
Achieving high-efficiency blue organic light-emitting diodes via pyridyl-unit incorporated thermally activated delayed fluorescence materials
Xiaoning
Li†
a,
Dian
Xie†
b,
Xiaojuan
Song
a,
Yujun
Xie
 *a,
Dongge
Ma
*b,
Ben Zhong
Tang
*ac and
Zhen
Li
*abd
*a,
Dongge
Ma
*b,
Ben Zhong
Tang
*ac and
Zhen
Li
*abd
aInstitute of Molecular Aggregation Science, Tianjin University, Tianjin, 300072, China. E-mail: xieyujun@tju.edu.cn
bThe State Key Laboratory of Luminescent Materials and Devices, South China University of Technology, Guangzhou, 510640, China. E-mail: msdgma@scut.edu.cn
cGuangdong Basic Research Center of Excellence for Aggregate Science, School of Science and Engineering, The Chinese University of Hong Kong (Shenzhen), Longgang, Shenzhen, Guangdong 518172, China. E-mail: tangbenz@cuhk.edu.cn
dHubei Key Lab on Organic and Polymeric Opto-Electronic Materials, Department of Chemistry, Wuhan University, Wuhan, 430072, China. E-mail: lizhen@whu.edu.cn
First published on 1st October 2025
Abstract
Thermally activated delayed fluorescence (TADF) materials, which enable 100% exciton utilization via reverse intersystem crossing (RISC), play a pivotal role in advancing organic light-emitting diodes (OLEDs). However, designing blue TADF emitters remains challenging due to the inherent trade-off between short emission wavelength and small singlet–triplet splitting. In this study, a strategy for developing such emitters is proposed by introducing pyridyl groups into the triindocarbazole donor to form intramolecular C–H⋯N hydrogen bonds. Compared with the phenyl-substituted counterpart TDBA-Ph, the pyridyl-incorporated TDBA-Pd exhibits a blueshifted emission (453 versus 460 nm in toluene) and a deeper HOMO energy (−5.63 eV vs. −5.58 eV). Both TDBA-Ph and TDBA-Pd exhibit high luminescence efficiencies of 94% and 88%, respectively, accompanied by rapid RISC rates of 1.85 × 106 s−1 and 1.57 × 106 s−1. Non-doped OLEDs based on these emitters demonstrate impressive performance: TDBA-Ph and TDBA-Pd exhibit the maximum luminance (Lmax) of 25![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 600 and 42498 cd m−2, and the maximum external quantum efficiency (EQEmax) of 12.4% and 11.7%, respectively. In the doped devices, TDBA-Ph and TDBA-Pd show a significantly enhanced EQEmax of 23.0% and 21.4%, respectively. Notably, both doped and nondoped devices display alleviated efficiency roll-off. This work confirms that introducing an sp2-hybridized nitrogen atom into a donor moiety represents a viable approach for constructing high-efficiency blue TADF emitters.
600 and 42498 cd m−2, and the maximum external quantum efficiency (EQEmax) of 12.4% and 11.7%, respectively. In the doped devices, TDBA-Ph and TDBA-Pd show a significantly enhanced EQEmax of 23.0% and 21.4%, respectively. Notably, both doped and nondoped devices display alleviated efficiency roll-off. This work confirms that introducing an sp2-hybridized nitrogen atom into a donor moiety represents a viable approach for constructing high-efficiency blue TADF emitters.
