Synthetic hydrogel platform for three-dimensional culture of embryonic stem cell-derived motor neurons†
Daniel D.
McKinnon
a,
April M.
Kloxin
b and
Kristi S.
Anseth
*a
aDepartment of Chemical and Biological Engineering, the BioFrontiers Institute, and the Howard Hughes Medical Institute, University of Colorado at Boulder, Jennie Smoly Caruthers Biotechnology Building, 3415 Colorado Ave, 596 UCB, Boulder, CO 80303, USA. E-mail: kristi.anseth@colorado.edu; Fax: +1-303-492-4341; Tel: +1-303-735-5336
bDepartment of Chemical and Biomolecular Engineering and Department of Materials Science and Engineering, University of Delaware, 150 Academy Street, Colburn Laboratory, Newark, DE 19716, USA. E-mail: akloxin@udel.edu; Fax: +1-302-831-1048; Tel: +1-302-831-3009
First published on 24th January 2013
Abstract
Culturing mammalian neurons in three-dimensional (3D) microenvironments that more closely recapitulate critical biochemical and biophysical aspects of the developing or adult central nervous system (CNS) milieu remains a significant challenge in neurobiological studies and in regenerative medicine. Here, we aimed to exploit recent advances in poly(ethylene glycol) (PEG) hydrogel chemistries to define a synthetic niche capable of supporting the culture and axonal outgrowth of both aggregated and dissociated mouse embryonic stem cell-derived motor neurons (ESMNs). Using thiol-ene click chemistry to create peptide crosslinked PEG hydrogels, we identified a hydrogel formulation that promotes neuronal survival and axon outgrowth through cell–extracellular matrix interactions, such as those between the laminin-derived peptide YIGSR and its integrin, and that allows neurons to remodel their extracellular environment through matrix metalloproteinase (MMP)-mediated polymer network degradation. Our results demonstrate a 3D platform for culture of both aggregated and single mammalian motor nerve cells that not only permits cell survival over more than a week of culture, but also allows for the robust extension of motor axons. In addition, the optical transparency of the hydrogel allows simultaneous imaging of live cell functions, and as such, this material system should prove useful for studying fundamental aspects of neuronal development.
Introduction
Since the initial studies on the CNS pioneered by Santiago Ramon y Cajal1 at the turn of the twentieth century, there has been an ever-expanding interest in the culture and manipulation of nerve cells ex vivo. Specifically, developing culture platforms that mimic cellular microenvironments through which axons navigate to their final target destination in vivo remains a daunting task. Culturing mammalian nerve cells has met significant challenges due to the difficulty of recapitulating many aspects of the intriguing complexities of the CNS milieu and architecture. Nerve cells are traditionally cultured on two-dimensional substrates that do not fully mimic the chemical and mechanical properties of nascent or adult CNS. For example, the shear modulus of glass, a substrate commonly used to culture nerve cells, is 26 GPa, or roughly seven orders of magnitude stiffer than typical in vivo neural environments. The bovine spinal cord, which contains spinal motor neurons, the cell type used in these studies, has a shear modulus of only ∼50 Pa.2 Moreover, axon guidance cues are usually added to the culture media, but this approach does not reproduce the in vivo-like presentation of such molecular cues. In vivo, axon guidance cues are generally tethered to cell membranes with which individual axons interact as well as to the extracellular matrix.3,4 In addition, 3D spatial organization of CNS tissues (e.g., cortical layers) is essential to directing axon guidance and neural circuit assembly.Despite these challenges, several recent studies have raised the possibility of culturing mammalian nerve cells in 3D using synthetic extracellular matrix material systems. Hydrolytically degradable synthetic PEG hydrogels have been used to culture neural progenitor cells (NPCs), where axon extension was observed and correlated strongly with the extent of material degradation.5 Peptides and proteins have been engineered to assemble into hydrogels that promote neuronal differentiation and axon extension.6,7 Chemically modified natural material systems have been leveraged to harness some of the benefits of naturally derived macromolecules while enabling selective property tuning. For example, dorsal root ganglia (DRG) cells were shown to extend processes into an agarose gel that was spatially patterned with RGDS peptide to facilitate cell–matrix interactions.8 Methacrylated hyaluronic acid was used to encapsulate ventral mesencephalic progenitor cells, which preferentially differentiated into neuronal rather than glial lineage in the softest gels.9 These studies show that neurons respond to biophysical (e.g., modulus) and biochemical (e.g., RGDS) signals presented by their 3D microenvironments in vitro and demonstrate the utility of material-based approaches for directing neural cell function and fate. We sought to gain further control over the 3D cellular milieu by introducing a chemically and physically defined synthetic system that allows for the systematic variation of these signals and subsequent study of cellular response. In vivo spinal motor neurons function in a complex environment with cell bodies residing in the spinal cord and motor axons in the periphery, and we aimed to recapitulate aspects of this complexity with a tunable synthetic hydrogel.
While most studies on nerve cells rely on isolation of primary neurons from animals, the excision process results in axotomy (cutting) of neurites and relies on subsequent re-growth of axons. To circumvent this issue, pluripotent embryonic stem (ES) cells have been shown to give rise to distinct types and subtypes of neurons that display characteristic properties and functionalities of their in vivo counterparts.10–13 One of the best-understood types of nerve cells in regards to their diversification and connectivity are spinal cord motor neurons.14 Importantly, spinal cord motor neurons can be efficiently differentiated from ES cells using developmentally relevant signaling cues, namely retinoic acid (RA) and sonic hedgehog (Shh).10,15 In all aspects examined, ESMNs appear to be indistinguishable from their primary counterparts.10–13 Importantly, spinal motor neurons are one of the only neurons of the CNS to project into the periphery. Moreover, individual motor neuron subtype loss is an underlying mechanism of ALS and SMA neurodegenerative disorders.16 In this regard, transplantation of spinal motor neurons could be a feasible strategy for CNS repair. However, intraspinal transplantation into the adult spinal cord generally yields poor outcomes with respect to integration into neural circuits and axon extension.17,18
Here, we sought to take advantage of the versatility of thiol-ene reactions and the robustness of ESMN differentiation to define a synthetic niche that permits survival and axonal outgrowth of dissociated and aggregated ESMNs in 3D hydrogels. Specifically, we examined the combinatorial importance of hydrogel mechanical properties, presentation of biochemical cues, and mimicry of axon-ECM interactions in directing survival and axonal outgrowth. As many neurodegenerative disorders result in loss of individual subtypes of nerve cells, studies similar to ours may lead to provision of novel insights into engineering of transplantable biomaterial matrices for future delivery of neural cellular substrates for cell-replacement therapies.
