copolymer suitable for supramolecular self-assembly and external triggered disassembly. Reprinted from ref. 78. | ||
On one hand, when the pH value is decreased to mildly acidic conditions (pH = 4) the acetal protecting group is cleaved and the whole block is transformed into the hydrophilic PHEMA, which causes disruption of the micelle due to the dramatic change in the hydrophilic/lipophilic ratio. On the other hand, the PNIPAM block turns hydrophobic when the temperature is increased above LCST and results in a double-hydrophobic block copolymer, which does not maintain its micelle structure any longer. Last but not least, the addition of DDT causes the reductive cleavage of the disulfide linkage. As a consequence of the separation into two blocks, the hydrophobic PHEMA block precipitates while the hydrophilic PNIPAM stays in solution. Until now the stimuli responsive functional groups have been investigated only separately. The special attraction originating from this polymer is the concerted stimulation of two or three responsive moieties at once. Again the application dictates the proper combination of stimuli-responsive groups. And indeed the behaviour of the block copolymers, resulting from the combinatorial interaction of multiple stimuli-responsive groups, can be depicted in a different manner, as shown in Fig. 9.
 | ||
| Fig. 9 Schematic representation of the polymeric response towards the external stimuli. Reprinted from ref. 78. | ||
While temperature stimulation leads to the precipitation of the polymer, the block copolymer exhibits no phase transition behaviour at all when increasing the temperature and adjusting the pH value to mild acidic conditions at the same time. This can be explained by the increase of the polarity due to the irreversible deprotection of the acetals. The enhanced hydrophilicity can stabilize the hydrophobic tendency of the PNIPMA block at higher temperatures and prevent precipitation or in other words, the deprotection of the acetals inhibits the temperature dependency of the polymer. Similar behaviour could be observed when treating the copolymer with DDT. The micelle disrupted, the PHEMA block precipitated in water and after filtration of the aqueous solution the LCST, which could be still observed, was dedicated to the PNIPAM polymer only. But the combination of all three stimuli provided an unexpected surprise: after the cleavage of both the disulfide linkage and the acetal protecting group, the temperature dependent behaviour of the aqueous solution should be determined by the LCST of the pure PNIPAM, even though PHEMA is water soluble, it features no LCST. But in fact no LCST could be detected at all, due to the hydrogen bonding between PHEMA and PNIPAM.84 This example showed that the application of just one stimulus leads to a very slow or incomplete release kinetic, but the combination of two stimuli accelerates the release of the encapsulated guest molecules, making drug delivery much more efficient.
A second example was published by Dong et al. in 2013.85 Although the polymer is not as complex as the responsive polymer mentioned in the previous example, it still can be considered as a multi-responsive material. The polymeric system is based on poly(dimethylaminoethyl methacrylate) (PDMAEMA), which is pH- and temperature responsive, and the third responsive group is introduced by quaternization of 13.9 mol% of the dimethylaminoethyl functions with 1-(bromomethyl)pyrene, which renders the pyrene modified part of the system hydrophobic. When the polymer is dissolved in water it forms micelles, with the hydrophobic pyrene compartments forming the core and the dimethylaminoethyl (DMAE) moieties forming the shell. They found that the DMAE moieties became hydrophobic, which resulted in shrinking of the micelle when the temperature is increased above LCST. However, the micelle dissociates when the solution is irradiated with UV light below LCST, due to the photo-induced solvolysis of pyrenylmethyl groups from the quaternized amine, which leads to an increase of the hydrophilicity of the whole polymer. Although the pH- and the temperature responsive behaviour is fully reversible, the light responsive disruption of the micelle is totally irreversible, i.e. after applying the light stimulus, the micelle cannot be formed again and the polymer turns into a double responsive polymer.