Introduction
Organic light-emitting diodes (OLEDs) have emerged as a revolutionary technology, distinguished by their self-emissive nature, low power consumption, and thin/flexible form factors.1–7 The developmental trajectory of OLED technology is deeply intertwined with the innovations in luminescent materials, as breakthroughs in each generation of materials have consistently enabled a significant leap in OLED performance.8–11 According to the spin-statistics rule in quantum mechanics, electrical excitation induces electron–hole recombination, generating singlet and triplet excitons in a 1:3 ratio.12,13 Thermally activated delayed fluorescence (TADF) materials have emerged as third-generation luminescent materials, offering remarkable advantages in achieving 100% exciton utilization efficiency. This is enabled by efficient reverse intersystem crossing (RISC), a process that recycles a triplet exciton into a singlet one.14–25 Structurally, TADF emitters typically adopt an orthogonal donor–acceptor (D–A) configuration to attain the spatially separated highest occupied molecular orbital (HOMO) and lowest unoccupied molecular orbital (LUMO), thereby minimizing the singlet–triplet splitting (ΔEST).26–31 While this approach has proven highly effective for constructing green and red TADF emitters, substantial challenges remain in the design of blue counterparts.32–37
The D–A architecture of TADF emitters inherently gives rise to intramolecular charge transfer (CT) states. For blue and deep-blue TADF emitters (with emission peaks at ∼460 nm or shorter wavelengths), enhancing the 1CT energy typically requires the reduction of the π-conjugation between D–A fragments. This brings the 1CT energy closer to the singlet local excited state (1LE) energy. However, this approach conversely weakens the 1CT state, which tends to increase the ΔEST and reduce RISC efficiency.38–43 Consequently, achieving both a small ΔEST and rapid RISC kinetics while maintaining blue emission remains a critical challenge. Despite these hurdles, blue TADF emitters with high luminescence efficiency are of paramount importance for full-color displays, as they can enhance operational stability, reduce energy consumption, and lower manufacturing costs.44–47 From the perspective of molecular design, lowering the HOMO energy of the donor unit represents a feasible strategy to increase the transition energy, thereby achieving blueshifted emission. Among various approaches, incorporating sp2-hybridized nitrogen atoms into the phenyl group has emerged as an effective method to attain this objective.48–51 On the one hand, functional groups containing these atoms (e.g., pyridyl, pyrimidine, and pyrazine) exhibit stronger electron-withdrawing abilities than analogous structures based on aromatic hydrocarbons. This property enables them to reduce the electron density and conjugation degree of the conjugated system, thereby lowering the HOMO energy. On the other hand, the lone-pair electrons on the nitrogen atom can form intramolecular C–H⋯N hydrogen bonds that act as a “conformational lock”, which blocks molecular torsion to reduce the CT state and mitigate vibrational relaxation losses. Collectively, these two factors promote systemic hypsochromic shifts in the material's photophysical properties. For example, by adjusting the nitrogen atom's position, Qi et al. found that hydrogen bond formation within the acceptor moiety of the phenyl(pyridyl)-methanone unit induces emission at 536 nm. In contrast, when a hydrogen bond forms between the D–A moieties of carbazole and phenyl(pyridyl)-methanone, the emission undergoes a significant blueshift to 514 nm.52 Wang et al. reported that by introducing one and two nitrogen atoms into the central phenyl group of a boron/nitrogen-embedded multiple-resonance TADF emitter, the emission wavelengths were blueshifted from 472 nm to 440 nm and 409 nm,53 respectively.
Currently, extensive research efforts have been devoted to designing TADF emitters incorporating sp2-hybridized nitrogen atoms. However, most of these studies focus on introducing such atoms either to form intramolecular hydrogen bonds between D–A moieties or to serve as components of the acceptor unit.54 A significant gap remains in exploring how incorporating these atoms into the donor unit regulates electronic structures. In this study, by replacing the phenyl group in triindocarbazole (TAT) with a pyridyl group (Scheme 1), the resulting compound TDBA-Pd exhibits a blueshifted emission at 453 nm in toluene and a deeper HOMO energy of −5.63 eV, compared to the parent compound TDBA-Ph (460 nm and −5.58 eV). Theoretical calculations further confirm the presence of intramolecular C–H⋯N hydrogen bonds between the nitrogen atom and adjacent hydrogen atoms, alongside reduced electron density. TDBA-Ph and TDBA-Pd exhibit high luminescence efficiencies of 94% and 88%, respectively, as well as rapid RISC rates of 1.85 × 106 s−1 and 1.57 × 106 s−1. Non-doped OLED devices based on these emitters exhibit the maximum luminance (Lmax) and external quantum efficiency (EQEmax) of 25600 cd m−2 and 12.4% for TDBA-Ph, and 42498 cd m−2 and 11.7% for TDBA-Pd, respectively. In doped devices, TDBA-Ph and TDBA-Pd show electroluminescence (EL) at 483 and 474 nm, with a significantly enhanced EQEmax of 23.0% and 21.4%, respectively. Notably, both emitters display minimal efficiency roll-off at high luminance.
