 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A transient in situ infrared spectroscopy study on methane oxidation over supported Pt catalysts
Sheedeh
Fouladvand
*,
Magnus
Skoglundh
and
Per-Anders
Carlsson
Competence Centre for Catalysis, Chalmers University of Technology, SE-412 96 Göteborg, Sweden. E-mail: sheedeh@chalmers.se
First published on 8th May 2014
Abstract
Catalysts with platinum dispersed on alumina, ceria and mixed alumina–ceria have been prepared by incipient wetness impregnation, characterized with transmission electron microscopy and X-ray diffraction, and evaluated for total oxidation of methane under both stationary and transient gas compositions (oxygen pulsing). Further, in situ diffuse reflectance Fourier transformed infrared spectroscopy has been used to monitor the evolution of surface species during precise transient gas composition changes using high-speed gas switching valves. The results show that platinum has a sufficiently strong interaction with all the supports so as to form small platinum particles. The smallest sizes are observed for the Pt/Ce sample. The alumina containing samples show, in contrast to the Pt/Ce sample, a decreased methane conversion with increasing oxygen concentration and a clear kinetic bistability between increasing and decreasing oxygen concentrations. The bistable kinetics is likely connected to oxidation and reduction of platinum occurring for different stoichiometric gas mixtures depending on the history of the system, for which an oxidation of the platinum particles effectively inhibits the dissociative adsorption of methane leading to a low reaction rate. It is shown for the alumina containing samples that the adverse effects of oxygen excess can be circumvented by the use of periodic operation so that the average methane conversion is improved. Further, the Pt/Ce sample seems to exhibit additional active sites at the platinum–ceria interface explaining the generally higher conversion of methane for this sample.
1 Introduction
The transport sector within the European Union is responsible for around a quarter of the emissions of greenhouse gases, where road transportation causes about one fifth of the total emissions of carbon dioxide (CO2).1 This has become a driving force for introducing alternative fuels while phasing out conventional diesel and petrol. At present, natural gas is increasingly used as an alternative fuel. As natural gas consists mainly of methane (CH4), technologies developed for combustion of natural gas can, in the longer perspective, also utilize the so-called biomethane produced from renewable biological sources.Natural gas is considered as a clean fuel. The low sulfur content results in low emissions of sulfur oxides2 and the formation of particulate matter and NOx emissions are lower in lean-burn natural gas engines than in diesel engines.3 Further, the high H/C ratio of methane yields significantly more useful energy per emitted CO2 as compared to conventional transport fuels. Yet, the global warming potential over a 20 year period has been reported to be 72 times higher for methane than for CO2,4 which stresses the importance of avoiding methane slip. In this connection, efficient exhaust after-treatment by catalysts designed for total oxidation of methane is the preferred technology to minimize the emission levels of non-combusted methane. Oxidation of methane is however challenging, especially at the low temperatures representative for modern engines, because methane is the least reactive hydrocarbon.5 To facilitate use of (bio)methane as a fuel, catalysts with high ability to dissociate and oxidize methane need to be developed.
The rate-limiting step in the catalytic oxidation of methane is activation and splitting of the first C–H bond, i.e. dissociative adsorption, which is associated with a high energy barrier.6 Oxide supported palladium has been found to be the most active catalyst for methane oxidation under oxidizing conditions.7,8 Unfortunately, palladium based catalysts are prone to poisoning by sulfur species and may suffer from water inhibition.7–9 The platinum counterparts are significantly more tolerant towards both sulfur and water8 but suffer instead from oxygen self-poisoning.5,10 Under net-reducing conditions, however, platinum catalysts have been reported to be more active.5,7,11 Self-poisoning can be circumvented by employing alternating exhaust gas composition, often referred to as periodic operation or transient control, which is a known strategy for enhancement of catalytic activity in such cases.12,13 Thereby, for example, by cycling between net-oxidizing and net-reducing reaction conditions, the effect of oxygen poisoning can be suppressed,12–15 which leads to improved overall activity for oxidation of methane. Another approach to promote methane oxidation over alumina and ceria supported platinum is to add SO2 to the feed. For Pt/Al2O3, this results in enhanced activity for methane oxidation,16 while for Pt/CeO2, the methane oxidation is first temporarily enhanced and then inhibited for prolonged sulfur exposure.16,17
Considering the support material, the traditional important role is to provide a high and stable surface area onto which the noble metals can be dispersed to form nm-sized particles.18 The support should also have a sufficiently strong interaction with the noble metal particles so as to stabilize the particles against sintering.18 For these reasons, alumina is commonly used as the support material in technical catalysts.18 Furthermore, the performance of the noble metal depends on the choice of support material. Besides influencing properties such as the oxidation state, strong interactions can also give rise to new active sites at the interface between the noble metal and the support.15,16,19,20 In the case of methane oxidation over ceria supported platinum, such interfacial sites have been considered to play an important role.15,16,20 Besides, cerium is well known to have the ability to readily change oxidation state (Ce3+ ↔ Ce4+), thereby functioning as an oxygen buffer.21 Therefore, ceria-doped alumina supports that benefit from the properties of both alumina and ceria have been developed.22
In the present study, the influence of the gas phase stoichiometry and periodic operation on the oxidation of methane over platinum supported on alumina, alumina doped with ceria and ceria is investigated. Specific attention is paid to the mechanistic understanding of the reaction under transient conditions by flow-reactor experiments combined with in situ Fourier transformed infrared spectroscopy measurements.
2 Experimental section
2.1 Catalyst preparation and characterization
Catalysts with 4 wt.% Pt supported on alumina (Pt/Al), alumina doped with ceria (Pt/AlCe) and ceria (Pt/Ce) were prepared by incipient wetness impregnation.17 The support material being alumina (Puralox SBa 200, Sasol), alumina doped with 20 wt.% ceria (Puralox SBa-200 Ce-20, Sasol) or ceria (99.5 H.S.A. 514, Rhône-Poulenc) was dispersed in an aqueous solution of tetraamineplatinum(II)nitrate (2.92 wt.% (NH3)4Pt(NO3)2, Johnson Matthey) as the platinum precursor. The pH of the solution was then adjusted to 11 by addition of diluted ammonia solution. The slurry was continuously stirred for 20 minutes, frozen in liquid nitrogen and then freeze-dried for 12 h. The samples were calcined in air at 550 °C for 1 h, with a heating rate of 5 °C min−1 from room temperature to 550 °C. Monolith substrates (L = 15 mm, ∅ = 12 mm) were cut out from a honeycomb cordierite structure with 400 cpsi and coated with 200 mg of the final powder catalyst (80% catalyst powder and 20% binder material) through a dip-coating process, see e.g.ref. 16. Böhmite (Disperal P2, Sasol) was used as the binder for the alumina containing samples and cerium acetate sol (23 wt.% CeO2 (ACT), Nyacol Nano Technologies) was used for the Pt/Ce sample. The monolith samples were finally calcined in air at 600 °C for 2 h.The BET surface area of the powder samples was measured using N2 physisorption (Micromeritics, Tristar). The specific surface area of the Pt/Al, Pt/AlCe and Pt/Ce samples was measured to be 169, 189 and 131 m2 g−1, respectively. The platinum particle size for the powder samples was studied by transmission electron microscopy (TEM), using an FEI Tecnai G2 TEM. Further characterization of the powder catalysts was performed by X-ray diffraction (XRD), using a Bruker XRD D8 Advance instrument with monochromatic CuKαl radiation, covering a 2θ range of 20–60°. The step size and the step time were 0.029° and 1 s, respectively. The sample rotation speed during the XRD measurement was 60 rpm.
2.2 Equipment for catalytic studies and in situ characterization
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 h−1 and a residence time of 0.2 s. The reactor outlet gas phase components were analysed by mass spectrometry (Prisma, Pfeiffer) following the mass to charge ratio of 2 (H2), 15 (CH4), 18 (H2O), 28 (CO), 32 (O2), 40 (Ar), and 44 (CO2).
000 h−1 and a residence time of 0.2 s. The reactor outlet gas phase components were analysed by mass spectrometry (Prisma, Pfeiffer) following the mass to charge ratio of 2 (H2), 15 (CH4), 18 (H2O), 28 (CO), 32 (O2), 40 (Ar), and 44 (CO2).