Background
Neurons are known to be delicate cell types requiring specific growth factors, appropriately functionalized surfaces, and complex growth media to remain viable and extend axons.19 This sensitivity presents a particular challenge for the 3D culture or delivery of primary neural cells. For example, a large portion of NPCs undergo cell death upon encapsulation5 within a polymer system that has successfully sustained many primary cell types, including chondrocytes,20 osteoblasts,21 smooth muscle cells,22 fibroblasts,23 and mesenchymal stem cells.24 Loss of viability during encapsulation is not necessarily an issue for progenitor cells, where rapid divisions can repopulate the hydrogel; however, for post-mitotic primary neurons, such as ESMNs, viability loss presents a serious problem for the encapsulation and delivery of a significant number of cells. Recent advances in thiol-ene chemistry25 have led to a step-growth hydrogel system that allows the facile incorporation of cell-degradable crosslinks and adhesive peptides, which we hypothesized could be used to rescue neural cell viability and aid in maintaining their function.26 Additionally, this photoinitiated polymerization requires no change in pH, temperature, or chemical environment, and the mild reaction conditions combined with the diverse chemistry motivated us to exploit this system to encapsulate spinal motor neurons.As illustrated in Fig. 1, network formation is initiated by photocleavage of a low concentration of visible light photoinitiator (lithium phenyl-2,4,6-trimethylbenzoylphosphonate, LAP), which is water soluble and known to be cytocompatible over a wide range of initiation conditions.27 The photocleaved fragment of LAP abstracts a proton from a thiol-terminated macromolecule to generate a thiyl radical, which then reacts rapidly and specifically with a norbornene-terminated macromer, forming a thioether bond. The thiyl radical is then regenerated when the reacted norbornene chain transfers to another thiol-terminated macromer. The thiol-ene gels thus form through a radical-mediated step growth process.28
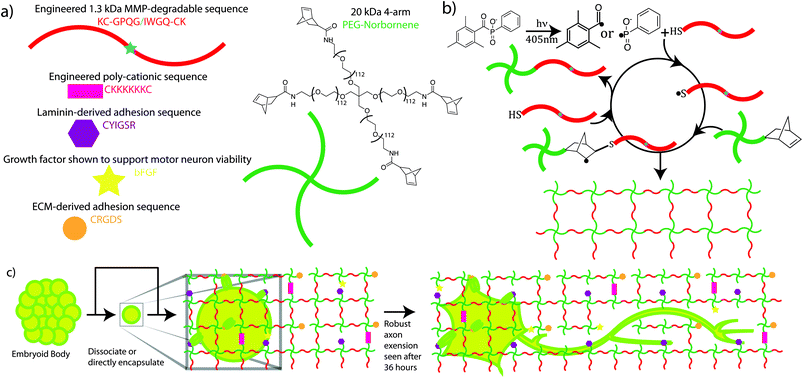 | ||
| Fig. 1 Schematic illustrating ESMN encapsulation in degradable peptide crosslinked PEG hydrogels functionalized with ECM-mimic peptides, a cationic peptide, and bFGF. Gels were formed using a thiol-ene click chemistry and LAP as a visible-light photoinitiator. (a) Chemical structures of network components are shown along with schematic representations. (b) The mechanism for gel formation relies on radical-mediated thiol-ene chemistry to produce step growth networks. Free thiols quickly and efficiently initiate chain transfer minimizing radical damage to other species. (c) Motor neurons are encapsulated in the gel formulations and interact with pendent YIGSR and RGDS groups through integrin-mediated binding. Within 36 hours MMPs expressed by the neurons erode defects through the network and allow axons to extend. The diagram is not to scale but has been enlarged to show all of the gel components. The mesh size of the network is on the order of ∼10 nm while an axon is ∼1 micron in diameter. | ||
While radicals are known to cause cell and protein damage, previous studies have shown that the thiol-ene reaction requires fewer radicals to generate a network, and the radicals have shorter lifetimes than more traditional photoinitiated chain polymerizations, leading to higher cell viability and protein activity.29,30 Further, thiol-ene reactions are not oxygen inhibited, expanding their utility for bulk polymerization in the presence of cells. For example, a thiol-ene PEG system was shown to support roughly twice the viability of the MIN6 pancreatic β-cell line relative to a methacrylate PEG system.30 In addition, the activity of a model protein, lysozyme, markedly decreased when encapsulated in an acrylate-based gel, but was virtually unchanged in a thiol-ene system.29 In addition to the mild polymerization conditions, thiol-ene gels also form near ideal network structures that present a consistent biophysical environment throughout the hydrogel whose modulus can be easily tuned by varying the pre-polymerization functional group concentrations.31–33 Furthermore, these networks allow for the facile introduction of different crosslinkers or pendant peptides either during or after gelation.34 Thus, the cellular environment can be modified initially or sequentially with developmentally relevant and biologically active thiol-containing molecules. Collectively, these prior results suggested that a thiol-ene hydrogel might be suitable for encapsulating a delicate cell type like motor neurons.
In vitro, ESMNs are typically plated either as EBs or dissociated cells on glass slides coated with poly-lysine and laminin, which supports cell adhesion and axon outgrowth. However, while this surface treatment is critical to dissociated ESMN adhesion, survival, and axon outgrowth, EBs will remain viable for weeks in suspension culture, on tissue culture polystyrene (TCPS), or on treated glass and once adhered will exhibit much more robust axon outgrowth than single cells. After 3 days of culture on poly-lysine and laminin treated glass in media supplemented by neurotrophins, only roughly 20% of dissociated ESMNs survive as compared with nearly all cells cultured in EBs.10 This dramatic difference is likely due to the tissue like structures formed within EBs during differentiation and the existence of Oligo2+ neural progenitors supporting the viability of ESMNs.10,35 For in vivo experimentation, EBs are often deployed because of this robustness.12 These observations served as a starting point for engineering a synthetic 3D platform for the culture of ESMNs. We postulated that EBs could likely be encapsulated without any survival-promoting matrix interactions, but dissociated ESMNs would require similar signals to those shown to be necessary in 2D. However, while ESMNs maintain higher viability, require fewer signals, and extend longer axons when cultured in EBs, their complex structure creates new challenges associated with deconvoluting the relative contributions of different biophysical and biological cues. A synthetic 3D culture platform for dissociated ESMNs would enable the study of their responses to differing biological cues without the confounding effects of cell–cell contact or naturally derived polymers, and might prove useful to improve upon methods to efficiently differentiate specific motor neuron subtypes.