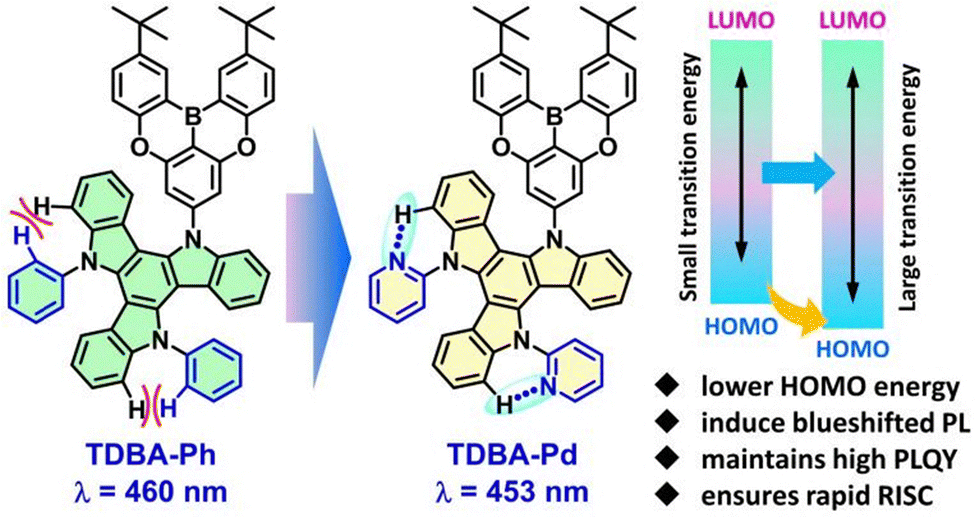 | ||
| Scheme 1 Molecular design concept of TDBA-Ph and TDBA-Pd involves incorporating a pyridyl group into the donor unit, which lowers the HOMO energy to induce a blueshifted PL, while the high PLQY and rapid RISC efficiencies are maintained. | ||
Results and discussion
Synthesis, thermal properties, and electrochemical properties
The synthesis of TDBA-Ph and TDBA-Pd was performed via the Pd-catalysed Buchwald–Hartwig amination reaction of phenyl/pyridyl substituted TAT with triarylborane (BO) units (Scheme S1). Synthesis procedures are described in detail in the SI. Their chemical structures were fully characterized by 1H and 13C nuclear magnetic resonance (NMR) spectroscopy (Fig. S1–S8) and high-quality MALDI-TOF mass spectrometry. The thermal properties of two emitters were studied using thermogravimetric analysis (TGA) and differential scanning calorimetry (DSC) at a heating rate of 10 °C min−1 under a nitrogen atmosphere. As depicted in Fig. S9, both TDBA-Ph and TDBA-Pd demonstrate excellent thermal stability, with thermal decomposition temperatures (Td95%, corresponding to 5% weight loss) of 465 °C and 458 °C, respectively. These values guarantee their suitability for the thermal evaporation process during OLED device fabrication. Notably, the DSC test revealed no detectable glass transition temperature (Tg) for either compound, suggesting high morphological stability and resistance to phase transition during device fabrication. The electrochemical properties of TDBA-Ph and TDBA-Pd were investigated via cyclic voltammetry (CV) in CH2Cl2 solution using ferrocene as the standard substance (Fig. S10).Substituting a phenyl group with a pyridyl group slightly increases the half-wave potential of the oxidation curve, indicating a reduction in the electron-donating ability of the donor moiety. Based on their quasi-reversible redox curves, TDBA-Ph and TDBA-Pd exhibit HOMO energies of −5.58 eV and −5.63 eV, respectively, which align with theoretical expectations. The energy gaps (Eg) of the two emitters were calculated from the onset values of their UV-vis absorption spectra, being 2.94 eV and 2.99 eV, respectively. Using the equation of ELUMO = EHOMO + Eg with their HOMO and Eg values, the LUMO energies of both TDBA-Ph and TDBA-Pd are calculated to be −2.64 eV. This identical LUMO energy is attributed to their shared triarylborane acceptor unit.