2.3 Experimental procedures
3. Results
3.1 Analysis of X-ray diffractograms and TEM micrographs
In Fig. 1, the results from the XRD and TEM analyses of the as prepared Pt/Al, Pt/AlCe and Pt/Ce samples are shown. Also, the X-ray diffractograms for the pristine support materials are included as a reference. The diffractograms for the Pt/Al (panel a) and Pt/AlCe (panel b) samples show two sharp peaks at 2θ = 39.8 and 46.2°, which can be assigned to (111) and (200) reflections from metallic Pt, respectively (JCPDS 04-0802). By using the Scherrer equation and the half width of the measured peaks at 2θ = 39.8 and 46.2°, the average diameter of the Pt particles is calculated to be 40 and 27 nm, respectively. The diffractogram from the Pt/Ce sample is dominated by peaks characteristic for the fluorite structure of CeO2. The only exception is the tiny peak at 2θ = 39.8° representing Pt (111).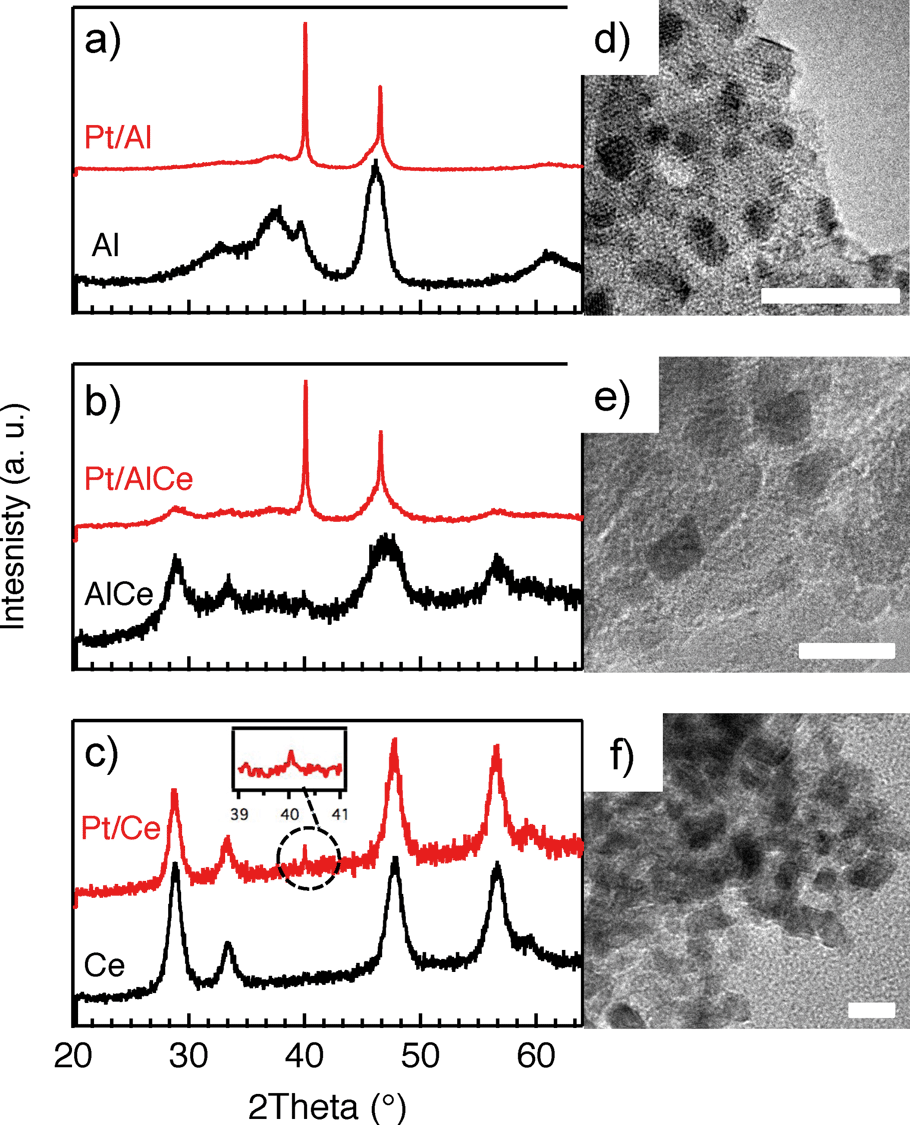 | ||
| Fig. 1 Normalized powder X-ray diffractograms of a) Pt/Al, b) Pt/AlCe and c) Pt/Ce. The diffractograms for the pure support materials are included as reference. TEM images of d) Pt/Al, e) Pt/AlCe and f) Pt/Ce. Scale bars measure 10 nm. | ||
Moving to the analysis of the TEM images, it is clear that in these images the majority of the Pt particles for the alumina and ceria supported samples are below 2 nm in diameter. In a previous study,16 analysis of the Pt particle size distributions for Pt/Al and Pt/Ce samples synthesized with the same preparation method as here showed that 99% of the Pt particles were below 3 nm for Pt/Al (with only a few larger particles up to 20 nm in diameter) and most of the Pt particles were 1–3 nm for Pt/Ce. The presence of ceria in the samples complicates platinum imaging due to low contrast between ceria and platinum and thus the statistics was based on a lower number of particles, however, still adequate. In the present TEM images, the smallest Pt particles are observed for the Pt/Ce sample. Some larger Pt particles were also discerned although not visible in the images shown here.
3.2 Continuous gas flow reactor experiments
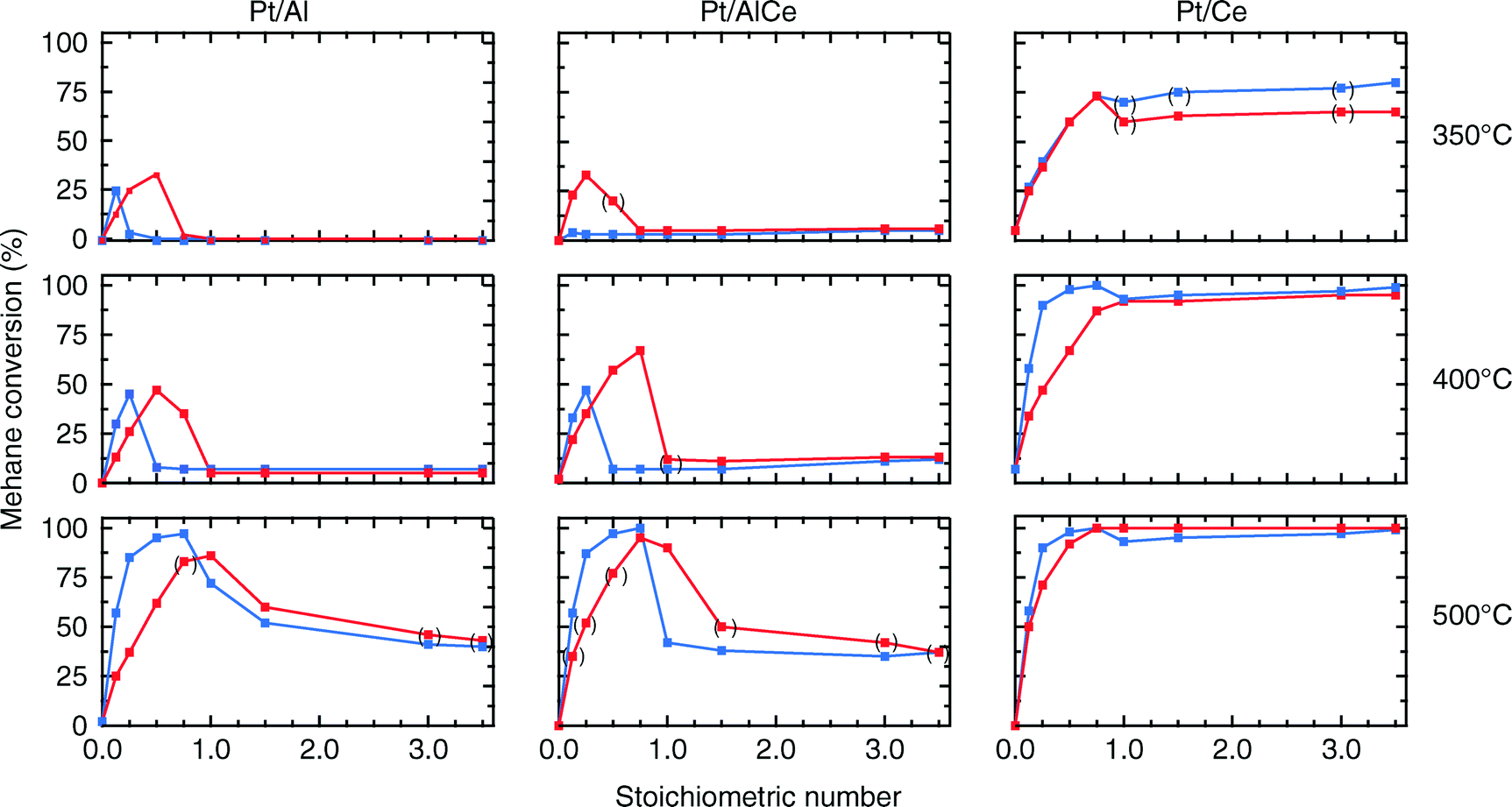 | ||
