
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
[Pd(C∧N)(X)(PPh3)] palladacycles react with 2,4,6-trifluorophenyl boronic acid to give stable transmetallation products of the type [Pd(C∧N)(2,4,6-F3C6H2)(PPh3)]†
Anant R.
Kapdi
a,
Gopal
Dhangar
a,
Jose Luis
Serrano
b,
Jose
Pérez
b,
Luis
García
b and
Ian J. S.
Fairlamb
*c
aDepartment of Chemistry, Institute of Chemical Technology, Matunga, Mumbai-400019, India
bDepartamento de Ingeniería Minera, Geológica y Cartográfica. Universidad Politécnica de Cartagena. Área de Química Inorgánica, Regional Campus of International Excellence “Campus Mare Nostrum”, 30203 Cartagena, Spain
cDepartment of Chemistry, University of York, Heslington, York, North Yorkshire YO10 5DD, UK. E-mail: ian.fairlamb@york.ac.uk
First published on 15th July 2014
Abstract
Direct transmetallation between palladacyclic complexes and arylboronic acid occurs to give isolable transmetallation products. In THF, the reaction occurs <0.5 h. Prolonged reaction leads to the generation of a dinuclear Pd complex bearing bridging μ-hydroxo and μ-acetoxy ligands. Insight into precatalyst activation for Suzuki–Miyaura cross-couplings mediated by palladacycles has been gained, where acetate and N-imidate anions activate a neutral arylboronic acid.
Cross-coupling reactions mediated by Pd allow rapid access to an eclectic array of organic products.1 Such reactions are arguably the go-to-transformation in synthetic chemistry for the synthesis of biaryls.2 Suzuki–Miyaura cross-coupling (SMCC) of an organohalide and organoboron-containing species, mediated by a Pd catalyst and exogenous base, are ubiquitous reactions, and one of the most widely applied.3 In recent years much has been learnt about the reaction mechanisms of SMCCs, especially the involvement of Pd-hydroxo species in activating neutral arylboronic acid species (by an oxo-palladium pathway).4 In addition, the involvement of higher order Pd species has been demonstrated, including some evidence for a heterogeneous SMCC reaction.5
The improvement of SMCC catalysts often derives from altering oxidative addition, transmetallation or reductive elimination steps. Controlling and understanding the precatalyst activation step is however, absolutely critical – an aspect that is poorly understood. Indeed, there are limited detailed studies6 concerning the mechanism of the precatalyst reduction step, i.e. how is the active catalyst species generated in SMCCs?
In previous studies we7 and others6b,8 have examined palladacycles as precatalysts for SMCCs and related cross-couplings. In our catalytic work,7a involving [Pd(phbz)(X)(PR3)] palladacycles (phbz = N-phenylbenzaldimine; X = acetate or N-imidate; R = aryl; Scheme 1), a reaction with arylboronic acid in the absence of base to release a common catalyst species of the type Pd0(PPh3)n was noted. The process involves arylation of the palladacyclic ligand backbone yielding an imine (the reductive elimination organic product), which is hydrolysed to 2-phenylbenzaldehyde either in the reaction or during work-up (depending on the conditions used). Similar observations were made by Bedford and co-workers for other palladacycles in the presence of base.8a
 | ||
| Scheme 1 Proposed activation of arylboronic acids by palladacyclic complexes. | ||
Indirect evidence by electrospray ionisation mass spectrometry (ESI-MS) showed that arylated PdII species could be present under the SMCC reaction conditions7a however, in all the studies to date no direct evidence has been gathered for the initial transmetallation product, [Pd(C∧N)(aryl)(PR3)] (where C∧N is the palladacyclic backbone). To address this gap in the field, in this paper we present the first direct evidence for transmetallation of [Pd(C∧N)(X)(PR3)] complexes with an arylboronic acid.
