 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Heterogeneous catalysis for sustainable biodiesel production via esterification and transesterification
Adam F.
Lee
*,
James A.
Bennett
,
Jinesh C.
Manayil
and
Karen
Wilson
European Bioenergy Research Institute, Aston University, Aston Triangle, Birmingham B4 7ET, UK. E-mail: a.f.lee@aston.ac.uk; Tel: +44 (0)121 2044036
First published on 24th June 2014
Abstract
Concern over the economics of accessing fossil fuel reserves, and widespread acceptance of the anthropogenic origin of rising CO2 emissions and associated climate change from combusting such carbon sources, is driving academic and commercial research into new routes to sustainable fuels to meet the demands of a rapidly rising global population. Here we discuss catalytic esterification and transesterification solutions to the clean synthesis of biodiesel, the most readily implemented and low cost, alternative source of transportation fuels to meet future societal demands.
 Adam F. Lee | Adam Lee is Professor of Sustainable Chemistry and an EPSRC Leadership Fellow in the European Bioenergy Research Institute, Aston University. He holds a BA (Natural Sciences) and PhD from the University of Cambridge, and following postdoctoral research at Cambridge and Lecturer/Senior Lecturer roles at the Universities of Hull and York respectively, held Chair appointments at Cardiff, Warwick and Monash universities. His research addresses the rational design of nanoengineered materials for clean catalytic technologies, with particular focus on sustainable chemical processes and energy production, and the development of in situ methods to provide molecular insight into surface reactions, for which he was awarded the 2012 Beilby Medal and Prize by the Royal Society of Chemistry. |
 James Bennett | Dr James Andrew Bennett obtained his Master and PhD at the University of Leicester, where he investigated the use of perfluoroalkyl moieties to allow heterogenisation of homogeneous catalysts over zirconium phosphonate supports. He then worked at the University of Birmingham, researching biogenic heterogeneous catalysts composed of transition metal nanoparticles supported on bacterial biomass, using waste sources of metals and biomass to produce "green" catalyst materials. He is currently working with Professors Karen Wilson and Adam Lee at the European Bioenergy Research Institute at Aston University, developing environmentally sustainable catalysts derived from industrial waste for pyrolysis oil upgrading. |
 Jinesh C. Manayil | Dr Jinesh Manayil obtained his MSc in Chemistry from Mahatma Gandhi University in 2004, prior to a MTech in Industrial Catalysis from Cochin University of Science and Technology in 2007. He subsequently undertook postgraduate research in catalytic and ion-exchange applications of layered double hydroxides, receiving his PhD in 2012 from the Central Salt and Marine Chemicals Research Institute (CSIR-CSMCRI), India under the supervision of Dr Kannan Srinivasan. He is currently a Research Associate with Professors Karen Wilson and Adam Lee at the European Bioenergy Research Institute at Aston University, where he is developing solid acid-base catalysts for biomass mass conversion. |
 Karen Wilson | Karen Wilson is Professor of Catalysis and Research Director of the European Bioenergy Research Institute at Aston University, where she holds a Royal Society Industry Fellowship. Her research interests lie in the design of heterogeneous catalysts for clean chemical synthesis, particularly the design of tunable porous materials for sustainable biofuels and chemicals production from renewable resources. She was educated at the Universities of Cambridge and Liverpool, and following postdoctoral research at Cambridge and the University of York, was appointed a Lecturer and subsequently Senior Lecturer at York, prior to appointment as a Reader in Physical Chemistry at Cardiff University. |
1. Introduction
Sustainability, in essence the development of methodologies to meet the needs of the present without compromising those of future generations, has become a watchword for modern society, with developed and developing nations and multinational corporations promoting international research programmes into sustainable food, energy, materials, and even city planning. In the context of energy, despite significant growth in proven and predicted fossil fuel reserves over the next two decades, notably heavy crude oil, tar sands, deepwater wells, and shale oil and gas, there are great uncertainties in the economics of their exploitation via current extraction methodologies, and crucially, an increasing proportion of such carbon resources (estimates vary between 65–80%1–3) cannot be burned without breaching the UNFCC targets for a 2 °C increase in mean global temperature relative to the pre-industrial level.4,5 There is clearly a tightrope to walk between meeting rising energy demands, predicted to climb 50% globally by 20406 and the requirement to mitigate current CO2 emissions and hence climate change. Similar considerations apply to ensuring a continued supply of organic materials for applications including polymers, plastics, pharmaceuticals, optoelectronics and pesticides, which underpin modern society, and for which significant future growth is anticipated, tracking the predicted four-fold rise in global GDP and associated requirements for advanced consumer products by 2050.7 The quest for sustainable resources to meet the demands of a rapidly rising world population represents one of this century's grand challenges.8,9 Heterogeneous catalysis has a rich history of facilitating energy efficient selective molecular transformations and contributes to 90% of chemical manufacturing processes and to more than 20% of all industrial products.10,11 In a post-petroleum era, catalysis will be central to overcoming the engineering and scientific barriers to economically feasible routes to alternative source of both energy and chemicals, notably bio-derived and solar-mediated via artificial photosynthesis (Scheme 1).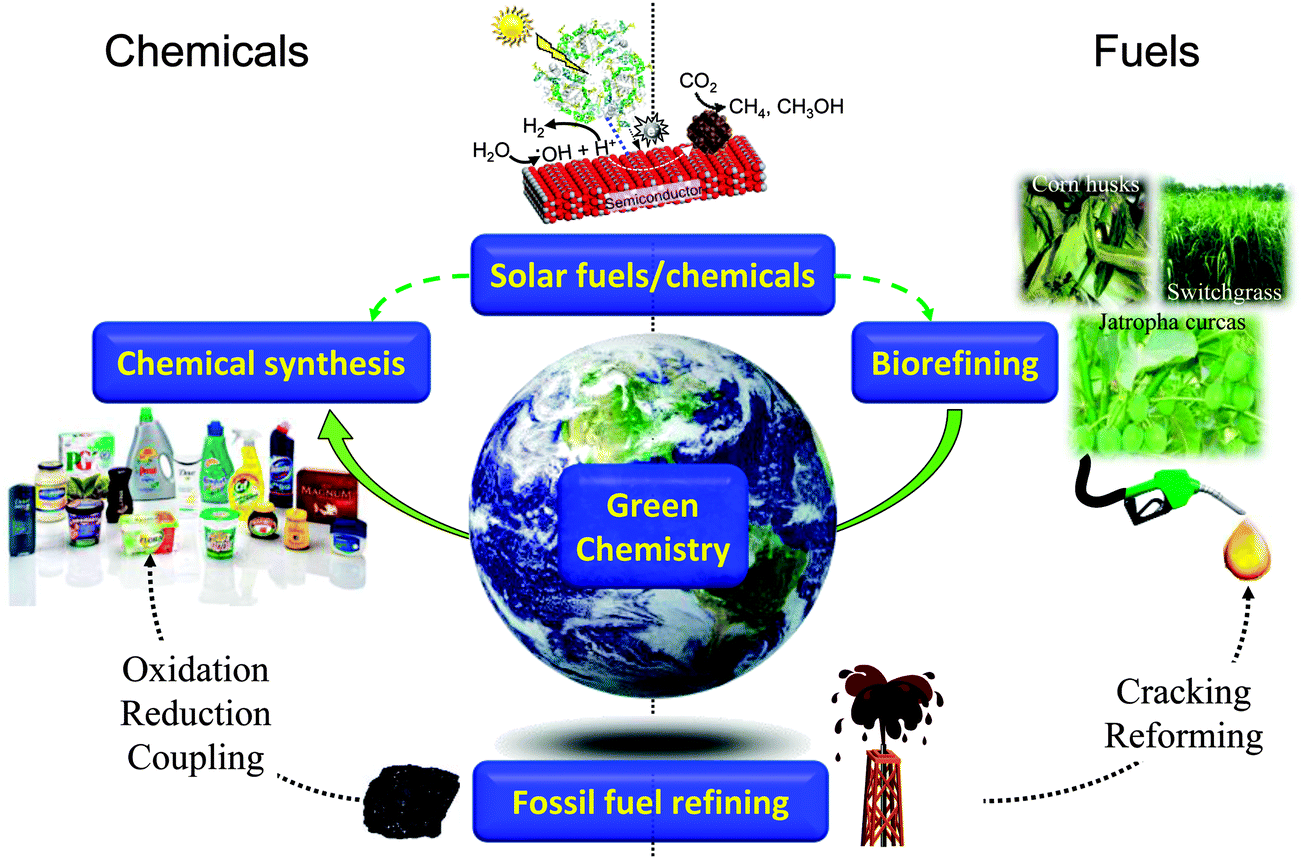 | ||
| Scheme 1 Current and future roles for heterogeneous catalysis in the production of sustainable chemicals and fuels. | ||
While many alternative sources of renewable energy have the potential to meet future demands for stationary power generation, biomass offers the most readily implemented, low cost solution to a drop-in transportation fuel for blending with/replacing conventional diesel12via the biorefinery concept, illustrated for carbohydrate pyrolysis/hydrodeoxygenation (HDO)13,14 or lipid transesterification15,16 to alkanes and biodiesel respectively in Scheme 2. First-generation bio-fuels derived from edible plant materials received much criticism over the attendant competition between land usage for fuel crops versus traditional agricultural cultivation.17 Deforestation practices, notably in Indonesia, wherein vast tracts of rainforest and peat land have been cleared to support palm oil plantations, have also provoked controversy.18 To be considered sustainable, second generation bio-based fuels and chemicals are sought that use biomass sourced from non-edible components of crops, such as stems, leaves and husks or cellulose from agricultural or forestry waste. Alternative non-food crops such as switchgrass or Jatropha curcas,19 which require minimal cultivation and do not compete with traditional arable land or drive deforestation, are other potential candidate biofuel feedstocks. There is also growing interest in extracting bio-oils from aquatic biomass, which can yield 80–180 times the annual volume of oil per hectare than that obtained from plants.20 Around 9% of transportation energy needs are predicted to be met via liquid biofuels by 2030.21
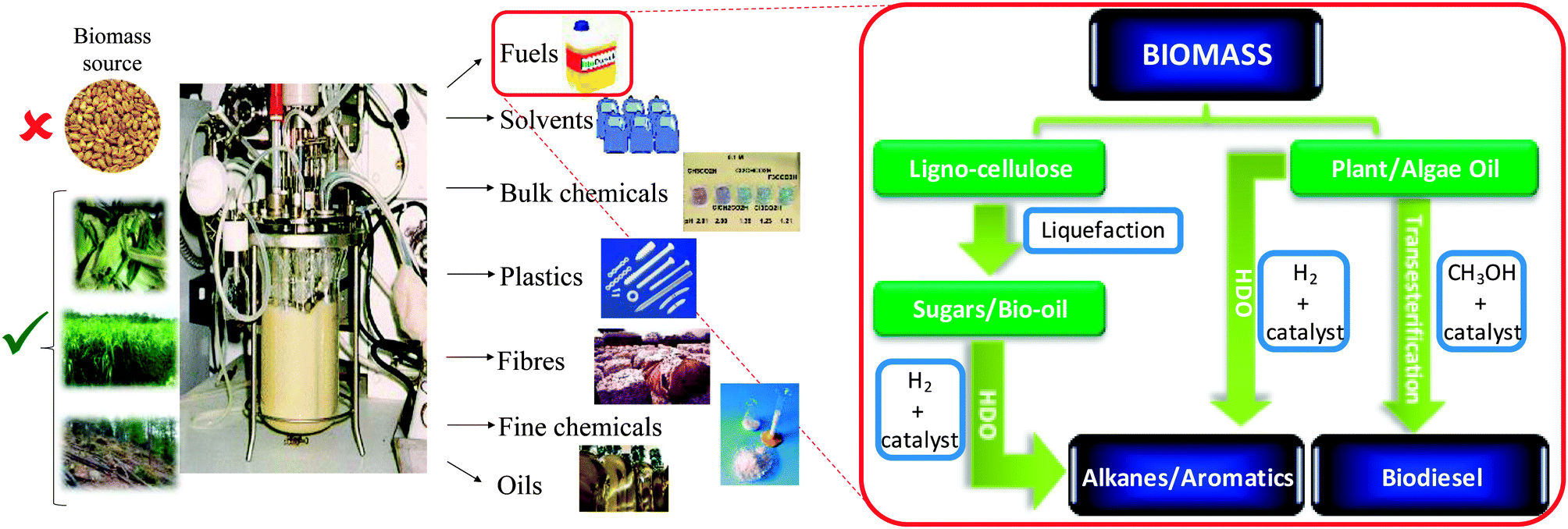 | ||
| Scheme 2 Biorefinery routes for the co-production of chemicals and transportation fuels from biomass. | ||
Biodiesel is a clean burning and biodegradable fuel which, when derived from non-food plant or algal oils or animal fats, is viewed as a viable alternative (or additive) to current petroleum-derived diesel.22 Commercial biodiesel is currently synthesised via liquid base catalysed transesterification of C14–C20 triacylglyceride (TAG) components of lipids with C1–C2 alcohols23–26 into fatty acid methyl esters (FAMEs) which constitute biodiesel as shown in Scheme 3, alongside glycerol as a potentially valuable by-product.27 While the use of higher (e.g. C4) alcohols is also possible,28 and advantageous in respect of producing a less polar and corrosive FAME29 with reduced cloud and pour points,30 the current high cost of longer chain alcohols, and difficulties associated with separating the heavier FAME product from unreacted alcohol and glycerol, remain problematic. Unfortunately, homogeneous acid and base catalysts can corrode reactors and engine manifolds, and their removal from the resulting biofuel is particularly problematic and energy intensive, requiring aqueous quench and neutralisation steps which result in the formation of stable emulsions and soaps.12,31,32 Such homogeneous approaches also yield the glycerine by-product, of significant potential value to the pharmaceutical and cosmetic industries, in a dilute aqueous phase contaminated by inorganic salts. The utility of solid base and acid catalysts for biodiesel production has been widely reported,15,25,33–41 wherein they offer improved process efficiency by eliminating the need for quenching steps, allowing continuous operation,42 and enhancing the purity of the glycerol by-product. Technical advances in catalyst and reactor design remain essential to utilise non-food based feedstocks, and thereby ensure that biodiesel remains a key player in the renewable energy sector for the 21st century. In this review, we highlight the contributions of tailored solid acid and base catalysts to catalytic biodiesel synthesis via TAG transesterification to FAMEs and free fatty acid (FFA) esterification.
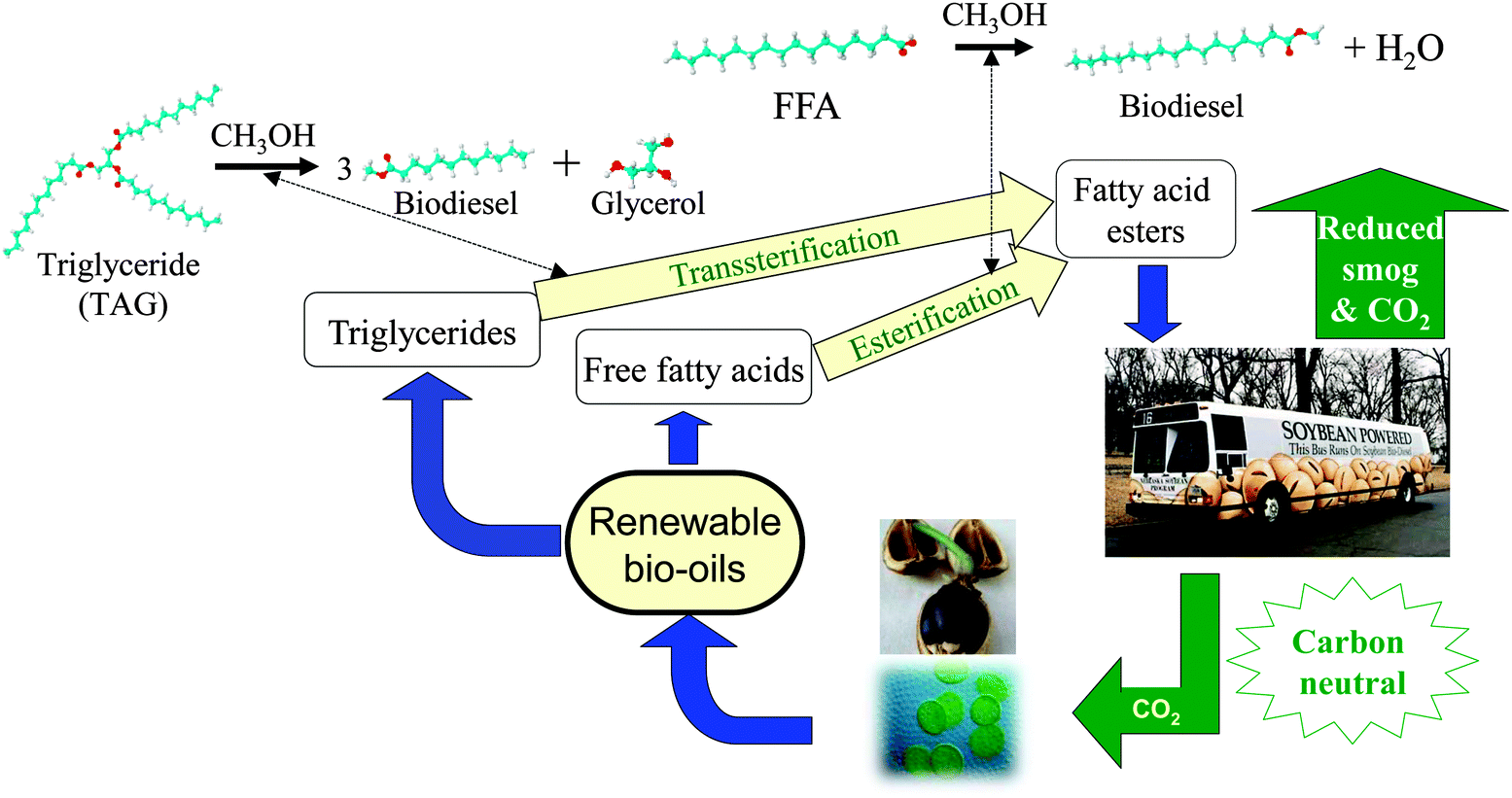 | ||
| Scheme 3 Biodiesel production cycle from renewable bio-oils via catalytic transesterification and esterification. | ||
2. Feedstocks for biodiesel
The feedstock sources employed for biodiesel synthesis have remained little changed since the first engine tests with vegetable oils in the late 1800s,43 and are normally classified as either first or second generation,44,45 the latter oft referred to as a source of ‘advanced biofuels’. First generation biodiesel is derived from edible vegetable oils such as soya, palm,46 oil seed rape47 and sunflower,48 however the attendant poor yields (typically 3000–5000 L hectare−1 year−1) and socio-political concern over the diversion of such food crops for fuels has led to their fall from favour within Europe and North America. Second generation biodiesel is normally considered to be that obtained from non-edible oils such as castor,49 Jatropha50 and neem,51 microalgae,44,52 animal fats (e.g. tallow and yellow grease),53 or waste oils including organic components of municipal waste:54 these offer lower greenhouse gas emissions,45e.g. 150 gCO2 MJ−1 for African biodiesel from Jatropha exported to the EU with attendant use of residual seedcake as a fertiliser versus 220 gCO2 MJ−1 for Mexico biodiesel from Jatropha without attendant methane capture;55 improved environmental and energy life cycles;56 and superior biodiesel yields (upto 100![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 L hectare−1 year−1 for microalgae). Commercial biodiesel is require to meet a range of national and international standards, the most widely conformed to being the American standard ASTM D6751,57 and the European standard EN 14214:58 the high free fatty acid of some non-edible oils can lower the FAME content below accepted standards,59 whereas feedstocks like Brassica carinata and Jatropha curcas have comparable or even higher oil content than many edible oils.15
000 L hectare−1 year−1 for microalgae). Commercial biodiesel is require to meet a range of national and international standards, the most widely conformed to being the American standard ASTM D6751,57 and the European standard EN 14214:58 the high free fatty acid of some non-edible oils can lower the FAME content below accepted standards,59 whereas feedstocks like Brassica carinata and Jatropha curcas have comparable or even higher oil content than many edible oils.15
Interest in biodiesel production soared following the global oil crisis of the 1970s, resulting in the United States, European Union, Brazil, China, India, and South Africa convening a UN International Biodiesel Forum for biodiesel development. Today, the United States, European Union and Brazil, alongside Malaysia, remain leading forces in the biodiesel market. Current industrial production is dominated by the utilisation of edible vegetable oils such as soybean (7.08 million), palm (6.34 million), rapeseed (6.01 million), castor, coconut and Jatropha curcas oil. The primary cost of biodiesel lies in the raw material, and since the market is dominated by food grade oils,59 which are significantly more expensive than petroleum-derived diesel, economic viability remains to be proven. Use of the surplus from edible oil production may assist countries to meet the demands for biodiesel production without negatively impacting upon food requirements.60 Feedstock selection is a strong function of local availability. Soybean oil, which is widely used in the United States and South America, is the third largest feedstock for biodiesel after rapeseed oil in Europe and palm oil in Asian countries, such as Malaysia and Indonesia, which also use sunflower and coconut oil, with Jatropha curcas oil widespread across South East Asia.61 Soybean and rapeseed oils account for about 85% of global biodiesel production,62 with 75% of total biodiesel produced in Europe. Competition for land to produce biodiesel feedstocks is problematic, hence maximising the yield of oil from a given feedstock is critical. Edible soybean seed consists of 20% oil versus rapeseed at 40%, whereas non-edible Jatropha and Karanja seeds contain around 40% and 33% oil respectively.60 Adoption of soybean (as in the US) as a global biodiesel feedstock would be problematic, not only due to competition for its use as a food crop, but also the high quantities of waste, associated with its low oil yield, although this could be mitigated by the introduction of the oil seed cake as a major animal feed. The oil yield from non-edible Jatropha is particularly noteworthy since it can grow in poor quality soil and waste land, avoiding competition with arable land for food crops, however harvesting of the toxic seeds is labour intensive.63 Around 15 million tons of waste cooking/frying oils is disposed of annually worldwide. Such low cost feedstocks, could meet a significant portion of current biodiesel demands, however chemical changes occurring during cooking which increase their FFA and moisture content must be taken into consideration.64 Recent studies suggest that the production cost of biodiesel could be halved through waste cooking oils in comparison with virgin oils.65 However because of its high melting point and viscosity, and less predictable supply, waste cooking oil has been less extensively investigated than vegetable oils.31 Algal biomass has received considerable recent attention, since lipids from algae can be used for biodiesel production via conventional transesterification technologies. Microalgae are fast-growing and produce higher oil yields than plant counterparts. The high oil content of different microalgae favours their commercialisation as a promising feedstock: one acre of microalgae can produce 5000 gallons of biodiesel annually compared to only 70 gallons from an equivalent area of soybean,52 and algae can flourish on land unusable for plant cultivation and without fresh water. Algal oil yields vary with the species, nutrient supply and harvest time,66 however the properties of the resulting FAMEs are not superior to those derived from plant oils, and further research into algal oils rich in saturated long chain fatty acids is required in order to improve the quality of the final biodiesel.67
The choice of oil feedstock in turn influences the biodiesel composition and hence fuel properties,43,68 notably acid value, oxidation stability, cloud point, cetane number and cold filter plugging point. Oils from plants usually comprise five major fatty acids components: palmitic (16:0); stearic (18:0); oleic (18:1); linoleic (18:2); and linolenic (18:3). Table 1 illustrates their distribution and associated physicochemical properties for some common feedstocks. High FFA oils not only compromise base catalysed transesterification and hence biodiesel yields, but can corrode engines and ancillary machinery; the acceptable acid range is between 0.5–3%.60 The cetane number (CN), a measure of diesel ignition quality, is higher for biodiesel (46–52) than that of conventional diesel (40–55), with the international standard specified in ASTM D6751 and EN 14214 at 47 and 51 respectively. Cetane number varies with the degree of oil unsaturation and chain length. Esters of palmitic and stearic acid possess CNs higher than 80, while that of oleate is 55–58, with CN generally decreasing with increasing unsaturation (e.g. CN = 40 for linoleic and 25 for linolenic acid), falling to 48-5 for soybean- and 52–55 for rapeseed-derived biodiesel.69 Fatty acid chain composition also influences NOx emissions, with biodiesel containing esters of saturated fatty acids emitting less NOx than petroleum diesel, and emissions increasing with the degree of unsaturation but decreasing with fatty acid chain length. NOx emissions of hydrogenated FAMEs derived from soybean oil is lower than from conventional diesel.70
| Feedstock | Composition/wt% fatty acid | Density/g cm3 | Flash point/°C | Acid value mg KOH g−1 | Heating value/MJ kg−1 | |
|---|---|---|---|---|---|---|
| Edible oils | Soybean | C16:0, C18:1, C18:2 | 0.91 | 254 | 0.2 | 39.6 |
| Rapeseed | C16:0, C18:0, C18:1, C18:2 | 0.91 | 246 | 2.92 | 39.7 | |
| Sunflower | C16:0, C18:0, C18:1, C18:2 | 0.92 | 274 | — | 39.6 | |
| Palm | C16:0, C18:0, C18:1, C18:2 | 0.92 | 267 | 0.1 | — | |
| Peanut | C16:0, C18:0, C18:1, C18:2, C20:0,C22:0 | 0.90 | 271 | 3 | 39.8 | |
| Corn | C16:0, C18:0, C18:1, C18:2, C18:3 | 0.91 | 277 | — | 39.5 | |
| Camelina | C16:0, C18:0, C18:1, C18:2, C18:3, C20:0, C20:1, C20:3 | 0.91 | — | 0.76 | 42.2 | |
| Cotton | C16:0, C18:0, C18:1, C18:2, C18:3 | 0.91 | 234 | — | 39.5 | |
| Non-edible oils | Jatropha curcas | C16:0, C16:1, C18:0, C18:1, C18:2 | 0.92 | 225 | 28 | 38.5 |
| Pongamina pinnata | C16:0, C18:0, C18:1, C18:2, C18:3 | 0.91 | 205 | 5.06 | 34 | |
| Palanga | C16:0, C18:0, C18:1, C18:2 | 0.90 | 221 | 44 | 39.25 | |
| Tallow | C14:0, C16:0, C16:1, C17:0, C18:0, C18:1, C18:2 | 0.92 | — | — | 40.05 | |
| Poultry | C16:0, C16:1, C18:0, C18:1, C18:2, C18:3 | 0.90 | — | — | 39.4 | |
| Used cooking oil | Depends on fresh cooking oil | 0.90 | — | 2.5 | — | |