| Fig. 2 Steady-state conversion of 0.1% methane over Pt/Al (left panels), Pt/AlCe (middle panels) and Pt/Ce (right panels) monolith catalysts for different feed gas compositions, defined by the stoichiometric number; [O2]/2[CH4], at 350 (top panels), 400 (middle panels) and 500 °C (bottom panels). The red and blue lines denote increasing and decreasing oxygen concentration, respectively. The points in parentheses denote the steps where steady-state conditions were not fully achieved during the designed step-time interval. | ||
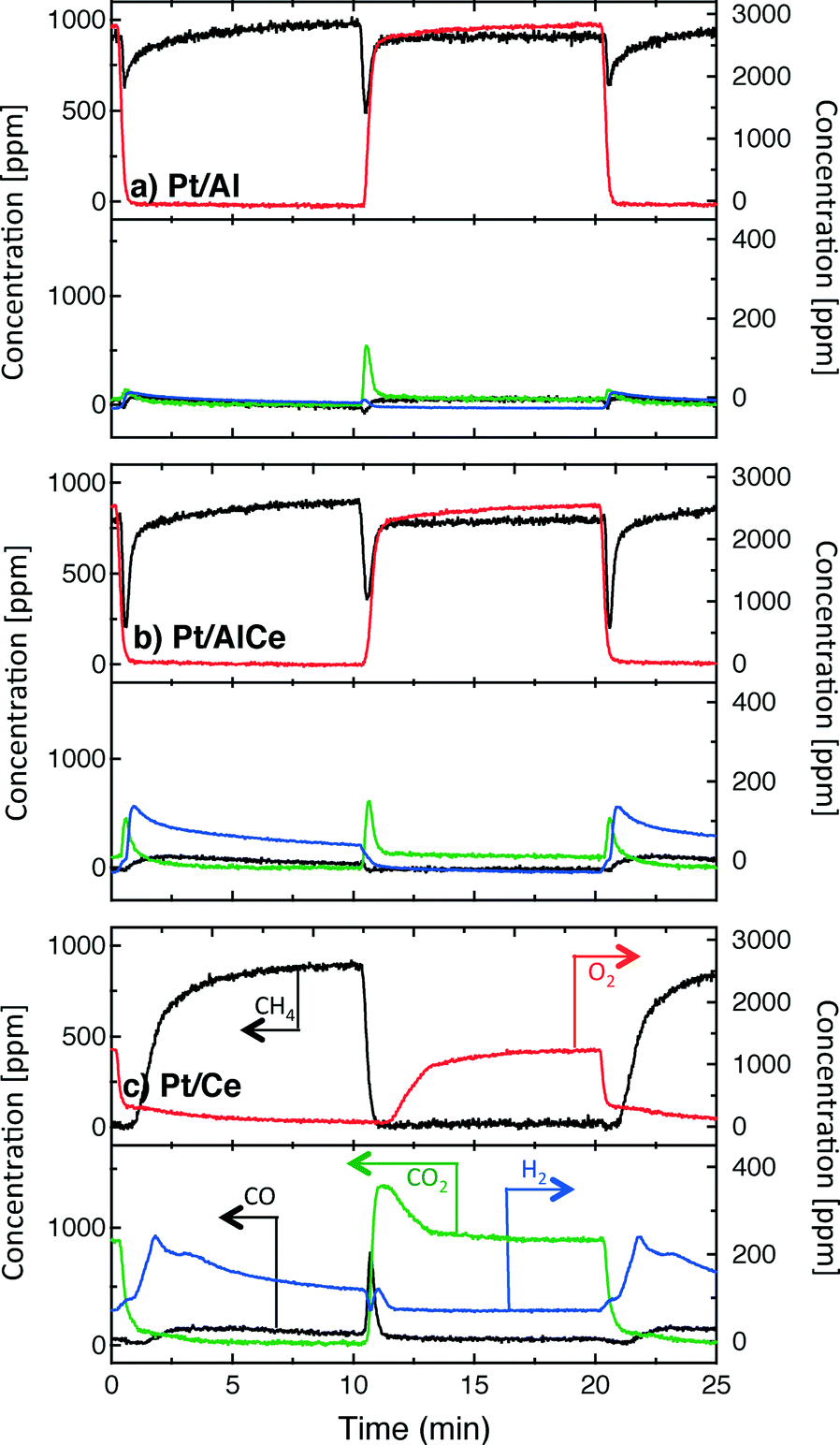 | ||
| Fig. 3 Reactor outlet gas concentrations during transient oxidation of 1000 ppm methane over the Pt/Al, Pt/AlCe and Pt/Ce monolith catalysts during periodic variation of the feed gas composition between S = 1.5 (3000 ppm O2) for 10 min and S = 0 (0 ppm O2) for 10 min at an inlet gas temperature of 400 °C. | ||
A sudden increase in methane conversion is clearly observable at the switches of feed composition for all catalysts except Pt/CeO2 at the lean/rich switch, for it has already reached nearly full conversion during the lean phase. Considering the reaction products, it seems that the total oxidation is promoted with higher ceria content of the catalyst during the lean phases and partial oxidation is more facilitated during the rich phases.
3.3 In situ DRIFT spectroscopy experiments
The DRIFTS results for the isothermal oxygen pulse-response experiments for Pt/Al (left panels), Pt/AlCe (middle panels) and Pt/Ce (right panels) at 400 °C are shown in Fig. 4. The top panels show the color coded intensities (red corresponds to high intensity) of the IR spectra in the interval 1000–4000 cm−1versus time and the insets show a magnification of the 1750–2100 cm−1 region for an oxygen-free phase. The bottom panels show the time evolution of the integrated IR band centered around 2350 cm−1 corresponding to gaseous carbon dioxide (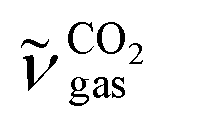 ). By representing the data in color maps, the dynamics of the evolution of IR absorption bands during the different phases of the experiment is clearly displayed.
). By representing the data in color maps, the dynamics of the evolution of IR absorption bands during the different phases of the experiment is clearly displayed.