Our starting point was to take advantage of the important contribution made by Osakada and co-workers,9 who showed that the employment of an excess 2,4,6-trifluorophenyl moiety retards reductive elimination from diarylpalladium(II) complexes containing strongly basic triethylphosphine ligands.
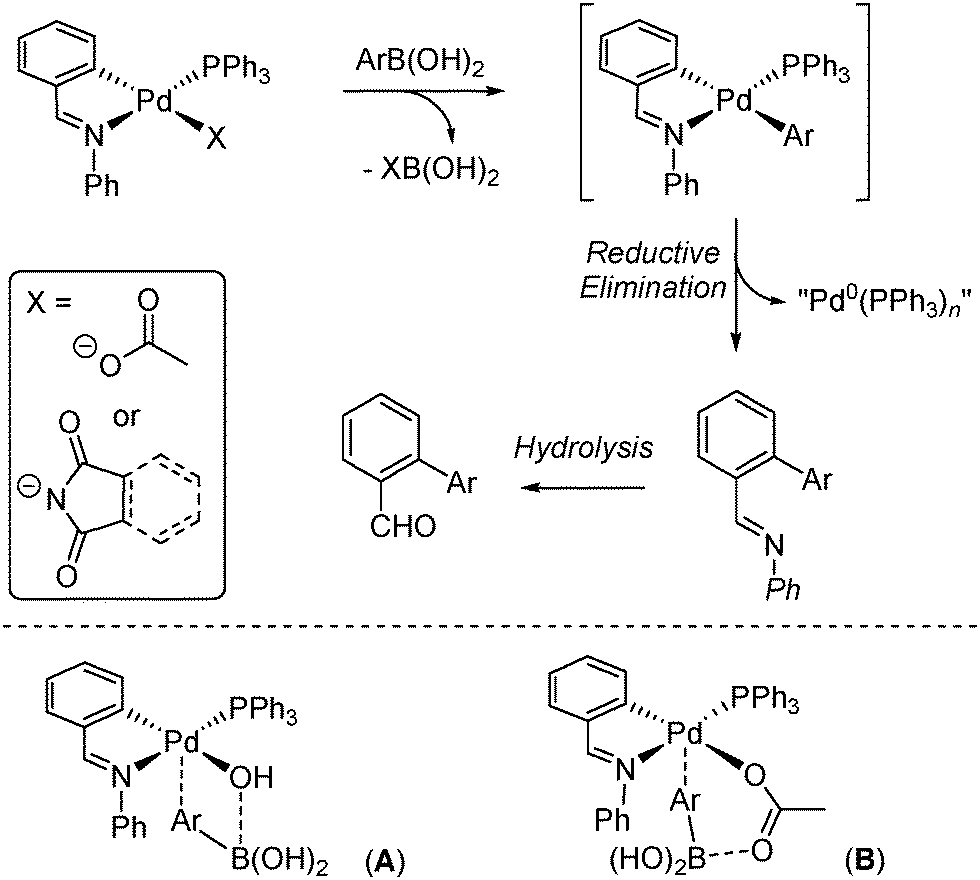
In the first experiment, 2,4,6-trifluorophenylboronic acid 1 was reacted with [Pd(phbz)(OAc)(PPh3)] in THF10 at 25 °C for 0.5 h. The ratio of the palladacycle and 1 was varied (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :6, 1:2 and 1:1), and in all experiments one new product dominated (mixed with the precursor only in the 1:1 reaction) as a white solid (ca. 70% yield), which contained one new phosphorus chemical environment, e.g.31P{1H} NMR δ 19.86 (s), considerably shifted from that of 41.80 (s) displayed by [Pd(phbz)(OAc)(PPh3)]. The IR (typical internal vibrational modes of 2,4,6-trifluorophenyl group at 1390, 1100 and 990 cm−1)11 and 19F NMR spectroscopic data, with two resonances at δ −84.50 (2F)/−120.30 (1F), in combination with the ESI-MS data (M+ = 680), suggested that the product was [Pd(phbz)(2,4,6-F3C6H2)(PPh3)]. The structure of [Pd(phbz)(2,4,6-F3C6H2)(PPh3)] was verified by single crystal X-ray diffraction (Fig. 1, left structure).12a Interestingly, the 2,4,6-trifluorophenyl moiety is positioned cis to the carbon found within the palladacycle, which would therefore be receptive to reductive elimination.