Oxidation stability also depends upon the degree of unsaturation of fatty acid chains within the oil feedstock, since double bonds are prone to oxidation. Biodiesel produced from feedstocks containing linoleic (C18, two C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C double bonds) and linolenic acid (C18, three CC double bonds), with one or two bis-allylic positions, are highly susceptible to oxidation. The relative rates of oxidation for linoleates and linolenates are respectively 41 and 98 times higher than that of the monounsaturated oleate.71 The viscosity of biodiesel also increases with chain length and saturation of fatty acids within the feedstock,72 influencing the fuel lubricity and flow properties. Low viscosity biodiesel can be obtained from low molecular weight triglycerides, however such biodiesel cannot be used directly as a fuel due to its poor cold temperature flow properties. The kinematic viscosities of the two most common biodiesels are 4.0–4.1 mm2 s−1 from soybean oil and 4.4 mm2 s−1 from rapeseed oil. The lubricity of biodiesel increases with chain length, and the presence of double bonds and alcohol groups. Hence, monoglycerides and trace glycerol increase biodiesel lubricity. The high lubricity of biodiesel can be utilised through blending with conventional, low-sulfur diesel to improve overall fuel lubricity.73 Cold point (CP) and pour point (PP) determine the flow properties of biodiesel, and also depend on the fatty acid composition of the feedstock. CP is the temperature at which a fuel begins to solidify, and PP is the temperature at which the fuel can no longer flow. For conventional diesel, CP and PP values are −16 °C and −27 °C respectively. Biodiesel derived from soybean possesses CP and PP values of around 0 °C to −2 °C, while the CP for rapeseed oil-derived biodiesel is −3 °C. These values are very high in comparison to conventional diesel, rendering biodiesel ill-suited for cold countries.70 Other common feedstocks, such as palm oil, jatropha oil, animal fat and waste cooking oil have even higher CP values of around 15 °C. In contrast, biodiesel derived from cuphea oil enriched with saturated, medium-chain C8–C14 fatty acids exhibits improved properties including a lower CP of −9 to −10 °C,74 comparable to conventional diesel. Genetic engineering of the parent plants or microalgae offers a route to optimise the fatty acid composition of feedstock oils to deliver fuels with the desired physicochemical properties.75
C double bonds) and linolenic acid (C18, three CC double bonds), with one or two bis-allylic positions, are highly susceptible to oxidation. The relative rates of oxidation for linoleates and linolenates are respectively 41 and 98 times higher than that of the monounsaturated oleate.71 The viscosity of biodiesel also increases with chain length and saturation of fatty acids within the feedstock,72 influencing the fuel lubricity and flow properties. Low viscosity biodiesel can be obtained from low molecular weight triglycerides, however such biodiesel cannot be used directly as a fuel due to its poor cold temperature flow properties. The kinematic viscosities of the two most common biodiesels are 4.0–4.1 mm2 s−1 from soybean oil and 4.4 mm2 s−1 from rapeseed oil. The lubricity of biodiesel increases with chain length, and the presence of double bonds and alcohol groups. Hence, monoglycerides and trace glycerol increase biodiesel lubricity. The high lubricity of biodiesel can be utilised through blending with conventional, low-sulfur diesel to improve overall fuel lubricity.73 Cold point (CP) and pour point (PP) determine the flow properties of biodiesel, and also depend on the fatty acid composition of the feedstock. CP is the temperature at which a fuel begins to solidify, and PP is the temperature at which the fuel can no longer flow. For conventional diesel, CP and PP values are −16 °C and −27 °C respectively. Biodiesel derived from soybean possesses CP and PP values of around 0 °C to −2 °C, while the CP for rapeseed oil-derived biodiesel is −3 °C. These values are very high in comparison to conventional diesel, rendering biodiesel ill-suited for cold countries.70 Other common feedstocks, such as palm oil, jatropha oil, animal fat and waste cooking oil have even higher CP values of around 15 °C. In contrast, biodiesel derived from cuphea oil enriched with saturated, medium-chain C8–C14 fatty acids exhibits improved properties including a lower CP of −9 to −10 °C,74 comparable to conventional diesel. Genetic engineering of the parent plants or microalgae offers a route to optimise the fatty acid composition of feedstock oils to deliver fuels with the desired physicochemical properties.75
3. Solid base catalysed biodiesel synthesis
Base catalysts are generally more active than acids in transesterification, and hence are particularly suitable for high purity oils with low FFA content. Biodiesel synthesis using a solid base catalyst in continuous flow, packed bed arrangement would facilitate both catalyst separation and co-production of high purity glycerol, thereby reducing production costs and enabling catalyst re-use. Diverse solid base catalysts are known, notably alkali or alkaline earth oxides, supported alkali metals, basic zeolites and clays such as hydrotalcites, and immobilised organic bases.763.1 Alkaline earth oxides
Basicity in alkaline earth oxides is believed to arise from M2+–O2− ion pairs present in different coordination environments.77 The strongest base sites occur at low coordination defect, corner and edge sites, or on high Miller index surfaces. Such classic heterogeneous base catalysts have been extensively tested for TAG transesterification78 and there are numerous reports on commercial and microcrystalline CaO applied to rapeseed, sunflower or vegetable oil transesterification with methanol.79,80 Promising results have been obtained, with 97% oil conversion achieved at 75 °C,80 however concern remains over Ca2+ leaching under reaction conditions and associated homogeneous catalytic contributions,81 a common problem encountered in metal catalysed biodiesel production which hampers commercialisation.82 While Ca and Mg are the more widely used alkaline earth metals in solid base catalysis, strontium oxides have also found application in biodiesel production. Pure strontium oxide possesses the highest base site density of the alkali earth oxides as determined by CO2 temperature programmed desorption (TPD),83 and a comparable base strength to that of BaO (26.5 < H−). Despite the lower surface area of SrO compared to Mg and Ca oxides (19, 14 and 3 m2 g−1 respectively), it showed the highest activity for hempseed oil transesterification, although it is questionable whether such low area/highly soluble materials could ever be commercially viable.Alkali-doped CaO and MgO have also been investigated for TAG transesterification,84–86 with their enhanced basicity attributed to the genesis of O− centres following the replacement of M+ for M2+ and associated charge imbalance and concomitant defect generation. In the case of Li-doped CaO, the electronic structure of surface lithium ions (as probed by XPS) evolves discontinuously as a function of concentration and phase. Maximal activity was observed upon formation of a saturated Li+ monolayer, with the phase to bulk-like LiNO3 at higher loadings suppressing TAG conversion coincident with loss of strong base sites.86 However, leaching of alkali promoters remains problematic.87
It is widely accepted that the catalytic activity of alkaline earth oxide catalysts is very sensitive to their preparation, and corresponding surface morphology and/or defect density. For example, Parvulescu and Richards demonstrated the impact of the different MgO crystal facets upon the transesterification of sunflower oil by comparing nanoparticles88versus (111) terminated nanosheets.89 Chemical titration revealed that both morphologies possess two types of base sites, with the nanosheets exhibiting well-defined, medium-strong basicity consistent with their uniform exposed facets and which confer higher FAME yields during sunflower oil transesterification (albeit scale-up of the nanosheet catalyst synthesis may be costly and non-trivial). Subsequent synthesis, screening and spectroscopic characterisation of a family of size-/shape-controlled MgO nanoparticles prepared via a hydrothermal synthesis, revealed small (<8 nm) particles terminate in high coordination (100) facets, and exhibit both weak polarisability and poor activity in tributyrin transesterification with methanol.90 Calcination drives restructuring and sintering to expose lower coordination stepped (111) and (110) surface planes, which are more polarisable and exhibit much higher transesterification activities under mild conditions. A direct correlation was therefore observed between the surface electronic structure and associated catalytic activity, revealing a pronounced structural preference for (110) and (111) facets (Fig. 1). In situ aberration corrected-transmission electron microscopy and XPS implicates coplanar anion vacancies as the active sites in tributyrin transesterification with the density of surface defects predicting activity.90,91
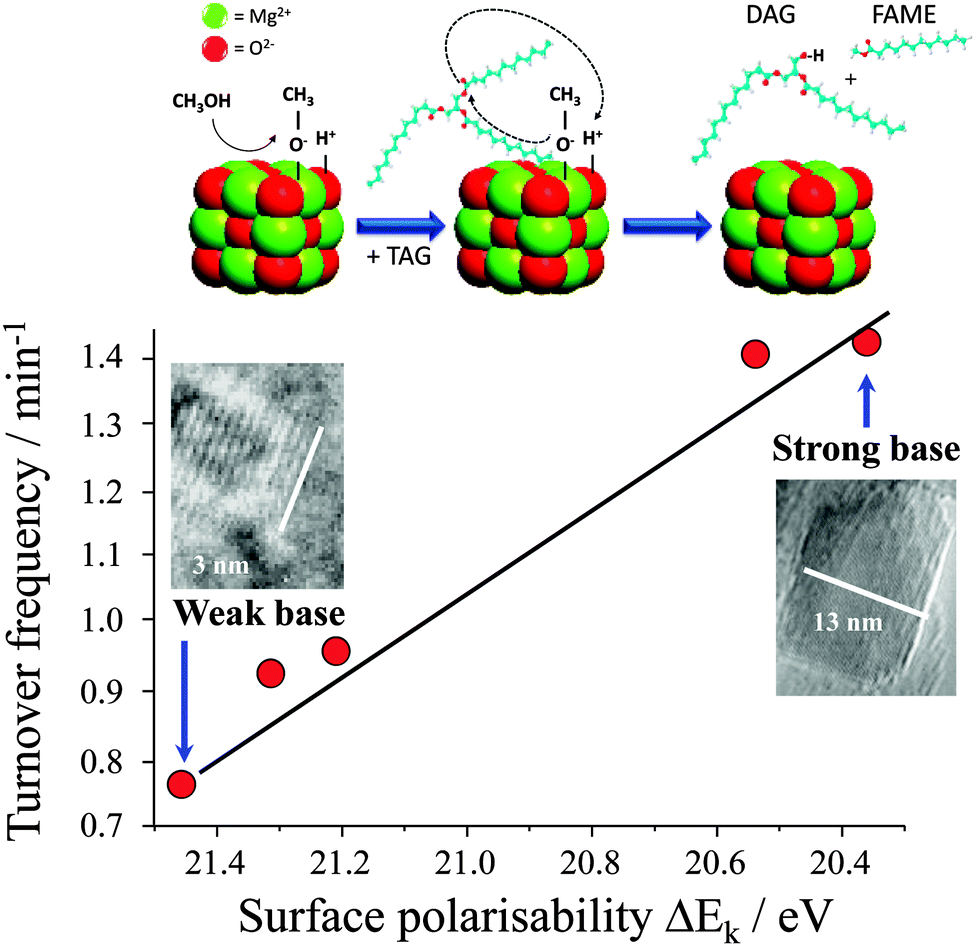 | ||
| Fig. 1 Relationship between surface polarisability of MgO nanocrystals and their turnover frequency towards tributyrin transesterifcation. Adapted from ref. 90 with permission from The Royal Society of Chemistry. | ||
Cesium doping via co-precipitation under supercritical conditions confers even greater activity towards tributyrin transesterification with methanol,85 due to the genesis of additional, and stronger, base sites associated with a new ordered mixed oxide phase which EXAFS analysis recently identified as Cs2Mg(CO3)2(H2O)4,92 resulting in superior performance compared with MgO and even homogeneous Cs2CO3 catalysts (Fig. 2). Unfortunately, surface carbon deposition and loss of this high activity Cs2Mg(CO3)2(H2O)4 phase due to partial Cs dissolution results in on-stream deactivation of Cs-doped MgO, although recalcination could help to regenerate activity.
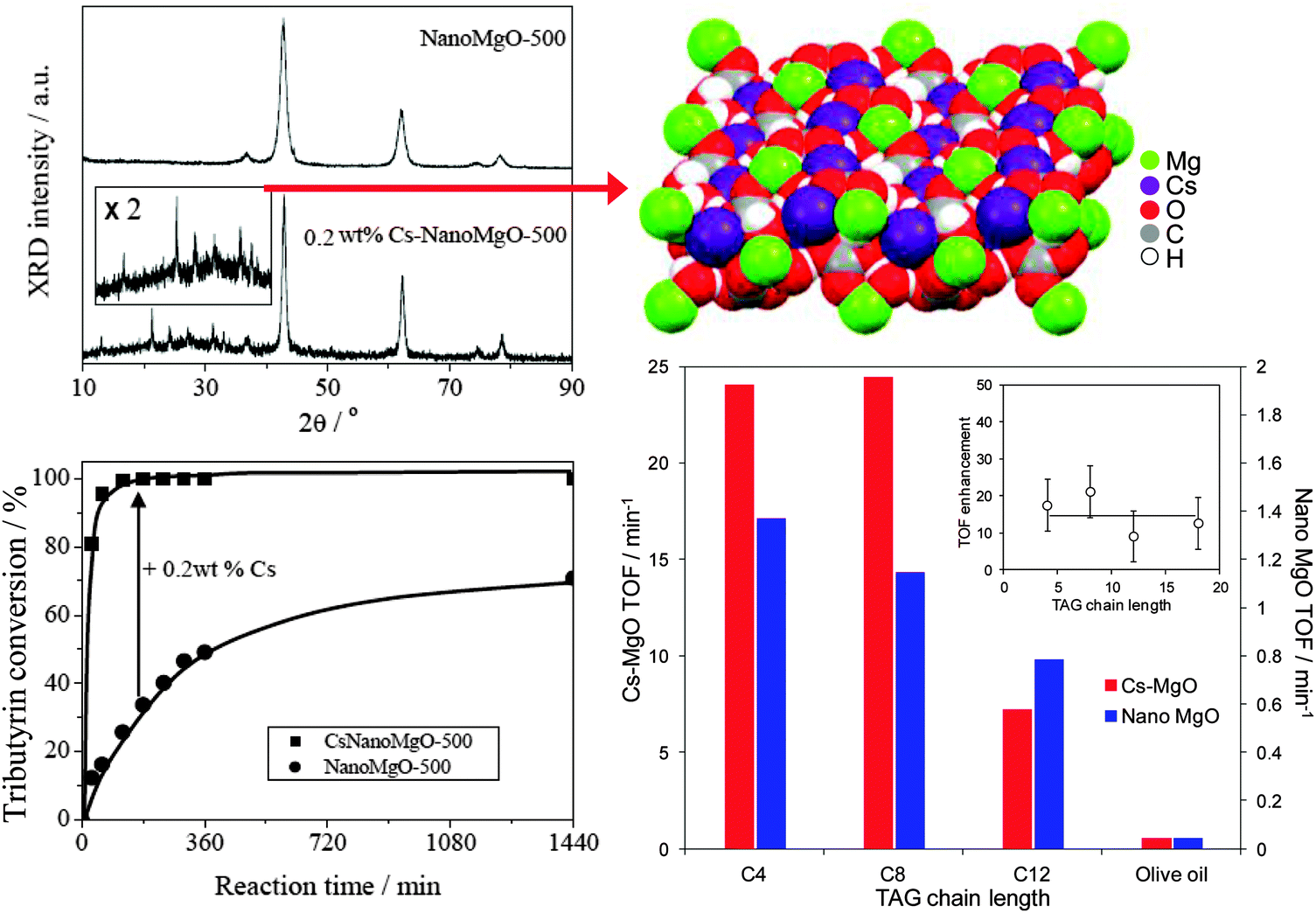 | ||
| Fig. 2 Formation of crystalline Cs2Mg(CO3)2(H2O)4 phase within co-precipitated Cs-doped MgO and resulting synergy in the transesterification of short and long chain TAGs with methanol compared with undoped nanocrystalline MgO. Adapted from ref. 85 with kind permission from Springer Science and Business Media and ref. 92 with permission from John Wiley and Sons. | ||
Alkaline earth metal oxides may be incorporated into metal oxides to form composite oxides93 which are also suitable as solid base catalysts for biodiesel production. The activity of such composites is similar to that of the parent alkaline earth (typically CaO), but they exhibit greater stability and are less prone to dissolution, facilitating separation from the reaction media. Calcination temperature strongly influences the resulting catalytic activity towards transesterification. For example, a Ca–Al composite oxide containing Ca12Al14O33 and CaO thermally processed between 120 °C and 1000 °C showed maximal activity after a 600 °C treatment due to changes in specific surface area and crystallinity. CaO was only observed in samples prepared >600 °C, accompanied by the formation of crystalline Ca12Al14O33. Synergy between these two phases greatly improved the transesterification activity, however calcination at temperatures significantly above 600 °C induced crystallite sintering and concomitant loss of surface area and activity. Unfortunately the catalyst synthesis employed sodium precursors, hence alkali contamination of these catalysts cannot be discounted, and which in any event were employed at high loadings (6 wt%) and without recycle tests.
Calcium also forms a mixed oxide with MoO3.94 Supporting both oxides on SBA-15 mesoporous silica afforded a transesterification catalyst with improved stability relative to CaO due the presence of acidic MoO3 sites on the SBA-15. The impact of Ca:Mo ratio and calcination temperatures was explored, with a Ca:Mo ratio of 6:1 maximising activity for soybean oil conversion, boosting FAME yields from 48 to 83% over extremely long reaction times in excess of 50 h. Raising the calcination temperature from 350 °C to 550 °C induced CaO and MoO3 crystallisation, with a corresponding rise in activity; higher temperature calcination did not promote further crystallisation and was not beneficial for transesterification.
Alkaline earth oxides may be used to support acidic or amphoteric materials to form materials with mixed acid–base character. Transesterification of soybean oil over CaO supported SnO2 prepared via impregnation was highly dependent on calcination temperature and the Ca:Sn ratio.95 The interaction between acidic SnO2 and basic CaO resulted in a highly SnO2 phase and associated active sites. Calcination above 350 °C was required to initiate decomposition of the Ca precursor, with temperatures >650 °C driving complete conversion to Ca oxides. Optimal performance was obtained for high calcination temperatures, which maximised the CaO content. Further heating again led to particle sintering/agglomeration and decreased reactivity. Supported CuO can also produce biodiesel from hempseed oil,83 with 10 wt% CuO/SrO offering 20% higher FAME yields under optimised conditions than other alkaline earth oxides. The CuO could also undergo chemical reduction during transesterification to form an active catalyst for the selective hydrogenation of polyunsaturated hydrocarbons for further biodiesel upgrading. It should be noted that the catalyst loadings employed in this study of 4–12 wt% would likely prove prohibitive in any commercial process, and that small but significant (29 ppm) quantities of leached Ca may have contributed to the observed performance.
Composites of Sr and Al were prepared by Farzaneh et al. and evaluated for soybean oil transesterification with methanol.96 The dominant crystalline phase was Sr3Al2O6, giving rise to medium and high strength base sites with corresponding CO2 desorption peak maxima of 388 °C and 747 °C respectively. The Sr–Al oxide also possessed a higher density of base sites compared to solid bases such as CaO/Al2O3, reflected in an eight-fold higher CO2 adsorption capacity. These superior base properties enhanced the activity of the strontium composite for soybean transesterification to FAMEs, resulting in comparable conversions at a lower catalyst loading and shorter reaction time than for a MgAl hydrotalcite and CaO/Al2O3. While oil conversions fell noticeably with repeated re-use, there was no evidence of alkaline earth dissolution, and the resulting biodiesel fuel met ASTM and EN standards.
3.2 Alkali doped materials
As shown in Fig. 1, lithium doped CaO can enhance tributyrin transesterification. Li doping has also been exploited over SiO2, wherein 800 °C calcination results in a lithium orthosilicate solid base catalyst, Li4SiO4.97 Although the basic strength of Li4SiO4, determined by Hammett indicators, was less than that of CaO, both materials exhibited similar initial activity towards soybean transesterification, with the lithium orthosilicate more stable and maintaining activity after prolonged exposure to air, in contrast to CaO. The superior stability of the Li4SiO4 catalyst was further demonstrated by its water and carbon dioxide tolerance, both of which poison conventional alkaline earth catalysts.Sodium silicate, Na2SiO3, is also active for biodiesel production from rapeseed and jatropha oils under both conventional98 and microwave assisted conditions,99 with a 98% FAME yield after one hour reaction under mild conditions. Although this catalyst displayed good recyclability, TAG conversions fell steadily to <60% after four re-uses, attributed to water adsorption and Si–O–Si bond cleavage and sodium leaching.98 The same catalyst was evaluated using microwave heating for only five minutes at a range of powers between 100–500 W (Fig. 3).99 At low power only 18% rapeseed oil conversion was obtained. Higher powers heated the reaction mixture (to ∼175 C for 400 W) in turn boosting FAME yields from both oils to ∼90%, highlighting the use of microwave heating to accelerate biodiesel production. Recycle studies again showed slow in situ deactivation due to particle agglomeration, water adsorption of water, and associated loss of basicity due to sodium leaching into methanol during both transesterification and washing procedures between recycles. Despite some recent successes in the scale-up of microwave-assisted (homogeneously catalysed) biodiesel production (see Section 6),28,100 it remains unlikely that such heating solutions can deliver the high throughput demanded for commercial processes.
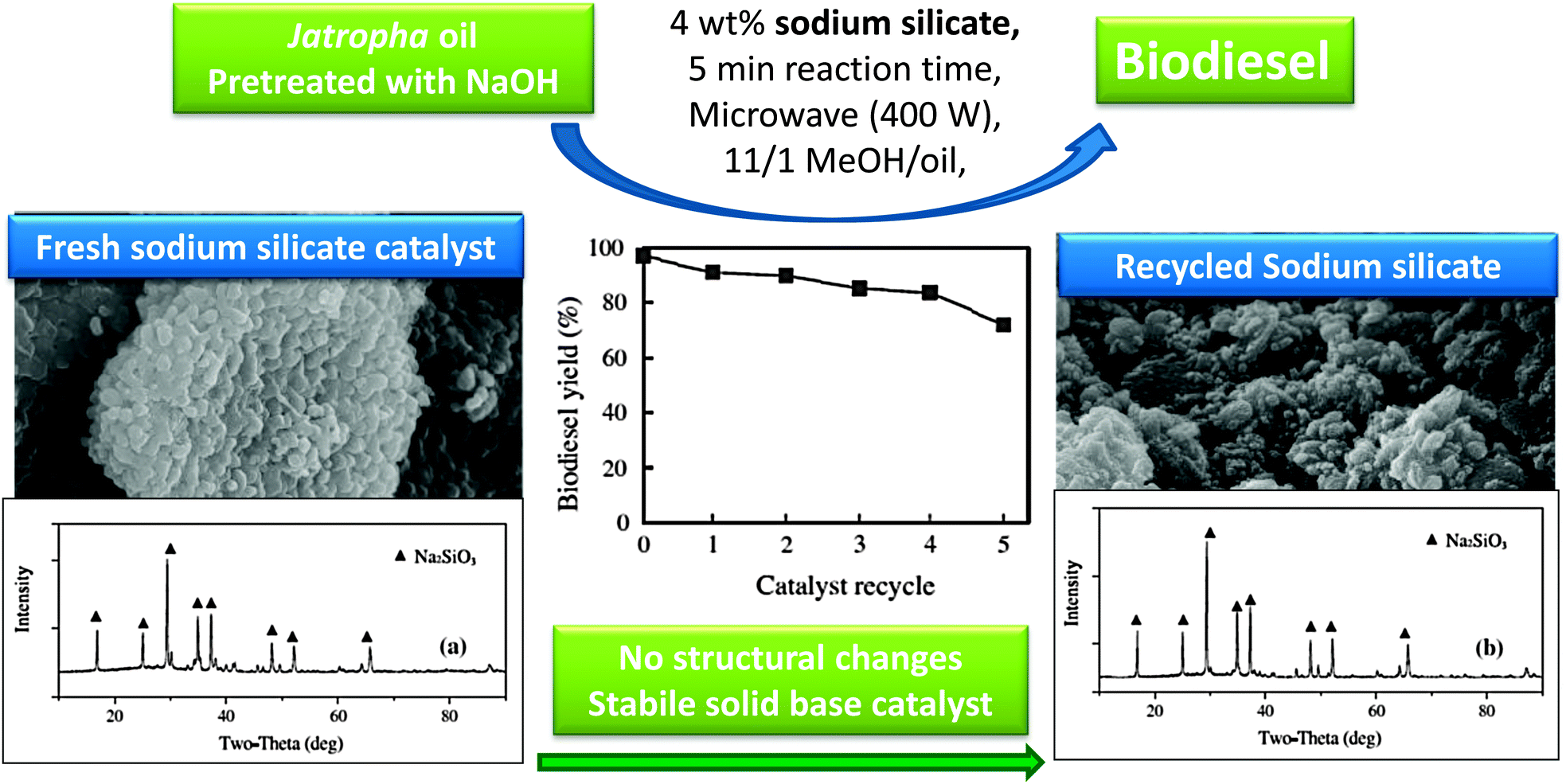 | ||
| Fig. 3 Demonstration of the structural stability and catalytic activity of sodium silicate as a solid base for biodiesel production. Adapted from ref. 99. Copyright (2014), with permission from Elsevier. | ||
Activated carbon can be used as an amphoteric support for basic alkaline metal salts such as K2CO3,101 which is known to be an active homogeneous catalyst for oil transesterification and biodiesel production.102 A study of K2CO3 supported over a range of support materials, such as MgO, activated carbon and SiO2, demonstrated that K2CO3 on basic carriers gave higher activity for rapeseed oil transesterification than when using acidic carriers (unsurprisingly due to self-neutralisation!).102 K2CO3/MgO was shown to be highly stable, with spent catalysts showing minimal loss of performance over six re-uses (though requiring 400 °C reactivation between cycles), and exhibiting negligible structural changes or potassium leaching. Kraft lignin is a low cost, renewable by-product of the Kraft wood pulping process, and possesses high carbon and low ash content and is therefore a popular precursor for activated carbons. Li et al. used K2CO3 in a one-pot method to prepare activated carbon and transform this into a solid base catalyst, namely K2CO3 on Kraft Lignin activated carbon (LKC), for biodiesel production.101 Thermal activation had a significant impact on the resulting catalytic activity, with higher calcination temperatures increasing the surface area and pore volume 100-fold and hence FAME production, however temperatures above 800 °C induced K2CO3 decomposition and poorer performance. Optimal reaction conditions of 65 °C, 3 wt% loading and a K/KLC ratio of 0.6, enabled a 98% FAME yield from rapeseed oil transesterification, which fell to 82% after four recycles as a result of progressive particle agglomeration and potassium leaching into the biodiesel. Wu et al. supported a range of potassium salts on mesoporous silicas for use as solid base biodiesel catalysts.103 A K2SiO3 impregnated catalyst proved superior to K2CO3 and KAc impregnated catalysts due to its higher base site density (1.94 versus 1.81 and 1.72 mmol g−1 respectively). Aluminium addition to the SBA-15 framework improved the morphology, increasing the surface area and pore volume, and CO2 desorption temperature indicative of a more strongly basic support; this observation is rather counter-intuitive, since Al-doping of SBA-15 is usually employed to promote the formation of Brönsted and Lewis acid sites of moderate acidity.104 A 30% K2SiO3/AlSBA-15 catalyst was used for the transesterification of Jatropha oil with MeOH at 60 °C, giving 95% conversion for a relatively low MeOH/oil molar ratio of 9:1. This catalyst was recycled five times with only a 6% drop in conversion, but the filtered catalyst required regenerative washing with a methanol–n-hexane mixture and re-calcination to avoid a significant drop in FAME yield to 47% after the fifth recycle. The magnitude of this activity loss indicates significant K leaching. In a related study, Xie et al. immobilised tetraalkylammonium hydroxides onto SBA-15 for soybean oil transesterification.105 The resulting SBA-15-pr-NR3OH catalyst gave 99% conversion to FAMEs under methanol reflux. Covalent linking of the tetraalkylammonium hydroxide to the silica surface prevented in situ leaching, resulting in only a 1% fall in FAME yield after five recycles and appears a promising methodology for biodiesel production at mild-moderate temperatures under which the covalently linked propyl backbone is thermally stable.