To rationally design a hydrogel capable of supporting the viability and axon outgrowth of dissociated ESMNs in three dimensions, we began with standard 2D culture platform of poly-lysine and laminin coated glass. Positive charge, which has been shown to promote axon outgrowth,36 is usually satisfied with poly-lysine in 2D culture. However, cationic polymers are often implicated in cytotoxicity,37 so we synthesized a hexalysine (CKKKKKKC) sequence to serve in the place of poly-lysine. Further, full laminin protein was replaced by two peptide sequences CRGDS and CYIGSR that have previously been shown to engage the appropriate integrin proteins and promote neuronal adhesion and axon outgrowth.38 Finally bFGF, which exists naturally in its reduced form, has been shown to increase ESMN viability and contains 4 free thiols for facile coupling to the hydrogel.39,40 We form these biofunctional hydrogels by coupling norbornene-terminated PEG macromers and the cysteine-containing, cell-cleavable peptide sequence26,41,42 (KCGPQG↓IWGQCK) using photo-initiated thiol-ene click chemistry, resulting in cytocompatible hydrogel networks that can be locally remodeled by the encapsulated cells (Fig. 1).
Results and discussion
Initial outgrowth of motor axons was observed from the majority of encapsulated cells after 12 hours of culture within 3D hydrogels (Fig. 2b). Embryoid bodies also began budding axons, but early extension was difficult to visualize owing to the size and brightness differential of the approximately thousand-cell aggregates in comparison to the nascent axons (data not shown).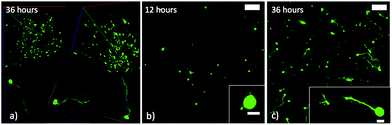 | ||
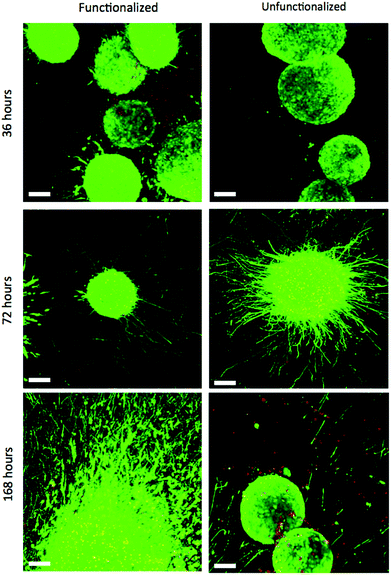
| Fig. 2 (a) ESMNs exhibit robust axonal outgrowth in three-dimensions when encapsulated in the engineered PEG hydrogel (scale bars 100 and 10 μm within inset). (b) Representative image of ESMNs 12 hours post-encapsulation. Small axonal buds are seen in the inset image. (c) Representative image of ESMNs 36 hours post-encapsulation. | ||
Single ESMNs assumed characteristic polarized shapes of in vivo motor neurons with a single axon emanating from the cell body 36 hours post-encapsulation (Fig. 2a and 2c). The gels functionalized with integrin binding peptides, cationic hexalysine peptide, and bFGF maintained dissociated ESMN viability at 21 ± 1% (Fig. 3 and 4). This gel formulation also resulted in robust axon outgrowth, with 83 ± 9% of eGFP positive cells extending axons an average of 148 ± 14 μm. In contrast, encapsulation of dissociated ESMNs in unfunctionalized gels lacking biological cues resulted in drastically reduced viability of 4 ± 2% (Fig. 3 and 4). The encapsulated EBs, however, did not show a preference for the functionalized material over the unfunctionalized with respect to cell viability as virtually 100% of the cells remained viable after 36 hours in both systems (Fig. 5). Their axon extension qualitatively appeared more robust in the functionalized gel; however, quantification was not possible due to axon fasciculation and the high density of cell bodies.
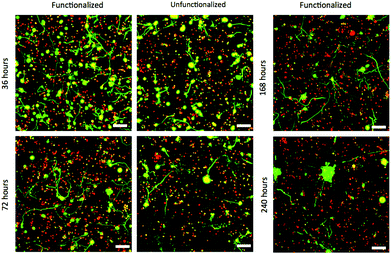 | ||
| Fig. 3 Confocal Z-stacks Live (green)/Dead (red) stain of dissociated ESMNs in functionalized and unfunctionalized hydrogels at different time points. The unfunctionalized gels were not imaged after 168 hours. By 240 hours, all live cells existed in multicellular aggregates (scale bar 100 μm). | ||
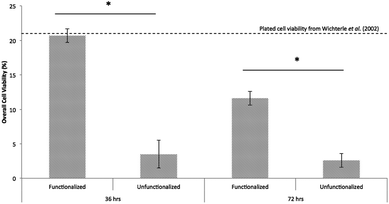 | ||
| Fig. 4 Hydrogels functionalized with integrin binding peptides, bFGF, and charged peptide maintain significantly higher dissociated ESMN viability than those lacking these functionalities (mean cells per condition = 422.2, p < 0.001). The viability of ESMNs encapsulated in the functional hydrogel is nearly that of cells plated on laminin-coated coverslips.10 | ||
 | ||
| Fig. 5 Confocal Z-stacks Live (green)/Dead (red) stain of encapsulated EBs in functionalized and unfunctionalized gels at different time points. The image of the EBs in the unfunctionalized gel after 168 hours is not representative, but shows that a subset of EBs failed to extend axons (scale bar 100 μm). | ||
By 72 hours, 12 ± 1% of the dissociated cells in the functionalized gels remained viable while only 3 ± 1% of those in the unfunctionalized gels did (Fig. 3 and 4). The surviving cells continued to elongate their axons, although not quantifiable owing to eGFP aggregates within the cells and axon extension beyond the field of view. At 72 hours, the cells within EBs still remained nearly 100% viable and extended axons millimeters through the gel (Fig. 5). By this point, every EB in the functionalized gel and many in the unfunctionalized gel exhibited robust axon outgrowth, but interestingly, in the unfunctionalized gel a minority of EBs, approximately 20%, did not extend axons.