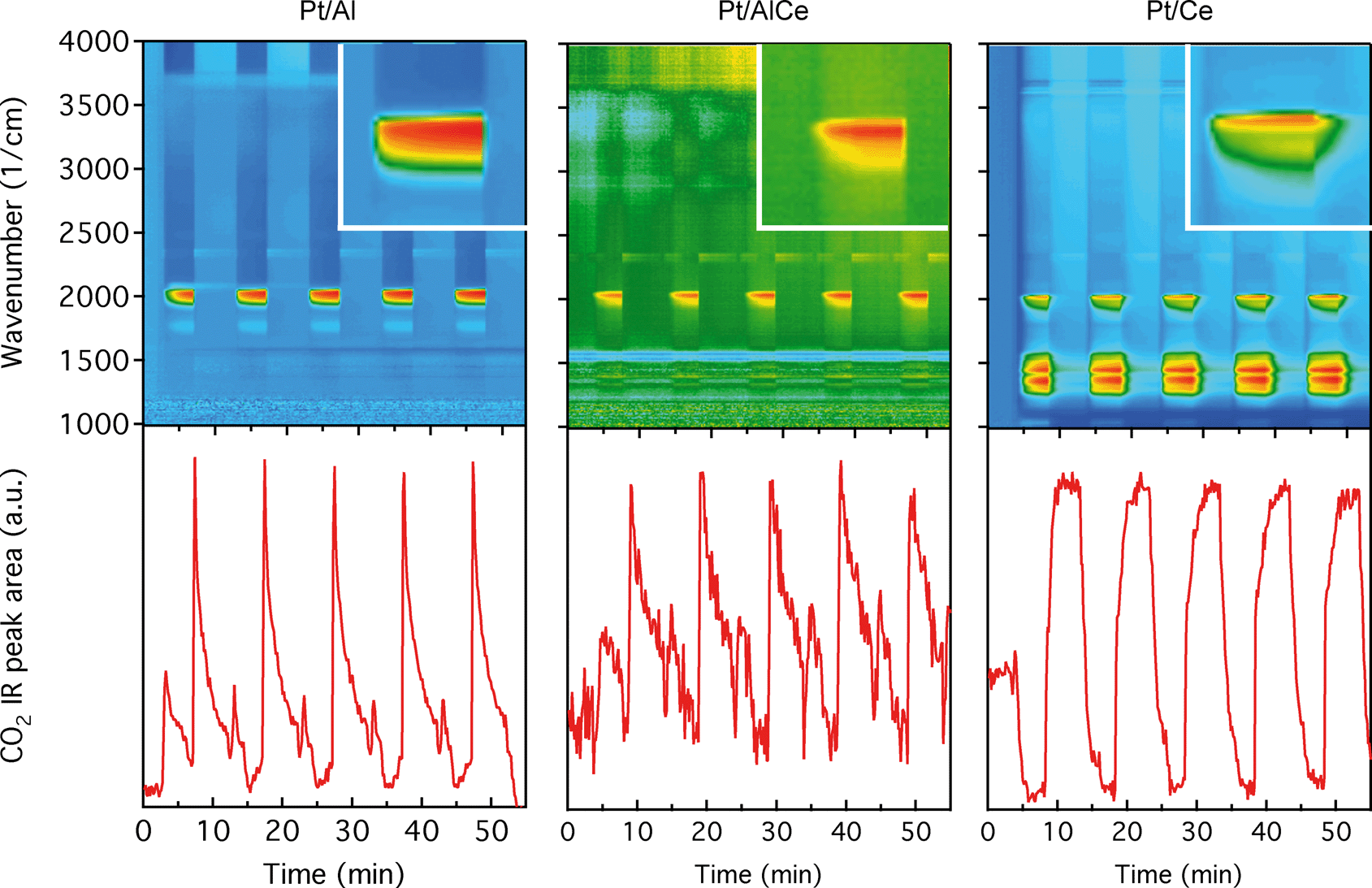 | ||
| Fig. 4 Transient oxidation of 0.1% methane over the Pt/Al (left panels), Pt/AlCe (middle panels) and Pt/Ce (right panels) powder catalysts during periodic variation of the feed gas composition between S = 1.5 (0.3% O2) for 5 min and S = 0 (0% O2) for 5 min at 400 °C. The top panels show the color coded intensities (red corresponds to high intensity) of the IR bands (log1/R, arbitrary units) in the region 1000–4000 cm−1versus time. The bottom panels show the IR peak area for gaseous carbon dioxide versus time. | ||
For the Pt/Al sample, it is clear that CO starts to form almost a minute after the switch to the oxygen-free phase, e.g., at t = 13 min. Looking closely at the CO profile, it can be seen that adsorbed CO on the Pt surface first gives rise to IR bands at higher wavenumbers and the appearance of absorption bands at lower wavenumbers is delayed. Moreover, the CO bands disappear rapidly at the switch to the lean phases. The color map for the Pt/AlCe sample in Fig. 4 shows a slightly different behaviour, as the delay in the formation of the CO bands at low wavenumbers is more pronounced. Pt/Ce, as a more complex system, exhibits a more interesting dynamics. Similar to the Pt/AlCe sample, a pronounced delay in the formation of CO absorption bands at low wavenumbers is observed. Interestingly, the CO species that form earlier on the surface disappear later after the switch to the lean phases. Some changes are also observed in the carbonate region, i.e. 1000–1700 cm−1 for the Pt/Ce sample. These species start to form slightly earlier than the CO species and it also takes a longer time for them to decline after the switch to lean phases. The double band for gaseous CO2 centered around 2350 cm−1 is generally stronger during the lean phases. The progress in the CO2 IR band (integrated peak area) over time is plotted in the bottom panels in Fig. 4. The CO2 band shows a sharp increase at the switches from rich to lean phases followed by a gradual decrease over the lean phases for the Pt/Al sample. However, switching to the rich phases is accompanied by a temporary decrease and increase in CO2 production, after which the CO2 signal follows a continuous decline until the end of the rich phase. Considering the results for the Pt/AlCe sample shown in Fig. 4, the same overall trend for CO2 production is observed as for the Pt/Al sample. However, the CO2 production over the Pt/Ce sample is different as seen in Fig. 4. At the switches from rich to lean conditions, the CO2 production increases rapidly and reaches a steady level until the switch to the rich phase takes place and the CO2 production drops and reaches a steady-state minimum level. To better understand the changes that the formed surface species undergo, a number of IR spectra are shown in Fig. 5(a–c). The spectra chosen to be shown in this figure are the ones revealing noticeable changes of the position/intensity of the IR bands during the cycle. The major changes in the IR spectra in Fig. 5(a–c) take place during the rich phase, where CO peaks start to appear within the 1700–2200 cm−1 region. For Pt/Al, switching to the rich phase is accompanied by formation of a peak at 2036 cm−1 (t = 32.9 min) representing linearly bonded CO on platinum (![[small nu, Greek, tilde]](https://www.rsc.org/images/entities/char_e0e1.gif) COlin).24 As the rich phase proceeds, this peak simultaneously shifts towards lower wavenumbers (2024 cm−1 at t = 36.7 min) and becomes more intense. A closer look shows a shoulder next to the (COlin) peak at lower wavenumbers, which changes similarly to the main (COlin) peak in peak-shift and intensity. By switching to the lean phase at t = 37.3 min, the peak position moves back to higher wavenumbers and gradually diminishes during the lean phase. The absorption bands within the 1810–1880 cm−1 region is characteristic for formation of bridge-bonded CO on platinum during the lean phase. However, the Pt/AlCe sample in Fig. 5(b) shows three distinct IR bands within the linearly adsorbed CO region. In the beginning of the rich phase, an IR band representative of linearly bonded CO to Pt starts to appear at 2058 cm−1 (t = 33.1 min), which slightly shifts towards 2047 cm−1 (t = 33.7 min) and becomes more intense until the end of the rich phase. Resuming the experiment by exposing the catalyst to lean conditions, a blue-shift is observed concomitant with a decline in intensity. The other two IR bands in the CO absorption region are observed at 2085 and 1976 cm−1 during the rich phase. Three CO peaks are also observed within the linearly bonded CO region for the Pt/Ce sample in Fig. 5(c). The sharpest one, which forms at 2028 cm−1, in contrast to the trend for the other two catalysts, experiences a blue-shift to 2045 cm−1 at t = 36.7 min simultaneously as the peak increases in intensity during the rich phase. With the beginning of the lean phase, the band undergoes a red-shift together with decreasing intensity of the peak. At t = 36.7 min, the shoulder at 1982 cm−1 is associated with CO adsorbed on platinum interacting with Ce3+ ions at the metal/support interface.25 The weak peak forming around 1750 cm−1 indicates the presence of CO adsorbed through both carbon and oxygen, the so-called tilted CO.26 This adsorbed state suggests involvement of the support. Cerium cations are reported to affect how CO is adsorbed by affecting the back-bonding through the metal.26 The OCO stretching vibrations of carbonate and formate species on the surface of the support give rise to peaks in the 1000–1700 cm−1 region.15,27 As seen in Fig. 5(c), the peaks in this region become more intense during the rich phase and less intense upon the switch to the lean phase. These IR bands that are changing in intensity depending on the stoichiometric number are assigned on a low-to-high wavenumber order in Table 1.