Despite its importance in the context of second generation biofuels, waste biomass has been less extensively investigated in catalyst preparation. Most such studies have focused on the synthesis of carbonaceous solid acid catalysts2,106–109 as discussed later. In contrast, rice husk ash modified with Li via a simple solid state preparative route, has been exploited as a solid base catalyst by for soybean oil transesterification with methanol.106 These materials exhibited high basicity (H− > 15.0), comparable to that of CaO, and consequent high activity, but superior air stability than CaO which deactivated due to hydration; the Li rice husk catalyst showed only a modest drop in oil conversion from 97% to 82% upon re-use. As with any material derived from a biogenic source the question of compositional variability arises, particularly in regard to residual heavy metals in the ash, which is likely to hamper catalyst reproducibility.110
3.3 Transition metal oxides
Solid bases usually afford higher rates of transesterification than solid acids, hence a range of transition metal oxides of varying Lewis base character have been explored in biodiesel production. MnO and TiO are mild bases with good activity for biodiesel production,111 and have been applied for the simultaneous transesterification of triglycerides and esterification of FFAs under continuous flow conditions using low grade feedstocks with high fatty acid contents (up to 15%). Soap formation, caused by leaching of metal from the catalyst surface under high FFA concentrations, was an order of magnitude less than that observed with conventional homogeneous base catalysts. Unfortunately, this study did not characterise the Mn or Ti oxidation state in either fresh or spent materials to confirm the nature of any catalytic centre. Zirconium has also been shown to activate and stabilise solid base catalysts for biodiesel production.101,112,113 Mixed oxides of CaO and ZrO2 prepared via co-precipitation showed increased surface area and stability with increasing Zr:Ca ratios (Fig. 4). However, the transesterification activity remained dependent upon the Ca content, decreasing at lower CaO loadings.112 Sodium zirconate, a potential CO2 adsorbent,84,114 has shown promise in biodiesel production,113 with 98% conversion of soybean oil to FAME after 3 h at 65 °C. Deactivation observed upon repeated decanting and recycling was attributed to surface poisoning, with methanol washing between cycles facilitating 84% conversion after five recycles. This material's affinity for carbon dioxide and large crystallite size/low surface area (∼1 m2 g−1) may render it air-sensitive and prone to further sintering. Zirconia was employed as a support for a range of sodium-containing bases, such as NaOH, NaH2PO4, C4H5O6Na (monosodium tartrate) and potassium sodium tartrate were doped on ZrO2 to prepare a series of catalysts with varying basic strength and total basicity for the microwave assisted transesterification of soybean oil with methanol.101 Catalytic activity was dependent upon basicity, increasing at higher Na:Zr ratios. The potassium sodium tartrate doped zirconia exhibited the strongest basicity and highest conversions, reaching 54% for Na:Zr = 1 and a 1:10 catalyst:soybean oil mass ratio at 60 °C under 600 W microwave power. Increasing the Na:Zr ratio to 2 improved conversion to 92%. Optimal conversions were obtained for catalysts calcined at 600 °C, possibly due to tartrate decomposition at higher temperatures, although this catalyst was recyclable via filtration and re-calcination.
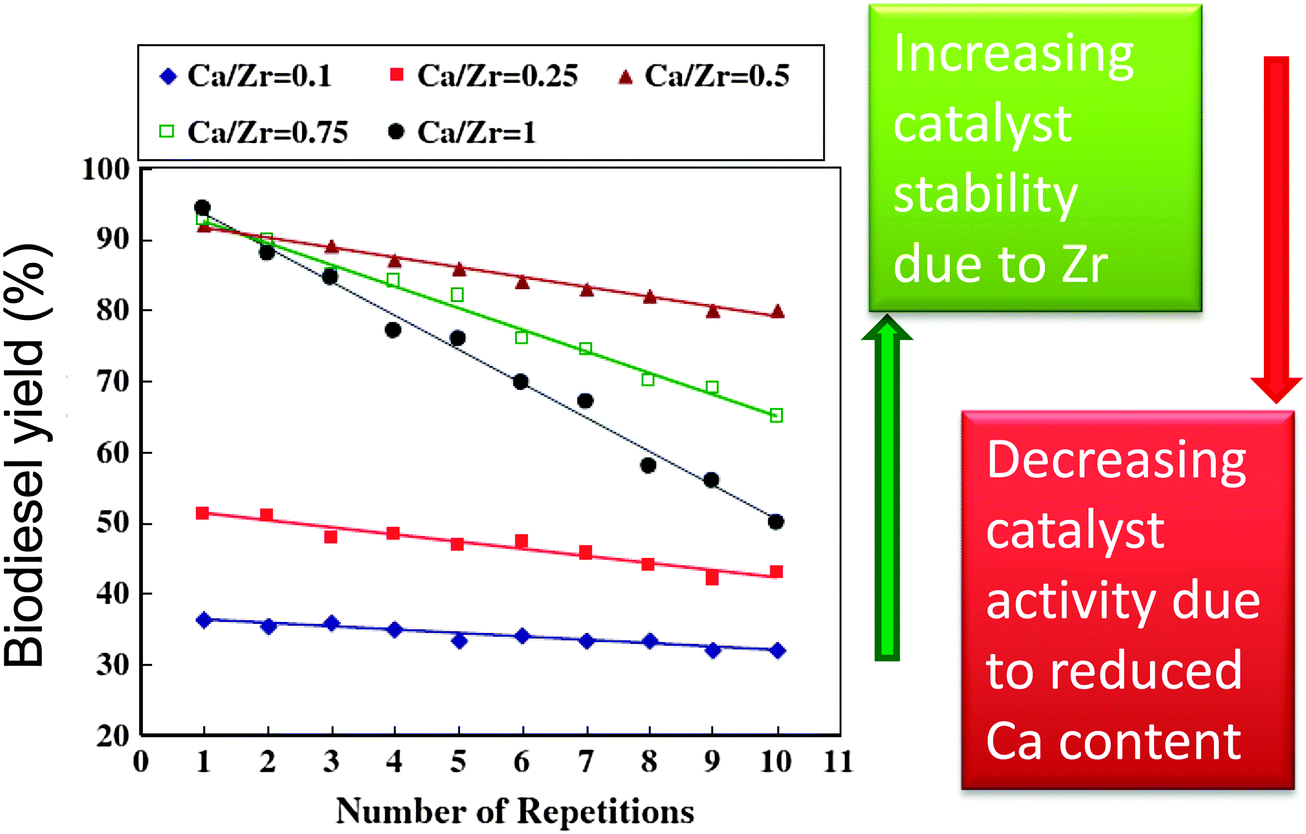 | ||
| Fig. 4 Effect of Zr-doping on CaO solid base catalysts for biodiesel production. Adapted from reference 112. Copyright (2012), with permission from Elsevier. | ||
Porosity was introduced to a titania-based catalyst through the construction of sodium titanate nanotubes as solid base catalysts for soybean oil transesterification with methanol.115 The catalyst exhibited a range of active sites of varying basicity, however the high sodium content (10 wt%) is a cause for concern due to the high probability of leaching in situ and associated homogeneous chemistry. The pore distribution was bimodal, consisting of 3 nm wide tubular mesopores and ∼40 nm voids between the aggregated nanotubes. Biodiesel yields of >97% were obtained for 1–2 wt% of catalyst at 65 °C. However, a large excess of methanol to oil was required (40:1 molar ratio), and while this material could be re-used several times, it was less active than that of CaO and MgO lacking such a nanoporous architecture.
3.4 Hydrotalcites
Hydrotalcites are another class of solid base catalysts that have attracted attention because of their high activity and robustness in the presence of water.116,117 Hydrotalcites ([M(II)1−xM(III)x(OH)2]x+(An−x/n)·mH2O) adopt a layered double hydroxide structure with brucite-like (Mg(OH)2) hydroxide sheets containing octahedrally coordinated M2+ and M3+ cations, separated by interlayer An− anions to balance the overall charge,118 and are conventionally synthesised via co-precipitation from their nitrates using alkalis as both pH regulators and a carbonate source. Mg–Al hydrotalcites have been applied to TAG transesterification of poor and high quality oil feeds,119 such as refined and acidic cottonseed oil (possessing 9.5 wt% FFA) and animal fat feed (45 wt% water), delivering 99% conversion within 3 h at 200 °C. It is important to note that many catalytic studies employing hydrotalcites for transesterification are suspect due to their use of Na or K hydroxide/carbonate solutions to precipitate the hydrotalcite phase. Complete removal of alkali residues from the resulting hydrotalcites is inherently difficult, resulting in ill-defined homogeneous contributions to catalysis arising from leached Na or K.120,121 This problem has been overcome by the development of alkali-free precipitation routes employing NH3OH and NH3CO3, which offer well-defined, thermally activated and rehydrated Mg–Al hydrotalcites with compositions spanning x = 0.25–0.55.116 Spectroscopic measurements reveal that increasing the Mg:Al ratio enables systematic enhancement of the surface charge and accompanying base strength, with a concomitant increase in the rate of tributyrin transesterification under mild reaction conditions (Fig. 5). Despite their high intrinsic activity, one limitation of co-precipitated pure hydrotalcites is their low surface areas, although delamination122,123 and grafting124 methodologies offer avenues to circumvent this.
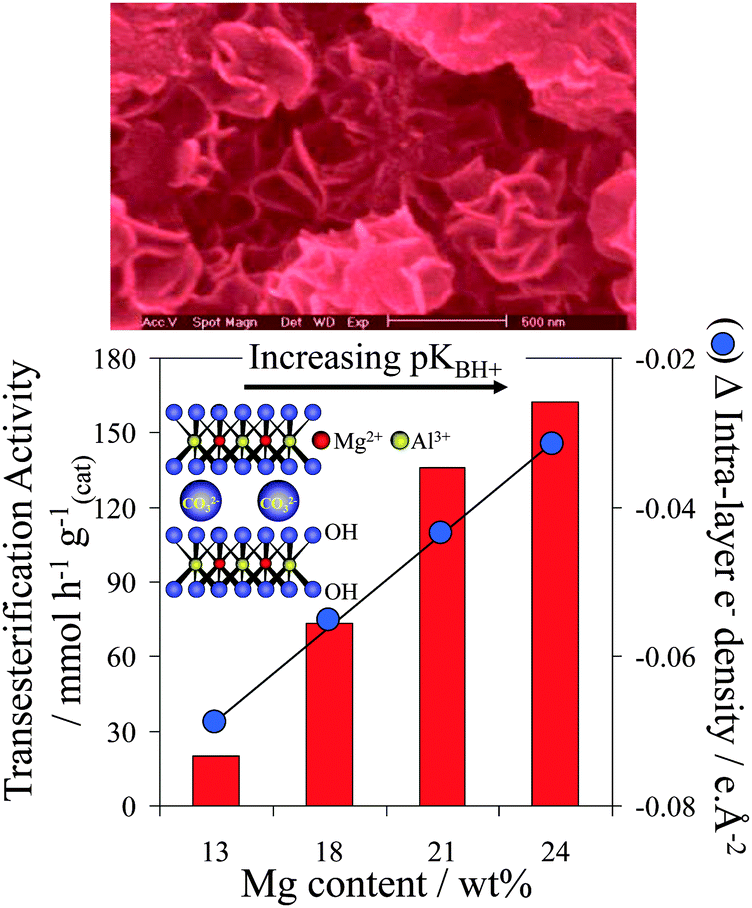 | ||
| Fig. 5 Impact of Mg:Al hydrotalcite surface basicity on their activity towards tributyrin transesterification. Adapted from ref. 117. Copyright (2005), with permission from Elsevier. | ||
Since conventionally-prepared hydrotalcites are microporous, they are poorly suited to transesterification of bulky C16–C18 TAGs which are the principal components of bio-oils. One solution has therefore been to utilise catalysts possessing a bimodal pore distribution, wherein micropores provide a high surface density of base sites while a complementary meso- or macropore network affords rapid transport of TAGs from the bulk reaction media to these active sites, and removal of FAME and glycerol products back out from the porous catalyst. Ordered, hierarchical materials possessing such bimodal pore architectures can be prepared by combining hard and soft templating approaches, exemplified by the methodology developed by Géraud and co-workers, wherein co-precipitation of the divalent and trivalent metal cations occurs within the interstices of an infiltrated polystyrene (PS) colloidal crystal.125,126 This approach has been adopted to incorporate macroporosity into an alkali-free Mg–Al hydrotalcite, and thus create a hierarchical macroporous–microporous hydrotalcite solid base catalyst.127 The resulting macropores act as rapid access conduits to transport heavy TAG oil components to active base sites present at the surface of (high aspect ratio) hydrotalcite nanocrystallites, thereby promoting triolein transesterification compared with that achievable over a Mg–Al microporous hydrotalcite of identical chemical composition (Fig. 6). Spiking experiments confirm that transesterification of the bulky C18 triolein by the hierarchical hydrotalcite catalyst is less hindered by reactively-formed glycerol than when using a conventional microporous hydrotalcite (wherein glycerol completely suppresses biodiesel production). In contrast the more mobile model C4 TAG, trubutyrin possesses an infinite dilution diffusion coefficient of 0.074 cm2 s−1 in methanol versus 0.037 cm2 s−1 for the triolein in methanol. Future scalability of such hierarchical catalysts will require either improved extraction protocols to enable re-use of the colloidal PS template, or the development of alternative polymeric templates derived from sustainable resources, such as polylactic or poly(lactide-co-glycolide) nanospheres.128
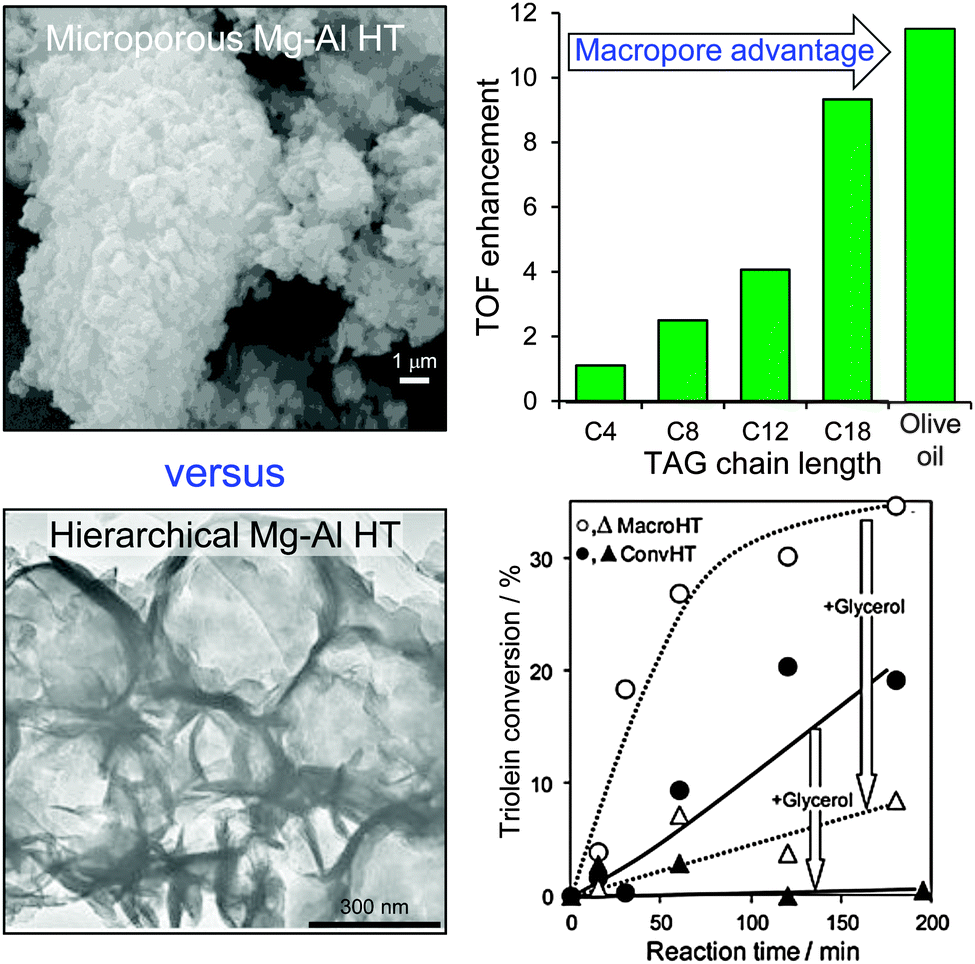 | ||
| Fig. 6 Superior catalytic performance of a hierarchical macroporous–microporous Mg–Al hydrotalcite solid base catalyst for TAG transesterification to biodiesel versus a conventional microporous analogue. Adapted from ref. 128 with permission from The Royal Society of Chemistry. | ||
In terms of sustainability, it is important to find low cost routes to the synthesis of solid base catalysts that employ earth abundant elements. Dolomitic rock, comprising alternating Mg(CO3)–Ca(CO3) layers, is structurally very similar to calcite (CaCO3), with a high natural abundance and low toxicity, and in the UK is sourced from quarries working Permian dolomites in Durham, South Yorkshire and Derbyshire.129 In addition to uses in agriculture and construction, dolomite finds industrial applications in iron and steel production, glass manufacturing and as fillers in plastics, paints, rubbers, adhesives and sealants. Catalytic applications for powdered, dolomitic rock offer the potential to further valorise this readily available waste mineral, and indeed dolomite has shown promise in biomass gasification130 as a cheap, disposable and naturally occurring material that significantly reduces the tar content of gaseous products from gasifiers. Dolomite has also been investigated as a solid base catalyst in biodiesel synthesis,131 wherein fresh dolomitic rock comprised approximately 77% dolomite and 23% magnesian calcite. High temperature calcination induced Mg surface segregation, resulting in MgO nanocrystals dispersed over CaO/(OH)2 particles, while the attendant loss of CO2 increases both the surface area and basicity. The resulting calcined dolomite proved an effective catalyst for the transesterification of C4, C8 and TAGs with methanol and longer chain C16–18 components present within olive oil, with TOFs for tributyrin conversion to methyl butanoate the highest reported for any solid base. The slower transesterification rates for bulkier TAGs were attributed to diffusion limitations in their access to base sites. Calcined dolomite has also shown promise in the transesterification of canola oil with methanol, achieving 92% FAME after 3 h reaction with 3 wt% catalyst.132
Doping of (calcined) Malaysian dolomite with ZnO and SnO2 resulted in respective three- and four-fold increases in the catalyst surface area and active base density, and a concomitant rise in base strength.133 The SnO2 doped dolomite gave >99.9% conversion under optimised conditions with a low methanol:oil molar ratio and catalyst loading.
Other waste materials employed for biodiesel production include waste water scale (obtained from residential kitchens in China), which upon 1000 °C calcination yielded a solid base material mixture of CaO, MgO, Fe2O3, Al2O3, and SiO2 as a stable and active catalyst for soybean transesterification with methanol.134 This composition is similar to that of Red Mud mineral waste, recently shown to be an active ketonisation catalyst.135,136 This waste to resource approach of catalyst design is highly desirable in terms of green credentials and the biofuel ideology.
In summary, a host of inorganic solid base catalysts have been developed for the low temperature transesterification of triglyceride components of bio-oil feedstocks, offering activities far superior to those achieved via alternative solid acid catalysts to date. However, leaching of alkali and alkaline-earth elements and associated catalyst recycling remains a challenge, while improved resilience to water and fatty acid impurities in plant, algal and waste oil feedstocks is required in order to eliminate additional esterification pre-treatments.
4. Solid acid catalysed biodiesel synthesis
A wide range of inorganic and polymeric solid acids are commercially available, however their application for the transesterification of oils into biodiesel has only been recently explored, in part reflecting their lower activity compared with base-catalysed routes,32 in turn necessitating higher reaction temperatures to deliver suitable conversions. Despite their generally poorer activity, solid acids have the advantage that they are less sensitive to FFA contaminants then their solid base analogues, and hence can operate with unrefined feedstocks containing high acid contents.32 In contrast to solid bases, which require feedstock pretreatment to remove these fatty acid impurities, solid acids are able to esterify FFAs through to FAME in parallel with transesterification of major TAG components, without saponification, and hence enable a reduction in the number of processing steps to biodiesel.137–1394.1 Mesoporous silicas
Mesoporous silicas from the SBA family140 have been examined for biodiesel synthesis, and include materials grafted with sulfonic acid groups141,142 or SO4/ZrO2 surface coatings.143 Phenyl and propyl sulfonic acid SBA-15 catalysts are particularly attractive materials with activities comparable to Nafion and Amberlyst resins in palmitic acid esterification.144 Phenylsulfonic acid functionalised silica are reportedly more active than their corresponding propyl analogues, in line with their respective acid strengths, but are more difficult to prepare. Unfortunately, conventionally synthesised sulfonic acid-functionalised SBA-15 silicas possess pore sizes below ∼6 nm and long, isolated parallel channels, and suffer correspondingly slow in-pore diffusion and catalytic turnover in FFA esterification. However, poragens such as trimethylbenzene,145 triethylbenzene and triisopropylbenzene146 can induce swelling of the Pluronic P123 micelles used to produce SBA-15, enabling ordered mesoporous silicas with diameters spanning 5–30 nm. This methodology was recently applied to prepare a range of large pore SBA-15 materials employing trimethylbenzene as the poragen, resulting in the formation of highly-ordered periodic mesostructures with pore diameters of ∼6, 8 and 14 nm.127 These silicas were subsequently functionalised by mercaptopropyl trimethoxysilane (MPTS) and oxidised with H2O2 to yield expanded PrSO3-SBA-15 catalysts which were effective in both palmitic acid esterification with methanol and tricaprylin and triolein transesterification with methanol under mild conditions. For both reactions, turnover frequencies dramatically increased with pore diameter, and all sulfonic acid heterogeneous catalysts significantly outperformed a commercial Amberlyst resin. These rate enhancements are attributed to superior mass-transport of the bulky free fatty acid and triglycerides within the expanded PrSO3-SBA-15. Similar observations have been made over poly(styrenesulfonic acid)-functionalised, ultra-large pore SBA-15 in the esterification of oleic acid with butanol.147 Mesopore expansion accelerates reactant/product diffusion to/from active sites, but there are limits to the extent to which this can be achieved without concomitant loss of pore ordering, which hampers mesoscopic modelling.148The two dimensional, micron-length channels characteristic of the SBA-15 p6mm structure are known to hamper rapid molecular exchange with the bulk reaction media, and hence three dimensional interconnected channels associated with the Ia![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) d structure of KIT-6 mesoporous silica offer one solution to improving the in-pore accessibility of sulfonic acid sites. Superior molecular transport within the interconnected cubic structure of KIT-6 has been shown to facilitate biomolecule immobilisation.149 This diversity of mesoporous silica architectures enabled the impact of pore connectivity upon FFA esterification to be quantified.150 A family of pore-expanded propylsulfonic acid KIT-6 analogues, PrSO3H-KIT-6, prepared via MPTS grafting and subsequent oxidation, have been screened for FFA esterification with methanol under mild conditions. Such a conventionally-prepared material exhibited 40 and 70% TOF enhancements for propanoic and hexanoic acid esterification respectively over an analogous PrSO3H-SBA-15 catalyst of comparable (5 nm) pore diameter, attributed to faster mesopore diffusion. However, pore accessibility remained rate-limiting for esterification of the longer chain lauric and palmitic acids. Pore expansion of the KIT-6 mesopores up to 7 nm via hydrothermal ageing doubled the resulting TOFs for lauric and palmitic acid esterification with respect to an unexpanded PrSO3H-SBA-15 (Fig. 7). It should be noted that the absolute conversions of FFAs over such tailored, inorganic solid acid catalysts remain significantly lower than those for commercial polymer alternatives which possess superior acid site densities (e.g. 4.7 mmol g−1 for Amberlyst-15151versus <1 mmol g−1 for PrSO3H-SBA-15 and PrSO3H-KIT-6150).
d structure of KIT-6 mesoporous silica offer one solution to improving the in-pore accessibility of sulfonic acid sites. Superior molecular transport within the interconnected cubic structure of KIT-6 has been shown to facilitate biomolecule immobilisation.149 This diversity of mesoporous silica architectures enabled the impact of pore connectivity upon FFA esterification to be quantified.150 A family of pore-expanded propylsulfonic acid KIT-6 analogues, PrSO3H-KIT-6, prepared via MPTS grafting and subsequent oxidation, have been screened for FFA esterification with methanol under mild conditions. Such a conventionally-prepared material exhibited 40 and 70% TOF enhancements for propanoic and hexanoic acid esterification respectively over an analogous PrSO3H-SBA-15 catalyst of comparable (5 nm) pore diameter, attributed to faster mesopore diffusion. However, pore accessibility remained rate-limiting for esterification of the longer chain lauric and palmitic acids. Pore expansion of the KIT-6 mesopores up to 7 nm via hydrothermal ageing doubled the resulting TOFs for lauric and palmitic acid esterification with respect to an unexpanded PrSO3H-SBA-15 (Fig. 7). It should be noted that the absolute conversions of FFAs over such tailored, inorganic solid acid catalysts remain significantly lower than those for commercial polymer alternatives which possess superior acid site densities (e.g. 4.7 mmol g−1 for Amberlyst-15151versus <1 mmol g−1 for PrSO3H-SBA-15 and PrSO3H-KIT-6150).