Conclusion and future perspective
Within the last few years tremendous efforts were devoted to the area of stimuli-responsive materials, driven by the need for precisely controllable material properties. Due to the high degree of control, external stimuli as triggers that induce changes of the physical properties of polymeric materials proved to be a very promising approach. While in the beginning the focus was laid on polymers bearing only one responsive moiety, the focus shifted over the years to multiple sensitive functions combined within one polymer. This development is not only due to the improved diversity of multi-stimuli materials, but is also evidence for the significant progress in polymer synthesis over the last few years. Developments of controlled polymerization techniques, especially within the area of controlled radical polymerization, led to the precise design of well-defined polymeric architectures with high degrees of functionality, which were not accessible by any previous method. Despite the manifold contributions that were made in respect of multi-responsive polymers, the number of responsive groups is usually limited to only two. Clearly two stimuli are more advanced than one stimulus, and hence, new possibilities are available. However, as mentioned previously for biomedical applications, multi-stimuli responsive polymers are highly desirable and are motivated by examples in nature.Multi-responsive materials are not only important in life science, but also equally essential for new developments in information technology. Especially in the context of the orthogonal and independent addressing of the responsive groups, parallel writing of information seems to be feasible. Accordingly a dramatic increase of memory density can be envisioned. To achieve these goals, the utilization and accurate control of several stimuli responsive groups combined into one polymer is mandatory. This requires new polymer chemistries. Next, several responsive groups show only partial reversibility or undergo irreversible changes upon stimulation. It would be highly desirable to have access to fully reversible responsive groups in these applications as well. And this requires the development of new chemistries. Accordingly, the area of multi stimuli-responsive polymers is a very interdisciplinary research area. Hence, joint efforts will lead to novel materials with properties beyond today's imagination. New inventions and more detailed research of multi-responsive polymers are crucial to achieve the self-defined goals of control, accuracy and sustainability.
Notes and references
- D. Roy, J. N. Cambre and B. S. Sumerlin, Prog. Polym. Sci., 2010, 35, 278–301 CrossRef CAS PubMed , http://www.sciencedirect.com/science/article/pii/S0079670009001063.
- M. A. C. Stuart, W. T. S. Huck, J. Genzer, M. Muller, C. Ober, M. Stamm, G. B. Sukhorukov, I. Szleifer, V. V. Tsukruk, M. Urban, F. Winnik, S. Zauscher, I. Luzinov and S. Minko, Nat. Mater., 2010, 9, 101–113, DOI:10.1038/nmat2614.
- F. Liu and M. W. Urban, Prog. Polym. Sci., 2010, 35, 3–23 CrossRef CAS PubMed , http://www.sciencedirect.com/science/article/pii/S0079670009001002.
- F. D. Jochum, F. R. Forst and P. Theato, Macromol. Rapid Commun., 2010, 31, 1456–1461 CrossRef CAS PubMed.
- H. Fu, D. M. Policarpio, J. D. Batteas and D. E. Bergbreiter, Polym. Chem., 2010, 1, 631–633, 10.1039/c0py00060d.
- S. Uchiyama, N. Kawai, A. P. de Silva and K. Iwai, J. Am. Chem. Soc., 2004, 126, 3032–3033 CrossRef CAS PubMed.
- (a) B. Jeong and A. Gutowska, Trends Biotechnol., 2002, 20, 305–311 CrossRef CAS , http://linkinghub.elsevier.com/retrieve/pii/S0167779902019625; (b) N. Zhang, S. Salzinger and B. Rieger, Macromolecules, 2012, 45, 9751–9758 CrossRef CAS; (c) Z. Ding, G. Chen and A. S. Hoffman, J. Biomed. Mater. Res., 1998, 39, 498–505, DOI:10.1002/(SICI)1097-4636(19980305)39:3<498::AID-JBM22>3.0.CO;2–5.
- (a) D. Schmaljohann, 2006 Supplementary Non-Thematic Collection, 2006, 58, 1655–1670, http://www.sciencedirect.com/science/article/pii/S0169409X06001839; (b) D. E. Meyer, B. C. Shin, G. A. Kong, M. W. Dewhirst and A. Chilkoti, Proceeding of the International Symposium on Tumor Targeted Delivery Systems, 2001, vol. 74, pp. 213–224, http://www.sciencedirect.com/science/article/pii/S0168365901003194 Search PubMed.
- (a) X.-Y. Liu, F. Cheng, Y. Liu, H.-J. Liu and Y. Chen, J. Mater. Chem., 2010, 20, 360–368, 10.1039/b915313f; (b) D. E. Bergbreiter, J. Polym. Sci., Part A: Polym. Chem., 2001, 39, 2351–2363, DOI:10.1002/pola.1212.
- (a) B. Xue, L. Gao, Y. Hou, Z. Liu and L. Jiang, Adv. Mater., 2013, 25, 273–277, DOI:10.1002/adma.201202799; (b) T. Zhao, F.-Q. Nie and L. Jiang, J. Mater. Chem., 2010, 20, 2176–2181, 10.1039/b918951c.