COlin).24 As the rich phase proceeds, this peak simultaneously shifts towards lower wavenumbers (2024 cm−1 at t = 36.7 min) and becomes more intense. A closer look shows a shoulder next to the (COlin) peak at lower wavenumbers, which changes similarly to the main (COlin) peak in peak-shift and intensity. By switching to the lean phase at t = 37.3 min, the peak position moves back to higher wavenumbers and gradually diminishes during the lean phase. The absorption bands within the 1810–1880 cm−1 region is characteristic for formation of bridge-bonded CO on platinum during the lean phase. However, the Pt/AlCe sample in Fig. 5(b) shows three distinct IR bands within the linearly adsorbed CO region. In the beginning of the rich phase, an IR band representative of linearly bonded CO to Pt starts to appear at 2058 cm−1 (t = 33.1 min), which slightly shifts towards 2047 cm−1 (t = 33.7 min) and becomes more intense until the end of the rich phase. Resuming the experiment by exposing the catalyst to lean conditions, a blue-shift is observed concomitant with a decline in intensity. The other two IR bands in the CO absorption region are observed at 2085 and 1976 cm−1 during the rich phase. Three CO peaks are also observed within the linearly bonded CO region for the Pt/Ce sample in Fig. 5(c). The sharpest one, which forms at 2028 cm−1, in contrast to the trend for the other two catalysts, experiences a blue-shift to 2045 cm−1 at t = 36.7 min simultaneously as the peak increases in intensity during the rich phase. With the beginning of the lean phase, the band undergoes a red-shift together with decreasing intensity of the peak. At t = 36.7 min, the shoulder at 1982 cm−1 is associated with CO adsorbed on platinum interacting with Ce3+ ions at the metal/support interface.25 The weak peak forming around 1750 cm−1 indicates the presence of CO adsorbed through both carbon and oxygen, the so-called tilted CO.26 This adsorbed state suggests involvement of the support. Cerium cations are reported to affect how CO is adsorbed by affecting the back-bonding through the metal.26 The OCO stretching vibrations of carbonate and formate species on the surface of the support give rise to peaks in the 1000–1700 cm−1 region.15,27 As seen in Fig. 5(c), the peaks in this region become more intense during the rich phase and less intense upon the switch to the lean phase. These IR bands that are changing in intensity depending on the stoichiometric number are assigned on a low-to-high wavenumber order in Table 1.
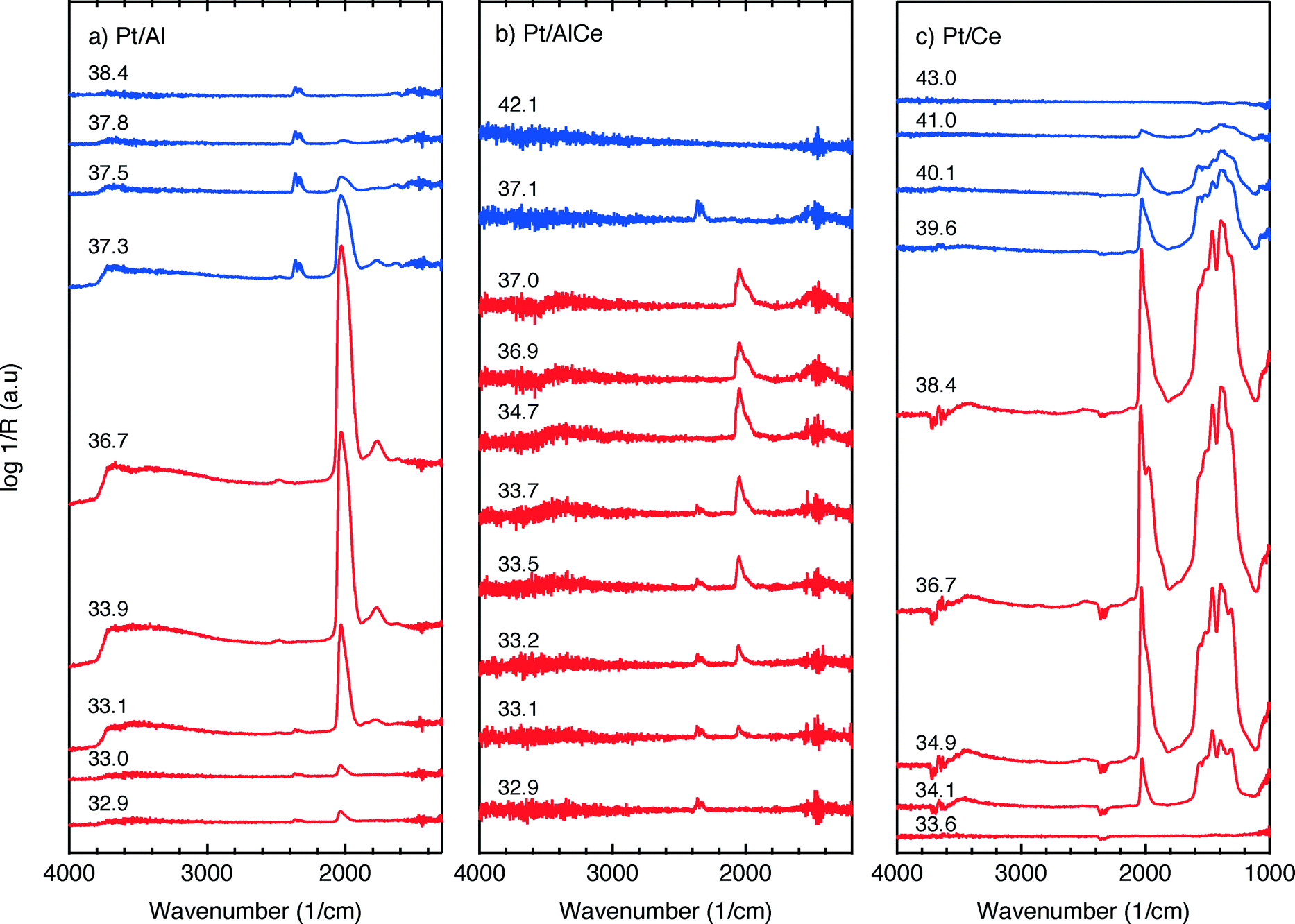 | ||
| Fig. 5 Evolution of IR bands in the wavenumber region 1000–4000 cm−1 for (a) Pt/Al, (b) Pt/AlCe and (c) Pt/Ce catalysts exposed to 0.1% methane while changing the oxygen concentration from 3000 ppm (S = 1.5) to 0% (S = 0) (red lines) and then back to 3000 ppm (blue lines) at an inlet gas temperature of 400 °C. The insert numbers indicate the time (min) in the dynamic experiment where the spectra were recorded. | ||
| Wavenumber (cm−1) | Species |
|---|---|
| 1052 | Unidentate carbonate28 |
| 1078 | Methoxy species on reduced ceria (Ce+3 sites)27 |
| 1218 | Bidentate carbonate29 |
| 1310 | Inorganic carboxylate,29 bidentate carbonate30 |
| 1365 | Bulk carbonate30 |
| 1396 | Unidentate carbonate,29 bulk carbonate30 |
| 1424 | Bulk carbonate30 |
| 1454 | Unidentate carbonate,29 bulk carbonate30 |
| 1550 | Inorganic carboxylate29 |
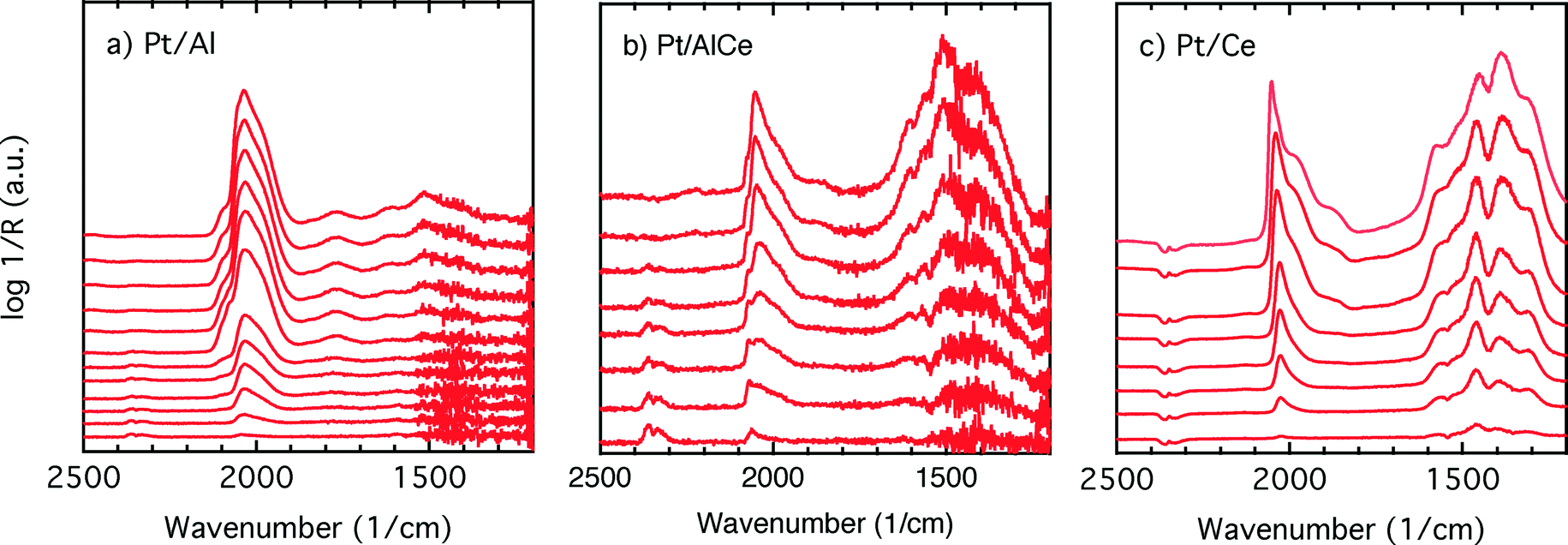 | ||
| Fig. 6 Evolution of IR bands in the wavenumber region 1200–2500 cm−1 for (a) Pt/Al, (b) Pt/AlCe and (c) Pt/Ce catalysts exposed to 0.05% CO after initial pretreatment in 0.1% methane and 0.3% oxygen for 30 min at an inlet gas temperature of 400 °C. | ||
4. Discussion
For all samples, the preparation procedure results in Pt particles that are smaller than about 2 nm and the smallest particles are observed for the Pt/Ce sample (cf.Fig. 1d–f). A few larger Pt particles are present in the alumina containing samples, which give rise to the two sharp diffraction peaks observed at 2θ = 39.8 and 46.2° in Fig. 1a and b. The particles below about 2 nm are difficult to resolve by XRD, even with modern step-scanning techniques.31 It has been reported that ceria can promote noble metal dispersion22,32 and Pt supported on ceria exhibits stronger metal–support interactions than Pt supported on alumina.33 Thus it is expected that the platinum in the ceria containing catalysts is at least as highly dispersed as in the Pt/Al sample. For all samples, most of the Pt particles are sufficiently small to obey various phenomena, e.g., edge effects, electron transfer and spill-over processes between