Computational investigation
The molecular geometry and electronic properties of the emitters were investigated via theoretical simulations using density functional theory (DFT) calculations at the B3LYP/6-31G level. As depicted in Fig. 1a, both TDBA-Ph and TDBA-Pd display highly twisted conformations, characterized by large dihedral angles between the TAT donor and BO acceptor moieties (with α values of 71.3° and 70.8°, respectively). This structural feature is advantageous for the spatial separation of the HOMO and LUMO. Additionally, the distorted molecular framework effectively suppresses intermolecular interactions. The HOMO and LUMO of TDBA-Ph and TDBA-Pd are primarily localized on the donor and acceptor moieties, respectively. The incorporation of the pyridyl group exerts a negligible effect on the spatial distributions of their frontier molecular orbitals. However, it causes a deepening of the HOMO energy, which decreases from −4.78 eV in TDBA-Ph to −4.84 eV in TDBA-Pd. Given that their LUMO energies are comparable (−1.85 and −1.82 eV), this adjustment results in a larger energy gap for TDBA-Pd (2.99 eV) compared to TDBA-Ph (2.96 eV), Consequently, the higher S1 energy of TDBA-Pd (2.58 eV, versus 2.50 eV of TDBA-Ph) indicates that its emission is bluer. The theoretical analysis is consistent with our expectations, validating the rationality of our molecular design strategy. Based on the excited-state energies, TDBA-Ph and TDBA-Pd exhibit small ΔEST values of 0.03 eV and 0.04 eV, respectively. Such tiny energy differences provide a critical prerequisite for achieving efficient TADF. The molecular surface electrostatic potentials (ESP) of the emitters further demonstrate the lower electron density of TDBA-Pd, especially on the TAT moiety (Fig. 1b). The rotational barrier of the emitters was calculated by evaluating the energy difference between the geometry after rotation of D–A moieties (α) and that at the equilibrium configuration (Fig. 1c). Compared to TDBA-Ph, TDBA-Pd exhibits a slightly smaller rotational barrier, which can be attributed to the reduced steric hindrance caused by its lower electron density. | ||
| Fig. 1 (a) Optimized geometry of TDBA-Ph and TDBA-Pd with the dihedral angle between donor and acceptor moieties (α), the torsion angle of phenyl/pyridyl groups (β/γ), and the distance of C–H (dC–H) and N–H (dN–H) being labelled. Calculated HOMO/LUMO contours and the energies, and S1/T1 energies. (b) Molecular surface electrostatic potentials (ESP, measuring scale × 10−2, red and blue indicate negative and positive electrostatic potential, respectively). (c) The rotation energy barrier calculated by evaluating the energy difference between the geometry after rotation (twist angle of α) and that at the equilibrium configuration. | ||
The formation of intramolecular C–H⋯N hydrogen bonds in TDBA-Pd was confirmed by the significantly short N–H distances (dN–H, 2.46 and 2.49 Å) between pyridyl nitrogen atoms and adjacent hydrogen atoms, as compared to C–H distances (dC–H, 2.73 and 2.66 Å) at corresponding positions in TDBA-Ph (Fig. 1a). Consequently, the twist angles of pyridyl groups (β/γ: 50.1°/56.3°) are notably smaller than those of the phenyl groups (β/γ: 60.2°/60.4°). The intramolecular non-covalent interactions induced by hydrogen bonds in the emitters were further analysed via the reduced density gradient (RDG) calculated using the Multiwfn program.55,56 The presence of C–H⋯N hydrogen bonds in TDBA-Pd was validated by the vivid visualization of the RDG isosurface (Fig. 2a). Meanwhile, the spikes in the range of −0.01 to 0.02 a.u. in the sign(λ2)ρ function in the RDG scattering diagram (Fig. 2b) confirm the enhanced non-covalent interactions in TDBA-Pd compared to those in TDBA-Ph. These results validate that substituting a phenyl group with a pyridyl group can promote blueshifted emission by lowing HOMO energy, while strengthening the intramolecular non-covalent interactions via hydrogen bonds.