- P. Schattling, F. D. Jochum and P. Theato, Chem. Commun., 2011, 47, 8859 RSC.
- (a) H. G. Schild, Prog. Polym. Sci., 1992, 17, 163–249 CrossRef CAS , http://www.sciencedirect.com/science/article/pii/007967009290023R; (b) Y. Zhang, S. Furyk, L. B. Sagle, Y. Cho, D. E. Bergbreiter and P. S. Cremer, J. Phys. Chem. C, 2007, 111, 8916–8924 CrossRef CAS PubMed; (c) H. Ringsdorf, J. Venzmer and F. M. Winnik, Macromolecules, 1991, 24, 1678–1686 CrossRef CAS; (d) Y.-H. Lien, T.-M. Wu, J.-H. Wu and J.-W. Liao, J. Nanopart. Res., 2011, 13, 5065–5075, DOI:10.1007/s11051-011-0487-8; (e) N. Haberkorn, K. Nilles, P. Schattling and P. Theato, Polym. Chem., 2011, 2, 645–650, 10.1039/c0py00314j.
- I. Dimitrov, B. Trzebicka, A. H. E. Müller, A. Dworak and C. B. Tsvetanov, Prog. Polym. Sci., 2007, 32, 1275–1343 CrossRef CAS PubMed , http://www.sciencedirect.com/science/article/pii/S0079670007000871.
- O. Confortini and F. E. Du Prez, Macromol. Chem. Phys., 2007, 208, 1871–1882 CrossRef CAS.
- (a) R. Hoogenboom, Angew. Chem., Int. Ed., 2009, 48, 7978–7994 CrossRef CAS PubMed; (b) C. Diehl and H. Schlaad, Macromol. Biosci., 2009, 9, 157–161, DOI:10.1002/mabi.200800213; (c) N. Zhang, R. Luxenhofer and R. Jordan, Macromol. Chem. Phys., 2012, 213, 973–981, DOI:10.1002/macp.201200015.
- (a) J.-F. Lutz, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 3459–3470 CrossRef CAS; (b) G. Pasparakis and C. Alexander, Angew. Chem., Int. Ed., 2008, 47, 4847–4850, DOI:10.1002/anie.200801098; (c) A. O. Saeed, J. P. Magnusson, E. Moradi, M. Soliman, W. Wang, S. Stolnik, K. J. Thurecht, S. M. Howdle and C. Alexander, Bioconjugate Chem., 2011, 22, 156–168 CrossRef CAS PubMed; (d) N. N. Shahidan, R. Liu, F. Cellesi, C. Alexander, K. M. Shakesheff and B. R. Saunders, Langmuir, 2011, 27, 13868–13878 CrossRef CAS PubMed.
- (a) R. Hoogenboom, H. M. L. Thijs, M. J. H. C. Jochems, B. M. van Lankvelt, M. W. M. Fijten and U. S. Schubert, Chem. Commun., 2008, 5758–5760, 10.1039/b813140f; (b) K. N. Plunkett, X. Zhu, J. S. Moore and D. E. Leckband, Langmuir, 2006, 22, 4259–4266 CrossRef CAS PubMed.
- (a) Y. Katsumoto and N. Kubosaki, Macromolecules, 2008, 41, 5955–5956 CrossRef CAS; (b) T. Hirano, Y. Okumura, H. Kitajima, M. Seno and T. Sato, J. Polym. Sci., Part A: Polym. Chem., 2006, 44, 4450–4460, DOI:10.1002/pola.21546.
- (a) H. Feil, Y. H. Bae, J. Feijen and S. W. Kim, Macromolecules, 1993, 26, 2496–2500 CrossRef CAS; (b) S. Huber and R. Jordan, Colloid Polym. Sci., 2008, 286, 395–402, DOI:10.1007/s00396-007-1781-y.
- (a) F. Meersman, J. Wang, Y. Wu and K. Heremans, Macromolecules, 2005, 38, 8923–8928 CrossRef CAS; (b) A. K. McClellan and M. A. McHugh, Polym. Eng. Sci., 1985, 25, 1088–1092, DOI:10.1002/pen.760251707.