:6, 1:2 and 1:1), and in all experiments one new product dominated (mixed with the precursor only in the 1:1 reaction) as a white solid (ca. 70% yield), which contained one new phosphorus chemical environment, e.g.31P{1H} NMR δ 19.86 (s), considerably shifted from that of 41.80 (s) displayed by [Pd(phbz)(OAc)(PPh3)]. The IR (typical internal vibrational modes of 2,4,6-trifluorophenyl group at 1390, 1100 and 990 cm−1)11 and 19F NMR spectroscopic data, with two resonances at δ −84.50 (2F)/−120.30 (1F), in combination with the ESI-MS data (M+ = 680), suggested that the product was [Pd(phbz)(2,4,6-F3C6H2)(PPh3)]. The structure of [Pd(phbz)(2,4,6-F3C6H2)(PPh3)] was verified by single crystal X-ray diffraction (Fig. 1, left structure).12a Interestingly, the 2,4,6-trifluorophenyl moiety is positioned cis to the carbon found within the palladacycle, which would therefore be receptive to reductive elimination.
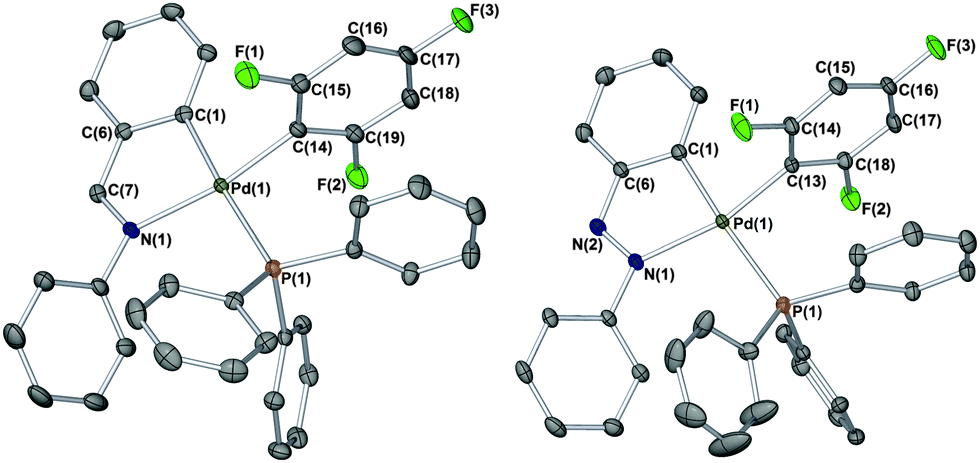 | ||
| Fig. 1 X-ray structures of [Pd(phbz)(2,4,6-F3C6H2)(PPh3)] (left) and [Pd(phazb)(2,4,6-F3C6H2)(PPh3)] (right). Selected bond angles (°) and lengths (Å): C(1)–Pd(1)–C(14) 88.49(6); C(1)–Pd(1)–N(1) 80.40(5); C(1)–Pd(1)–P(1) 175.05(4); C(14)–Pd(1)–N(1) 168.86(5); C(14)–Pd(1)–P(1) 88.55(4); N(1)–Pd(1)–P(1) 102.58(3); Pd(1)–C(1) 2.0377(14); Pd(1)–C(14) 1.9945(13); Pd(1)–N(1) 2.1534(11) and Pd(1)–P(1) 2.3756(4) for [Pd(phbz)(2,4,6-F3C6H2)(PPh3)]. C(1)–Pd(1)–C(13) 89.38(8); C(1)–Pd(1)–N(1) 78.02(8); C(1)–Pd(1)–P(1) 170.65(6); C(13)–Pd(1)–N(1) 165.92(8); C(13)–Pd(1)–P(1) 88.78(6); N(1)–Pd(1)–P(1) 104.66(5); Pd(1)–C(1) 2.020(2); Pd(1)–C(13) 2.005(2); Pd(1)–N(1) 2.1427(17) and Pd(1)–P(1) 2.3746(6) for [Pd(phazb)(2,4,6-F3C6H2)(PPh3)]. | ||
The structure around Pd can be described as nearly planar, and its deviation from the planar coordination has been quantified by measures of improper torsion angles: = 0.00 and −2.88°, which means C(1)–N(1)–Pd(1)–C(14) defining a plane and P(1) slightly above that plane.13 The angle between the planes C(1)–C(5)–C(7)–N(1)–Pd(1) and C(14)–C(15)–C(16)–C(17)–C(18)–C(19) is 86.20(13)°.