the Pt and the support material that may affect the catalytic properties.34 Considering the support materials, it can be seen that the XRD pattern for pure alumina–ceria mainly follows the corresponding pattern from the pure ceria support with some influence from the alumina, which is noticeable as a peak shift compared to pristine alumina and ceria supports at 2θ = 47°. This suggests that the properties of both alumina and ceria should be considered when interpreting the results for the Pt/AlCe catalyst. Concerning catalytic activity, the present results clearly show that the Pt/Ce catalyst is significantly more active for methane oxidation than the Pt/Al and Pt/AlCe samples (cf.Fig. 2 and 3). The superior activity of Pt supported on ceria compared to alumina has been reported in previous studies.15,35 For high S-values at 400 °C under steady-state conditions (cf.Fig. 2) and during the dynamic conditions used here (Fig. 3), the Pt/AlCe sample seems to be slightly more active than the Pt/Al counterpart. This suggests that inclusion of ceria in alumina based support materials may be used to enhance the activity for methane oxidation, at least in applications with unsteady gas compositions. In the remaining discussion, we will first comment on the observed hysteresis before we analyze in detail the influence of unsteady feed gas conditions and role of the support material in the oxidation of methane.Hysteresis phenomena are common in many catalytic systems. Generally, the mechanism behind hysteresis behavior depends on the system at hand and the governing parameter, e.g., reactant pressure and temperature. A well-known example is the bistable kinetics of CO oxidation on platinum observed when, e.g., increasing and decreasing the relative reactant ratio β = pCO/(pCO + pO2). The kinetic bistability is in this case caused primarily by the different sticking probabilities of CO and oxygen on the Pt surface in combination with a high reaction rate.36 However, the literature on bistable kinetics in methane oxidation on platinum is rarer. Related to hysteresis, step-wise changes in methane conversion as a function of oxygen pressure have been observed previously.7,11,37 Burch et al. observed a hysteresis in methane conversion between heating and cooling in temperature programmed reaction (TPReaction) experiments in both stoichiometric and oxygen deficit gas mixtures. The hysteresis was attributed to a favorable adsorbate composition that once established upon heating the catalyst can be maintained during cooling.5 In the present study, the hysteresis is observed when changing the S-value at (nearly) isothermal conditions (cf.Fig. 2). The maximum measured catalyst temperature rise was around 10 °C and the adiabatic temperature rise is calculated to be around 34 °C for the actual methane conversion. Although still not straightforward, this type of experiment may be easier to interpret as primarily only oxygen adsorption is impacted (of course this has a corresponding impact on coupled processes) as compared to the TPReaction experiment where the rate of all temperature dependent surface processes changes exponentially. By employing kinetic Monte Carlo simulations of methane oxidation on platinum, Zhdanov et al.10 predicted a similar hysteresis behavior as observed here for the alumina samples at 350 and 400 °C (cf.Fig. 2). The model shows a sharp transition from a state with low activity to a highly active state, which was attributed to a shift from a surface entirely covered by platinum oxide to a surface covered by chemisorbed oxygen. Validation of these predictions would require in situ structural characterization techniques; however, the results seem plausible, especially when considering the results of previous in situ ED-XANES spectroscopy studies on methane oxidation over Pt/alumina during transient oxygen supply, where it was shown that oxide formation on platinum drastically lowers the activity for methane oxidation.14,38 The idea of platinum oxidation/reduction as the mechanism behind the hysteresis may also explain the more complicated behavior at 500 °C, for which no clear hysteresis is observed, as at this high temperature platinum oxides are less stable.39 Furthermore, no clear hysteresis is observed for the Pt/Ce sample. This may be due to the stronger interaction between platinum and ceria imposing an oxidized character of the generally smaller Pt particles that cannot be reduced by methane. The generally high activity for the Pt/Ce sample is likely due to other effects originating from the strong interaction between platinum and ceria, which will be discussed below.