- (a) S. Furyk, Y. Zhang, D. Ortiz-Acosta, P. S. Cremer and D. E. Bergbreiter, J. Polym. Sci., Part A: Polym. Chem., 2006, 44, 1492–1501, DOI:10.1002/pola.21256; (b) P. J. Roth, F. D. Jochum, F. R. Forst, R. Zentel and P. Theato, Macromolecules, 2010, 43, 4638–4645 CrossRef CAS.
- (a) C. Pietsch, R. Hoogenboom and U. S. Schubert, Polym. Chem., 2010, 1, 1005–1008 RSC; (b) R. Hoogenboom, H. M. L. Thijs, D. Wouters, S. Hoeppener and U. S. Schubert, Soft Matter, 2008, 4, 103–107 RSC; (c) Q. Zhang, P. Schattling, P. Theato and R. Hoogenboom, Polym. Chem., 2012, 3, 1418–1426 RSC.
- (a) Y. Shiraishi, R. Miyamoto and T. Hirai, Org. Lett., 2009, 11, 1571–1574 CrossRef CAS PubMed; (b) A. Albini, E. Fasani and D. Faiardi, J. Org. Chem., 1987, 52, 155–157 CrossRef CAS.
- F. D. Jochum and P. Theato, Polymer, 2009, 50, 3079–3085 CrossRef CAS PubMed.
- Z. Mahimwalla, K. Yager, J.-i. Mamiya, A. Shishido, A. Priimagi and C. Barrett, Polym. Bull., 2012, 69, 967–1006, DOI:10.1007/s00289-012-0792-0.
- (a) C. Pietsch, U. S. Schubert and R. Hoogenboom, Chem. Commun., 2011, 8750–8765, 10.1039/c1cc11940k; (b) T. Hirakura, Y. Nomura, Y. Aoyama and K. Akiyoshi, Biomacromolecules, 2004, 5, 1804–1809 CrossRef CAS PubMed; (c) M. Rini, A.-K. Holm, E. T. J. Nibbering and H. Fidder, J. Am. Chem. Soc., 2003, 125, 3028–3034 CrossRef CAS PubMed.
- (a) F. Wilkinson, J. Hobley and M. Naftaly, J. Chem. Soc., Faraday Trans., 1992, 88, 1511–1517 RSC; (b) Y. Huang, W. Liang, J. K. S. Poon, Y. Xu, R. K. Lee and A. Yariv, Appl. Phys. Lett., 2006, 88, 181102–181103 CrossRef; (c) A. K. Chibisov and H. Gorner, Phys. Chem. Chem. Phys., 2001, 3, 424–431, 10.1039/B007713P.
- Y. Zhao, J. Mater. Chem., 2009, 19, 4887–4895, 10.1039/b819968j.
- H. Logtenberg and W. R. Browne, Org. Biomol. Chem., 2013, 11, 233–243, 10.1039/c2ob26723c.
- M. Mazurowski, M. Gallei, J. Li, H. Didzoleit, B. Stühn and M. Rehahn, Macromolecules, 2012, 45, 8970–8981 CrossRef CAS.
- D. J. Phillips and M. I. Gibson, Chem. Commun., 2012, 48, 1054–1056, 10.1039/c1cc16323j.
- K. Oyaizu and H. Nishide, Adv. Mater., 2009, 21, 2339–2344 CrossRef CAS.
- T. Suga, H. Ohshiro, S. Sugita, K. Oyaizu and H. Nishide, Adv. Mater., 2009, 21, 1627–1630 CrossRef CAS.
- (a) B. Nagel, A. Warsinke and M. Katterle, Langmuir, 2007, 23, 6807–6811 CrossRef CAS PubMed; (b) T. Tatsuma, K.-i. Saito and N. Oyama, J. Chem. Soc., Chem. Commun., 1994, 1853–1854, 10.1039/c39940001853.
- (a) S. P. Nunes, A. R. Behzad, B. Hooghan, R. Sougrat, M. Karunakaran, N. Pradeep, U. Vainio and K.-V. Peinemann, ACS Nano, 2011, 5, 3516–3522 CrossRef CAS PubMed; (b) H. Jia, A. Wildes and S. Titmuss, Macromolecules, 2012, 45, 305–312 CrossRef CAS; (c) R. Liu, P. Liao, J. Liu and P. Feng, Langmuir, 2011, 27, 3095–3099 CrossRef CAS PubMed.
- S. Dai, P. Ravi and K. C. Tam, Soft Matter, 2008, 4, 435–449 RSC.