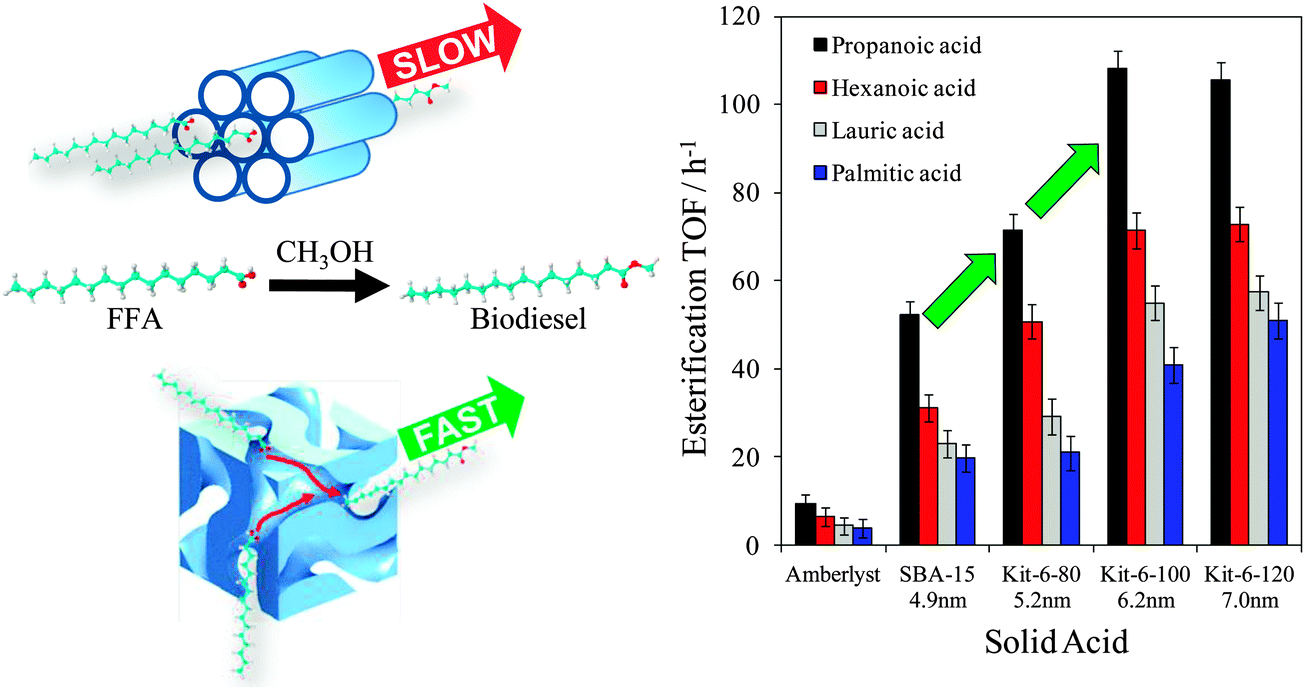 | ||
| Fig. 7 Superior performance of interconnected, mesoporous propylsulfonic acid KIT-6 catalysts for biodiesel synthesis via free fatty acid esterification with methanol versus non-interconnected mesoporous SBA-15 analogue. Adapted from ref. 151. Copyright 2012 American Chemical Society. | ||
Propylsulfonic acid functionalised SBA-15 (SBA-15-PrSO3H) has also been evaluated for oleic acid esterification with methanol,152 showing good stability in boiling water, with the mesopore structure allowing facile diffusion of the acid to active sites. This catalyst exhibited similar activity to phenylethylsulfonic acid functionalised silica gel, and was superior to dry Amberlyst-15, reflecting the higher surface area and pore volume of the SBA-15-PrSO3H relative to the more strongly acidic phenylethyl mesoporous silica. The SBA-15-PrSO3H could be recycled by simple ethanol washing and drying at 80 °C, and maintained an esterification rate of 2.2 mmol min−1 gcat−1. Simultaneous esterification and transesterification of vegetable oils with methanol has performed with Ti-doped SBA-15.153 A range of oils including soybean, rapeseed, crude palm, waste cooking oil and crude Jatropha oil (CJO), and palm fatty acid distillates were successfully converted to biodiesel by the Ti-SBA-15 catalyst at 200 °C. The mesoporous framework gave improved accessibility to the weakly Lewis acidic Ti4+ sites, affording higher activity than microporous titanosilicate and TiO2 supports. The Ti-SBA-15 was tolerant of common oil impurities, performing well in the presence of 5 wt% water or 30 wt% FFA. High catalyst loadings of 15 wt% relative to CJO permitted recycling without loss in conversion, although catalyst regeneration between recycles necessitated washing with acetone and subsequent 500 °C calcination.
Most solid acid catalysts employed in biodiesel synthesis are microporous or mesoporous,32,34,154 properties which the preceding sections highlights are not desirable for accommodating sterically-challenging C16–C18 TAGs or FFAs for biodiesel synthesis. Incorporation of secondary mesoporosity into a microporous H-β-zeolite to create a hierarchical solid acid significantly accelerated microalgae oil esterification with methanol by lowering diffusion barriers.155 Templated mesoporous solids are widely used as catalyst supports,156,157 with SBA-15 silica popular candidates for reactions pertinent to biodiesel synthesis as described above.142,144,158 However, such surfactant-templated supports possessing long, isolated parallel and narrow channels to not afford efficient in-pore diffusion of bio-oil feedstocks, with resultant poor catalytic turnover. Further improvements in pore architecture are hence required to optimise mass-transport of heavier, bulky TAGs and FFAs common in plant and algal oils. Simulations demonstrate that in the Knudsen diffusion regime,159 where reactants/products are able to diffuse enter/exit mesopores but experience moderate diffusion limitations, hierarchical pore structures may significantly improve catalyst activity. Materials with interpenetrating, bimodal meso-macropore networks have been prepared using microemulsion160 or co-surfactant161 templating routes and are particularly attractive for liquid phase, flow reactors wherein rapid pore diffusion is required. Liquid crystalline (soft) and colloidal polystyrene nanospheres (hard) templating methods have been combined to create highly organised, macro-mesoporous aluminas162 and ‘SBA-15 like’ silicas163 (Scheme 4), in which both macro- and mesopore diameters can be independently tuned over the range 200–500 nm and 5–20 nm respectively.
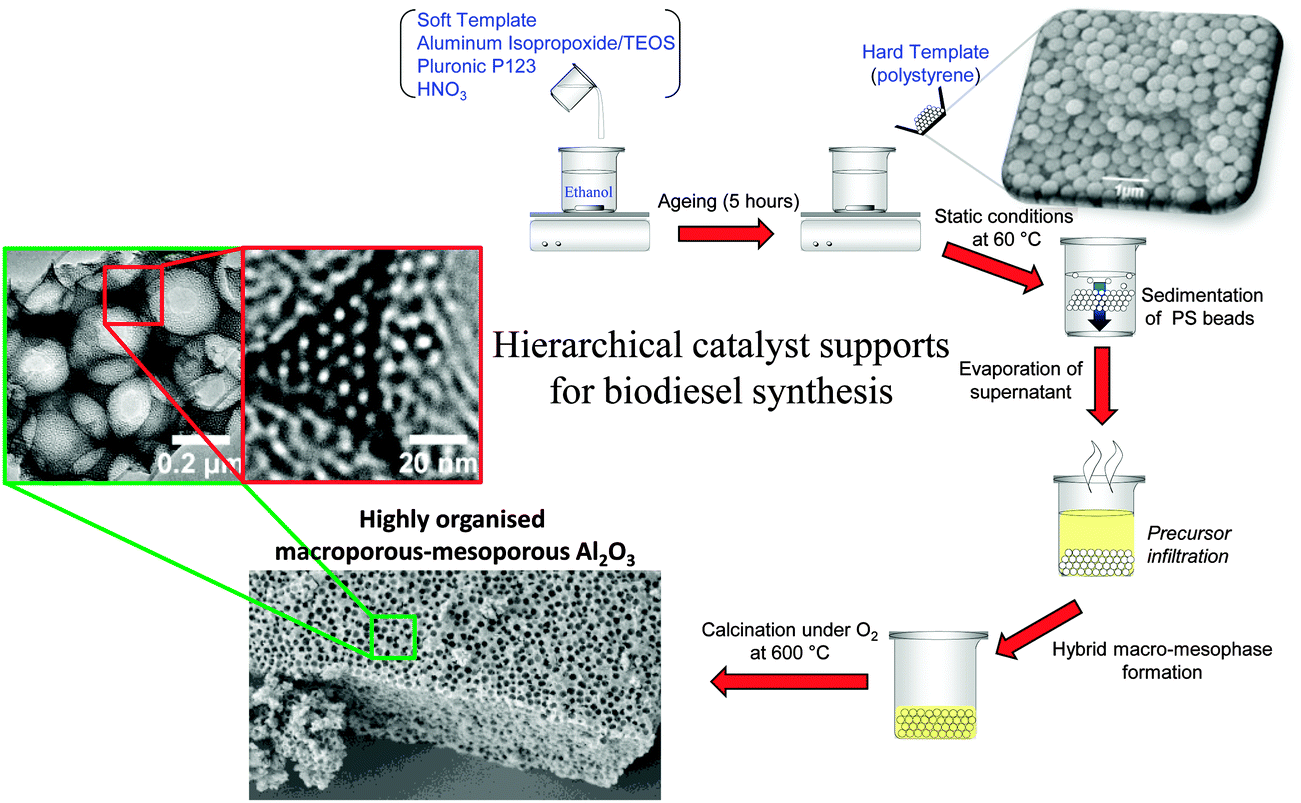 | ||
| Scheme 4 Liquid crystal and polystyrene nanosphere dual surfactant/physical templating route to hierarchical macroporous–mesoporous silicas. | ||
The resulting hierarchical pore network of a propylsulfonic acid functionalised macro-mesoporous SBA-15, illustrates how macropore incorporation confers a striking enhancement in the rates of tricaprylin transesterification and palmitic acid esterification with methanol, attributed to the macropores acting as transport conduits for reactants to rapidly access PrSO3H active sites located within the mesopores.
ZnO is a heterogeneous photocatalyst which has been used for the degradation of organic pollutants in water and air under UV irradiation164–167 and for the photoepoxidation of propene by molecular oxygen.168 ZnO/SiO2 has also been trialled in biodiesel production from crude Mexican Jatropha curcas oil via a two-step process169 in which fatty acids were photocatalytically esterified with MeOH under high energy UVC light unrepresentative of the solar spectrum at ground level. Thermally activated transesterification was subsequently performed employing homogeneous NaOH. Porosimetry and IR studies showed no room temperature CO2 or H2O adsorption suggesting this catalyst should be stable for low temperature esterification. ZnO/SiO2 gave >95% FFA conversion after 8 h of UV irradiation (Fig. 8), with activity constant even after 10 successive runs, although loss of solid catalyst between recycles resulted in a final conversion of only ∼20% per run, albeit using very high catalyst loadings. Reaction was proposed to occur via FFA adsorption at Lewis acidic Zn2+ and MeOH at lattice oxygen, followed by photon adsorption by ZnO and the reaction of photogenerated holes to form H+ and CH3O˙ radicals, with photogenerated electrons reacting with adsorbed acids to form ˙HOOCR radicals; protons and free radicals then reacted to generate intermediates and products. No spectroscopic or chromatographic evidence was presented in support of this elaborate mechanism. Despite the advantages afforded by the ZnO/SiO2 photocatalyst for low temperature FFA esterification, the use of a conventional soluble base in the transesterification step and consequent washing and saponification issues remains problematic, and scale-up of such photocatalysed batch processes to deliver a significant volume of biodiesel will require new photoreactor designs. ZnO/SiO2 materials are also active for the thermally-driven esterification of FFAs (although no details were provided on the nature of these fatty acids) within Jatropha curcas crude oils, wherein activity was proportional to acid site density.170
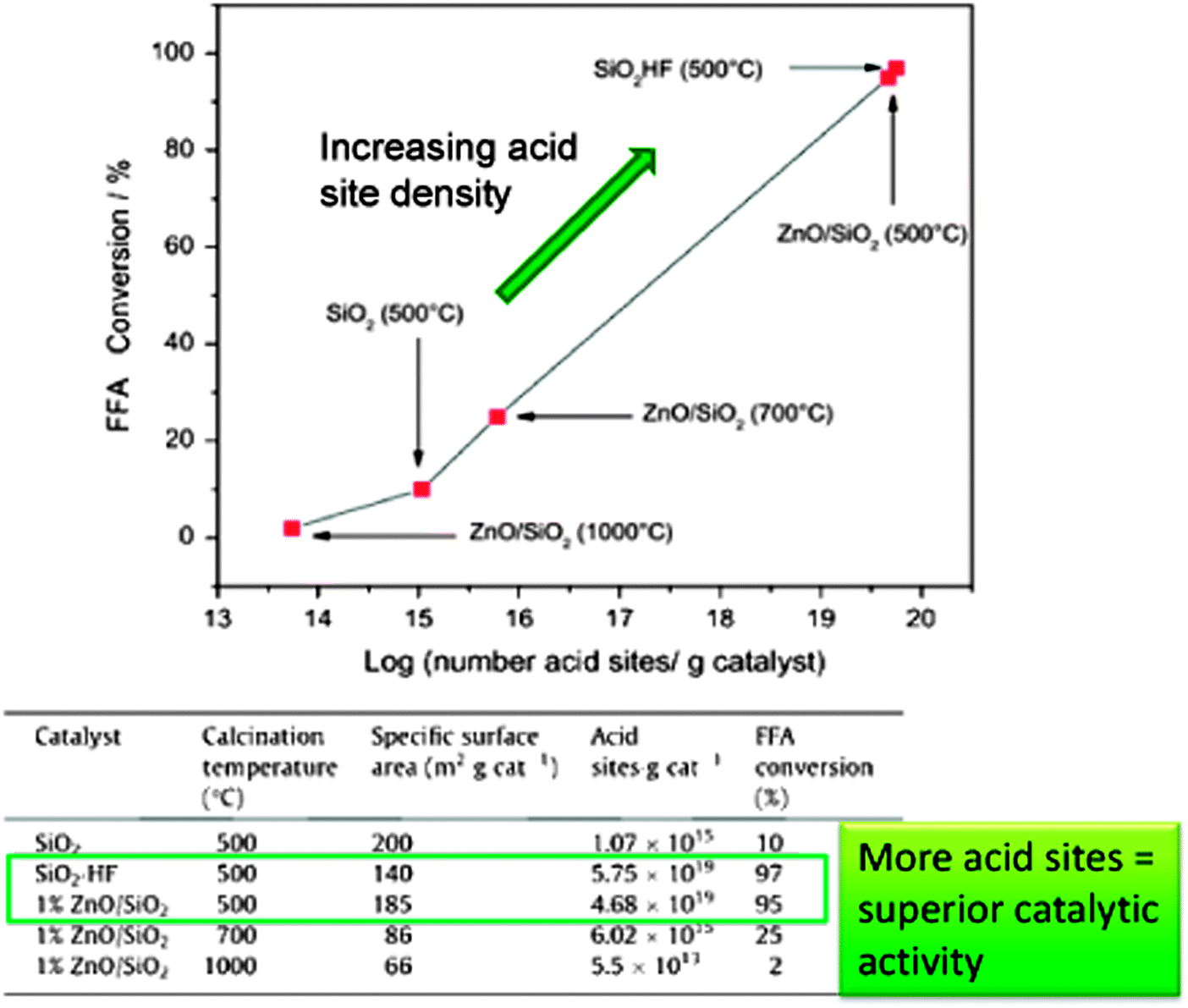 | ||
| Fig. 8 Relationship between acid site density and catalytic performance in FFA esterification. Adapted from ref. 171. Copyright (2014), with permission from Elsevier. | ||
In summary, recent developments in tailoring the structure and surface functionality of mesoporous silicas has led to a new generation of tunable solid acid catalysts well-suited to the esterification of short and long chain FFAs, and transesterification of diverse TAGs, with methanol under mild reaction conditions. A remaining challenge is to extend the dimensions and types of pore-interconnectivities present within the host silica frameworks, and to find alternative low cost soft and hard templates to facilitate synthetic scale-up of these catalysts for multi-kg production. Surfactant template extraction is typically achieved via energy-intensive solvent reflux, which results in significant volumes of contaminated waste and long processing times, while colloidal templates often require high temperature calcination which prevents template recovery/re-use and releases carbon dioxide. Preliminary steps towards the former have been recently taken, employing room temperature ultrasonication in a small solvent volume to deliver effective extraction of the P123 Pluronic surfactant used in the preparation of SBA-15 in only 5 min, with a 99.9% energy saving and 90% solvent reduction over reflux methods, and without compromising textural, acidic or catalytic properties of the resultant Pr-SO3H-SBA-15 in hexanoic acid esterification (Fig. 9).171
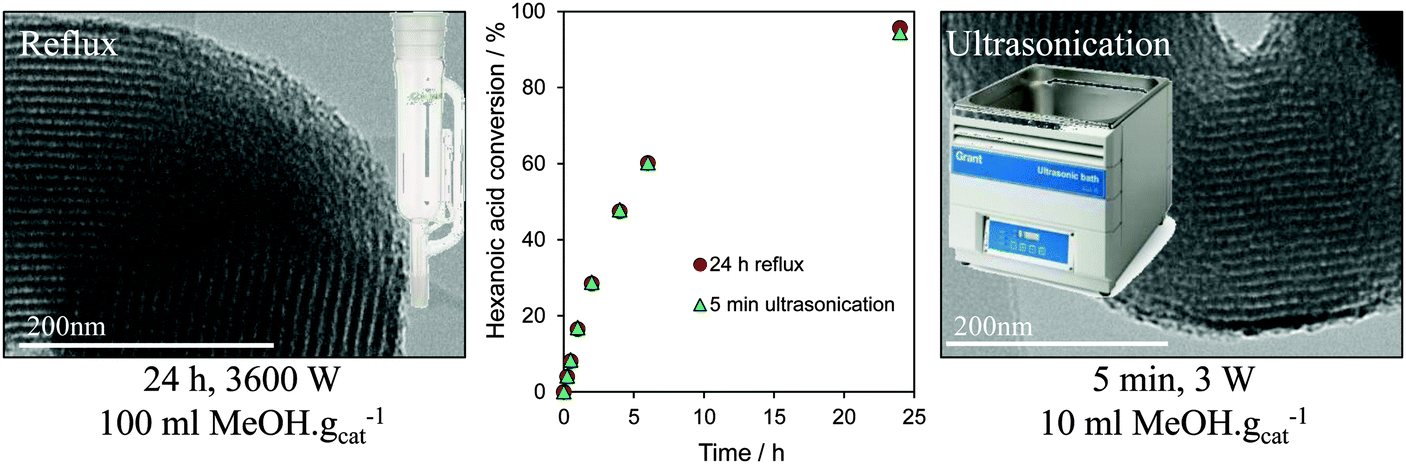 | ||
| Fig. 9 Surfactant template extraction via energy/atom efficient ultrasonication delivers a one-pot PrSO3H-SBA-15 solid acid catalyst with identical structure and reactivity to that obtained by conventional, inefficient reflux. Adapted from ref. 172 with permission from The Royal Society of Chemistry. | ||
4.2 Heteropolyacids
Heteropolyacids are another interesting class of well-defined acid catalysts, capable of exhibiting superacidity (pKH+ > 12) and possessing flexible structures.172 In their native form, heteropolyacids are unsuitable as heterogeneous catalysts for biodiesel applications due to their high solubility in polar media.173 Dispersing such polyoxometalate clusters over traditional high area oxide supports can modulate their acid site densities,174,175 but does little to improve their solubility during alcoholysis. Ion-exchanging larger cations into Keggin type phospho- and silicotungstic acids can increase their chemical stability. For example, Cs salts of phosphotungstic acid CsxH(3−x)PW12O40 and CsyH(4−y)SiW12O40 are virtually insoluble in water, with proton substitution accompanied by a dramatic increase in surface area of the resulting crystallites.137,176 As a consequence of these enhanced structural properties, albeit at the expense of losing acidic protons, both CsxH(3−x)PW12O40 and CsyH(y−x)SiW12O40 are active for palmitic acid esterification to methyl palmitate and tributyrin transesterification (Fig. 10). For CsxH(3−x)PW12O40, optimum esterification and transesterification activity was obtained for x = 2.1–2.4, a similar degree of Cs doping to that maximising palmitic acid esterification for CsyH(4−y)SiW12O40 catalysts (y = 2.8–3.4). These optimal compositions reflect a maximum in the density of accessible surface acid sites within the insoluble Cs-doped catalysts. For CsyH(4−y)SiW12O40, wherein C4 and C8 TAG transesterification were compared, the absolute reaction rates were faster for the shorter chain triglyceride, attributed to slow in-pore diffusion of the longer chain oil. Absolute TOFs for tributyrin transesterification over the optimised Cs-doped catalyst were greater than for the homogeneous H4SiW12O40 polyoxometalate clusters, a consequence of the greater hydrophobicity of the CsxSiW12O40 salts compared with the parent H4SiW12O40, which thus afford enhanced activity for the more lipophilic C8 TAG. Optimising the heterogeneous catalytic activity of CsyH4−ySiW12O40 requires a balance between the retention of acidic protons and generation of stable mesopores to facilitate molecular diffusion. Cs ion-exchange generates interparticle voids large enough to accommodate short-chain TAGs and longer saturated FFAs. Oil/fatty acid and biodiesel polarity and associated mass transport to/from active acid sites is obviously critical in regulating reactivity, and an area where improved materials design in conjunction with molecular dynamics simulations will offer further avenues for high-performance heteropolyacid catalysts.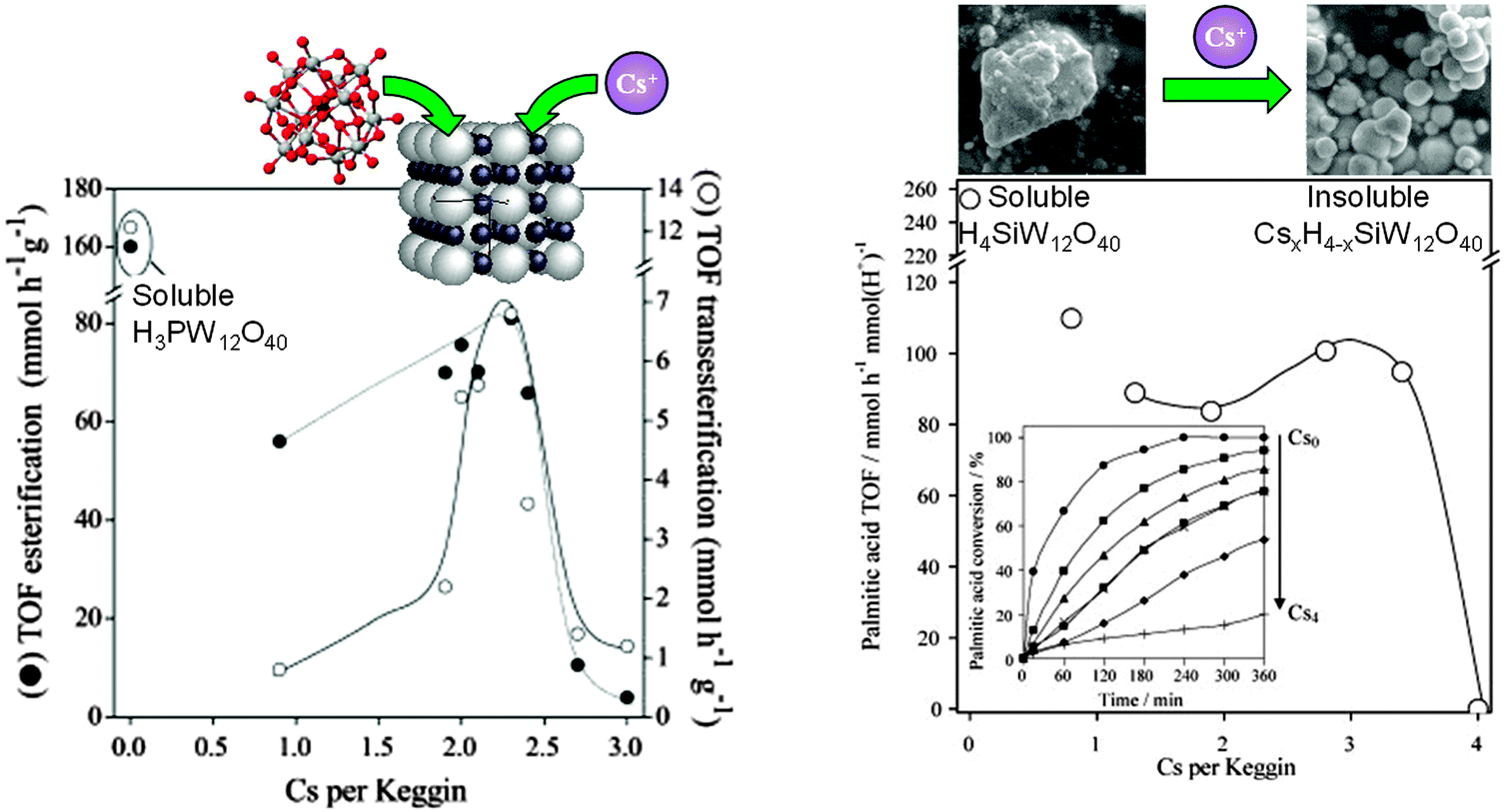 | ||
| Fig. 10 Impact of Cs ion-exchange into (left) both CsxH(3−x)PW12O40 for palmitic acid esterification and tributyrin transesterification with methanol; and (right) and CsyH(y−x)SiW12O40 for palmitic acid esterification, benchmarked against parent fully protonated, soluble clusters. Adapted from ref. 138 and 177. Copyright (2007 and 2009), with permission from Elsevier. | ||
Duan et al. have prepared H3PW12O40 supported on magnetic iron oxide particles (MNP-HPA) via an acid–base interaction and tested them in palmitic acid esterification with methanol under mild conditions.177 The magnetic nanoparticles were first coated in a protective SiO2 layer and then functionalised with aminopropyl groups, with the heteropolyacid immobilised by reaction with the amine. Water tolerance was imbued by the addition of nonyl chains to the catalyst surface which lowered the acid loading but improved palmitic acid conversion to 90% at 65 °C. Magnetic separation enabled catalyst recycling without activity loss (Fig. 11), while the presence hydrophobic/oleophilic nonyl groups improved diffusion of the reagent to the active sites, enhancing TOFs compared to the parent MNP-HPA. However, the water tolerance of these materials was limited, with only 1 wt% water reducing FFA conversion to 34%.