 | ||
| Fig. 2 (a) Calculated reduced density gradient (RDG) isosurface based on the optimized geometry of TDBA-Ph and TDBA-Pd, and the standard coloring method and chemical explanation of sign(λ2)ρ on the RDG isosurface. (b) The calculated RDG scattering diagram. | ||
Photophysical properties
The UV-vis absorption and fluorescence spectra of the emitters were measured in toluene at room temperature. As shown in Fig. 3a and b, TDBA-Ph and TDBA-Pd exhibit similar absorption profiles, each comprising three distinct absorption bands with varying intensities. The strong absorption bands at approximately 314 nm and 380 nm are ascribed to π–π* transitions of the TAT and BO moieties,57,58 respectively, while the relatively weaker absorption in the 400–420 nm range originates from the intramolecular CT state between the donor and acceptor units. Their fluorescence spectra both display blue emission, with TDBA-Ph featuring a peak wavelength of 460 nm and a full width at half maximum (FWHM) of 60 nm, and TDBA-Pd showing a blueshifted peak at 453 nm with a FWHM of 63 nm. This result aligns with theoretical predictions of an increased energy gap for TDBA-Pd, while the slightly narrower FWHM of TDBA-Ph is attributed to reduced CT character arising from its larger rotational barrier. Additionally, the minimal overlap between the absorption and fluorescence spectra of both emitters indicates effective suppression of self-quenching, thus enabling high photoluminescence quantum yields (PLQY). | ||
| Fig. 3 UV-vis absorption (Abs), fluorescence (FL), and cryogenic phosphorescence (PH) of (a) TDBA-Ph (b) TDBA-Pd in toluene (5 × 10−5 M). Fluorescence spectra of (c) TDBA-Ph and (d) TDBA-Pd in solvents of hexane (Hex), toluene (Tol), tetrahydrofuran (THF), and dichloromethane (DCM). (e) Fluorescence and (f) time-resolved fluorescence decay curves of TDBA-Ph and TDBA-Pd in the DBFPO film at 20 wt%, the insets show the molecular structure of DBFPO and prompt decay curves. | ||
To characterize the strength of the CT state, the absorption and fluorescence spectra of the emitters in various solvents were measured. With increasing solvent polarity, no significant shifts were observed in the absorption wavelengths (Fig. S11), whereas the fluorescence spectra exhibited pronounced redshifts accompanied by broadening of the FWHM (Table S1). Specifically, the fluorescence of TDBA-Ph and TDBA-Pd was redshifted by 120 nm and 112 nm, respectively, when transitioning from n-hexane to toluene, tetrahydrofuran (THF), and dichloromethane (DCM). The Lippert–Mataga analysis was performed to investigate the excited-state dipole moments (μe) of the two emitters. Using the slope derived from the plot of the Stokes shifts (Δν) against the solvent polarity (Δf), similar μe was determined for TDBA-Ph and TDBA-Pd, with values of 3.24 D and 3.36 D (Fig. S12), respectively.
Cryogenic temperature measurements at 77 K revealed dual phosphorescence peaks for TDBA-Ph and TDBA-Pd at 442/469 nm and 445/475 nm, respectively. From the onset of the fluorescence and phosphorescence spectra, the singlet (S1) and triplet (T1) excited state energies were calculated to be 2.95/3.05 eV for TDBA-Ph and 2.88/2.90 eV for TDBA-Pd (Table 1). Correspondingly, experimental ΔEST values of 0.07 eV and 0.15 eV were determined, confirming their potential for TADF. The excellent agreement between experimentally derived ΔEST values and theoretical simulations further validates the rationality of the molecular design. Upon progressively increasing the water fraction in the THF solution, the PL intensity of both emitters decreases, whereas their emission wavelength first redshifts and then blueshifts (Fig. S13). This phenomenon indicates a strong aggregate tendency of the emitters. In the solid state, TDBA-Ph and TDBA-Pd exhibit slightly redshifted emissions at 463 nm and 455 nm, respectively. Corresponding FWHM values are 54 and 62 nm, while their prompt fluorescence lifetimes (τp) are measured to be 15.5 and 5.8 ns (Fig. S14). Despite these characteristic photophysical properties, both emitters maintain relatively low PLQYs: 25.4% for TDBA-Ph and 27.4% for TDBA-Pd. Additionally, no delayed fluorescence lifetimes (τd) were detected for either emitter, which is attributed to the compact π-stacking in the solid-state.