- G. Li, S. Song, L. Guo and S. Ma, Polym. Sci., Part A: Polym. Chem., 2008, 46, 5028–5035 CrossRef CAS.
- F. Liu and M. W. Urban, Macromolecules, 2008, 41, 6531–6539 CrossRef CAS.
- (a) G. Vancoillie, S. Pelz, E. Holder and R. Hoogenboom, Polym. Chem., 2012, 3, 1726–1729, 10.1039/c1py00380a; (b) Z. Xu, K. M. A. Uddin and L. Ye, Macromolecules, 2012, 45, 6464–6470 CrossRef CAS; (c) G. Pasparakis, M. Vamvakaki, N. Krasnogor and C. Alexander, Soft Matter, 2009, 5, 3839–3841, 10.1039/b911341j; (d) Q. Jin, L.-P. Lv, G.-Y. Liu, J.-P. Xu and J. Ji, Polymer, 2010, 51, 3068–3074 CrossRef CAS PubMed , http://www.sciencedirect.com/science/article/pii/S0032386110003873; (e) B. Elmas, S. Senel and A. Tuncel, React. Funct. Polym., 2007, 67, 87–96 CrossRef CAS PubMed , http://www.sciencedirect.com/science/article/pii/S1381514806001775.
- F. Yuen and K. C. Tam, Soft Matter, 2010, 6, 4613–4630 RSC.
- (a) H. Ritter, O. Sadowski and E. Tepper, Angew. Chem., Int. Ed., 2003, 42, 3171–3173 CrossRef CAS PubMed; (b) O. Kretschmann, S. W. Choi, M. Miyauchi, I. Tomatsu, A. Harada and H. Ritter, Angew. Chem., 2006, 118, 4468–4472 CrossRef.
- (a) D. Han, O. Boissiere, S. Kumar, X. Tong, L. Tremblay and Y. Zhao, Macromolecules, 2012, 45, 7440–7445 CrossRef CAS; (b) Z. Guo, Y. Feng, Y. Wang, J. Wang, Y. Wu and Y. Zhang, Chem. Commun., 2011, 47, 9348–9350, 10.1039/c1cc12388b; (c) Y. Zhao, K. Landfester and D. Crespy, Soft Matter, 2012, 8, 11687–11696, 10.1039/c2sm26440d; (d) S. Lin and P. Theato, Macromol. Rapid Commun., 2013, 34, 1118–1133, DOI:10.1002/marc.201300288.
- J. Y. Quek, P. J. Roth, R. A. Evans, T. P. Davis and A. B. Lowe, Polym. Sci., Part A: Polym. Chem., 2013, 51, 394–404, DOI:10.1002/pola.26397.
- Q. Yan, R. Zhou, C. Fu, H. Zhang, Y. Yin and J. Yuan, Angew. Chem., Int. Ed., 2011, 50, 4923–4927, DOI:10.1002/anie.201100708.
- F. D. Jochum, L. Zur Borg, P. J. Roth and P. Theato, Macromolecules, 2009, 42, 7854–7862 CrossRef CAS.
- D. Kungwatchakun and M. Irie, Makromol. Chem., Rapid Commun., 1988, 9, 243–246 CrossRef CAS.
- X. Tang, X. Liang, L. Gao, X. Fan and Q. Zhou, Polym. Sci., Part A: Polym. Chem., 2010, 48, 2564–2570 CrossRef CAS.
- (a) R. Kröger, H. Menzel and M. L. Hallensleben, Macromol. Chem. Phys., 1994, 195, 2291–2298 CrossRef; (b) X. Tao, Z. Gao, T. Satoh, Y. Cui, T. Kakuchi and Q. Duan, Polym. Chem., 2011, 2, 2068–2073 RSC; (c) A. Dirani, X. Laloyaux, A. E. Fernandes, B. Mathy, O. Schicke, O. Riant, B. Nysten and A. M. Jonas, Macromolecules, 2012, 45, 9400–9408 CrossRef CAS; (d) N. Ishii, J.-i. Mamiya, T. Ikeda and F. M. Winnik, Chem. Commun., 2011, 47, 1267–1269, 10.1039/c0cc04009f.