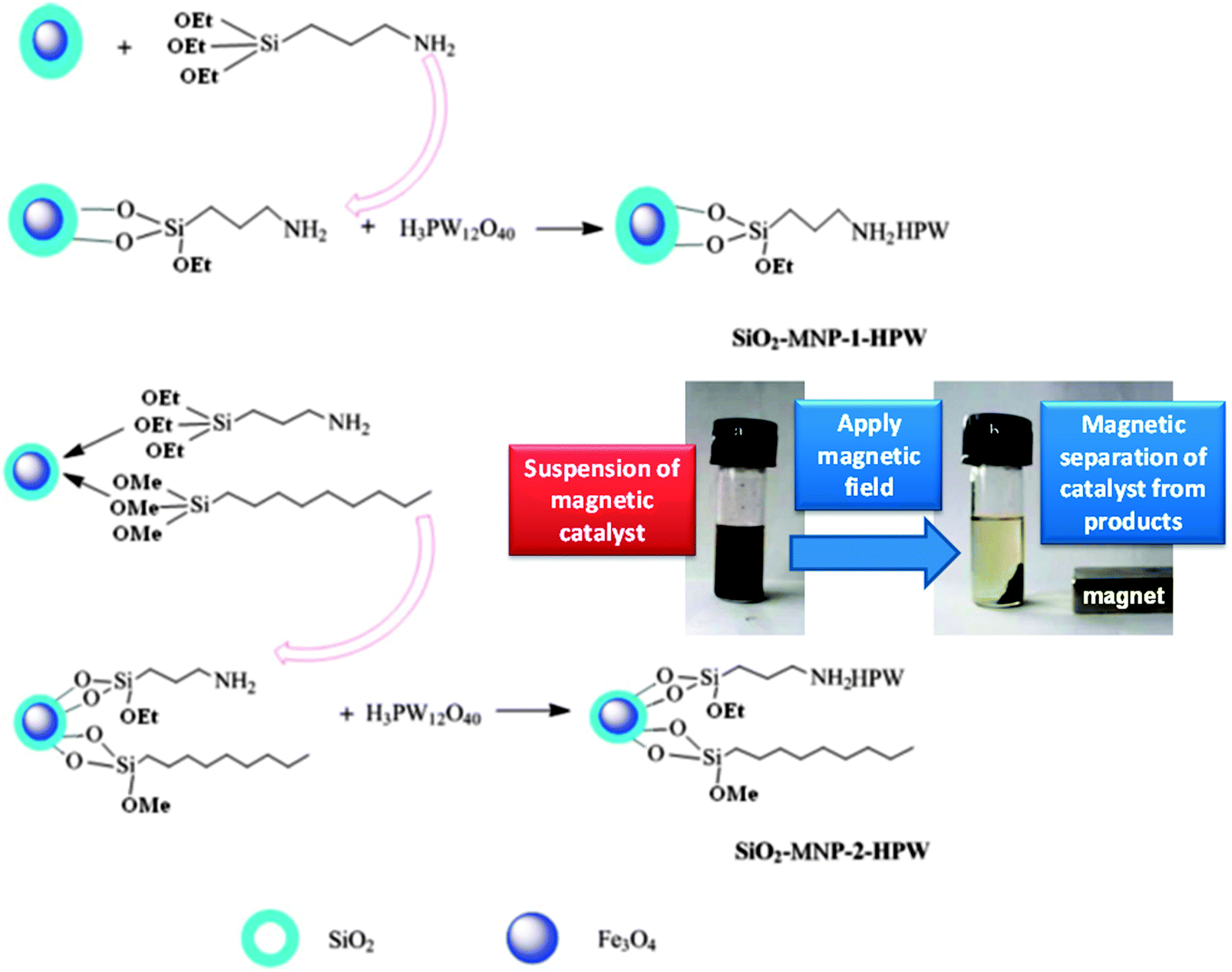 | ||
| Fig. 11 Preparation of water-tolerant heteropolyacid on magnetic nanoparticles for palmitic acid esterification. Reprinted from ref. 178 with permission from The Royal Society of Chemistry. | ||
Mesostructured silicas have also been employed as supports for HPAs, for example 12-tungstophosphoric acid (TPA) dispersed over mesoporous MCM-48 is a promising solid acid catalyst for oleic acid esterification with methanol.178 This catalyst gave 95% conversion to biodiesel with modest alcohol:acid molar ratios, but very high catalyst loadings (30 wt% TPA). Leaching studies employing insensitive colorimetric tests, suggested good catalyst water stability, with minimal loss of W from MCM-48 detectable by atomic absorption (rather than more sensitive ICP), and retention of the majority of acid sites post-reaction (1.50 mmol g−1). No explanation was advanced for this extremely surprising water tolerance of TPA, which usually exhibits a high solubility in methanol; entrapment of primary Keggin units within the 3 nm diameter MCM-48 pores seems improbable, and any physical barrier to their dissolution would also likely hinder FFA and FAME access to TPA acid sites. The principal disadvantage of heteropolyacids for esterification and transesterification reactions in short-chain alcohols thus remains their limited water tolerance, which to date can only be overcome through advanced catalyst design and the sacrifice of their high acid strength and site density.
4.3 Acidic polymers and resins
While inorganic frameworks such as SBA-15 or ZrO2 are popular supports for solid acid catalysis, their hydrophilic nature can hinder diffusion of organic reagents. This problem can be avoided by the use of hydrophobic and oleophilic supports, such as mesoporous organic polymers. Sulfonated mesoporous polydivinylbenzene (PDVB) is one such solid acid catalyst,179 which exhibits absorption capacities for sunflower oil and methanol three times those of H3PO40W12, sulfonated-ZrO2, SBA-15-SO3H or Amberlyst 15, and consequent superior performance in tripalmitin transesterification, giving an 80% yield of methyl palmitate after 12 h reaction. PDVB-SO3H proved easily recyclable, with only a modest drop in yield after three recycles, ascribed to a combination of its high surface area, large pore volume, high acid site density, and hydrophobic/oleophilic pore network. Liu et al. utilised an aminophosphonic acid resin based on a polystyrene backbone in the microwave-assisted esterification of stearic acid with EtOH.180 FAME yields of 90% were obtained after microwave heating to (notionally) 80 °C for 7 h at a catalyst loading of 9 wt%, with slower reaction and a lower limiting conversion of 88% resulting from conventional heating. Kinetic analysis suggested a pseudohomogeneous mechanism in which microwave radiation excited the polar reactants in the solution phase in addition to the solid catalyst. This resin was structurally stable as determined by XRD, TGA and SEM, and recyclable with 87% acid conversion after five uses (Fig. 12).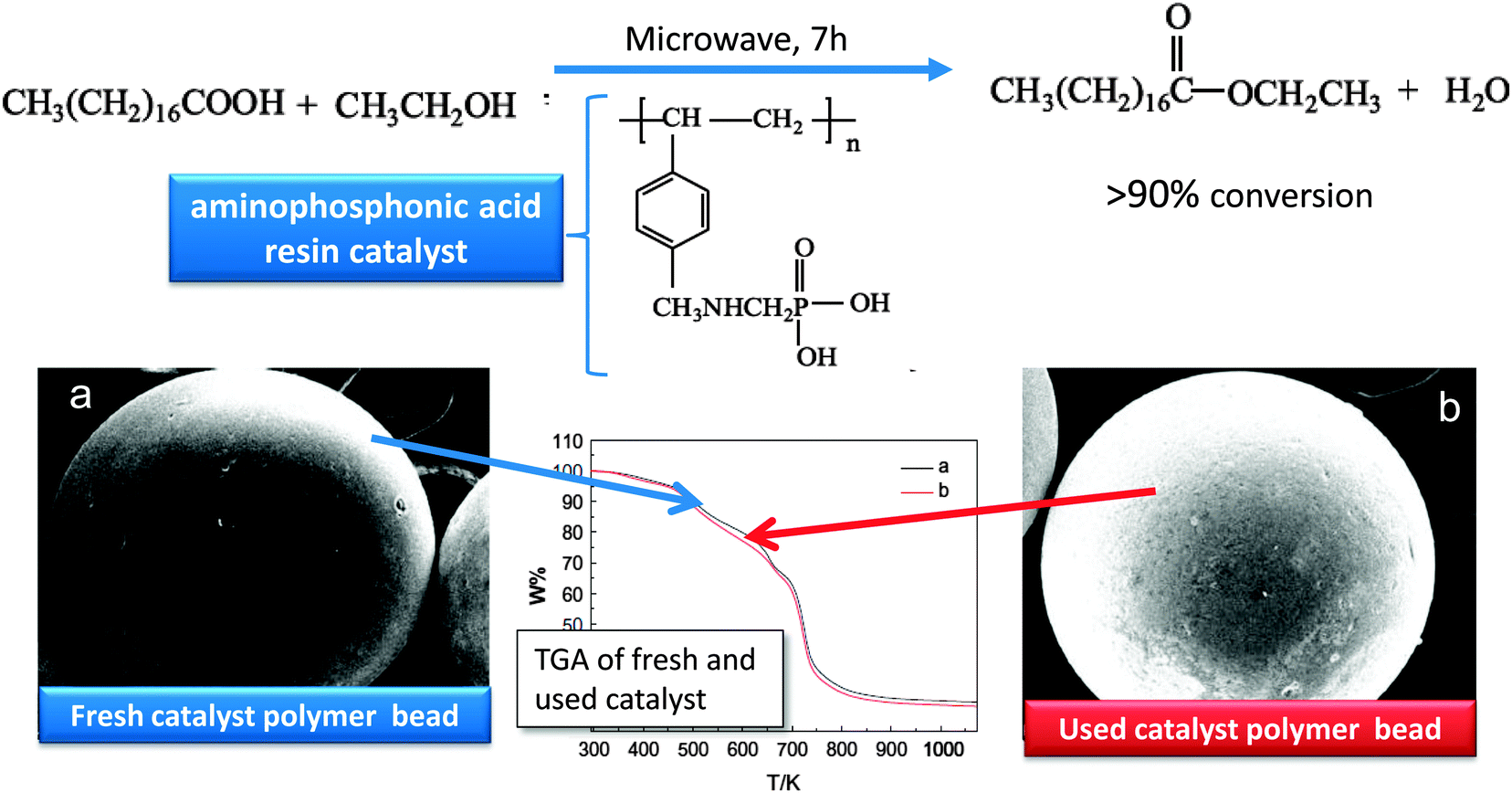 | ||
| Fig. 12 Stability of a solid acid resin catalyst for stearic acid esterification. Adapted from ref. 181. Copyright (2013), with permission from Elsevier. | ||
The acid exchange resin, Relite CFS, was tested under batch and continuous modes for the simultaneous esterification and transesterification of oleic acid and soybean oil with methanol,181 evidencing good activity with 80% FAME obtained after 150 min at 100 °C. Unfortunately this resin was deactivated via exchange with metals such as iron present in the feedstream causing catalyst discolouration of beads during continuous operation (Fig. 13); activity could be completely regenerated by suspending the resin in sulphuric acid for 24 h and a further lengthy washing and drying protocol. A copolymer of acidic ionic liquid oligomers and divinylbenzene (PIL) has also been utilised as a catalyst for simultaneous esterification and transesterification of FFA-containing triglyceride mixtures (waste cooking oil), possessing a high acid density of 4.4 mmol g−1, high pore volume and surface area of 323 m2 g−1, and 35 nm mean pore diameter.182 The latter and hydrophobic surface character permitted efficient substrate diffusion through the pore network. The PIL copolymer was more active than the acidic ionic liquid alone, giving >99% conversion of oleic acid with MeOH at only 1 wt% catalyst loading. PIL also achieved >99% yield in rapeseed transesterification with MeOH under the same reaction conditions, and proved able to convert high FFA content waste cooking oil into biodiesel with 99% yield in 12 h. The spent catalyst showed no structural changes or loss of acidic sulphur, and hence could be efficiently recycled with almost no loss in performance.
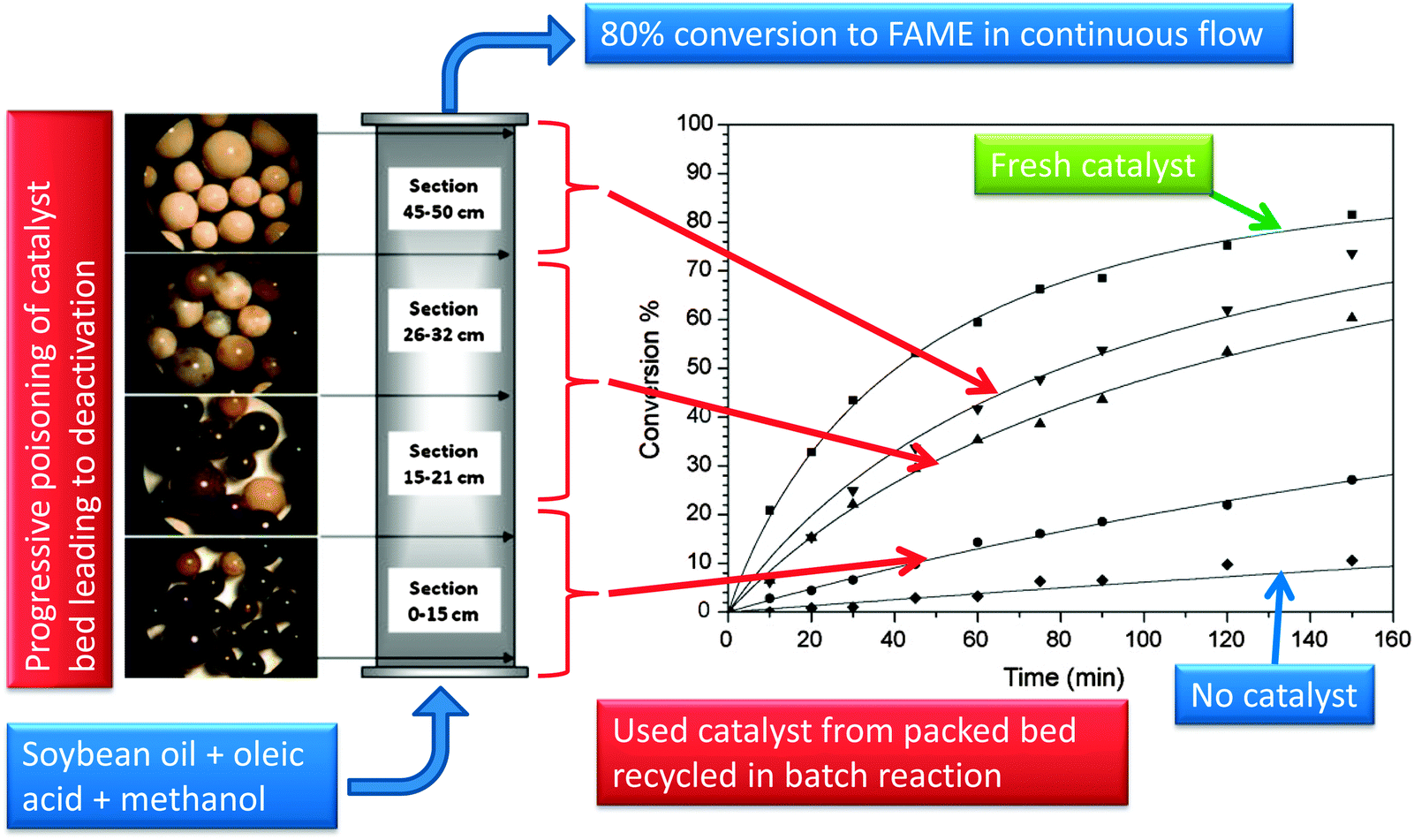 | ||
| Fig. 13 Deactivation of an acid resin catalyst during continuous esterification/transesterification of FFA and oil mixtures. Adapted from ref. 182. Copyright (2010), with permission from Elsevier. | ||
4.4 Waste carbon-derived solid acids
As discussed earlier in this review, many studies have investigated the development of carbon catalysts prepared from second generation biomass such as non-edible crop waste,2,106,107 algal residues108 and even waste products from biodiesel production.109 Sulfonated carbonaceous materials show promising activity for FFA esterification, generally affording higher rates of biodiesel production than commercial resins such Amberlyst with which they are often compared.Residue of the non-edible seed Calophyllum inophyllum has been carbonised to make a biomass-derived solid acid catalyst via sulfonation.107 The resulting catalysts, comprising randomly oriented, amorphous aromatic sheets of low surface area (0.2 to 3.4 m2 g−1) and variable acid densities (0.6 to 4.2 mmol g−1 dependent on the S wt%), were tested in the simultaneous esterification and transesterification of Calophyllum inophyllum seed oil. Esterification activity was greatly proportional to the S loading, but also influenced by the balance of hydrophobic/hydrophilic sites on the carbon which affected diffusion and adsorption of oleo substrates. This balance, and related surface properties, varied with the carbonisation and sulphonation conditions employed; short carbonisation times lead to smaller sheets with higher SO3H densities and increased activity, but also increased S leaching and concomitant deactivation. Rice husk char was sulfonated with concentrated sulfonic acid under various conditions, and evaluated in the esterification of oleic acid with MeOH.2 All catalysts were amorphous, with a maximum SO3H density of 0.7 mmol g−1. High conversions were obtained at 110 °C in 2 h for a low alcohol:oil molar ratio of 4:1, with the catalyst recyclable and still delivering 84% methyl oleate after seven re-uses despite losing 23% of the initial S through leaching.
Peanut shells processed in a similar manner to that above also yield a strong Brönsted solid acid catalyst, with an acid strength superior to H-ZSM-5 (Si/Al = 75).183 This catalyst gave >90% conversion of cottonseed oil in methanol transesterification at a methanol:oil molar ratio of only 9:1. Recycling and re-use studies employed centrifugation to separate the catalyst, with subsequent acetone washes leading to a 50% reduction in acid site density, although regeneration was achievable by prolonged treatment with 1 M H2SO4 solution. Despite the environmental compatibility of waste biomass-derived solid acid catalysts, active site retention over prolonged use remains a critical challenge if they are to find implementation in continuous biodiesel production; leaching of sulphate or sulfonic acid groups into the product stream would both shorten catalyst lifetime and degrade fuel quality.
Microalgae are an exciting, potential feedstock for biodiesel production, but following extraction of algal oils, the residue is typically burned or discarded. Fu et al.108 has partially carbonised and sulfonated such residue to create a solid acid catalyst for the esterification of oleic acid and transesterification of triolein with methanol at 80 °C (Fig. 14). Although the resulting catalyst comprised disordered, non-porous aromatic carbon sheets with a very low surface area, the sulfonic acid density of 4.25 mmol g−1 afforded an active catalyst with a stable FFA conversion >98% over six sequential oleic acid esterification cycles. The corresponding FAME yield for triolein transesterification was only 22%, but likewise stable across numerous recycles. However, such catalysts were prone to deactivation by adsorbed methanol and hence required regenerative sulphuric acid and hot water washes between recycles. A similar approach was adopted for the waste glycerol by-product of biodiesel production, whereby the polyol was converted in situ by partial carbonisation and sulfonation into a solid acid catalyst.109 High catalyst loadings, reaction temperatures (160 °C) and MeOH:oil ratios (>45) were required to achieve 99% conversion of Karanja oil to FAME, with conversion dropping to only 5% after five recycles, although no analysis of the spent catalyst or leaching studies were reported. Leaching of acid sites was however addressed by Deshmane et al.,184 who investigated sulfonated carbon catalysts prepared from sugar and polyacrylic acid for oleic acid esterification. These catalysts were deactivated by the formation of irregularly-shaped, 1 μm colloidal carbon aggregates, comprised of sulfonated polycyclic hydrocarbons, during the hydrothermal, sulfonation or pulverisation preparative steps, which subsequently leaching into the esterification reaction mixture.
 | ||
| Fig. 14 Microalgae as a source of bio-oils/fatty acids for biodiesel production, and waste, biomass residue for the synthesis of solid acid catalysts to drive such biodiesel production. | ||
The kinetics of palm oil fatty acid esterification with MeOH over carbonised, sulfonated microcrystalline cellulose (CSMC) have also been compared with those of homogeneous sulphuric acid catalysts,185 compensating for the phase equilibrium and reaction equilibrium to provide an accurate kinetic reaction model; this approach ensured the biphasic nature of the water–alcohol–oil reaction mixture was correctly represented instead of assuming a pseudo-homogeneous model. Methanol and FFA adsorption over the CSMC was believed a key step in the heterogeneous process, and hence adsorption equilibrium constants were calculated for these molecules along with water and FAME. Unsurprisingly, the free fatty acid was found to adsorb preferentially in the presence of low concentrations of the other molecules. At the start of the esterification reaction, FFA and alcohol were fully miscible, but water and FAME production led to the evolution of two phases; one comprising aqueous methanol and catalyst, and the other methyl ester and unreacted FFA. Mass-transport between these phases is essential, but likely the rate-limiting step. Kinetics of both homogeneously and heterogeneously catalysed biphasic systems were modelled with high conversions favoured by the limited solubility of water in the organic phase, and the use of hydrophobic catalysts which displace water from reaction sites.
A major drawback of the preceding sulfonated carbons is their low surface area, which can be alleviated through the use of carbon nanotubes. Poonjarernsilp and co-workers prepared solid acid catalysts by sulfonating single-walled carbon nanohorns (SWCNHs)186 which possessed surface areas of 210 m2 g−1 and could be further improved by high temperature calcination to open up micropores. The resulting oxidised nanohorns (ox-SWCNs) had surface areas of 1000 m2 g−1 and superior pore volumes. However the subsequent sulfonation step required to introduce surface acidity, somewhat lowered the final surface area and pore volume, and drastically altered the pore size distribution, eliminating all the meso- and macropores to leave a narrow range of 2–10 nm pores. Despite the improved morphology of the sulfonated ox-SWCNs relative to the SWCNs, the former had a lower acid site density and was consequently less active in palmitic acid the esterification with methanol; the best yield was obtained for SO3H-SWCNHs, which gave 93% methyl palmitate after 5 h with a catalyst:MeOH:FFA ratio of 0.15:0.15:5 g. Recycling tests showed a progressive decrease in methyl palmitate yield associated with a loss of acid sites.
4.5 Miscellaneous solid acids
A range of additional solid acids have also been investigated, including ferric hydrogen sulphate [Fe(HSO4)3],187 supported tungsten oxides (WO3/SnO2),188 supported partially substituted heteropolytungstates,189 and bifunctional catalysts, such as Mo-Mn/Al2O3-15 wt% MgO,190 designed to incorporate the benefits of both acid and base catalysis. The iron catalyst had a low surface area of 4–5 m2 g−1, and required higher operating temperatures than other solid acids to achieve good biodiesel yields (94% at 205 °C),187 but was easily recycled by simple washing and drying to remove adsorbed products, maintaining activity over 5 cycles with no evidence of metal leaching. WO3/SnO2 was water tolerant and showed good conversion of soybean oil to FAME at a lower reaction temperature (110 °C), but required high MeOH:oil ratios >30 to achieve a 78% yield,188 but was prone to on-stream deactivation upon recycling. Tungsten-containing HPAs supported on silica, alumina, and zirconia were also active in biodiesel production from 10 wt% oleic acid in soybean oil delivering FAME yields >75% at a high reaction temperature. Performance was unaffected by the presence of up to 25 wt% of the fatty acid blended with the oil. Cesium addition to the HPA suppressed leaching and thereby improved catalyst stability, resulting in only a 10% fall in biodiesel production after multiple recycles attributed to physical sample loss during product separation.
In an attempt to incorporate acid and base character in a single material, Farooq et al. prepared a Mo-Mn/γ-Al2O3-15 wt% MgO catalysts via wet impregnation of alumina with MgO, followed by impregnation of the γ-Al2O3-MgO with [(NH4)6Mo7O24]·4H2O and subsequently aqueous Mn(NO3)2.190 The resulting thermally processed catalyst possessed highly dispersed MoO3 and MnO acid sites, affording 75% biodiesel yield at 95 °C with a MeOH:oil molar ratio of 15. This bifunctional material could be repeatedly recycled with the yield falling by 20% after 10 uses, a modest deactivation that was attributed to poisoning by strongly adsorbed organics and leaching of the various active metals during transesterification.