While motor neuron differentiation is highly robust and reproducible, only 30%–50% of the cells taken through the differentiation protocol are GFP+ motor neurons.15 The remainder is composed of interneurons and Oligo2+ progenitors that can self-renew or further differentiate into oligodendrocytes and other neuroglial cells.10 Even after 36 hours in these gel cultures, there is a marked difference in viability in dispersed cells and EBs. This difference is likely caused by the supportive effects of these neuroglial cells. While thiol-ene chemistry is a relatively gentle method of cell encapsulation, the process is still stressful to cells, as small changes in osmolarity and other stressers exist over the short timescale required for encapsulation. Remarkably, however, the viability of ESMNs encapsulated in the functional hydrogel is nearly that of cells plated on laminin-coated coverslips. In standard 2D culture, only 5% of ESMNs survive without supportive neurotrophins (NT3, CNTF, BDNF, GDNF) and only 22% survive with these factors after 72 hours.10 While this is in sharp contrast to the essentially 100% viability of encapsulated EBs, it is congruent with development in vivo in which approximately half of all spinal motor neurons die while competing for a limited pool of neurotrophins.43,44 This indicates that cell–cell signaling in the EB is extremely important to cell viability within this in vitro system.
This conclusion is reinforced by confocal images of the dissociated motor neurons after 168 hours in culture. Dissociated ESMNs encapsulated in the functionalized hydrogel extended axons beyond the field of view of a 10× objective while cell viability drops to 11 ± 4% (Fig. 3). Interestingly, many of the live cells appeared in small 2–5 cell clusters, suggesting one or two motor neurons surrounded by Oligo2+ neural progenitors as evidenced by the one or two axons emanating from these aggregates and the fact that these aggregates increase in size over time and motor neurons are post-mitotic. This trend increased to the point where every live cell was found in an aggregate by 240 hours (Fig. 3). The existence of these aggregates lends credence to the idea that the Oligo2+ progenitors generated during the ESMN differentiation strongly promote ESMN survival. After 168 hours in culture, the EBs encapsulated in both the functionalized and unfunctionalized gels still remained nearly 100% viable, and every EB in the functionalized gel and many in the unfunctionalized gel extended axons from one end of the gel to another, which had swollen to over a centimeter in diameter from its original 5 mm. The entire gel was permeated by axons, even at large distances from encapsulated EBs (ESI Fig. 1†); yet, while in the minority, several EBs (∼10%) in the unfunctionalized gel failed to extend any axons.
These results indicate that, even at early time points, receptor signaling satisfied by integrin-binding ECM-mimic peptides, bFGF, and charge-coupled interactions are essential for the viability and axon outgrowth of dissociated ESMNs, but are not critical if cell–cell interactions are provided (i.e., cells within an EB). At later time points (>7 days in culture), these ECM-based signals were not sufficient to maintain cell viability and only clusters of cells survive. This observation is consistent with motor neuron behavior in vivo, however, as any cell that does not successfully form a mature synapse with a myotube will apoptose, with only half of the motor neurons generated making it to adulthood.16 Future studies will examine this phenomenon through an ESMN/myotube co-culture.
Next, we investigated whether motor axon outgrowth is dependent on the MMP-degradable cross-linker peptide KCGPQG↓IWGQCK. We replaced the MMP-degradable cross-linker with a 3.4 kDa PEG cross-linker, a macromer containing neither cell-degradable bonds nor cell-interactive domains. Such hydrogels demonstrated similar swollen moduli to those containing the MMP-degradable cross-linker. Upon replacement of the MMP-degradable cross-linker peptide with PEG-dithiol, many encapsulated ESMNs remained viable, but the vast majority of encapsulated ESMNs failed to extend axons in such a microenvironment (Fig. 6a and 6b). Fig. 7 presents results after quantification of several of these confocal images (N = 105 cells for degradable gels, N = 118 cells for non-degradable gels). In the non-degradable gel systems, 39 ± 7% of cells extended axons with an average length of 11 ± 4 μm, and no branching was observed. In contrast, in the MMP-degradable gels, 83 ± 9% of cells extended axons with an average length of 148 ± 14 μm. Additionally, individual motor axons branched in MMP-degradable gels, characteristic of their in vivo phenotype, and exhibited no branching in non-degradable gels. Furthermore, immunostaining of ESMNs with an antibody specific to MMP2 revealed a strong expression of MMP2 in the cell bodies and growth cones of ESMNs (Fig. 6c and 6d). Together, these findings reveal an identification of a peptide functionalized PEG hydrogel formulation which depends on cell–ECM interactions and endogenous MMP2 activity encoded by ESMNs to support ESMN survival and axon outgrowth.