The reaction of related [Pd(phbz)(N-imidate)(PPh3)] complexes with 1 under similar conditions also gave [Pd(phbz)(2,4,6-F3C6H2)(PPh3)] as the product (N-imidate = N-succinimidate – 56% yield; = N-maleimidate – 71% yield).12b
A similar series of reactions of [Pd(phbz)(OAc)(PPh3)] with either phenylboronic acid or p-fluoroboronic acid did not give [Pd(phbz)(aryl)(PPh3)] type products; in keeping with our previous observations, facile reduction was observed.7a
In an independent reaction, an alternative palladacycle containing a N-phenylazabenzene (phazb) backbone, namely [Pd(phazb)(OAc)(PPh3)], was reacted with 1 in a ratio of 1:6, cleanly affording a new product as a white solid (68%), which contained one new phosphorus chemical environment, e.g.31P{1H} NMR δ 19.85 (s). The IR and other NMR spectroscopic data mirrored that obtained for [Pd(phbz)(2,4,6-F3C6H2)(PPh3)], suggesting that an analogous complex [Pd(phazb)(2,4,6-F3C6H2)(PPh3)] was obtained. The ESI Accurate-Mass TOF LC/MS data accounted for the change in the orthometallated moiety to phazb, showing fragments with one unit increase with respect to those displayed by [Pd(phbz)(2,4,6-F3C6H2)(PPh3)]. The structure of [Pd(phazb)(2,4,6-F3C6H2)(PPh3)] was verified by single crystal X-ray diffraction (Fig. 1, right structure).12c Again, a near planar geometry is seen at PdII, which closely resembles the phbz analogue.
Finally, it is important to note that when reactions of [Pd(phbz)(AcO)(PPh3)] with 1 were left for prolonged periods (ca. 16–24 h), other minor phosphorus signals were also observed in the 31P NMR spectrum (at 24.26 ppm) of the crude reaction mixture. In one particular case, we observed the formation of a different set of crystals. The absence of phbz resonances in its 1H NMR spectrum, and shifted signals in the 19F NMR δ −87.30 (2F)/−119.26 (1F) were also noticeable changes. The structure of this new product was confirmed by X-ray diffraction as [Pd2(2,4,6-C6F3H2)2(PPh3)2(μ-OH)(μ-OAc)] (Fig. 2). A similar species was recently reported by Wei and co-workers14 in the catalytic borylation of aryl halides by electron-rich Pd catalysts, in the presence of n-Bu4NOAc. Therefore, this and related species could be present in SMCCs under working catalyst conditions.