The hysteresis phenomena suggest that the methane oxidation reaction can be enhanced by applying periodic operation. The results presented in Fig. 3 clearly show that this is the case for the Pt/Al and Pt/AlCe samples. For these samples, a temporary high methane conversion is achieved both when the oxygen feed is switched on and off. This is in accordance with previous studies on methane oxidation over Pt/alumina.14,38 At the introduction of oxygen, the temporary high methane conversion is similar for the two samples although the Pt/AlCe sample is generally slightly more active. However, when oxygen is switched off the temporary high methane conversion for Pt/Al is mainly due to dissociation of methane as only minor amounts of CO2 are formed. Contrary, for the Pt/AlCe sample, a clear production of CO2 is seen for this period. This suggests that the dynamic oxygen storage capacity of the Pt/AlCe system is not sufficiently high to prevent the oxygen poisoning of the platinum upon introduction of the excess oxygen conditions but is neither negligible since CO2 is formed after the oxygen is switched off. For the Pt/Ce system, no clear benefit of dynamic operation can be seen in the present experiments as the sample is highly active at the studied temperature. For lower temperatures, however, it is likely that dynamic operation can be used to enhance the activity for methane oxidation also for this system. Furthermore, during dynamic operation conditions, the oxidation of methane alternates between total and partial oxidation. This is most clear for the ceria containing samples. For stoichiometric partial oxidation, the expected concentration of hydrogen should be twice that of carbon monoxide. Interestingly, for the Pt/Ce catalyst the formed hydrogen largely exceed the stoichiometric formation of CO. Both water-gas shift (WGS) and methane steam reforming reactions may occur during rich phases and these reactions can be can be further enhanced by ceria.40,41 However, the low formation of CO2 in the present experiments seems not to fully support a significant hydrogen formation due to the WGS reaction. Concerning the Pt/Al sample, Becker et al.14,15 studied in similar experiments the white line area of the time-resolved ED-XANES spectra as a measure of the surface O/Pt ratio during the rich phase. It turned out that the O/Pt ratio was fairly constant, which implies that platinum oxide species are likely not the main source for oxygen during rich phases. Instead, the oxygen source for partial methane oxidation during the rich phases has been proposed to be either water formed during the previous lean phases or surface carbonates/hydroxyl species.14
To better understand the surface processes occurring in the different systems during dynamic operation, we turn to the in situ DRIFT spectroscopy experiments. Before continuing the discussion, we first comment on the actual temperature in the Harrick reaction cell. It has been shown that the set-point temperature and the actual temperature in the IR sampling volume of the bed may differ significantly, especially at high temperatures.42 However, in the present study, this is not critical since we do not use the IR data for direct comparison with kinetic data as measured in the flow-reactor at 400 °C. Rather the DRIFT spectroscopy experiments are qualitative and used for discussion of occurring surface processes. The general behavior in these experiments corresponds well with the flow-reactor results discussed above. During dynamic operation, temporary high activity is seen at the switches for the alumina containing samples (cf. lower panels in Fig. 4), and during transition from oxygen excess to oxygen-free conditions, the Pt/AlCe catalyst forms more CO2 than the Pt/Al sample. Also, during this transition, the coverage of CO adsorbed on the platinum particles increases on all samples reflecting the change from total oxidation to partial oxidation of methane. In the case of Pt/Ce, several absorption bands related to carbonates on ceria develop as well. Following the increase of linearly bonded CO on Pt at around 2050 cm−1, an initial red-shift is observed (cf.Fig. 5) after which the band broadens towards lower wavenumbers. Furthermore, it is apparent that this broadening is more delayed with increasing ceria content of the support material. Simultaneously, an increase in bridge-bonded CO on Pt at around 1850 cm−1 can be observed, at least for the Pt/Al sample. Upon introduction of oxygen, the CO adsorbed on platinum vanishes abruptly for the Pt/Al and Pt/AlCe samples due to the fast CO oxidation reaction. However, for the Pt/Ce catalyst the disappearance of adsorbed CO is significantly slower and seems to be related to the disappearance of the carbonates. This may suggest that carbonate species formed on bare ceria in the vicinity of the platinum particles transform into carbonyls on platinum, for example on Pt sites in close contact with ceria, which then reacts with oxygen. Such phenomena have been discussed previously for the same type of catalyst during the reversed WGS reaction.43 Also, upon introduction of oxygen, carbonates on ceria may be destabilized resulting in desorption of CO2.44 Both these effects can explain the more pronounced overshoot in CO2 formation in the dynamic experiment displayed in Fig. 3. In the remaining discussion, we focus on the evolution of the carbonyl bands and how these may reflect the different processes occurring on the catalysts during periodic operation.
Adsorption of CO on platinum oxide results in an IR absorption band at around 2110 cm−1 (ref. 45–47), while adsorption on metallic platinum gives rise to IR absorption bands at lower wavenumbers. During CO oxidation over a Pt/alumina catalyst under dynamic oxygen feed,39 it has been shown that in the high active state the platinum particles are partially oxidised. Upon switching off the oxygen supply, CO starts to accumulate on the platinum particles, which become reduced. During this process, a red-shift of the linearly adsorbed CO on platinum is initially observed, and as the CO saturation coverage is approached on the metallic platinum, a blue-shift is observed. Considering carefully the results presented in Fig. 5, it is clear that the linearly bonded CO on platinum for the alumina containing catalysts follows this behavior, i.e., first undergoes a red-shift followed by a blue-shift although not so well pronounced. The magnitude of the red-shift is about 5 and 10 cm−1 for the Pt/Al and Pt/AlCe samples, respectively. It is clearer in the CO adsorption experiment in Fig. 6 as CO is included in the feed in this experiment. In this case, the red-shift is 10 and 20 cm−1 for the Pt/Al and Pt/AlCe samples, respectively. The red-shifts presented here are somewhat lower as compared to the previous study,39 which likely are due to the significantly lower oxygen concentration used here. Despite this, the trends in the evolution of the linearly adsorbed CO on Pt support that low methane conversion over the Pt/Al and Pt/AlCe catalysts in the presence of excess oxygen is due to a low dissociation rate of methane on oxidised platinum particles. Interestingly, for the Pt/Ce sample, the IR band for the linearly adsorbed CO during the oxygen-free period in the dynamic experiment shows a blue-shift followed by a red-shift, while in the CO adsorption experiment only a blue-shift is seen. The absence of a red-shift may reflect that the chemical state of the Pt in the platinum particles is not significantly changed during the adsorption of CO or that carbon monoxide adsorbs on sites that do not change considerably. The blue-shift may be due to a gradual increase in the population of platinum sites in close contact with the support, i.e., slightly oxidised platinum sites.
The initial peak positions for the COlin(Pt) in the CO exposure experiments are 2040, 2060 and 2023 cm−1 for Pt/Al, Pt/AlCe and Pt/Ce, respectively (cf.Fig. 6). During CO saturation, the COlin(Pt) broadens and shoulders develop. The different peak positions and shoulders suggest that different types of CO adsorption sites present on the different catalysts become populated. The fundamental reasons may be several, e.g., size and morphology of the platinum particles and interactions between the platinum and the respective support. Thus, we cannot unambiguously deconvolve the COlin(Pt) band for precise assignments. However, some general comments can be made. IR absorption in the region 2060–090 is often considered to correspond to CO adsorbed on extended surfaces (facets),48 while absorption in lower wavenumber regions has been assigned to CO adsorbed on under-coordinated sites (edges and corners)48,49 or sites in the platinum–support interface.50 Our results agree with the idea that with increasing ceria content the interaction between platinum and the support becomes stronger which leads to smaller Pt particles where the abundance of under-coordinated sites is expected to be higher. These sites may be of particular importance for the methane oxidation.
5 Concluding remarks
The present study shows the importance of the support material in the total oxidation of methane over supported platinum catalysts. The results show that platinum has a sufficiently strong interaction with alumina, alumina–ceria and ceria to form nanoparticles of the platinum particles around 2 nm in diameter or below. The smallest Pt particles are observed for the Pt/Ce sample. For the alumina-containing samples, in contrast to the Pt/Ce sample, the oxidation of methane decreases with increasing oxygen concentration in the feed. Furthermore, for the former samples, a clear kinetic bistability between increasing and decreasing oxygen concentration of the feed is observed. This bistability is connected with oxidation and reduction of platinum proceeding for different feed stoichiometry depending on the history of the catalyst system. The oxidation of platinum results in suppressed reaction rate for methane oxidation by hindering the dissociative adsorption of methane. For the alumina-containing samples, the negative impact of oxygen can be circumvented by use of periodic operation which results in increased average conversion of methane. The Pt/Ce catalyst is considerably less sensitive to oxygen than the alumina-based catalysts. Our results support the presence of under-coordinated sites and boundary sites in platinum–ceria systems that may be of particular importance in the oxidation of methane.Acknowledgements
This work was financially supported by the Foundation for Strategic Research (SSF) and the Competence Centre for Catalysis, which is financially supported by Chalmers University of Technology, the Swedish Energy Agency and the member companies: AB Volvo, ECAPS AB, Haldor Topsøe A/S, Volvo Car Corporation, Scania CV AB, and Wärtsilä Finland Oy.References
- European Commission Directorate-General for Energy and Transport (DG TREN), http://ec.europa.eu/energy, 2010.