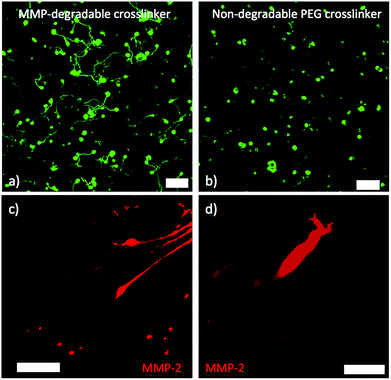 | ||
| Fig. 6 (a) Z-projection of motor neurons cultured for 36 hours in gel with MMP-degradable peptide crosslinker (scale bar 100 μm). (b) Z-projection of motor neurons cultured for 36 hours in gel with non-degradable PEG crosslinker. Motor neurons axon extension is limited (scale bar 100 μm). (c) MMP-2 immunostaining showing MMP-2 in growth cone of multiple axons. The cells bodies are contained in an embryoid body to the right of the frame (scale bar 50 μm). (d) Higher magnification image of one growth cone showing MMP-2 staining (scale bar 10 μm). | ||
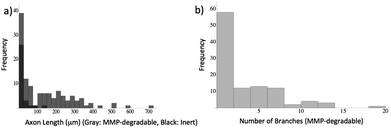 | ||
| Fig. 7 Quantification of motor neuron phenotype. Stacks are loaded into Simple Neurite Tracer48 and axon length and branching are quantified. (a) Histogram of axon length in both non-degradable (black) and MMP-degradable (gray) gels. (b) Number of major axon branches in MMP-degradable gels. None of the axons in the non-degradable gel branched. | ||
Crosslinking the hydrogel with an inert PEG macromer, rather than an MMP-degradable peptide sequence effectively eliminates axon outgrowth. Unlike naturally-derived and fibrous 3D scaffolds, step-growth PEG networks have a mesh size on the order of 10 nm.45 An axon is roughly one micron in diameter and is unable to reptate through such a polymer network unless significant defects are present. Furthermore, ESMNs were shown to express MMP-2 by immunostaining, which has been shown to cleave the peptide crosslinker KCGPQG↓IWGQCK.42 While studies in Drosophila have shown a direct requirement for MMP2 in fasciculation of motor axons, whether MMPs play a role in motor axon guidance remains unclear.46 Our studies provide evidence that motor axon outgrowth in 3D hydrogels relies on the presence of MMP-degradable peptide cross-linkers, removal of which results in reduced axonal outgrowth. Our results also provide evidence that the 3D hydrogel system defined here provides a permissive environment for in vivo-like branching of spinal motor axons shown to be essential for target connectivity.47
Finally, we examined whether the elastic modulus of the hydrogel matrix influences the extent of motor axon outgrowth of ESMNs by tracking axonal outgrowth as a function of varying crosslinking densities. We tested this hypothesis by varying the initial PEG-norbornene monomer concentrations from 3.5 to 6.3 wt% while keeping the pendant peptide concentrations constant. Such manipulations resulted in hydrogel matrices with shear elastic moduli that ranged from 350 Pa to 2220 Pa. When ESMNs were encapsulated into hydrogel formulations with the highest modulus of 2220 Pa, ESMNs underwent uniform cell death (Fig. 8). Interestingly, encapsulation of ESMNs in 780 Pa hydrogels allowed some survival, but motor axons failed to grow in these hydrogels. Finally, robust axon outgrowth was observed in 350 Pa hydrogels.
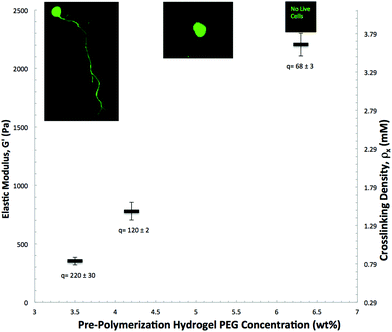 | ||
| Fig. 8 3D ESMN axon outgrowth is highly dependent on the initial gel crosslinking density. At 0.9 mmol crosslinker per liter, axons are able to penetrate through the polymer-network by locally cleaving the collagen-derived peptide crosslinks. However, increasing the crosslinking density to 1.5 mM prevents neurons from extending axons and further increasing the crosslinking density to 3.6 mM results in 100% cell mortality. Mass swelling ratios are shown below and representative cell images are shown close to each data point. | ||
Hydrogels with an elastic modulus below 350 Pa may provide an even more robust platform for axonal outgrowth. However, hydrogels with moduli below 350 Pa were difficult to manipulate post gelation, making consistent imaging and observations impossible. Despite this, the examined moduli range is consistent with the range of interest for neural microenvironments (50–250 Pa) in vivo.49 These in vivo environments, despite being very soft, are distinctly elastic rather than viscous, a trait accurately recapitulated in our hydrogel platform which has a storage modulus roughly one thousand times higher than the loss modulus (ESI Fig. 2†).
Experimental
ES cell culture
ES cells were differentiated into spinal motor neurons as previously described.15 Briefly, Hb9::GFP mouse embryonic stem cells were plated into ES cell medium (ES DMEM, ES FBS, glutamine, non-essential amino acids, nucleosides, 2-mercaptoethanol, LIF (Life Technologies)) at approximately 5 × 105 cells per gelatinized T25 flask. After 24 hours the media was replaced, and on day 2 of culture, ES cells were trypsinized and placed in suspension culture in motor neuron media (Advanced-DMEM/F12, Neurobasal, and Knockout Serum Replacement (Life Technologies)) at 5 × 105 cells per untreated 10 cm tissue culture dish. In suspension culture, the cells aggregated into embryoid bodies (EBs). Two days after initial seeding the EBs were split 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :4 and induced into motor neurons with 1 μM retinoic acid (RA) (Sigma) and smoothened agonist (SAG) (Millipore). After 3 days of exposure to RA and SAG, the EBs displayed strong expression of Hb9::GFP transgene.15
:4 and induced into motor neurons with 1 μM retinoic acid (RA) (Sigma) and smoothened agonist (SAG) (Millipore). After 3 days of exposure to RA and SAG, the EBs displayed strong expression of Hb9::GFP transgene.15
Peptide synthesis
CRGDS and KCGPQG↓IWGQCK were purchased (American Peptide Company). CKKKKKKC and CYIGSR were synthesized on a Protein Technologies Tribute Peptide Synthesizer using standard Fmoc chemistry and Rink Amide MBHA resin. Peptide cleavage solution was formed by dissolving 250 mg dithiothreitol (DTT) and 250 mg phenol in a solution of 95% trifluoroacetic acid (TFA), 2.5% triisopropylsilane (TIPS), and 2.5% deionized water. Synthesized peptides were cleaved in the solution for 2 hours. Cleaved peptides were precipitated in cold diethyl ether, recovered via centrifugation, desiccated overnight, and then purified by reverse-phase HPLC (Waters Delta Prep 4000) purification on a C18 column using a linear acetonitrile:water gradient. The collected fractions of purified peptides were identified by matrix-assisted laser desorption/ionization-time-of-flight (MALDI-TOF) mass spectrometry (ESI Fig. 3†).Monomer synthesis