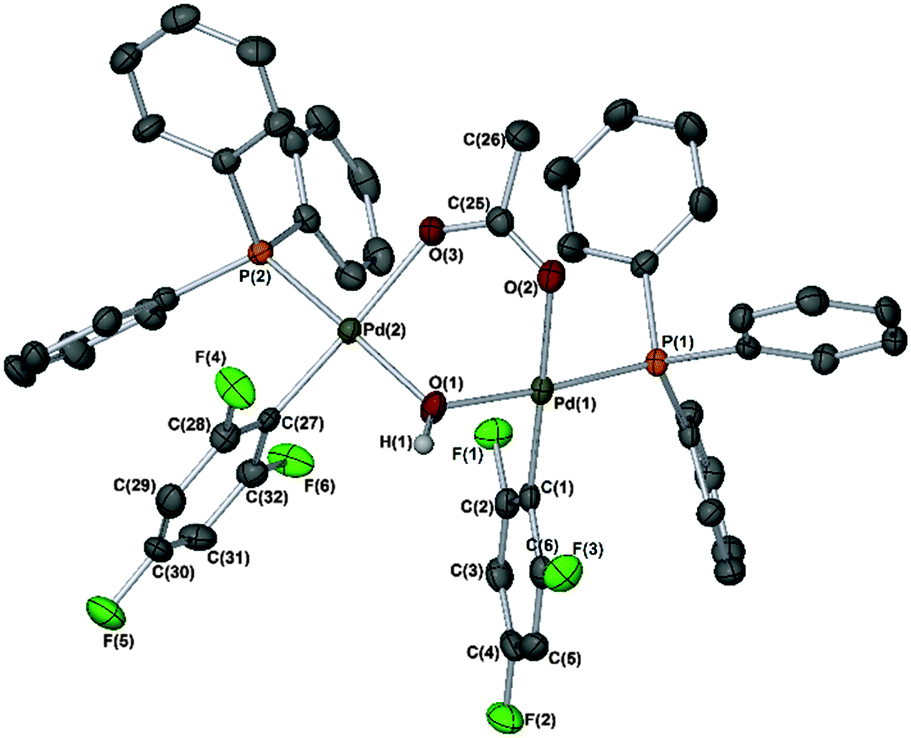 | ||
| Fig. 2 X-ray structure of a dinuclear PdII–hydroxo complex. Selected bond angles (°) and lengths (Å): C(1)–Pd(1)–O(1) 91.10(14); C(1)–Pd(1)–O(2) 178.02(13); C(1)–Pd(1)–P(1) 92.40(10); O(1)–Pd(1)–O(2) 88.55(12); O(1)–Pd(1)–P(1) 176.06(9); O(2)–Pd(1)–P(1) 87.88(8); Pd(1)–C(1) 1.996(4); Pd(1)–O(1) 2.058(3); Pd(1)–O(2) 2.087(3) and Pd(1)–P(1) 2.2303(10). C(27)–Pd(2)–O(1) 88.24(13); C(27)–Pd(2)–O(3) 176.18(13); C(27)–Pd(2)–P(2) 90.99(10); O(1)–Pd(2)–O(3) 92.76(11); O(1)–Pd(2)–P(2) 176.02(9); O(3)–Pd(2)–P(2) 88.25(7); Pd(2)–C(27) 1.984(4); Pd(2)–O(1) 2.072(3); Pd(2)–O(3) 2.124(3) and Pd(2)–P(2) 2.2417(10). | ||
In conclusion, direct evidence for transmetallation between several palladacycles of the type [Pd(C∧N)(X)(PPh3)] (where C∧N = N-phenylbenzaldimine and N-phenylazabenzene and X = acetate or N-imidate ligands) with 2,4,6-trifluorophenylboronic acid has been gathered. The reactions occur rapidly in THF in <0.5 h. Prolonged reaction leads to the generation of other species, one of which is a novel dinuclear Pd–hydroxo complex. The findings provide insight into the precatalyst activation step for SMCCs mediated by palladacycles. Our results indicate that acetate and N-imidate anions can activate arylboronic acids in palladacycles,15 which adds to the mechanistic debate about transmetallation in SMCCs. Moreover, mixing an organoboronic acid with the palladacyclic precatalyst in SMCCs can lead to a reaction taking place7a prior to adding exogenous base – the latter is a mandatory requirement for catalysis but not for catalyst activation.