5. Hydrophobicity studies
The hydrophilic nature of polar silica surfaces hinders their application for reactions involving apolar organic molecules. This is problematic for TAG transesterification (or FFA esterification) due to preferential in-pore diffusion and adsorption of alcohol versus fatty acid components. The presence of water in bio-oils (and an inevitable by-product of esterification) can significantly influence biodiesel production, however a major barrier to commercialisation is the development of an efficient, inexpensive and reusable heterogeneous catalyst that can perform at low temperature and pressure.191 Solid catalysts with ordered and large pores to minimise diffusion limitations, moderate to strong acid sites to overcome the presence of FFAs impurities, and a hydrophobic surface to nullify the effect of water are hence sought.32,192–196 While solid acid catalysts are of great interest in this regard due to their ability to catalyse both FFA esterification and TAG transesterification,144,197 sensitivity to water is a common cause of deactivation,198,199 and water-tolerant solid acids would be highly desirable.31,37,200 Surface hydrophobicity, and the relative adsorption/desorption rates of reactants/products, are critical parameters influencing (trans)esterification,201 and tuning catalyst polarity thus offers a route to control competitive adsorption and promote product desorption. Steric factors associated with long fatty acid alkyl chains can also influence reaction rates;202 Alonso and co-workers explored the relationship between fatty acid polarity/chain length (C2–C16) and transesterification rates over solid and liquid acid catalysts.203 Activity decreased with increasing chain length for a heterogeneous (SAC-13) catalyst, but remained constant when catalysed by H2SO4, highlighting the negative impact of hydrophilic surfaces on biodiesel production.203Surface hydroxyl groups favour H2O adsorption, which if formed during FFA esterification can drive the reverse hydrolysis reaction and lowering FAME yields. Surface modification via the incorporation of organic functionality into polar oxide surfaces, or dehydroxylation, can lower their polarity and thereby increase initial rates of acid catalysed transformations of liquid phase organic molecules.204 Surface polarity can also be tuned by incorporating alkyl/aromatic groups directly into the silica framework, for example polysilsesquioxanes can be prepared via the co-condensation of 1,4-bis(triethoxysilyl)benzene (BTSB), or 1,2-bis(trimethoxysilyl)-ethane (BTME), with TEOS and MPTS in the sol–gel process205,206 which enhances small molecule esterification207 and etherification.208 This approach has been adopted for the direct synthesis of Lewis acidic, zirconium-containing periodic mesoporous organosilicas (Zr-PMOs), in which zirconocene dichloride was employed as the zirconium source and BTEB was progressively substituted for TEOS.209 The resulting organosilanes were topologically similar to a purely inorganic Zr-SBA-15 material, but are strongly hydrophobic in nature. Although the one-pot metal doping protocol adopted resulted in relatively low densities of Zr incorporated into the final solid catalyst, hydrophobisation significantly enhanced the per acid site activity in the simultaneous esterification of FFAs and transesterification of TAGs in crude palm oil with methanol at 200 °C, with conversions approaching 90% after only 6 h (Fig. 15). As significant, the catalytic performance of the high organic content Zr-PMO materials was barely influenced by the addition of up to 20 wt% water to the feedstock, in contrast to the inorganic Zr-SBA-15 analogue which was completely poisoned by such water addition. The high water and fatty acid tolerance of these Zr-PMO catalysts renders them especially promising for biodiesel production from waste oil sources.
 | ||
| Fig. 15 (top) FAME yield and turnover frequency calculated for Zr-PMO materials in the methanolysis of crude palm oil highlighting the impact of catalyst hydrophobicity; and (bottom) FAME yield as a function of organic content for Zr-PMO materials in the presence of additional water in the crude palm oil reaction media evidencing superior water tolerance of hybrid solid acid catalysts. Reprinted from ref. 210. Copyright 2013 John Wiley and Sons. | ||
The incorporation of organic spectator groups (e.g. phenyl, methyl or propyl) during the sol–gel syntheses of SBA-15210 and MCM-41211 sulphonic acid silicas is also achievable via co-grafting or simple addition of the respective alkyl or aryltrimethoxysilane during co-condensation protocols. An experimental and computational study of sulphonic acid functionalised MCM-41 materials was undertaken in order to evaluate the effect of acid site density and surface hydrophobicity on catalyst acidity and associated performance.212 MCM-41 was an excellent candidate due to the availability of accurate models for the pore structure from kinetic Monte Carlo simulations,213 and was modified with surface groups to enable dynamic simulation of sulphonic acid and octyl groups co-attached within the MCM-41 pores. In parallel experiments, two catalyst series were investigated towards acetic acid esterification with butanol (Scheme 5). In one series, the propylsulphonic acid coverage was varied between θ(RSO3H) = 0–100% ML over the bare silica (MCM-SO3H). For the second octyl co-grafted series, both sulfonic acid and octyl coverages were tuned (MCM-Oc-SO3H). These materials allow the effect of lateral interactions between acid head groups and the role of hydrophobic octyl modifiers upon acid strength and activity to be separately probed.
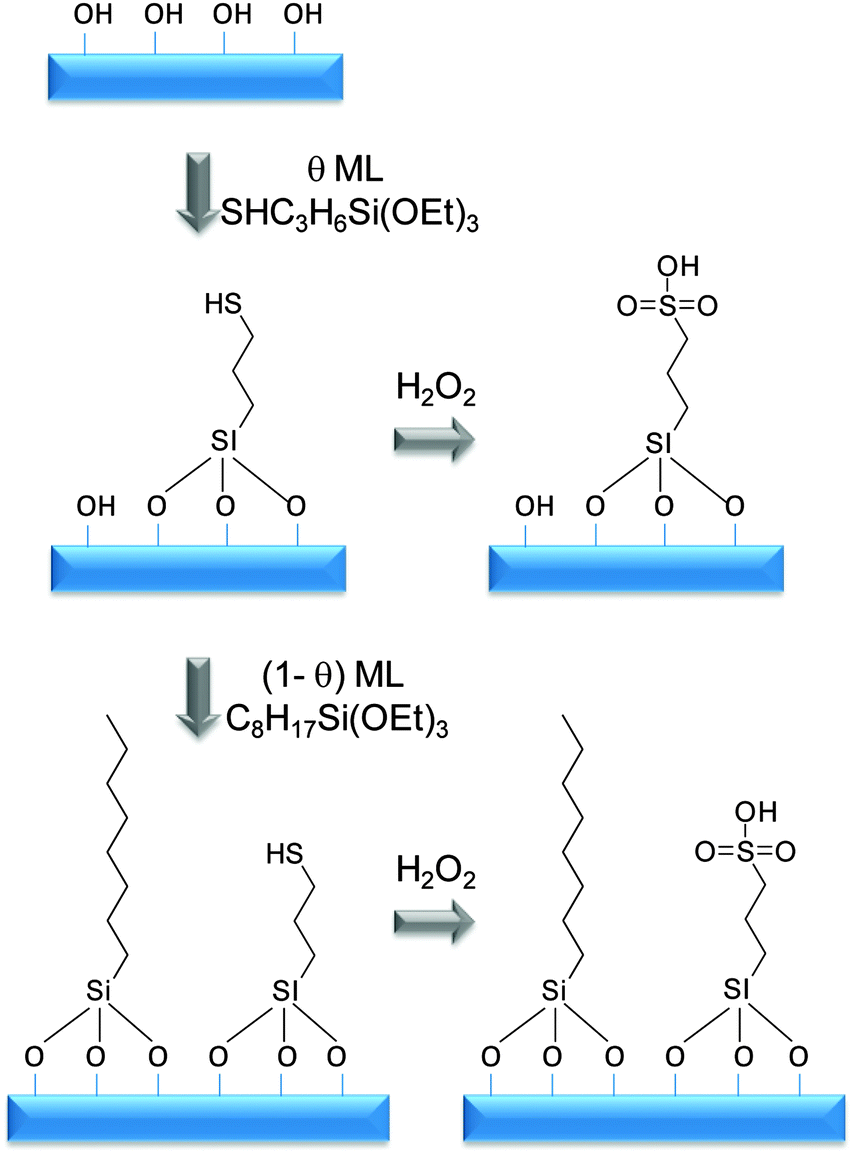 | ||
| Scheme 5 Protocol for the synthesis of sulfonic acid and octyl co-functionalised sulfonic acid MCM-41 catalysts. Adapted from ref. 213 with permission from The Royal Society of Chemistry. | ||
To avoid diffusion limitations, butanol esterification with acetic acid was selected as a model reaction (Fig. 16). Ammonia calorimetry revealed that the acid strength of polar MCM-SO3H materials increases from 87 to 118 kJ mol−1 with sulphonic acid loading. Co-grafted octyl groups dramatically enhance the acid strength of MCM-Oc-SO3H for submonolayer SO3H coverages, with ΔHads(NH3) rising to 103 kJ mol−1. The per site activity of the MCM-SO3H series in butanol esterification with acetic acid mirrors their acidity, increasing with SO3H content. Octyl surface functionalisation promotes esterification for all MCM-Oc-SO3H catalysts, doubling the turnover frequency of the lowest loading SO3H material. Molecular dynamic simulations indicate that the interaction of isolated sulphonic acid moieties with surface silanol groups is the primary cause of the lower acidity and activity of submonolayer samples within the MCM-SO3H series. Lateral interactions with octyl groups help to re-orient sulphonic acid headgroups into the pore interior, thereby enhancing acid strength and associated esterification activity.
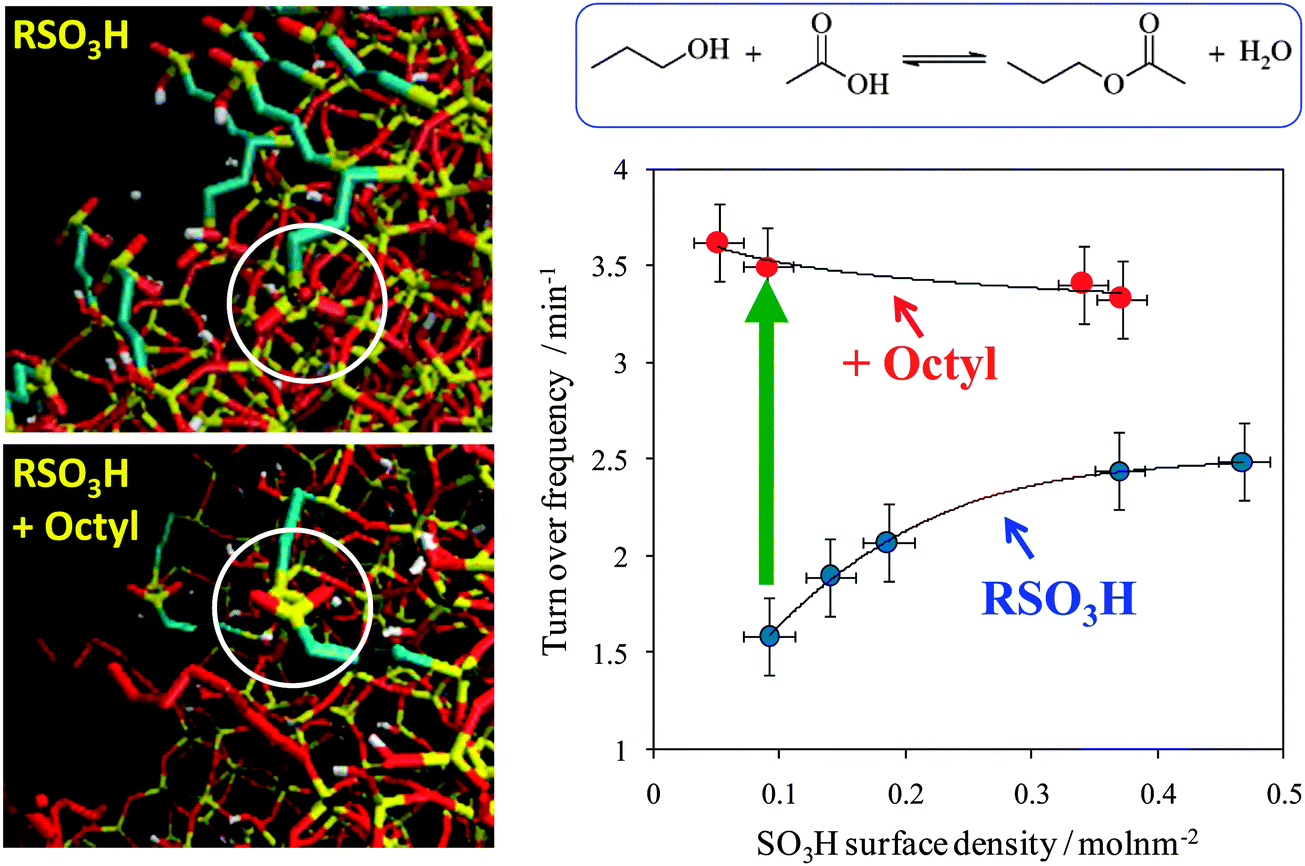 | ||
| Fig. 16 (left) Molecular dynamics simulations of MCM-SO3H and MCM-Oc-SO3H pore models highlighting the interaction between surface sulfonic acid and hydroxyl groups in the absence of co-grafted octyl chains; (right) influence of PrSO3H surface density and co-grafted octyl groups on catalytic performance in acetic acid esterification with butanol. Adapted from ref. 213 with permission from The Royal Society of Chemistry. | ||
In some cases, the introduction of hydrophobic functionalities may actually cap the active catalytic site. For example, post-modification of an arene-sulfonic acid SBA-15 by methoxytrimethylsilane deactivated the catalyst by capping the active sites with methyl groups and changing the textural properties, whereas methyl groups introduced via a one-pot synthesis did not affect activity towards the microwave-assisted transesterification of soybean oil with 1-butanol.214 Ethyl groups may also be introduced onto the surface of sulfonic acid modified SBA-15 to impart hydrophobicity. While such ethyl groups has no impact on overall conversions, they improved the initial rate of octanoic acid esterification by displacing reactively-formed water during the start of reaction.215
As discussed earlier in this review, hydrophobic solid acid catalysts with large pores are desirable to enhance in-pore mass transport of bulky bio-oils and fatty acids, and to minimise the impact of reactively-formed water during FFA esterification.37,216 Although many solid catalysts exist with potential in biodiesel production,154,217 research is increasingly focused on modifying surface hydrophobicity to achieve these goals. Hydrophobicity can be imparted to zeolites by incorporating organic species within their micropores; however, for transesterification involving long chain TAGs, large pore zeolites are preferable, with activity increasing with Si:Al ratio and surface hydrophobicity.195,218 Fe–Zn double metal cyanides (DMC), possessing only Lewis acid sites, were reported active for sunflower oil transesterification with methanol at 98% conversion. These catalysts exhibited good water tolerance, even in the presence of 20 wt% water in oil, possibly reflecting their surface hydrophobicity and higher coverage of adsorbed reactants.194 The hydrophobic nature of these catalysts was demonstrated by them in oil–water, water–toluene and water–CCl4 mixtures, wherein the catalyst remained suspended in the hydrophobic layer (Fig. 17).201,219 Fe–Zn DMC was compared against SZ and Al-MCM-41 for the esterification of long chain (C8–C18) FFAs, and the transesterification of soybean oil. SZ and Al-MCM-41 showed better conversion than DMC towards the fatty acids, but reverse was observed for the more hydrophobic soybean oil.201 Fe–Zn DMC possessed a hybrid structure containing both crystalline and amorphous phases; hydrophobicity ascribed to the presence of the latter phase.220
 | ||
| Fig. 17 Preferential dispersion of DMC in the nonpolar, organic phase, and SZ and Al-MCM-41 in the polar aqueous phase of (a) water–CCl4 and (b) water–toluene solvent mixtures. Reprinted with permission from ref. 202. Copyright 2010 American Chemical Society. | ||
Cesium-doped dodecatungstophosphoric acid (CsPW) has shown promise as a water-tolerant solid acid catalyst for the hydrolysis of ethyl acetate,221 and found subsequent employ in the transesterification of Eruca sativa Gars (ESG) oil.202 The authors claimed that CsPW exhibited excellent water-tolerance towards ESG transesterification, despite oil conversions falling by ∼90% upon the addition of only 1% water. Zn containing HPAs display more impressive credentials for transforming challenging feedstocks, with zinc dodecatungstophosphate nanotubes possessing Lewis and Brönsted acid sites effective for the for the simultaneous esterification and transesterification of palmitic acid, and transesterification of waste cooking oils with 26% FFA and 1% water.
The one-pot synthesis of a styrene modified sulfonic acid silica 15 was achieved by adding styrylethyl-trimethoxysilane during a conventional SBA-15 synthesis.222 Styryl groups polymerised on the silica surface imparted hydrophobicity. Subsequent acid functionalisation of these materials resulted in a polystyrene-modified sulfonic acid SBA-15, which was active for oleic acid esterification with n-butanol, and proved superior to SAC-13 and Amberlyst-15 due to the hydrophobic polystyrene coating and high surface area.223
Surface acidity has also been imparted to hydrophobic, mesoporous polydivinylbenzene (PDVB) by sulfonic acid grafting. Such materials were employed in tripalmitin transesterification with methanol, revealing that mesoporous PDVB with electron withdrawing –SO3H–SO2CF3 groups gave good activity with 91% yield maintained up to 5 re-uses. Contact angle measurements confirmed the hydrophobic nature and high oleophilicity of these materials. PDVB grafted with chlorosulfonic acid also generated hydrophobic solid acid catalysts for tripalmitin which were successfully transesterification whose performance (80% methyl palmitate yield) was superior to HPA, SBA-15-SO3H, Amberlyst 15, and mesoporous SO4–ZrO2. The same activity trend was observed for sunflower oil transesterification wherein all C16–C27 fatty acids were converted to FAMEs reflecting the higher adsorption capacity and hence reactivity of these PDVB acids.179,223,224 Polyaniline functionalised with methanosulfonic (MSA-Pani), camphorosulfonic (CSA-Pani) and lignosulfonic (LG-Pani) acids and polyaniline sulfate (S-Pani) also show promise in biodiesel synthesis with the LG-Pani catalyst possessing the greatest acid site density (3.62 mmolH+ g−1) and highest conversion due to the close proximity of hydrophobic centres to the active sites. Sulfonic acid containing ionic liquids have also been co-polymerised with divinyl benzene, to form a hydrophobic, solid acidic ionic liquid polymer (PIL) for the transesterification of rapeseed and waste cooking oils, outperforming homogeneous counterparts.182
Partial carbonisation and sulfonation of organic matter offers a route to combine acidity and hydrophobicity into carbon based mesoporous materials.225,226 Such solids are typically partially amorphous, but offer efficient transesterification of non-edible seed oils.107 It has proven difficult to introduce organic groups into the surface of ordered mesoporous carbons (OMCs) prepared through high temperature carbonisation, however surface pretreatment with H2O2 to introduce hydroxyl anchors enables their subsequent sulfonation and a resulting hydrophobic and stable acid catalyst for oleic acid esterification.227 Sulfonated single-walled carbon nanotubes (SO3H-SWCH) have also been investigated for palmitic acid esterification, exhibiting higher activity than other sulfonated carbons, such as oxidized SWCNHs (ox-SWCNHs), activated carbon (AC), and carbon black (CB), attributed to the stronger acidity of SO3H-SWCH and hydrophobicity of the carbon surface in the vicinity of acid sites,186 enabling it to even outperform liquid H2SO4. Another interesting class of porous hydrophobic catalysts are mesoporous titanosilicates which are active for biodiesel and biolubricant synthesis. Ti incorporation into the surface of mesoporous SBA-12 and SBA-16 generates Lewis acid sites which are active for esterification and transesterification. The high activity of these Lewis acid sites is comparable to that observed for Fe–Zn double metal cyanides.194 Solid state 29Si NMR studies show that Ti-SBA-16 is more hydrophobic than Ti-SBA-12. In biolubricant synthesis, for which surface hydrophobicity is crucial, Ti-SBA-16 is significantly more active than Ti-SBA-12.228
Lipase has also been immobilised on hydrophobic supports with a view to transesterifying water containing oils,229 wherein small amounts of water improved lipase activity.230 The application of lipase enzymes can be made more cost-effective by heterogenisation over a solid support, with hydrophobic supports both assisting lipase surface attachment and promoting FFA esterification and bio-oil transesterification. Burkholderia lipase supported on hydrophobic magnetic particles for olive oil transesterification gave 70% conversion to FAME even in the presence of up to 10% water and was readily recycled.231 FAME production from canola oil was also achieved using lipase immobilised on a hydrophobic, microporous styrene-divinylbenzene copolymer, wherein the support hydrophobicity mitigated the inhibitory effect of water and glycerol affording a 97% yield.232
Solid basic hydrotalcites also showed enhanced activity and reusability for soybean oil transesterification when dispersed over polyvinylalcohol (PVA) membranes, although increasing the hydrophobicity via polymer cross-linking lowered activity, presumably due to poor active site accessibility by the bulky substrate. Hydrophilicity versus hydrophobicity may be tuned over such membranes by succinic anhydride and acetic anhydride treatments, with a mix of hydrophilic and hydrophobic environments near the active hydrotalcite sites required for optimal transesterification.233An interesting contrast to the preceding systems (wherein water poisons FAME formation) was reported for CaO catalysed soybean transesterification, for which small amounts of water actually improve activity, attributed to an increase in the concentration of surface OH- active base sites.234 Mixed MgO–CaO also exhibited a surprising water tolerance in rapeseed oil transesterification, enabling 98% conversion with 2% water, with La2O3–CaO active even in the presence of 10% water.235,236
Periodic Mesoporous Organosilicas (PMOs) are a promising class of materials that can be used as catalyst supports for biodiesel production. PMOs are hybrid organic–inorganic materials with mesopore networks akin to SBA-15.236 Functionalisation of PMOs with catalytically active organic moieties is an emergent field of heterogeneous catalysis, and since the organic groups are dispersed throughout the framework (rather than confined to hydroxylated patches of the surface212), active sites and hydrophobic centres can be co-located in high concentrations. Methylpropyl sulfonic acid functionalised phenylene- and ethyl-bridged PMOs have been synthesised and tested for the transesterification of sunflower oil, canola oil, corn oil, refined olive oil and olive sludge.237 These functionalised PMOs gave comparable or better activity than SBA-15-PrSO3H under optimised conditions, with the ethyl-bridged PMO showing highest activity with a 98% yield. Water adsorption studies proved that the phenylene-bridged PMO was more hydrophobic than the ethyl-bridged variant, but less active, showing that a balance of hydrophobic versus hydrophilic mesostructural properties are necessary for optimum transesterification.
Heterogeneous catalysts with tunable hydrophobicity, acid/base character, and good thermal stability, whether based upon polymeric or inorganic frameworks, are hence promising new solutions to TAG transesterification and FFA esterification of high moisture content feedstocks.
6. Influence of reactor design and operating conditions
One other development likely to impact on the commercial exploitation of heterogeneous catalysts for biodiesel production is the design of innovative chemical reactors to facilitate continuous processing of viscous bio-oils. Although many industrial biodiesel production plants operate in batch mode at a significant scale (∼7000 tons year−1),238–240 there is a need to move towards heterogeneously catalysed, continuous flow reactors in order to avoid the separation issues of homogeneous catalysts and drawbacks of batch mode (notably increased capital investment required to run at large volumes and increased labour costs of a start/stop process)241 and increase the scale of operation (8000–125000 tons year−1).239,240 A range of process engineering solutions have been considered for the continuous esterification of FFAs, including the use of fixed bed242 or microchannel-flow reactors,243 pervaporation methods,244 and reactive distillation.245,246 Process intensification methods in biodiesel production have been reviewed in depth elsewhere.247,248
Reactive distillation combines chemical conversion and separation steps in a single stage. This simplifies the process flow sheets, reduces production costs, and extends catalyst lifetimes through the continuous removal of water from the system. However, this technique is only applicable if the reaction is compatible with the temperatures and pressures required for the distillation. Kiss et al. demonstrated this approach for the esterification of dodecanoic acid with a range of alcohols catalysed by sulphated zirconia.245 Their reactive distillation was 100% selective, permitted shorter residence times than comparable flow systems, and did not require excess alcohol. The latter is a major advantage over the overwhelming majority of conventional biodiesel syntheses wherein, since reaction between the triglyceride and alcohol is reversible, large alcohol excesses are normally required to achieve full conversion (the excess alcohol must then be separated and re-used to ensure economic process viability).