8 equivalents of 5-norbornene-2-carboxylic acid were dissolved in anhydrous DMF and activated with 7.5 equivalents of HBTU (Sigma). 16 equivalents of N-methylmorpholine (Sigma) were added as base. After activating for 5 minutes, this mixture was added to 1 equivalent of 20 kDa 4-arm PEG-amine (JenKem) dissolved in anhydrous DMF and reacted overnight at room temperature. PEG-norbornene was precipitated in diethyl ether, redissolved in DI water, and dialyzed against DI water for 24 h (2000 MWCO). The product was then lyophilized and used for experimentation (ESI Fig. 4†). LAP was synthesized as previously described.27Cell encapsulation
Gels were prepared with a total volume of 40 μL from stock solutions of 4-arm 20 kDa PEG-tetra-norbornene, MMP-degradable peptide (KCGPQG↓IWGQCK), ECM-mimic peptides (CRGDS and CYIGSR), cationic peptide (CKKKKKKC), bFGF (R&D Systems), and LAP.27 The PEG-norbornene was carefully added to the motor neuron medium, followed the MMP-degradable peptide crosslinker or the PEG-dithiol (Sigma), the adhesive peptides, the LAP, and the bFGF. For the unfunctionalized gel, peptides were replaces with equal concentrations of cysteine, and bFGF was replaced with PBS. The mixture was then triturated to evenly mix the polymers. To vary the stiffness, the macromolecular monomer solution weight percent was scaled accordingly, while the total volume was maintained at 40 μL. For gels containing dissociated cells, EBs were exposed to 1 mL of ice-cold trypsin per 10 cm dish for 3 minutes and triturated vigorously to yield single cells. Single neurons were then re-suspended at 10 million cells per mL, and 10 μL of the cell suspension was gently mixed into the prepared gel formulation and polymerized for 1 minute under 40 mW cm−2 405 nm blue light (Thor Labs Collimated 405 nm LED). Gels containing EBs were prepared in an identical manner, but after 1 minute the gel solution was mixed to prevent EB settling. Cell-laden gels were then placed in motor neuron medium supplemented with 10 ng mL−1 GDNF and N2 and B27 Supplement. Table 1 provides a summary of all gel formulations used.| PEG-tetra-norbornene | KCGPQGIWGQCK | HS-PEG-SH | CRGDS | CY/GSR | CKKKKKC | Cysteine | LAP | bFGF | ESMNs | |
|---|---|---|---|---|---|---|---|---|---|---|
| Soft functionalized (3.5 wt%) | 2.1 mM | 3.85 mM | 0 mM | 1.25 mM | 1.25 mM | 0.0125 mM | 0 mM | 0.625 mM | 10 nM | 2.5 M mL−1 |
| Medium functionalized (4.2 wt%) | 2.9 mM | 4.5 mM | 0 mM | 1.25 mM | 1.25 mM | 0.0125 mM | 0 mM | 0.625 mM | 10 nM | 2.5 M mL−1 |
| Stiff functionalized (6.3 wt%) | 3.8 mM | 7.2 mM | 0 mM | 1.25 mM | 1.25 mM | 0.0125 mM | 0 mM | 0.625 mM | 10 nM | 2.5 M mL−1 |
| Soft unfunctionalized (3.5 wt%) | 2.1 mM | 3.85 mM | 0 mM | 0 mM | 0 mM | 0 mM | 2.51 mM | 0.625 mM | 0 nM | 2.5 M mL−1 |
| Non-degradable functionalized (3.5 wt%) | 2.1 mM | 0 mM | 3.85 mM | 1.25 mM | 1.25 mM | 0.0125 mM | 0 mM | 0.625 mM | 10 nM | 2.5 M mL−1 |
Rheology
The moduli of the hydrogels were measured using a rheometer with an 8 mm parallel plate geometry (AR-G2, TA Instruments), irradiation attachment (UV Light Guide Accessory, TA Instruments), and mercury arc lamp light source with 400–500 nm filter and light guide (Omnicure S2000, Exfo). The samples were formed in situ using a modified version of a previously published protocol.50 Briefly, monomer solutions for each composition were prepared, and 5–10 μL of solution was pipetted on to the bottom plate. The top plate was lowered until the gap was completely filled with solution (approximately 50–100 microns). The irradiation intensity was set to ∼16 mW cm−2 with the Omnicure shutter fully open (SilverLine Control-cure Radiometer and International Light Radiometer). Owing to this, the irradiation time for sample polymerization was adjusted to ensure that the same irradiation dose used for cell encapsulation (time × intensity) was applied to all samples (time increased from 1 to 2.5 minutes). A dynamic time sweep (10% strain, 5 rad s−1 frequency) was used to monitor modulus evolution during polymerization to ensure that all samples reached their fully developed moduli (ESI Fig. 2†). Final moduli were recorded for 3 to 5 replicates of each composition. Moduli of polymerized samples were adjusted to their equilibrium swollen values using measured swelling ratios and rubber elasticity theory.51Confocal imaging
Gels were placed between a glass slide and a coverslip separated by a rubber gasket and were imaged using a 10× or 20× water immersion objective. A 488 nm laser was used to excite eGFP and calcein AM, and a 514 nm laser was used to excite ethidium homodimer-1. The Cy3 secondary antibody was excited using a 528 nm laser. A Z-stack of 100 images was taken through the entire gel in three different fields of view with approximately 20 μm between images.Staining
Gels were fixed in a PBS solution containing 4% paraformaldehyde (PFA) for 2 hours at 4 °C in the cold room and rinsed 3× with PBS. Gels were then swollen in a solution containing 10% horse serum in PBS overnight. Horse serum was then swollen out for 10 hours into PBS that was periodically changed. The primary antibody (Abcam Rabbit anti-MMP-2 Ab37150) diluted at 100× in 2% horse serum in PBS was then incubated overnight at 4 °C on a rocker. In the morning, gels were transferred into PBS solutions and the primary antibody was given 10 hours to swell out in PBS that was periodically changed. The secondary (Jackson ImmunoResearch Donkey Anti-Rabbit Cy3) was then diluted 1000× in 2% horse serum and swelled in overnight at 4 °C. Finally, the secondary was swelled out for 24 hours in PBS and the gels were imaged. For Live/Dead imaging, 2 μM of calcein AM and 4 μM of ethidium homodimer-1 stock solutions were diluted in PBS. Gels were incubated in this solution for 30 minutes on a shaker in a cell culture incubator and then imaged.SEM: Gels were swollen overnight in DI water and dehydrated in ethanol solutions of 20%, 40%, 60%, 80%, 90%, and 100% for 10 minutes each, with the 100% solution being repeated three times. The gels were then dried in hexamethyldisilazane (HDMS), placed under vacuum for 30 minutes, mounted on stubs, and sputter coated with gold.52 Samples were examined with a JEOL JSM-6480LV. Images of their surfaces can be found in the supporting in the ESI (Fig. 5†).