- (a) B. Mu and P. Liu, React. Funct. Polym., 2012, 72, 983–989, DOI:10.1016/j.reactfunctpolym.2010.12.006; (b) C. M. Schilli, M. Zhang, E. Rizzardo, S. H. Thang, Y. K. Chong, K. Edwards, G. Karlsson and A. H. E. Müller, Macromolecules, 2004, 37, 7861–7866 CrossRef CAS; (c) S. Kulkarni, C. Schilli, B. Grin, A. H. E. Müller, A. S. Hoffman and P. S. Stayton, Biomacromolecules, 2006, 7, 2736–2741 CrossRef CAS PubMed.
- V. Bütün, S. P. Armes and N. C. Billingham, Polymer, 2001, 42, 5993–6008 CrossRef , http://www.sciencedirect.com/science/article/pii/S0032386101000660.
- D. J. Phillips and M. I. Gibson, Biomacromolecules, 2012, 13, 3200–3208 CrossRef CAS PubMed.
- K. Sumaru, M. Kameda, T. Kanamori and T. Shinbo, Macromolecules, 2004, 37, 4949–4955 CrossRef CAS.
- N. Kuramoto, Y. Shishido and K. Nagai, Polym. Sci., Part A: Polym. Chem, 1997, 35, 1967–1972, DOI:10.1002/(SICI)1099-0518(19970730)35:10<1967::AID-POLA12>3.0.CO;2-F.
- Y.-Y. Yu, F. Tian, C. Wei and C.-C. Wang, Polym. Sci., Part A: Polym. Chem, 2009, 47, 2763–2773 CrossRef CAS.
- H. Akiyama and N. Tamaoki, Macromolecules, 2007, 40, 5129–5132 CrossRef CAS.
- H. Akiyama and N. Tamaoki, Polym. Sci., Part A: Polym. Chem, 2004, 42, 5200–5214 CrossRef CAS.
- D. S. Achilleos and M. Vamvakaki, Macromolecules, 2010, 43, 7073–7081 CrossRef CAS.
- J. Zhang, H.-J. Liu, Y. Yuan, S. Jiang, Y. Yao and Y. Chen, ACS Macro Lett., 2013, 2, 67–71 CrossRef CAS.
- S. Uchiyama, N. Kawai, A. P. de Silva and K. Iwai, J. Am. Chem. Soc., 2004, 126, 3032–3033, DOI:10.1021/ja039697p.
- J.-F. Lutz, Polym. Int., 2006, 55, 979–993 CrossRef CAS.
- F. S. Bates, Science, 1991, 251, 898–905 CrossRef CAS PubMed.
- (a) M. Nowakowska, S. Zapotoczny and A. Karewicz, Macromolecules, 2000, 33, 7345–7348 CrossRef CAS; (b) Q. Zhang, E. E. Remsen and K. L. Wooley, J. Am. Chem. Soc., 2000, 122, 3642–3651 CrossRef CAS.
- (a) Y. Q. Yang, L. S. Zheng, X. D. Guo, Y. Qian and L. J. Zhang, Biomacromolecules, 2011, 12, 116–122 CrossRef CAS PubMed; (b) E. S. Lee, K. Na and Y. H. Bae, Nano Lett., 2005, 5, 325–329 CrossRef CAS PubMed.
- (a) M. Changez, N.-G. Kang, H.-D. Koh and J.-S. Lee, Langmuir, 2010, 26, 9981–9985 CrossRef CAS PubMed; (b) H. M. Aliabadi, S. Elhasi, A. Mahmud, R. Gulamhusein, P. Mahdipoor and A. Lavasanifar, Int. J. Pharm., 2007, 329, 158–165 CrossRef CAS PubMed , http://www.sciencedirect.com/science/article/pii/S0378517306006934.
- D. Han, X. Tong, O. Boissière and Y. Zhao, ACS Macro Lett., 2012, 1, 57–61 CrossRef CAS.
- (a) P. De, S. R. Gondi and B. S. Sumerlin, Biomacromolecules, 2008, 9, 1064–1070 CrossRef CAS PubMed; (b) H.-i. Lee, J. Ah Lee, Z. Poon and P. T. Hammond, Chem. Commun., 2008, 3726–3728, 10.1039/b807561a; (c) X. André, M. Zhang and A. H. E. Müller, Macromol. Rapid Commun., 2005, 26, 558–563, DOI:10.1002/marc.200400510; (d) M. Fujiwara, T. Okano, T.-H. Nakashima, A. Nakamura and G. Sugihara, Colloid Polym. Sci., 1997, 275, 474–479, DOI:10.1007/s003960050106.