Any continuous flow reactor must be designed appropriately to harness the full potential of the integrated heterogeneous catalyst; plug flow is a desirable characteristic since it permits tight control over the product composition, and hence minimises downstream separation processes, and associated capital investment and running costs. Conventional plug flow reactors are ill-suited to slow reactions such as FFA esterification and TAG transesterification, since they require very high length:diameter ratios to achieve good mixing, and in any event are problematic due to their large footprints and pumping duties, and control difficulties. Oscillatory Baffled Reactors (OBRs) circumvent these problems by oscillating the reaction fluid through orifice plate baffles to achieve efficient mixing and plug flow,249 thereby decoupling mixing from the net fluid flow in a scalable fashion, enabling long reaction times on an industrial scale, and have been applied to homogeneously catalysed biodiesel synthesis.250 Vortical mixing in the OBR also offers an effective, controllable method of uniformly suspending solid particles and was recently utilised to entrain a PrSO3H-SBA-15 mesoporous silica within a glass OBR under an oscillatory flow for the continuous esterification of propanoic, hexanoic, lauric and palmitic acid (Fig. 18).42 Excellent semi-quantitative agreement was obtained between the kinetics of hexanoic acid esterification within the OBR and a conventional stirred batch reactor, with fatty acid chain length identified as a key predictor of solid acid activity. Continuous esterification within the OBR improved ester yields compared with batch operation due to water by-product removal from the catalyst reaction zone, evidencing the versatility of the OBR for heterogeneous flow chemistry and potential role as a new clean catalytic technology.
 | ||
| Fig. 18 Schematic of reactor flow and mixing characteristics within an OBR, and associated optical images of a PrSO3H-SBA-15 solid acid powder without oscillation (undergoing sedimentation) or with a 4.5 Hz oscillation (entrained within baffles). Adapted from ref. 42 with permission from The Royal Society of Chemistry. | ||
Phase equilibria considerations are very important in biodiesel production via TAG transesterification with methanol, since the reactant and alcohol are generally immiscible, whereas the FAME product is miscible, hampering mass transport and retarding reaction. Separation and purification of the product phase, a mixture of solid catalyst, unreacted oil, glycerol and biodiesel, adds further complexity and cost to production.251 These problems may be alleviated through the use of membrane reactors,252–256 wherein the reactor walls are made of a semi-permeable material designed to allow passage of the FAME/glycerol phase, while retaining the oil-rich/MeOH emulsion for further reaction. Xu et al. utilised a MCM-41 supported p-toluenesulfonic acid catalyst to pack a ceramic membrane tube for the transesterification of a recirculating soybean oil and methanol feed (Fig. 19a). A higher biodiesel yield was obtained with the membrane reactor than with a homogeneous p-toluenesulfonic acid catalyst under comparable conditions in batch mode (84 versus 66%). Catalyst re-used evidenced only a minor loss of activity (92% of original after the third cycle).254 Biodiesel yield was a strong function of circulation velocity; low velocities improved permeation efficiency, while high velocities enhanced reactant mixing intensity. Although membrane reactors offer efficient transesterification and separation, they require high catalyst volumes, for example a 202 cm3 continuous reactor employed 157 g of a microporous TiO2/Al2O3 membrane packed with potassium hydroxide supported on palm shell activated carbon to produce high quality methyl esters from palm oil (Fig. 19b).252
 | ||
| Fig. 19 Schematic of recirculating packed membrane reactors for continuous biodiesel production via (a) solid acid and (b) base catalysts. Reprinted from ref. 252 and 254. Copyright (2011 and 2014), with permission from Elsevier. | ||
Enzymatic catalysed biodiesel production has been reported in both continuous257,258 and batch modes.259 Nature has developed a range of lipase biocatalysts for the selective synthesis of FAME at low reaction temperature, which are tolerate to high FFA levels.260,261 Immobilisation on solid supports enables such biocatalysts to be used in continuous mode with low methanol:oil ratios.262 However, there are numerous shortcomings of biocatalysts including high enzyme costs, long residence times, and low biodiesel yields. Some enzymes can also be deactivated by short chain alcohols and the glycerol by-product;263 this problem can be overcome through the use of organic solvents to extract the alcohols and glycerol, but this adds further complexity and cost, and weakens the green credentials of biodiesel production. Enzymes must also operate in the presence of water in order to avoid denaturation, however this additional water must be subsequently removed from the resulting fuel to meet biodiesel standards (<0.05 vol% H2O), these drying steps introducing further costs. An alternative approach is the use of near-critical264 or supercritical CO2255,256 as a reaction medium to minimise enzyme inhibition by methanol, enhance oil solubility and diffusion, and assist catalyst/biodiesel separation via simple depressurisation. The associated strengths and weaknesses of supercritical biodiesel production are reviewed elsewhere.265
Ultrasound266,267 and microwaves268,269 have been explored as a means of eliminating heat and mass transfer limitations, and shortening residence times to achieve high biodiesel conversions. Ultrasound was used by Gude et al. in place of thermal heating for the transesterification of waste cooking oil,266 allowing efficient heating to a temperature of 60–65 °C and lowering reaction times to 1–2 min. Chand et al. observed similar improvements in heat transfer and reaction time applying ultrasonication to soybean oil transesterification.270 However, both groups employed a homogeneous NaOH catalyst, hindering product purification. Ultrasound was used with a heterogeneous catalyst for continuous biodiesel production from palm oil by Salamatinia et al.271 BaO and SrO catalysts were tested, and ultrasound again found to reduce the reaction times and catalyst loadings needed to achieve >95% FAME yields. Cost analysis of an ultrasonic process suggests it would be at least three times more expensive to run than a conventionally heated continuous biodiesel reactor.270 The origin of ultrasonic enhancements in respect of reaction mixing via e.g. cavitation or micro-streaming, remains a matter of debate.272 Microwaves have been coupled with continuous flow reactors for the transesterification of waste cooking oil, accelerating biodiesel production compared to conventional thermal heating, and hence higher throughput.269 The majority of microwave studies to date have focused on homogeneously catalysed processes, although some innovative combinations of waste derived (eggshell) solid catalysts and microwaves are emerging.273 Such microwave systems also require less solvent and catalyst. However, microwave penetration depth is a limiting factor268 which may restrict scale-up from laboratory reactor designs, and uncontrolled and irregular heat distribution can result in ‘hot spots’ and ‘cold spots’.267,268
7. Future directions
If sourced and produced in a sustainable fashion, biodiesel has the potential to play an important role in meeting renewable fuel targets. However, developments in materials design and construction are critical to achieve significant improvements in heterogeneously catalysed biodiesel production. Designer solid acid and base catalysts with tailored surface properties and pore networks offer process improvements over existing, commercial homogeneous catalysed production employing liquid bases, facilitating simple catalyst separation and fuel purification, coupled with continuous biodiesel synthesis. Tuning the surface hydrophobicity of heterogeneous catalysts can strongly influence oil transesterification and FFA esterification through the expulsion of water away from active catalytic centres, thus limiting undesired reverse hydrolysis processes, notably in high water content waste oils. Solid materials capable of simultaneous FFA esterification and TAG transesterification under mild conditions present a major challenge for catalytic scientists, although (insoluble) high area superacids represent a step in this direction. We predict that in the future, hierarchical solid acids may be employed to first hydrolyse non-edible oil feedstocks, and subsequently esterify the resulting FFAs to FAME. Synthesis of nanostructured (e.g. nanocrystalline) catalysts and the application of surface-initiated, controlled polymerisation to functionalise oxide surfaces with polymeric organic species to create hybrid organic–inorganic architectures with high active site loadings, will prove valuable in the quest for enhanced catalyst performance.Despite concerns over long term biodiesel use in high performance engines, the implementation of FAME containing longer chain (>C18) esters in heavy-duty diesel engines should prove less problematic to on short timecales. However, the widespread uptake and development of next-generation biodiesel fuels requires progressive government policies and incentive schemes to place biodiesel on a comparative footing with (heavily subsidised) fossil-fuels. Blending of biodiesel with pyrolysis oil derived from lignocellulosic waste is an attractive route to power low-medium scale Combined Heat and Power (CHP) engines. Increasing use of waste or low grade oil sources remains a challenge for existing heterogeneous catalysts, since the high concentration of impurities (acid, moisture, heavy metals) induce rapid on-stream deactivation, and necessitate improved upstream oil purification, or more robust catalyst formulations tolerant to such components. Feedstock selection is dominated by regional availability, however the drive to use non-edible oil sources in areas where they cannot be readily sourced will require close attention to the entire supply chain and emissions/costs associated with new transportation networks, and may favour genetic modification of plant and algal strains to adapt to non-native climates.
The viscosity and attendant poor miscibility of many oil feedstocks with light alcohols continues to hamper the use of new heterogeneous catalysts for continuous biodiesel production, from both a materials and engineering perspective. Future process optimisation and growth in biodiesel supply and demand needs a concerted effort between catalyst chemists, chemical engineers and experts in molecular simulation in order to take advantage of innovative reactor designs and develop catalysts and reactors in tandem. Alternative reactor technologies and process intensification via e.g. reactive distillation and oscillatory flow reactors will facilitate distributed biodiesel production. It is essential that technical advances in both materials chemistry and reactor engineering are pursued if biodiesel is to remain a key player in the renewable energy sector during the 21st century.
Acknowledgements
A.F.L. thanks the EPSRC for the award of a Leadership Fellowship (EP/G007594/4). K.W. thanks the Royal Society for the award of an Industry Fellowship.References
- S. Kretzmann, http://priceofoil.org/2013/11/26/new-analysis-shows-growing-fossil-reserves-shrinking-carbon-budget/.
- C. C. Authority, Reducing Australia's Greenhouse Gas Emissions – Targets and Progress Review Draft Report, Commonwealth of Australia, 2013.
- C. C. Secretariat, The critical decade 2013 Climate change science, risks and responses, Commonwealth of Australia, 2013.
- I. E. Agency, Prospect of limiting the global increase in temperature to 2 °C is getting bleaker, http://www.iea.org/newsroomandevents/news/2011/may/name,19839,en.html.
- I. E. Agency, Redrawing the Energy Climate Map, 2013.
- U. S. E. I. Administration, International Energy Outlook 2013, 2013.
- PwC, World in 2050. The BRICs and beyond: prospects, challenges and opportunities, 2013.
- N. Armaroli and V. Balzani, Angew. Chem., Int. Ed., 2007, 46, 52–66 CrossRef CAS PubMed.
- P. Azadi, O. R. Inderwildi, R. Farnood and D. A. King, Renewable Sustainable Energy Rev., 2013, 21, 506–523 CrossRef CAS PubMed.
- J. M. Thomas, Proceedings of the Royal Society A: Mathematical, Physical and Engineering Science, 2012, 468, 1884–1903 CrossRef CAS.
- G. A. Somorjai, H. Frei and J. Y. Park, J. Am. Chem. Soc., 2009, 131, 16589–16605 CrossRef CAS PubMed.
- A. Demirbas, Energy Policy, 2007, 35, 4661–4670 CrossRef PubMed.
- T. P. Vispute, H. Zhang, A. Sanna, R. Xiao and G. W. Huber, Science, 2010, 330, 1222–1227 CrossRef CAS PubMed.
- P. M. Mortensen, J. D. Grunwaldt, P. A. Jensen, K. G. Knudsen and A. D. Jensen, Appl. Catal., A, 2011, 407, 1–19 CrossRef CAS PubMed.
- R. Luque, L. Herrero-Davila, J. M. Campelo, J. H. Clark, J. M. Hidalgo, D. Luna, J. M. Marinas and A. A. Romero, Energy Environ. Sci., 2008, 1, 542–564 CAS.
- C. S. K. Lin, L. A. Pfaltzgraff, L. Herrero-Davila, E. B. Mubofu, S. Abderrahim, J. H. Clark, A. A. Koutinas, N. Kopsahelis, K. Stamatelatou, F. Dickson, S. Thankappan, Z. Mohamed, R. Brocklesby and R. Luque, Energy Environ. Sci., 2013, 6, 426–464 CAS.
- D. W. McLaughlin, Conserv. Biol., 2011, 25, 1117–1120 CrossRef PubMed.
- F. Danielsen, H. Beukema, N. D. Burgess, F. Parish, C. A. Brühl, P. F. Donald, D. Murdiyarso, B. E. N. Phalan, L. Reijnders, M. Struebig and E. B. Fitzherbert, Conserv. Biol., 2009, 23, 348–358 CrossRef PubMed.
- W. M. J. Achten, L. Verchot, Y. J. Franken, E. Mathijs, V. P. Singh, R. Aerts and B. Muys, Biomass Bioenergy, 2008, 32, 1063–1084 CrossRef CAS PubMed.
- T. M. Mata, A. A. Martins and N. S. Caetano, Renewable Sustainable Energy Rev., 2010, 14, 217–232 CrossRef CAS PubMed.
- BP, BP Energy Outlook 2030, 2011.
- G. Knothe, Top. Catal., 2010, 53, 714–720 CrossRef CAS.
- M. J. Climent, A. Corma, S. Iborra and A. Velty, J. Catal., 2004, 221, 474–482 CrossRef CAS PubMed.
- U. Constantino, F. Marmottini, M. Nocchetti and R. Vivani, Eur. J. Inorg. Chem., 1998, 1439–1446 CrossRef.
- K. Narasimharao, A. Lee and K. Wilson, J. Biobased Mater. Bioenergy, 2007, 1, 19–30 Search PubMed.
- M. R. Othman, Z. Helwani, Martunus and W. J. N. Fernando, Appl. Organomet. Chem., 2009, 23, 335–346 CrossRef CAS.
- Y. Liu, E. Lotero, J. G. Goodwin and X. Mo, Appl. Catal., A, 2007, 33, 138–148 CrossRef PubMed.
- J. Geuens, J. M. Kremsner, B. A. Nebel, S. Schober, R. A. Dommisse, M. Mittelbach, S. Tavernier, C. O. Kappe and B. U. W. Maes, Energy Fuels, 2007, 22, 643–645 CrossRef.
- J. Hu, Z. Du, Z. Tang and E. Min, Ind. Eng. Chem. Res., 2004, 43, 7928–7931 CrossRef CAS.
- G. Knothe, Fuel Process. Technol., 2005, 86, 1059–1070 CrossRef CAS PubMed.
- F. Ma and M. A. Hanna, Bioresour. Technol., 1999, 70, 1–15 CrossRef CAS.
- E. Lotero, Y. Liu, D. E. Lopez, K. Suwannakarn, D. A. Bruce and J. G. Goodwin, Ind. Eng. Chem. Res., 2005, 44, 5353–5363 CrossRef CAS.
- R. Luque, J. C. Lovett, B. Datta, J. Clancy, J. M. Campelo and A. A. Romero, Energy Environ. Sci., 2010, 3, 1706–1721 CAS.
- J.-P. Dacquin, A. F. Lee and K. Wilson, Thermochemical Conversion of Biomass to Liquid Fuels and Chemicals, The Royal Society of Chemistry, 2010, pp. 416–434 Search PubMed.
- K. Wilson and A. F. Lee, Catal. Sci. Technol., 2012, 2, 884 CAS.
- L. J. Konwar, J. Boro and D. Deka, Renewable Sustainable Energy Rev., 2014, 29, 546–564 CrossRef CAS PubMed.
- A. Islam, Y. H. Taufiq-Yap, C.-M. Chu, E.-S. Chan and P. Ravindra, Process Saf. Environ. Prot., 2013, 91, 131–144 CrossRef CAS PubMed.
- K. Ramachandran, T. Suganya, N. Nagendra Gandhi and S. Renganathan, Renewable Sustainable Energy Rev., 2013, 22, 410–418 CrossRef CAS PubMed.
- Y. M. Sani, W. M. A. W. Daud and A. R. Abdul Aziz, Appl. Catal., A, 2014, 470, 140–161 CrossRef CAS PubMed.
- I. M. Atadashi, M. K. Aroua, A. R. Abdul Aziz and N. M. N. Sulaiman, J. Ind. Eng. Chem., 2013, 19, 14–26 CrossRef CAS PubMed.
- Y. M. Sani, W. M. A. W. Daud and A. R. Abdul Aziz, J. Environ. Chem. Eng., 2013, 1, 113–121 CrossRef CAS PubMed.
- V. C. Eze, A. N. Phan, C. Pirez, A. P. Harvey, A. F. Lee and K. Wilson, Catal. Sci. Technol., 2013, 3, 2373–2379 CAS.
- S. P. Singh and D. Singh, Renewable Sustainable Energy Rev., 2010, 14, 200–216 CrossRef CAS PubMed.
- P. Schenk, S. Thomas-Hall, E. Stephens, U. Marx, J. Mussgnug, C. Posten, O. Kruse and B. Hankamer, BioEnergy Res., 2008, 1, 20–43 CrossRef PubMed.
- R. E. H. Sims, W. Mabee, J. N. Saddler and M. Taylor, Bioresour. Technol., 2010, 101, 1570–1580 CrossRef CAS PubMed.
- J. Kansedo, K. T. Lee and S. Bhatia, Biomass Bioenergy, 2009, 33, 271–276 CrossRef CAS PubMed.
- J. M. Encinar, J. F. Gonzalez, A. Pardal and G. Martinez, Fuel Process. Technol., 2010, 91, 1530–1536 CrossRef CAS PubMed.
- J. Calero, D. Luna, E. D. Sancho, C. Luna, F. M. Bautista, A. A. Romero, A. Posadillo and C. Verdugo, Fuel, 2014, 122, 94–102 CrossRef CAS PubMed.
- V. Scholz and J. N. da Silva, Biomass Bioenergy, 2008, 32, 95–100 CrossRef CAS PubMed.
- E. Akbar, Z. Yaakob, S. K. Kamarudin, M. Ismail and J. Salimon, Eur. J. Sci. Res., 2009, 29, 396–403 Search PubMed.
- A. Karmakar, S. Karmakar and S. Mukherjee, Renewable Sustainable Energy Rev., 2012, 16, 1050–1060 CrossRef CAS PubMed.
- Y. Chisti, Biotechnol. Adv., 2007, 25, 294–306 CrossRef CAS PubMed.
- H. N. Bhatti, M. A. Hanif, M. Qasim and R. Ataur, Fuel, 2008, 87, 2961–2966 CrossRef CAS PubMed.
- D. M. Kargbo, Energy Fuels, 2010, 24, 2791–2794 CrossRef CAS.
- D. Frieden, N. Pena, D. N. Bird, H. Schwaiger and L. Canella, Center for International Forestry Research (CIFOR), Bogor, Indonesia, 2011, p. 61 Search PubMed.
- L. Lardon, A. Hélias, B. Sialve, J.-P. Steyer and O. Bernard, Environ. Sci. Technol., 2009, 43, 6475–6481 CrossRef CAS.
- ASTM Standard D6751, Standard Specification for Biodiesel Fuel Blend Stock (B100) for Middle Distillate Fuels, ASTM International, West Conshohocken, PA, 2012, DOI: 0.1520/D6751-12, http://www.astm.org.
- ASTM Standard D6751, Standard Specification for Biodiesel Fuel Blend Stock (B100) for Middle Distillate Fuels, ASTM International, West Conshohocken, PA, 2012, DOI: 0.1520/D6751-12, http://www.astm.org.
- D. Y. C. Leung, X. Wu and M. K. H. Leung, Appl. Energy, 2010, 87, 1083–1095 CrossRef CAS PubMed.
- Y. C. Sharma and B. Singh, Renewable Sustainable Energy Rev., 2009, 13, 1646–1651 CrossRef CAS PubMed.
- A. Demirbas, Energy Convers. Manage., 2009, 50, 14–34 CrossRef CAS PubMed.
- A. Elbehri, A. Segerstedt and P. Liu, Biodiesel and sustainability challenge: A global assessment of sustainability issues, trends and policies for biofuels and related feedstocks, Food and Agriculture Organization of the United Nations, 2013 Search PubMed.
- R. Sarin, M. Sharma, S. Sinharay and R. K. Malhotra, Fuel, 2007, 86, 1365–1371 CrossRef CAS PubMed.
- M. K. Lam, K. T. Lee and A. R. Mohamed, Biotechnol. Adv., 2010, 28, 500–518 CrossRef CAS PubMed.
- J. C. Escobar, E. S. Lora, O. J. Venturini, E. E. Yáñez, E. F. Castillo and O. Almazan, Renewable Sustainable Energy Rev., 2009, 13, 1275–1287 CrossRef CAS PubMed.
- N. H. Tran, J. R. Bartlett, G. S. K. Kannangara, A. S. Milev, H. Volk and M. A. Wilson, Fuel, 2010, 89, 265–274 CrossRef CAS PubMed.
- G. Knothe, Green Chem., 2011, 13, 3048–3065 RSC.
- A. Karmakar, S. Karmakar and S. Mukherjee, Bioresour. Technol., 2010, 101, 7201–7210 CrossRef CAS PubMed.
- The Biodiesel Handbook, AOCS Publishing, 2005 Search PubMed.
- T. P. Durrett, C. Benning and J. Ohlrogge, Plant J., 2008, 54, 593–607 CrossRef CAS PubMed.
- E. N. Frankel, Lipid Oxidation, The Oily Press, Bridgewater, England, 2nd edn, 2005 Search PubMed.
- C. A. W. Allen, K. C. Watts, R. G. Ackman and M. J. Pegg, Fuel, 1999, 78, 1319–1326 CrossRef CAS.
- G. Knothe and K. R. Steidley, Energy Fuels, 2005, 19, 1192–1200 CrossRef CAS.
- G. Knothe, S. C. Cermak and R. L. Evangelista, Energy Fuels, 2009, 23, 1743–1747 CrossRef CAS.
- R. Radakovits, R. E. Jinkerson, A. Darzins and M. C. Posewitz, Eukaryotic Cell, 2010, 9, 486–501 CrossRef CAS PubMed.
- Y. Ono and T. Baba, Catal. Today, 1997, 38, 321–337 CrossRef CAS.
- H. Hattori, Chem. Rev., 1995, 95, 537–558 CrossRef CAS.
- M. C. G. Albuquerque, D. C. S. Azevedo, C. L. Cavalcante Jr, J. Santamaría-González, J. M. Mérida-Robles, R. Moreno-Tost, E. Rodríguez-Castellón, A. Jiménez-López and P. Maireles-Torres, J. Mol. Catal. A: Chem., 2009, 300, 19–24 CrossRef CAS PubMed.
- G. R. Peterson and W. P. Scarrah, J. Am. Oil Chem. Soc., 1984, 61, 1593–1597 CrossRef CAS.
- M. Verziu, S. M. Coman, R. Richards and V. I. Parvulescu, Catal. Today, 2011, 167, 64–70 CrossRef CAS PubMed.
- M. López Granados, D. Martin Alonso, A. C. Alba-Rubio, R. Mariscal, M. Ojeda and P. Brettes, Energy Fuels, 2009, 23, 2259–2263 CrossRef.
- M. Di Serio, R. Tesser, L. Casale, A. Dapos;Angelo, M. Trifuoggi and E. Santacesaria, Top. Catal., 2010, 53, 811–819 CrossRef CAS.
- M. Su, R. Yang and M. Li, Fuel, 2013, 103, 398–407 CrossRef CAS PubMed.
- C. S. MacLeod, A. P. Harvey, A. F. Lee and K. Wilson, Chem. Eng. J., 2008, 135, 63–70 CrossRef CAS PubMed.
- J. Montero, K. Wilson and A. Lee, Top. Catal., 2010, 53, 737–745 CrossRef CAS.
- R. S. Watkins, A. F. Lee and K. Wilson, Green Chem., 2004, 6, 335–340 RSC.
- D. M. Alonso, R. Mariscal, M. L. Granados and P. Maireles-Torres, Catal. Today, 2009, 143, 167–171 CrossRef CAS PubMed.
- M. Verziu, B. Cojocaru, J. Hu, R. Richards, C. Ciuculescu, P. Filip and V. I. Parvulescu, Green Chem., 2008, 10, 373–381 RSC.
- K. Zhu, J. Hu, C. Kübel and R. Richards, Angew. Chem., Int. Ed., 2006, 45, 7277–7281 CrossRef CAS PubMed.
- J. M. Montero, P. Gai, K. Wilson and A. F. Lee, Green Chem., 2009, 11, 265–268 RSC.
- J. M. Montero, D. R. Brown, P. L. Gai, A. F. Lee and K. Wilson, Chem. Eng. J., 2010, 161, 332–339 CrossRef CAS PubMed.
- J. J. Woodford, C. M. A. Parlett, J.-P. Dacquin, G. Cibin, A. Dent, J. Montero, K. Wilson and A. F. Lee, J. Chem. Technol. Biotechnol., 2014, 89, 73–80 CrossRef CAS.
- Y. L. Meng, B. Y. Wang, S. F. Li, S. J. Tian and M. H. Zhang, Bioresour. Technol., 2013, 128, 305–309 CrossRef CAS PubMed.
- W. Xie and L. Zhao, Energy Convers. Manage., 2014, 79, 34–42 CrossRef CAS PubMed.
- W. Xie and L. Zhao, Energy Convers. Manage., 2013, 76, 55–62 CrossRef CAS PubMed.
- E. Rashtizadeh, F. Farzaneh and Z. Talebpour, Bioresour. Technol., 2013, 154C, 32–37 Search PubMed.
- J.-X. Wang, K.-T. Chen, J.-S. Wu, P.-H. Wang, S.-T. Huang and C.-C. Chen, Fuel Process. Technol., 2012, 104, 167–173 CrossRef CAS PubMed.
- Y.-D. Long, F. Guo, Z. Fang, X.-F. Tian, L.-Q. Jiang and F. Zhang, Bioresour. Technol., 2011, 102, 6884–6886 CrossRef CAS PubMed.
- Y.-D. Long, Z. Fang, T.-C. Su and Q. Yang, Appl. Energy, 2014, 113, 1819–1825 CrossRef CAS PubMed.
- T. M. Barnard, N. E. Leadbeater, M. B. Boucher, L. M. Stencel and B. A. Wilhite, Energy Fuels, 2007, 21, 1777–1781 CrossRef CAS.
- X.-f. Li, Y. Zuo, Y. Zhang, Y. Fu and Q.-x. Guo, Fuel, 2013, 113, 435–442 CrossRef CAS PubMed.
- X. Liang, S. Gao, H. Wu and J. Yang, Fuel Process. Technol., 2009, 90, 701–704 CrossRef CAS PubMed.
- H. Wu, J. Zhang, Y. Liu, J. Zheng and Q. Wei, Fuel Process. Technol., 2014, 119, 114–120 CrossRef CAS PubMed.
- Y. Li, W. Zhang, L. Zhang, Q. Yang, Z. Wei, Z. Feng and C. Li, J. Phys. Chem. B, 2004, 108, 9739–9744 CrossRef CAS.
- W. Xie and M. Fan, Chem. Eng. J., 2014, 239, 60–67 CrossRef CAS PubMed.
- K.-T. Chen, J.-X. Wang, Y.-M. Dai, P.-H. Wang, C.-Y. Liou, C.-W. Nien, J.-S. Wu and C.-C. Chen, J. Taiwan Inst. Chem. Eng., 2013, 44, 622–629 CrossRef CAS PubMed.
- F. A. Dawodu, O. Ayodele, J. Xin, S. Zhang and D. Yan, Appl. Energy, 2014, 114, 819–826 CrossRef CAS PubMed.
- X. Fu, D. Li, J. Chen, Y. Zhang, W. Huang, Y. Zhu, J. Yang and C. Zhang, Bioresour. Technol., 2013, 146, 767–770 CrossRef CAS PubMed.
- B. L. A. Prabhavathi Devi, T. Vijai Kumar Reddy, K. Vijaya Lakshmi and R. B. N. Prasad, Bioresour. Technol., 2013, 153, 370–373 CrossRef PubMed.
- Y. Shen, P. Zhao and Q. Shao, Microporous Mesoporous Mater., 2014, 188, 46–76 CrossRef CAS PubMed.
- K. Gombotz, R. Parette, G. Austic, D. Kannan and J. V. Matson, Fuel, 2012, 92, 9–15 CrossRef CAS PubMed.
- A. Molaei Dehkordi and M. Ghasemi, Fuel Process. Technol., 2012, 97, 45–51 CrossRef CAS PubMed.
- N. Santiago-Torres, I. C. Romero-Ibarra and H. Pfeiffer, Fuel Process. Technol., 2014, 120, 34–39 CrossRef CAS PubMed.
- G. G. Santillán-Reyes and H. Pfeiffer, Int. J. Greenhouse Gas Control, 2011, 5, 1624–1629 CrossRef PubMed.
- P. Hernández-Hipólito, M. García-Castillejos, E. Martínez-Klimova, N. Juárez-Flores, A. Gómez-Cortés and T. E. Klimova, Catal. Today, 2014, 220–222, 4–11 CrossRef PubMed.