Statistics
Cell viability error is reported as the standard error from three image stacks taken from the same gel. Live cells were manually counted due to their complex shape and overlapping axons, and dead cells were counted using the Analyze Particles tool in ImageJ. More than 400 individual cells were counted for viability experiments. A t-test was performed between the functionalized and unfunctionalized gels at 36 hours and 72 hours to determine significance with p < 0.001. Axon length was measured using the Simple Neurite Trace plugin for Fiji48 (ESI Fig. 6†). The error for axon length is reported as the standard error with 105 cells counted for the degradable gel and 118 cells for the non-degradable gels. This experiment was repeated multiple times (>10), and all results were similar. To translate 3D pixel lengths into microns, axons were assumed to have an equal chance of traveling in the x, y, or z directions. The scale in the x- and y-directions was 1.08 microns per pixel and in the z-direction was 3.89 microns per pixel. Therefore, the axon extension in pixels was multiplied by (2/3 × 1.06 + 1/3 × 3.89) to convert to microns.Conclusions
Together, our findings emphasize the importance of biochemical and physical microenvironment characteristics in enabling motor axon outgrowth in 3D. In addition, this work highlights the potential of synthetic biomaterials as ECM mimics in the examination of nerve cell biology and axon guidance. Our studies allowed systematic characterization and identification of the chemical and physical properties sufficient for motor axon outgrowth in 3D hydrogel matrices. Such 3D hydrogel matrices may be used to investigate and identify the molecular mechanisms that drive directed axonal outgrowth, as in many cases genetic analysis is confounded by the presence of multiple receptor–ligand signaling systems that operate during axonal pathfinding.Motor neuron subtype loss is an underlying cause of ALS and SMA disorders. Successful transplantation of nerve cells into the adult CNS for ALS and SMA treatment remains one of the obstacles in utilization of stem cell-based cell-replacement therapy. Fundamental in vitro studies, such as this one, can provide a useful screening tool and may provide novel insights into the requirements for design of transplantable matrices capable of directing axonal outgrowth and suggest improved strategies for cellular delivery vehicles for treatment of neurodegenerative disease states.
Acknowledgements
The authors would like to thank Prof. Hynek Wichterle for the kind gift of Hb9::GFP ES cells, Dr Josh McCall and Dr Malar Azagarsamy for their helpful discussions on material properties and chemical synthesis, and Ravi Moghe for verifying the reproducibility of these experiments. These studies were supported by a grant from the NSF (DMR 1006711), the NIH Pharmaceutical Biotechnology Training Program (5 T32 GM 8732), and the Howard Hughes Medical Institute.Notes and references
- S. R. Cajal, Texture of the Nervous System of Man and the Vertebrates, 1999 Search PubMed.
- L. A. Flanagan, Y.-E. Ju, B. Marg, M. Osterfield and P. A. Janmey, NeuroReport, 2002, 13, 2411–2415 CrossRef.
- B. J. Dickson, Science, 2002, 298, 1959–1964 CrossRef CAS.
- C. Manitt, M. Colicos, K. Thompson, E. Rousselle, A. Peterson and T. Kennedy, J. Neurosci., 2001, 21, 3911–3922 CAS.
- M. Mahoney and K. S. Anseth, Biomaterials, 2006, 27, 2265–2274 CrossRef CAS.
- G. A. Silva, C. Czeisler, K. L. Niece, E. Beniash and S. I. Stupp, Science, 2004, 303, 1352–1355 CrossRef CAS.
- C. T. S. Wong Po Foo, J. S. Lee, W. Mulyasasmita, A. Parisi-Amon and S. C. Heilshorn, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 22067–22072 CrossRef.
- Y. Luo and M. S. Shoichet, Nat. Mater., 2004, 3, 249–253 CrossRef CAS.
- S. Seidlits, Z. Khaing, R. Petersen, J. Nickels, J. E. Vanscoy, J. B. Shear and C. E. Schmidt, Biomaterials, 2010, 31, 3930–3940 CrossRef CAS.
- H. Wichterle, I. Lieberam, J. Porter and T. M. Jessell, Cell, 2002, 110, 385–397 CrossRef CAS.
- G. B. Miles, D. C. Yohn, H. Wichterle, T. M. Jessell, V. F. Rafuse and R. M. Brownstone, J. Neurosci., 2004, 24, 7848–7858 CrossRef CAS.
- M. Peljto, J. S. Dasen, E. O. Mazzoni, T. M. Jessell and H. Wichterle, Cell Stem Cell, 2010, 7, 355–366 CrossRef CAS.
- J. A. Umbach, K. L. Adams, C. B. Gundersen and B. G. Novitch, PLoS One, 2012, 7, e36049 CAS.
- J. S. Dasen, A. De Camilli, B. Wang, P. W. Tucker and T. M. Jessell, Cell, 2008, 134, 304–316 CrossRef CAS.
- H. Wichterle and M. Peljto, Current Protocols in Stem Cell Biology, 2008 Search PubMed.
- K. C. Kanning, A. Kaplan and C. E. Henderson, Annu. Rev. Neurosci., 2010, 33, 409–440 CrossRef CAS.
- J. M. Harper, C. Krishnan, J. S. Darman, D. Deshpande, S. Peck, I. Shats, S. Backovic, J. Rothstein and D. A. Kerr, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 7123–7128 CrossRef CAS.
- D. Deshpande, Y. Kim, T. Martinez, J. Carmen, S. Dike, I. Shats, L. L. Rubin, J. Drummond, C. Krishnan, A. Hoke, N. Maragakis, J. Shefner, J. Rothstein and D. A. Kerr, Ann. Neurol., 2006, 60, 32–44 CrossRef CAS.
- C. E. Henderson, W. Camu, C. Mettling, A. Gouin, K. Poulsen, M. Karihaloo, J. Ruilamas, T. Evans, S. B. McMahon, M. P. Armanini, L. Berkemeier, H. S. Phillips and A. Rosenthal, Nature, 1993, 363, 266–270 CrossRef CAS.