This paper is dedicated to Professor Richard J. K. Taylor on the occasion of his 65th birthday. We thank Fundación Séneca (project 08670/PI/08) for financial support. This project is underpinned by work previously conducted and funded by the EPSRC (EP/D078776/1) and Royal Society. We would also like to thank Department of Science and Technology, India for DST Inspire faculty award (IFA12-CH-22) for A.R.K.
References
- (a) C-H and C-X Bond Functionalization. Transition Metal Mediation, ed. X. Ribas, RSC Catalysis, 2013 Search PubMed; (b) Organotransition metal chemistry, from bonding to catalysis, ed. J. F. Hartwig, University Science Books, Sausalito, 2010 Search PubMed; (c) Metal Catalyzed Cross-Coupling Reactions and More, ed. A. de Meijere, S. Brase and M. Oestreich, Wiley-VCH, 2014 Search PubMed.
- Palladium-Catalyzed Coupling Reactions: Practical Aspects and Future Developments, ed. Á. Molnár, John Wiley & Sons, 2013 Search PubMed.
- N. Miyaura and A. Suzuki, Chem. Rev., 1995, 95, 2457–2483 CrossRef CAS.
- (a) C. Amatore, A. Jutand and G. Le Duc, Chem. – Eur. J., 2011, 17, 2492–2503 CrossRef CAS PubMed; (b) B. P. Carrow and J. F. Hartwig, J. Am. Chem. Soc., 2011, 133, 2116–2119 CrossRef CAS PubMed; (c) A. F. Schmidt, A. A. Kurokhtina and E. V. Larina, Russ. J. Gen. Chem., 2011, 81, 1573–1574 CrossRef CAS . Aspects relating to this, and more, are detailed in a mini-review: ; (d) A. Lennox and G. C. Lloyd-Jones, Angew. Chem., Int. Ed., 2013, 52, 7362–7370 CrossRef CAS PubMed.
- (a) P. J. Ellis, I. J. S. Fairlamb, S. F. J. Hackett, K. Wilson and A. F. Lee, Angew. Chem., Int. Ed., 2010, 49, 1820–1824 CrossRef CAS PubMed; (b) A. F. Lee, P. J. Ellis, I. J. S. Fairlamb and K. Wilson, Dalton Trans., 2010, 39, 10473 RSC . For a recent discussion of Pdn species in cross-coupling, see: ; (c) C. Deraedt and D. Astruc, Acc. Chem. Res., 2014, 47, 494–503 CrossRef CAS PubMed.
- (a) F. dOrlye and A. Jutand, Tetrahedron, 2005, 61, 9670–9678 CrossRef CAS PubMed; (b) M. Beller, H. Fischer, W. A. Herrmann, K. Ölefe and C. Brossmer, Angew. Chem., Int. Ed. Engl., 1995, 35, 1848–1849 CrossRef; (c) A. H. M. de Vries, J. M. C. A. Mulders, J. H. M. Mommers, H. J. W. Henderickx and J. G. de Vries, Org. Lett., 2003, 5, 3285–3288 CrossRef CAS PubMed . For a review on palladacycles, see: ; (d) I. P. Beletskaya and A. V. Cheprakov, J. Organomet. Chem., 2004, 689, 4055–4082 CrossRef CAS PubMed . An interesting paper has been reported by Hii on the speciation of palladium(II) diacetate under various reaction conditions, see: ; (e) L. A. Adrio, B. N. Nguyen, G. Guilera, A. G. Livingston and K. K. Hii, Catal. Sci. Technol., 2012, 2, 316–323 RSC . For work on impurities found in palladium(II) diacetate, see: ; (f) S. E. Bajwa, T. E. Storr, L. E. Hatcher, T. J. Williams, C. G. Baumann, A. C. Whitwood, D. R. Allan, S. J. Teat, P. R. Raithby and I. J. S. Fairlamb, Chem. Sci., 2012, 3, 1656–1661 RSC , and references cited therein.