- M. Arotçaréna, B. Heise, S. Ishaya and A. Laschewsky, J. Am. Chem. Soc., 2002, 124, 3787–3793 CrossRef PubMed.
- J. E. Chung, M. Yokoyama, M. Yamato, T. Aoyagi, Y. Sakurai and T. Okano, J. Controlled Release, 1999, 62, 115–127 CrossRef CAS , http://www.sciencedirect.com/science/article/pii/S0168365999000292.
- C. Pietsch, U. Mansfeld, C. Guerrero-Sanchez, S. Hoeppener, A. Vollrath, M. Wagner, R. Hoogenboom, S. Saubern, S. H. Thang, C. R. Becer, J. Chiefari and U. S. Schubert, Macromolecules, 2012, 45, 9292–9302 CrossRef CAS.
- F. D. Jochum, P. J. Roth, D. Kessler and P. Theato, Biomacromolecules, 2010, 11, 2432–2439 CrossRef CAS PubMed.
- P. J. Roth, T. P. Davis and A. B. Lowe, Macromolecules, 2012, 45, 3221–3230 CrossRef CAS.
- C. Cheng, M. Schmidt, A. Zhang and A. D. Schlüter, Macromolecules, 2007, 40, 220–227 CrossRef CAS.
- X. Tang and C.-Y. Pan, J. Biomed. Mater. Res., Part A, 2008, 86, 428–438, DOI:10.1002/jbm.a.31515.
- F. Yuen and K. C. Tam, J. Appl. Polym. Sci., 2013, 127, 4785–4794, DOI:10.1002/app.38072.
- (a) S. Liu, N. C. Billingham and S. P. Armes, Angew. Chem., Int. Ed., 2001, 40, 2328–2331 CrossRef CAS; (b) J. V. M. Weaver, S. P. Armes and V. Butun, Chem. Commun., 2002, 2122–2123 RSC; (c) D. Wang, J. Yin, Z. Zhu, Z. Ge, H. Liu, S. P. Armes and S. Liu, Macromolecules, 2006, 39, 7378–7385 CrossRef CAS.
- D. Roy, J. N. Cambre and B. S. Sumerlin, Chem. Commun., 2009, 2106–2108 RSC.
- (a) A. Rösler, G. W. M. Vandermeulen and H.-A. Klok, Adv. Drug Delivery Rev., 2001, 53, 95–108 CrossRef , http://www.sciencedirect.com/science/article/pii/S0169409X01002228; (b) G. Pasparakis and M. Vamvakaki, Polym. Chem., 2011, 2, 1234–1248 RSC; (c) R. K. O'Reilly, C. J. Hawker and K. L. Wooley, Chem. Soc. Rev., 2006, 35, 1068–1083 RSC.
- A. Klaikherd, C. Nagamani and S. Thayumanavan, J. Am. Chem. Soc., 2009, 131, 4830–4838 CrossRef CAS PubMed.
- Y. Bae, S. Fukushima, A. Harada and K. Kataoka, Angew. Chem., Int. Ed., 2003, 42, 4640–4643 CrossRef CAS PubMed.
- H. Wei, X.-Z. Zhang, H. Cheng, W.-Q. Chen, S.-X. Cheng and R.-X. Zhuo, J. Controlled Release, 2006, 116, 266–274 CrossRef CAS PubMed , http://www.sciencedirect.com/science/article/pii/S0168365906004329.
- D. Klinger and K. Landfester, Macromolecules, 2011, 44, 9758–9772 CrossRef CAS.
- S. Lee, K. Saito, H.-R. Lee, M. J. Lee, Y. Shibasaki, Y. Oishi and B.-S. Kim, Biomacromolecules, 2012, 13, 1190–1196 CrossRef CAS PubMed.
- B. Yan, D. Han, O. Boissiere, P. Ayotte and Y. Zhao, Soft Matter, 2013, 9, 2011–2016, 10.1039/c2sm27469h.
- Y.-Y. Liu, J. Lü and Y.-H. Shao, Macromol. Biosci., 2006, 6, 452–458 CrossRef CAS PubMed.
- J. Dong, Y. Wang, J. Zhang, X. Zhan, S. Zhu, H. Yang and G. Wang, Soft Matter, 2013, 9, 370–373, 10.1039/c2sm27116h.
| This journal is © The Royal Society of Chemistry 2014 |