- S. J. Bryant and K. S. Anseth, J. Biomed. Mater. Res., 2001, 59, 63–72 CrossRef.
- J. A. Burdick and K. S. Anseth, Biomaterials, 2002, 23, 4315–4323 CrossRef CAS.
- B. K. Mann, A. S. Gobin, A. T. Tsai, R. H. Schmedlen and J. L. West, Biomaterials, 2001, 22, 3045–3051 CrossRef CAS.
- S. J. Bryant, C. R. Nuttelman and K. S. Anseth, J. Biomater. Sci. Polym. Ed., 2000, 11, 439–457 CrossRef CAS.
- C. R. Nuttelman, M. C. Tripodi and K. S. Anseth, Matrix Biol., 2005, 24, 208–218 CrossRef CAS.
- C. E. Hoyle, A. B. Lowe and C. N. Bowman, Chem. Soc. Rev., 2010, 39, 1355–1387 RSC.
- B. D. Fairbanks, M. P. Schwartz, A. E. Halevi, C. R. Nuttelman, C. N. Bowman and K. S. Anseth, Adv. Mater., 2009, 21, 5005–5010 CrossRef CAS.
- B. D. Fairbanks, M. P. Schwartz, C. N. Bowman and K. S. Anseth, Biomaterials, 2009, 30, 6702–6707 CrossRef CAS.
- C. E. Hoyle and C. N. Bowman, Angew. Chem., Int. Ed., 2010, 49, 1540–1573 CrossRef CAS.
- J. D. McCall and K. S. Anseth, Biomacromolecules, 2012, 13, 2410–2417 CrossRef CAS.
- C. Lin, A. Raza and H. Shih, Biomaterials, 2011, 32, 9685–9695 CrossRef CAS.
- M. Malkoch, R. Vestberg, N. Gupta, L. Mespouille, P. Dubois, A. F. Mason, J. L. Hedrick, Q. Liao, C. W. Frank, K. Kingsbury and C. J. Hawker, Chem. Commun., 2006, 2774–2776 RSC.
- T. Sakai, T. Matsunaga, Y. Yamamoto, C. Ito, R. Yoshida, S. Suzuki, N. Sasaki, M. Shibayama and U.-I. Chung, Macromolecules, 2008, 41, 5379–5384 CrossRef CAS.
- T. Matsunaga, T. Sakai, Y. Akagi, U.-I. Chung and M. Shibayama, Macromolecules, 2009, 42, 1344–1351 CrossRef CAS.
- C. A. DeForest, B. D. Polizzotti and K. S. Anseth, Nat. Mater., 2009, 8, 659–664 CrossRef CAS.
- T. C. Doetschman, H. Eistetter, M. Katz, W. Schmidt and R. Kemler, J. Embryol. Exp. Morphol., 1985, 27–45 CAS.
- M. Dadsetan, A. M. Knight, L. Lu, A. J. Windebank and M. J. Yaszemski, Biomaterials, 2009, 30, 3874–3881 CrossRef CAS.
- J. H. Jeong and T. G. Park, J. Controlled Release, 2002, 82, 159–166 CrossRef CAS.
- K.-I. Tashiro, G. C. Sephel, B. Weeks, M. Sasaki, G. R. Martin, H. K. Kleinman and Y. Yamada, J. Biol. Chem., 1989, 264, 16174–16182 CAS.
- S. A. Thompson, J. Biol. Chem., 1992, 267, 2269–2273 CAS.
- R. A. Hughes, M. Sendtner, M. Goldfarb, D. Lindholm and H. Thoenen, Neuron, 1993, 10, 369–377 CrossRef CAS.
- M. P. Lutolf, J. L. Lauer-Fields, H. G. Schmoekel, A. T. Metters, F. E. Weber, G. B. Fields and J. A. Hubbell, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 5413–5418 CrossRef CAS.
- H. Nagase and G. B. Fields, Pept. Sci., 1996, 40, 399–416 CrossRef CAS.
- R. W. Oppenheim, L. J. Houenou, J. E. Johnson, L.-F. H. Lin, L. Li, A. C. Lo, A. L. Newsome, D. M. Prevette and S. Wang, Nature, 1995, 373, 344–346 CrossRef CAS.
- P. Vanderhaeghen and H. J. Cheng, Cold Spring Harb. Perspect. Biol., 2010, 2, a001859–a001859 CrossRef.
- F. Brandl, F. Kastner, R. M. Gschwind, T. Blunk, J. Teßmar and A. Göpferich, J. Controlled Release, 2010, 142, 221–228 CrossRef CAS.
- C. M. Miller, A. Page-McCaw and H. T. Broihier, Development, 2008, 135, 95–109 CrossRef CAS.
- J. Livet, M. Sigrist, S. Stroebel, V. De Paola, S. R. Price, C. E. Henderson, T. M. Jessell and S. Arber, Neuron, 2002, 35, 877–892 CrossRef CAS.
- M. H. Longair, D. A. Baker and J. D. Armstrong, Bioinformatics, 2011, 27, 2453–2454 CrossRef CAS.
- Y. Lu, K. Franze, G. Seifert, C. Steinhauser, F. Kirchhoff, H. Wolburg, J. Guck, P. A. Janmey, E.-Q. Wei, J. Kas and A. Reichenbach, PNAS, 2006, vol. 103, pp. 17759–17764 Search PubMed.
- A. M. Kloxin, M. W. Tibbitt, A. M. Kasko, J. A. Fairbairn and K. S. Anseth, Adv. Mater., 2010, 22, 61–66 CrossRef CAS.
- C. N. Salinas and K. S. Anseth, Macromolecules, 2008, 41, 6019–6026 CrossRef CAS.
- F. Braet, R. De Zanger and E. Wisse, J. Microsc., 1997, 186, 84–87 CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Image of axons millimeters from nearest EB, time evolution rheology of the hydrogel, peptide MALDI spectra, PEG-norbornene NMR, SEM images of the hydrogel surfaces, and a simple neurite tracer example image are available in ESI. See DOI: 10.1039/c3bm00166k |
| This journal is © The Royal Society of Chemistry 2013 |