- D. G. Cantrell, L. J. Gillie, A. F. Lee and K. Wilson, Appl. Catal., A, 2005, 287, 183–190 CrossRef CAS PubMed.
- M. Di Serio, M. Ledda, M. Cozzolino, G. Minutillo, R. Tesser and E. Santacesaria, Ind. Eng. Chem. Res., 2006, 45, 3009–3014 CrossRef CAS.
- F. Cavani, F. Trifirò and A. Vaccari, Catal. Today, 1991, 11, 173–301 CrossRef CAS.
- N. Barakos, S. Pasias and N. Papayannakos, Bioresour. Technol., 2008, 99, 5037–5042 CrossRef CAS PubMed.
- J. M. Fraile, N. García, J. A. Mayoral, E. Pires and L. Roldán, Appl. Catal., A, 2009, 364, 87–94 CrossRef CAS PubMed.
- H. E. Cross and D. R. Brown, Catal. Commun., 2010, 12, 243–245 CrossRef CAS PubMed.
- T. Hibino and M. Kobayashi, J. Mater. Chem., 2005, 15, 653–656 RSC.
- J. M. Hidalgo, C. Jiménez-Sanchidrián and J. R. Ruiz, Appl. Catal., A, 2014, 470, 311–317 CrossRef CAS PubMed.
- J. J. Creasey, A. Chieregato, J. C. Manayil, C. M. A. Parlett, K. Wilson and A. F. Lee, Catal. Sci. Technol., 2014, 4, 861–870 CAS.
- E. Géraud, V. Prévot, J. Ghanbaja and F. Leroux, Chem. Mater., 2005, 18, 238–240 CrossRef.
- E. Géraud, S. Rafqah, M. Sarakha, C. Forano, V. Prevot and F. Leroux, Chem. Mater., 2007, 20, 1116–1125 CrossRef.
- J. J. Woodford, J.-P. Dacquin, K. Wilson and A. F. Lee, Energy Environ. Sci., 2012, 5, 6145–6150 CAS.
- M. N. V. Ravi Kumar, U. Bakowsky and C. M. Lehr, Biomaterials, 2004, 25, 1771–1777 CrossRef CAS PubMed.
- D. Highley, A. Bloodworth and R. Bate, Dolomite-Mineral planning factsheet, British Geological Survey, 2006 Search PubMed.
- D. Sutton, B. Kelleher and J. R. H. Ross, Fuel Process. Technol., 2001, 73, 155–173 CrossRef CAS.
- K. Wilson, C. Hardacre, A. F. Lee, J. M. Montero and L. Shellard, Green Chem., 2008, 10, 654–659 RSC.
- O. Ilgen, Fuel Process. Technol., 2011, 92, 452–455 CrossRef CAS PubMed.
- Z. A. Shajaratun Nur, Y. H. Taufiq-Yap, M. F. Rabiah Nizah, S. H. Teo, O. N. Syazwani and A. Islam, Energy Convers. Manage., 2014, 78, 738–744 CrossRef CAS PubMed.
- P. Zhang, Q. Han, M. Fan and P. Jiang, Fuel, 2014, 124, 66–72 CrossRef CAS PubMed.
- E. Karimi, I. F. Teixeira, L. P. Ribeiro, A. Gomez, R. M. Lago, G. Penner, S. W. Kycia and M. Schlaf, Catal. Today, 2012, 190, 73–88 CrossRef CAS PubMed.
- E. Karimi, A. Gomez, S. W. Kycia and M. Schlaf, Energy Fuels, 2010, 24, 2747–2757 CrossRef CAS.
- K. Narasimharao, D. R. Brown, A. F. Lee, A. D. Newman, P. F. Siril, S. J. Tavener and K. Wilson, J. Catal., 2007, 248, 226–234 CrossRef CAS PubMed.
- K. Suwannakarn, E. Lotero, K. Ngaosuwan and J. G. Goodwin, Ind. Eng. Chem. Res., 2009, 48, 2810–2818 CrossRef CAS.
- M. Kouzu, A. Nakagaito and J.-s. Hidaka, Appl. Catal., A, 2011, 405, 36–44 CrossRef CAS PubMed.
- D. Zhao, Q. Huo, J. Feng, B. F. Chmelka and G. D. Stucky, J. Am. Chem. Soc., 1998, 120, 6024–6036 CrossRef CAS.
- I. K. Mbaraka and B. H. Shanks, J. Catal., 2005, 229, 365–373 CrossRef CAS PubMed.
- J. A. Melero, L. F. Bautista, G. Morales, J. Iglesias and D. Briones, Energy Fuels, 2008, 23, 539–547 CrossRef.
- X.-R. Chen, Y.-H. Ju and C.-Y. Mou, J. Phys. Chem. C, 2007, 111, 18731–18737 CAS.
- I. K. Mbaraka, D. R. Radu, V. S. Y. Lin and B. H. Shanks, J. Catal., 2003, 219, 329–336 CrossRef CAS.
- D. Chen, Z. Li, Y. Wan, X. Tu, Y. Shi, Z. Chen, W. Shen, C. Yu, B. Tu and D. Zhao, J. Mater. Chem., 2006, 16, 1511–1519 RSC.
- L. Cao, T. Man and M. Kruk, Chem. Mater., 2009, 21, 1144–1153 CrossRef CAS.
- A. Martin, G. Morales, F. Martinez, R. van Grieken, L. Cao and M. Kruk, J. Mater. Chem., 2010, 20, 8026–8035 RSC.
- P. Zeigermann, S. Naumov, S. Mascotto, J. Kärger, B. M. Smarsly and R. Valiullin, Langmuir, 2012, 28, 3621–3632 CrossRef CAS PubMed.
- A. Vinu, N. Gokulakrishnan, V. V. Balasubramanian, S. Alam, M. P. Kapoor, K. Ariga and T. Mori, Chem. – Eur. J., 2008, 14, 11529–11538 CrossRef CAS PubMed.
- C. Pirez, J.-M. Caderon, J.-P. Dacquin, A. F. Lee and K. Wilson, ACS Catal., 2012, 2, 1607–1614 CrossRef CAS.
- D. E. López, J. G. Goodwin Jr, D. A. Bruce and E. Lotero, Appl. Catal., A, 2005, 295, 97–105 CrossRef PubMed.
- W. W. Mar and E. Somsook, J. Oleo Sci., 2013, 62, 435–442 CrossRef CAS.
- S.-Y. Chen, T. Mochizuki, Y. Abe, M. Toba and Y. Yoshimura, Appl. Catal., B, 2014, 148–149, 344–356 CrossRef CAS PubMed.
- J. A. Melero, J. Iglesias and G. Morales, Green Chem., 2009, 11, 1285–1308 RSC.
- A. Carrero, G. Vicente, R. Rodríguez, M. Linares and G. L. del Peso, Catal. Today, 2011, 167, 148–153 CrossRef CAS PubMed.
- J. Y. Ying, C. P. Mehnert and M. S. Wong, Angew. Chem., Int. Ed., 1999, 38, 56–77 CrossRef CAS.
- Y. Lu, Angew. Chem., Int. Ed., 2006, 45, 7664–7667 CrossRef CAS PubMed.
- S. Garg, K. Soni, G. M. Kumaran, R. Bal, K. Gora-Marek, J. K. Gupta, L. D. Sharma and G. M. Dhar, Catal. Today, 2009, 141, 125–129 CrossRef CAS PubMed.
- S. Gheorghiu and M.-O. Coppens, AIChE J., 2004, 50, 812–820 CrossRef CAS.
- X. Zhang, F. Zhang and K.-Y. Chan, Mater. Lett., 2004, 58, 2872–2877 CrossRef CAS PubMed.
- J.-H. Sun, Z. Shan, T. Maschmeyer and M.-O. Coppens, Langmuir, 2003, 19, 8395–8402 CrossRef CAS.
- J.-P. Dacquin, J. r. m. Dhainaut, D. Duprez, S. b. Royer, A. F. Lee and K. Wilson, J. Am. Chem. Soc., 2009, 131, 12896–12897 CrossRef CAS PubMed.
- J. Dhainaut, J.-P. Dacquin, A. F. Lee and K. Wilson, Green Chem., 2010, 12, 296–303 RSC.
- R. Hong, T. Pan, J. Qian and H. Li, Chem. Eng. J., 2006, 119, 71–81 CrossRef CAS PubMed.
- M. L. Curri, R. Comparelli, P. D. Cozzoli, G. Mascolo and A. Agostiano, Mater. Sci. Eng., C, 2003, 23, 285–289 CrossRef.
- G. P. Fotou and S. E. Pratsinis, Chem. Eng. Commun., 1996, 151, 251–260 CrossRef CAS.
- S. Chakrabarti and B. Dutta, J. Hazard. Mater., 2004, 112, 269–278 CrossRef CAS PubMed.
- H. Yoshida, S. Takashi, C. Murata and T. Hattori, J. Catal., 2003, 220, 226–232 CrossRef CAS.
- G. Corro, U. Pal and N. Tellez, Appl. Catal., B, 2013, 129, 39–47 CrossRef CAS PubMed.
- G. Corro, F. Bañuelos, E. Vidal and S. Cebada, Fuel, 2014, 115, 625–628 CrossRef CAS PubMed.
- C. Pirez, K. Wilson and A. F. Lee, Green Chem., 2014, 16, 197–202 RSC.
- N. Mizuno and M. Misono, Chem. Rev., 1998, 98, 199–218 CrossRef CAS PubMed.
- I. V. Kozhevnikov, Chem. Rev., 1998, 98, 171–198 CrossRef CAS PubMed.
- A. D. Newman, D. R. Brown, P. Siril, A. F. Lee and K. Wilson, Phys. Chem. Chem. Phys., 2006, 8, 2893–2902 RSC.
- A. D. Newman, A. F. Lee, K. Wilson and N. A. Young, Catal. Lett., 2005, 102, 45–50 CrossRef CAS.
- L. Pesaresi, D. R. Brown, A. F. Lee, J. M. Montero, H. Williams and K. Wilson, Appl. Catal., A, 2009, 360, 50–58 CrossRef CAS PubMed.
- X. Duan, Y. Liu, Q. Zhao, X. Wang and S. Li, RSC Adv., 2013, 3, 13748–13755 RSC.
- S. Singh and A. Patel, J. Cleaner Prod., 2014, 72, 46–56 CrossRef CAS PubMed.
- P. Xia, F. Liu, C. Wang, S. Zuo and C. Qi, Catal. Commun., 2012, 26, 140–143 CrossRef CAS PubMed.
- W. Liu, P. Yin, X. Liu, W. Chen, H. Chen, C. Liu, R. Qu and Q. Xu, Energy Convers. Manage., 2013, 76, 1009–1014 CrossRef CAS PubMed.
- R. Tesser, M. Di Serio, L. Casale, L. Sannino, M. Ledda and E. Santacesaria, Chem. Eng. J., 2010, 161, 212–222 CrossRef CAS PubMed.
- X. Liang, Ind. Eng. Chem. Res., 2013, 52, 6894–6900 CrossRef CAS.
- D. Zeng, S. Liu, W. Gong, G. Wang, J. Qiu and H. Chen, Appl. Catal., A, 2014, 469, 284–289 CrossRef CAS PubMed.
- C. A. Deshmane, M. W. Wright, A. Lachgar, M. Rohlfing, Z. Liu, J. Le and B. E. Hanson, Bioresour. Technol., 2013, 147, 597–604 CrossRef CAS PubMed.
- D. D. Chabukswar, P. K. K. S. Heer and V. G. Gaikar, Ind. Eng. Chem. Res., 2013, 52, 7316–7326 CrossRef CAS.
- C. Poonjarernsilp, N. Sano and H. Tamon, Appl. Catal., B, 2014, 147, 726–732 CrossRef CAS PubMed.
- F. H. Alhassan, R. Yunus, U. Rashid, K. Sirat, A. Islam, H. V. Lee and Y. H. Taufiq-Yap, Appl. Catal., A, 2013, 456, 182–187 CrossRef CAS PubMed.
- W. Xie and T. Wang, Fuel Process. Technol., 2013, 109, 150–155 CrossRef CAS PubMed.
- R. Sheikh, M.-S. Choi, J.-S. Im and Y.-H. Park, J. Ind. Eng. Chem., 2013, 19, 1413–1419 CrossRef CAS PubMed.
- M. Farooq, A. Ramli and D. Subbarao, J. Cleaner Prod., 2013, 59, 131–140 CrossRef CAS PubMed.
- A. Talebian-Kiakalaieh, N. A. S. Amin and H. Mazaheri, Appl. Energy, 2013, 104, 683–710 CrossRef CAS PubMed.
- S. Yan, C. DiMaggio, S. Mohan, M. Kim, S. Salley and K. Y. S. Ng, Top. Catal., 2010, 53, 721–736 CrossRef CAS.
- L. Peng, A. Philippaerts, X. Ke, J. Van Noyen, F. De Clippel, G. Van Tendeloo, P. A. Jacobs and B. F. Sels, Catal. Today, 2010, 150, 140–146 CrossRef CAS PubMed.
- P. S. Sreeprasanth, R. Srivastava, D. Srinivas and P. Ratnasamy, Appl. Catal., A, 2006, 314, 148–159 CrossRef CAS PubMed.
- Z. Helwani, M. R. Othman, N. Aziz, J. Kim and W. J. N. Fernando, Appl. Catal., A, 2009, 363, 1–10 CrossRef CAS PubMed.
- S. Miao and B. H. Shanks, Appl. Catal., A, 2009, 359, 113–120 CrossRef CAS PubMed.
- I. Jiménez-Morales, J. Santamaría-González, P. Maireles-Torres and A. Jiménez-López, Appl. Catal., B, 2011, 105, 199–205 CrossRef PubMed.
- Q. Shu, J. Gao, Z. Nawaz, Y. Liao, D. Wang and J. Wang, Appl. Energy, 2010, 87, 2589–2596 CrossRef CAS PubMed.
- I. M. Atadashi, M. K. Aroua, A. R. Abdul Aziz and N. M. N. Sulaiman, Renewable Sustainable Energy Rev., 2012, 16, 3456–3470 CrossRef CAS PubMed.
- D. Kusdiana and S. Saka, Bioresour. Technol., 2004, 91, 289–295 CrossRef CAS.
- J. K. Satyarthi, D. Srinivas and P. Ratnasamy, Energy Fuels, 2010, 24, 2154–2161 CrossRef CAS.
- Y. Liu, E. Lotero and J. G. Goodwin Jr, J. Catal., 2006, 243, 221–228 CrossRef CAS PubMed.
- D. M. Alonso, M. L. Granados, R. Mariscal and A. Douhal, J. Catal., 2009, 262, 18–26 CrossRef CAS PubMed.
- K. Wilson, A. Rénson and J. H. Clark, Catal. Lett., 1999, 61, 51–55 CrossRef CAS.
- B. Rác, P. Hegyes, P. Forgo and Á. Molnár, Appl. Catal., A, 2006, 299, 193–201 CrossRef PubMed.
- Q. Yang, J. Liu, J. Yang, M. P. Kapoor, S. Inagaki and C. Li, J. Catal., 2004, 228, 265–272 CrossRef CAS PubMed.
- Q. Yang, M. P. Kapoor, N. Shirokura, M. Ohashi, S. Inagaki, J. N. Kondo and K. Domen, J. Mater. Chem., 2005, 15, 666–673 RSC.
- G. Morales, G. Athens, B. F. Chmelka, R. van Grieken and J. A. Melero, J. Catal., 2008, 254, 205–217 CrossRef CAS PubMed.
- R. Sánchez-Vázquez, C. Pirez, J. Iglesias, K. Wilson, A. F. Lee and J. A. Melero, ChemCatChem, 2013, 5, 994–1001 CrossRef.
- D. Margolese, J. A. Melero, S. C. Christiansen, B. F. Chmelka and G. D. Stucky, Chem. Mater., 2000, 12, 2448–2459 CrossRef CAS.
- I. Díaz, C. Márquez-Alvarez, F. Mohino, J. N. Pérez-Pariente and E. Sastre, J. Catal., 2000, 193, 283–294 CrossRef.
- J.-P. Dacquin, H. E. Cross, D. R. Brown, T. Duren, J. J. Williams, A. F. Lee and K. Wilson, Green Chem., 2010, 12, 1383–1391 RSC.
- C. Schumacher, J. Gonzalez, P. A. Wright and N. A. Seaton, J. Phys. Chem. B, 2005, 110, 319–333 CrossRef PubMed.
- D. Zuo, J. Lane, D. Culy, M. Schultz, A. Pullar and M. Waxman, Appl. Catal., B, 2013, 129, 342–350 CrossRef CAS PubMed.
- L. Sherry and J. A. Sullivan, Catal. Today, 2011, 175, 471–476 CrossRef CAS PubMed.
- J. A. Melero, R. van Grieken and G. Morales, Chem. Rev., 2006, 106, 3790–3812 CrossRef CAS PubMed.
- A. P. S. Chouhan and A. K. Sarma, Renewable Sustainable Energy Rev., 2011, 15, 4378–4399 CrossRef CAS PubMed.
- A. Macario, G. Giordano, B. Onida, D. Cocina, A. Tagarelli and A. M. Giuffrè, Appl. Catal., A, 2010, 378, 160–168 CrossRef CAS PubMed.
- D. Srinivas and J. Satyarthi, Catal. Surv. Asia, 2011, 15, 145–160 CrossRef CAS.
- F. Yan, Z. Yuan, P. Lu, W. Luo, L. Yang and L. Deng, Renewable Energy, 2011, 36, 2026–2031 CrossRef CAS PubMed.
- T. Nakato, M. Kimura, S.-I. Nakata and T. Okuhara, Langmuir, 1998, 14, 319–325 CrossRef CAS.
- A. Drelinkiewicz, Z. Kalemba-Jaje, E. Lalik and R. Kosydar, Fuel, 2014, 116, 760–771 CrossRef CAS PubMed.
- G. Morales, R. van Grieken, A. Martín and F. Martínez, Chem. Eng. J., 2010, 161, 388–396 CrossRef CAS PubMed.
- I. Noshadi, R. Kumar, B. Kanjilal, R. Parnas, H. Liu, J. Li and F. Liu, Catal. Lett., 2013, 143, 792–797 CrossRef CAS PubMed.
- L. Geng, G. Yu, Y. Wang and Y. Zhu, Appl. Catal., A, 2012, 427–428, 137–144 CrossRef CAS PubMed.
- R. Liu, X. Wang, X. Zhao and P. Feng, Carbon, 2008, 46, 1664–1669 CrossRef CAS PubMed.
- B. Chang, J. Fu, Y. Tian and X. Dong, J. Phys. Chem. C, 2013, 117, 6252–6258 CAS.
- M. Kotwal, A. Kumar and S. Darbha, J. Mol. Catal. A: Chem., 2013, 377, 65–73 CrossRef CAS PubMed.
- L. Deng, T. Tan, F. Wang and X. Xu, Eur. J. Lipid Sci. Technol., 2003, 105, 727–734 CrossRef CAS.
- M. Iso, B. Chen, M. Eguchi, T. Kudo and S. Shrestha, J. Mol. Catal. B: Enzym., 2001, 16, 53–58 CrossRef CAS.
- C.-H. Liu, C.-C. Huang, Y.-W. Wang, D.-J. Lee and J.-S. Chang, Appl. Energy, 2012, 100, 41–46 CrossRef CAS PubMed.
- N. Dizge, B. Keskinler and A. Tanriseven, Biochem. Eng. J., 2009, 44, 220–225 CrossRef CAS PubMed.
- L. Guerreiro, P. M. Pereira, I. M. Fonseca, R. M. Martin-Aranda, A. M. Ramos, J. M. L. Dias, R. Oliveira and J. Vital, Catal. Today, 2010, 156, 191–197 CrossRef CAS PubMed.
- X. Liu, H. He, Y. Wang, S. Zhu and X. Piao, Fuel, 2008, 87, 216–221 CrossRef CAS PubMed.
- S. Yan, H. Lu and B. Liang, Energy Fuels, 2007, 22, 646–651 CrossRef.
- Q. Yang, J. Liu, L. Zhang and C. Li, J. Mater. Chem., 2009, 19, 1945–1955 RSC.
- B. Karimi, H. M. Mirzaei and A. Mobaraki, Catal. Sci. Technol., 2012, 2, 828–834 CAS.
- M. Bender, Bioresour. Technol., 1999, 70, 81–87 CrossRef CAS.
- T. Sakai, A. Kawashima and T. Koshikawa, Bioresour. Technol., 2009, 100, 3268–3276 CrossRef CAS PubMed.
- E. F. Aransiola, T. V. Ojumu, O. O. Oyekola, T. F. Madzimbamuto and D. I. O. Ikhu-Omoregbe, Biomass Bioenergy, 2014, 61, 276–297 CrossRef CAS PubMed.
- M. B. Tasić, O. S. Stamenković and V. B. Veljković, Energy Convers. Manage., 2014, 84, 405–413 CrossRef PubMed.
- Y. Cheng, Y. Feng, Y. Ren, X. Liu, A. Gao, B. He, F. Yan and J. Li, Bioresour. Technol., 2012, 113, 65–72 CrossRef CAS PubMed.
- A. A. Kulkarni, K.-P. Zeyer, T. Jacobs and A. Kienle, Ind. Eng. Chem. Res., 2007, 46, 5271–5277 CrossRef CAS.
- Ó. de la Iglesia, R. Mallada, M. Menéndez and J. Coronas, Chem. Eng. J., 2007, 131, 35–39 CrossRef PubMed.
- A. A. Kiss, A. C. Dimian and G. Rothenberg, Energy Fuels, 2007, 22, 598–604 CrossRef.
- C. Buchaly, P. Kreis and A. Górak, Ind. Eng. Chem. Res., 2011, 51, 891–899 CrossRef.
- Z. Qiu, L. Zhao and L. Weatherley, Chemical Engineering and Processing: Process Intensification, 2010, 49, 323–330 CrossRef CAS PubMed.
- G. L. Maddikeri, A. B. Pandit and P. R. Gogate, Ind. Eng. Chem. Res., 2012, 51, 14610–14628 CrossRef CAS.
- X. Ni, M. R. Mackley, A. P. Harvey, P. Stonestreet, M. H. I. Baird and N. V. Rama Rao, Chem. Eng. Res. Des., 2003, 81, 373–383 CrossRef CAS.
- A. N. Phan, A. P. Harvey and V. Eze, Chem. Eng. Technol., 2012, 35, 1214–1220 CrossRef CAS.
- R. G. Nelson and S. A. Hower, Sixth national bioenergy conference, 1994.
- S. Baroutian, M. K. Aroua, A. A. Raman and N. M. Sulaiman, Bioresour. Technol., 2011, 102, 1095–1102 CrossRef CAS PubMed.
- H. Falahati and A. Y. Tremblay, Fuel, 2012, 91, 126–133 CrossRef CAS PubMed.
- W. Xu, L. Gao, S. Wang and G. Xiao, Bioresour. Technol., 2014, 159, 286–291 CrossRef CAS PubMed.
- P. Cao, A. Y. Tremblay, M. A. Dubé and K. Morse, Ind. Eng. Chem. Res., 2007, 46, 52–58 CrossRef CAS.
- P. Cao, A. Y. Tremblay and M. A. Dubé, Ind. Eng. Chem. Res., 2009, 48, 2533–2541 CrossRef CAS.
- P. Lozano, J. M. Bernal and M. Vaultier, Fuel, 2011, 90, 3461–3467 CrossRef CAS PubMed.
- X. Wang, X. Liu, C. Zhao, Y. Ding and P. Xu, Bioresour. Technol., 2011, 102, 6352–6355 CrossRef CAS PubMed.
- D. Lv, W. Du, G. Zhang and D. Liu, Process Biochem., 2010, 45, 446–450 CrossRef CAS PubMed.
- A. Bajaj, P. Lohan, P. N. Jha and R. Mehrotra, J. Mol. Catal. B: Enzym., 2010, 62, 9–14 CrossRef CAS PubMed.
- L. A. Nelson, T. A. Foglia and W. N. Marmer, J. Am. Oil Chem. Soc., 1996, 73, 1191–1195 CrossRef CAS.
- Y. Watanabe, Y. Shimada, A. Sugihara, H. Noda, H. Fukuda and Y. Tominaga, J. Am. Oil Chem. Soc., 2000, 77, 355–360 CrossRef CAS PubMed.
- K. Bélafi-Bakó, F. Kovács, L. Gubicza and J. Hancsók, Biocatal. Biotransform., 2002, 20, 437–439 CrossRef.
- M. Lee, D. Lee, J. Cho, S. Kim and C. Park, Appl. Biochem. Biotechnol., 2013, 171, 1118–1127 CrossRef CAS PubMed.
- K. T. Tan and K. T. Lee, Renewable Sustainable Energy Rev., 2011, 15, 2452–2456 CrossRef CAS PubMed.
- V. G. Gude and G. E. Grant, Appl. Energy, 2013, 109, 135–144 CrossRef CAS PubMed.
- V. L. Gole and P. R. Gogate, Chemical Engineering and Processing: Process Intensification, 2012, 53, 1–9 CrossRef CAS PubMed.
- A. Mazubert, C. Taylor, J. Aubin and M. Poux, Bioresour. Technol., 2014, 161, 270–279 CrossRef CAS PubMed.
- W. A. Wali, A. I. Al-Shamma'a, K. H. Hassan and J. D. Cullen, J. Process Control, 2012, 22, 1256–1272 CrossRef CAS PubMed.
- P. Chand, V. R. Chintareddy, J. G. Verkade and D. Grewell, Energy Fuels, 2010, 24, 2010–2015 CrossRef CAS.
- B. Salamatinia, H. Mootabadi, S. Bhatia and A. Z. Abdullah, Fuel Process. Technol., 2010, 91, 441–448 CrossRef CAS PubMed.
- H. A. Choudhury, S. Chakma and V. S. Moholkar, Ultrason. Sonochem., 2014, 21, 169–181 CrossRef CAS PubMed.
- P. Khemthong, C. Luadthong, W. Nualpaeng, P. Changsuwan, P. Tongprem, N. Viriya-empikul and K. Faungnawakij, Catal. Today, 2012, 190, 112–116 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2014 |