- P. Gèlin and M. Primet, Appl. Catal., B, 2002, 39, 1–37 CrossRef.
- D. L. Mowery and R. L. McCormick, Appl. Catal., B, 2001, 34, 287–297 CrossRef CAS.
- P. Forster, V. Ramaswamy, P. Artaxo, T. Berntsen, R. Betts, D. W. Fahey, J. Haywood, J. Lean, D. C. Lowe, G. Myhre, J. Nganga, R. Prinn, G. Raga, M. Schulz and R. V. Dorland, The Physical Science Basis. Contribution of Working Group I to the Fourth Assessment Report of the Intergovernmental Panel on Climate Change, Cambridge University Press, Cambridge, UK and New York, NY, USA, 2007, ch. 2, p. 212 Search PubMed.
- R. Burch and P. K. Loader, Appl. Catal., B, 1994, 5, 149–164 CrossRef CAS.
- R. Burch and M. J. Hayes, J. Mol. Catal. A: Chem., 1995, 100, 13–33 CrossRef CAS.
- R. Burch, P. K. Loader and F. J. Urbano, Catal. Today, 1996, 27, 243–248 CrossRef CAS.
- P. Gélin, L. Urfels, M. Primet and E. Tena, Catal. Today, 2003, 83, 45–57 CrossRef.
- J. K. Lampert, M. S. Kazi and R. J. Farrauto, Appl. Catal., B, 1997, 14, 211–223 CrossRef CAS.
- V. P. Zhdanov, P.-A. Carlsson and B. Kasemo, J. Chem. Phys., 2007, 126, 234705 CrossRef CAS PubMed.
- S. H. Oh, P. J. Mitchell and R. M. Siewert, J. Catal., 1991, 132, 287–301 CrossRef CAS.
- P.-A. Carlsson, E. Fridell and M. Skoglundh, Catal. Lett., 2007, 115, 1–7 CrossRef CAS PubMed.
- P.-A. Carlsson, M. Nordström and M. Skoglundh, Top. Catal., 2009, 52, 1962–1966 CrossRef CAS.
- E. Becker, P.-A. Carlsson, H. Grönbeck and M. Skoglundh, J. Catal., 2007, 252, 11–17 CrossRef CAS PubMed.
- E. Becker, P.-A. Carlsson and M. Skoglundh, Top. Catal., 2009, 52, 1957–1961 CrossRef CAS.
- D. Bounechada, S. Fouladvand, L. Kylhammar, T. Pingel, E. Olsson, M. Skoglundh, J. Gustafson, M. D. Michiel, M. A. Newton and P.-A. Carlsson, Phys. Chem. Chem. Phys., 2013, 15, 8648–8661 RSC.
- L. Kylhammar, P.-A. Carlsson and M. Skoglundh, J. Catal., 2011, 284, 50–59 CrossRef CAS PubMed.
- R. M. Heck, R. J. Farauto and S. T. Gulati, Catalytic Air Pollution Control: Commercial Technology, John Wiley & Sons, 2009 Search PubMed.
- S. Fouladvand, S. Schernich, J. Libuda, H. Grönbeck, T. Pingel, E. Olsson, M. Skoglundh and P.-A. Carlsson, Top. Catal., 2012, 56, 410–415 CrossRef.
- L. Kylhammar, P.-A. Carlsson and M. Skoglundh, J. Catal., 2011, 284, 50–59 CrossRef CAS PubMed.
- A. Trovarelli, Catal. Rev.: Sci. Eng., 1996, 38, 439–520 CAS.
- R. Ramírez-López, I. Elizalde-Martinez and L. Balderas-Tapia, Catal. Today, 2010, 150, 358–362 CrossRef PubMed.
- C. Wang-Hansen, C. J. Kamp, M. Skoglundh, B. Andersson and P.-A. Carlsson, J. Phys. Chem. C, 2011, 115, 16098–16108 CAS.
- S. Guerrero, J. T. Miller, A. J. Kropf and E. E. Wolf, J. Catal., 2009, 262, 102–110 CrossRef CAS PubMed.
- P. Panagiotopoulou, J. Papavasiliou, G. Avgouropoulos, T. Ioannides and D. I. Kondarides, Chem. Eng. J., 2007, 134, 16–22 CrossRef CAS PubMed.
- A. Yee, S. J. Morrisson and H. Idriss, J. Catal., 2000, 191, 30–45 CrossRef CAS.
- G. Jacobs, L. Williams, U. Graham, G. A. Thomas, D. E. Sparks and B. H. Davis, Appl. Catal., A, 2003, 252, 107–118 CrossRef CAS.
- K.-R. Hwang, S.-K. Ihm and J.-S. Park, Fuel Process. Technol., 2010, 91, 729–736 CrossRef CAS PubMed.
- C. Li, Y. Sakata, T. Arai, K. Domen, K.-I. Maruya and T. Onishi, J. Chem. Soc., Faraday Trans. 1, 1989, 85, 929–943 RSC.
- F. Bozon-Verduraz and A. Bensalem, J. Chem. Soc., Faraday Trans., 1994, 90, 653–657 RSC.
- J. R. Anderson and K. C. Pratt, Introduction to Characterization and Testing of Catalysts, Academic Press, 1985 Search PubMed.
- J. Kašpar, P. Fornasiero and M. Graziani, Catal. Today, 1999, 50, 285–298 CrossRef.
- P. Meriaudeau, J. Dutel, M. Dufaux and C. Naccache, Metal-Support and Metal-Additive Effects in Catalysis Proceedings of an International Symposium, 1982, pp. 95–104 Search PubMed.
- I. Chorkendorff and H. Niemantsverdriet, Concepts of Modern Catalysis and Kinetics, Wiley-VCH, Weinheim, 2003 Search PubMed.
- P.-A. Carlsson and M. Skoglundh, Appl. Catal., B, 2011, 101, 669–675 CrossRef CAS PubMed.
- P.-A. Carlsson, V. P. Zhdanov and B. Kasemo, Appl. Surf. Sci., 2005, 239, 424–431 CrossRef CAS PubMed.
- J. G. Firth and H. B. Holland, Trans. Faraday Soc., 1969, 65, 1121 RSC.
- E. Becker, P.-A. Carlsson, L. Kylhammar, M. A. Newton and M. Skoglundh, J. Phys. Chem. C, 2011, 115, 944–951 CAS.
- P.-A. Carlsson, L. Österlund, P. Thormählen, A. Palmqvist, E. Fridell, J. Jansson and M. Skoglundh, J. Catal., 2004, 226, 422–434 CrossRef CAS PubMed.
- J. Barbier Jr. and D. Duprez, Appl. Catal., B, 1994, 4, 105–140 CrossRef.
- R. Craciun, B. Shereck and R. J. Gorte, Catal. Lett., 1998, 51, 149–153 CrossRef CAS.
- H. Li, M. Rivallan, F. Thibault-Starzyk, A. Travert and F. Meunier, Phys. Chem. Chem. Phys., 2013, 15, 7321–7327 RSC.
- A. Goguet, F. C. Meunier, D. Tibiletti, J. P. Breen and R. Burch, J. Phys. Chem. B, 2004, 108, 20240–20246 CrossRef CAS.
- F. C. Meunier, D. Tibiletti, A. Goguet, D. Reid and R. Burch, Appl. Catal., B, 2005, 289, 104–112 CrossRef CAS PubMed.
- M. Primet, J. M. Basset, M. V. Mathieu and M. Prettre, J. Catal., 1973, 29, 213 CrossRef CAS.
- M. Primet, J. Catal., 1984, 88, 273–282 CrossRef CAS.
- N. Sheppard and T. Nguyen, Adv. Infrared Raman Spectrosc., 1978, 5, 67 CAS.
- P. Hollins, Surf. Sci. Rep., 1992, 16, 51–94 CrossRef CAS.
- M. J. Kappers and J. H. van der Maas, Catal. Lett., 1991, 10, 365–374 CrossRef CAS.
- A. Yee, S. J. Morrison and H. Idriss, J. Catal., 2000, 191, 30–45 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2014 |