- (a) J. L. Serrano, L. Garcia, J. Perez, E. Perez, J. Garcia, G. Sanchez, P. Sehnal, S. De Ornellas, T. J. Williams and I. J. S. Fairlamb, Organometallics, 2011, 30, 5095–5109 CrossRef CAS; (b) I. J. S. Fairlamb, S. Grant, P. McCormack and J. Whittall, Dalton Trans., 2007, 859–865 RSC; (c) I. J. S. Fairlamb, A. R. Kapdi, A. F. Lee, G. Sanchez, G. Lopez, J. L. Serrano, L. Garcia, J. Perez and E. Perez, Dalton Trans., 2004, 3970–3981 RSC.
- (a) R. B. Bedford, C. S. J. Cazin, M. B. Hursthouse, M. E. Light, K. J. Pike and S. Wimperis, J. Organomet. Chem., 2001, 633, 173–181 CrossRef CAS; (b) R. B. Bedford, C. S. J. Cazin and D. Holder, Coord. Chem. Rev., 2004, 248, 2283–2321 CrossRef CAS PubMed.
- (a) Y. Nishihara, H. Onodera and K. Osakada, Chem. Commun., 2004, 192–193 RSC; (b) K. Osakada, Y. Nishihara and H. Onodera, Organometallics, 2005, 24, 190–192 CrossRef CAS.
- The purity of the THF did not appreciably affect the outcome of the reaction, in that [Pd(phbz)(2,4,6-F3C6H2)(PPh3)] was the dominant and major product after 0.5 h reaction time.
- G. López, G. García, G. Sánchez, C. de haro, M. D. Santana, J. Casabó, M. Mejías and E. Molins, J. Chem. Soc., Dalton Trans., 1991, 3311–3315 RSC.
- (a) In different sowings involving the crystallisation of [Pd(phbz)(2,4,6-F3C6H2)(PPh3)] from CH2Cl2, crystals of trans-[PdCl(2,4,6-F3C6H2)(PPh3)] were formed, the structure of which was confirmed by single crystal X-ray diffraction (see ESI†). This is adequately explained by adventitious HCl promoting the decomposition of [Pd(phbz)(2,4,6-F3C6H2)(PPh3)]; (b) Again, the crystallisation of the complex obtained by reaction of [Pd(phbz)(N-maleimidate)(PPh3)] with 1 from the same CH2Cl2 produced crystals of trans-[PdCl(2,4,6-F3C6H2)(PPh3)]; (c) In the same batch the crystallisation of [Pd(phazb)(2,4,6-F3C6H2)(PPh3)] from CH2Cl2 also yielded crystals of trans-[PdCl(2,4,6-F3C6H2)(PPh3)]. The fact that in three separate instances these identical crystals were obtained gives additional support to the formation of a common compound formed by different synthetic routes.
- J. Pérez, L. García, E. Pérez, J. L. Serrano, J. F. Martínez, G. Sánchez, G. López, A. Espinosa, M. Liu and F. Sanz, New J. Chem., 2003, 27, 1490–1496 RSC.
- C. S. Wei, G. H. M. Davies, O. Soltani, J. Albrecht, Q. Gao, C. Pathirana, Y. Hsiao, S. Tummala and M. D. Eastgate, Angew. Chem., Int. Ed., 2013, 52, 5822–5826 CrossRef CAS PubMed.
- It is important to note that [ArPd(OAc)(PPh3)2] does not react with aryl boronic acid, see: C. Amatore, A. Jutand and G. Le Duc, Chem. – Eur. J., 2012, 18, 6616–6625 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, characterisation data and single crystal X-ray diffraction data. CCDC 1005721–1005724. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c4cc04203d |
| This journal is © The Royal Society of Chemistry 2014 |