| Emitter | λ abs [nm] | λ FL [nm] | FWHMa [nm] | λ PH [nm] | S1c [eV] | T1c [eV] | ΔESTc [eV] | λ FL [nm] | PLQYe [%] | τ p [ns] | τ d [μs] | k r [107 s−1] | k d [106 s−1] | k ISC [107 s−1] | k RISC [106 s−1] | E HOMO/ELUMOh (eV) |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| a Absorption (abs), fluorescence (FL) wavelengths and related FWHM in toluene at room temperature (5 × 10−5 M). b Cryogenic phosphorescence (PH) wavelength in toluene. c S1 and T1 energies are calculated by the onset of the fluorescence and phosphorescence spectra; ΔEST = ES1 – ET1. d Fluorescence wavelength in the DBFPO film at 20 wt%. e Measured in the DBFPO film using the integral sphere under a nitrogen atmosphere. f Prompt lifetime (τp) and delayed lifetime (τd) in the DBFPO film. g Calculated using PLQY, τp, and τd. h Calculated by cyclic voltammetry, EHOMO = −[Eox − E(Fc/Fc+) + 4.8] eV, ELUMO = EHOMO + Eg. | ||||||||||||||||
| TDBA-Ph | 314/380 | 460 | 60 | 442/469 | 2.54 | 2.43 | 0.11 | 483 | 94 | 15.7 | 0.95 | 3.39 | 1.05 | 2.67 | 1.85 | −5.58/−2.64 |
| TDBA-Pd | 314/380 | 453 | 63 | 445/475 | 2.45 | 2.36 | 0.09 | 473 | 88 | 12.9 | 0.78 | 5.58 | 1.28 | 1.41 | 1.57 | −5.63/−2.64 |
The photophysical properties of the emitters in the thin-film state were investigated by doping them into dibenzo[b,d]furan-2,8-diylbis(diphenylphosphine oxide) (DBFPO) at a 20 wt% concentration, with specific data listed in Table 1. Compared to their fluorescence spectra in toluene and solid-state, the thin-film spectra of TDBA-Ph and TDBA-Pd exhibit a redshift of approximately 20 nm, with emission peaks at 483 nm and 473 nm (Fig. 3e) and FWHM values of 57 nm and 59 nm, respectively. This significant redshift can be primarily attributed to the enhanced solid-state solvation effect within the polar DBFPO host matrix. Integrating sphere tests show their PLQY reach 94% and 88%, respectively. Such high PLQYs provide a critical prerequisite for fabricating efficient OLEDs, while the slightly inferior PLQY of TDBA-Pd was ascribed to the reduced CT state caused by the reduced electron density of the donor unit moiety. Their transient fluorescence decay curves exhibit double-exponential characteristics. The τp and τd values of TDBA-Ph and TDBA-Pd are 15.7/12.9 ns and 0.95/0.78 μs, respectively (Fig. 3f). The relatively short τd values contribute to the short residence of triplet excitons and the efficient RISC in the rigid DBFPO matrix. Temperature-dependent transient decay tests (Fig. S15 and Table S2) further confirm their TADF properties: as the temperature increases from 100 K to 300 K, the τd components gradually increase according to the integrated area in the transient PL spectra. Based on PLQY, τp, and τd of DBFPO films, the radiative rates (kr) of TDBA-Ph and TDBA-Pd are calculated to be 3.39 × 107 s−1 and 5.58 × 107 s−1, while their RISC rates (kRISC) reach 1.85 × 106 s−1 and 1.57 × 106 s−1 (Table 1). The high kRISC values are advantageous for reducing OLED efficiency roll-off and enhancing device stability.
Electroluminescence properties
Considering their excellent thermal stability and photophysical properties in the solid-state, nondoped OLED devices based on TDBA-Ph and TDBA-Pd were preliminarily fabricated. The device structures, energy level diagrams, and chemical structures of the functional materials used are shown in Fig. 4a, specifically: indium tin oxide (ITO)/1,4,5,8,9,11-hexaazatriphenylene hexacarbonitrile (HATCN, 10 nm)/4,4′-(cyclohexane-1,1-diyl)bis(N,N-di-p-tolylaniline) (TAPC, 60 nm)/tris(4-(9H-carbazol-9-yl)phenyl)amine (TCTA, 5 nm)/emissive layer (EML, 20 nm)/3,3′-(5′-(3-(pyridin-3-yl)phenyl)-[1,1′:3′,1′′-terphenyl]-3,3′′-diyl)dipyridyl (TmPyPB, 40 nm)/LiF (1 nm)/Al. Herein, ITO and Al serve as the anode and cathode, and HATCN and LiF act as the hole injection layer and electron injection layer, respectively. TAPC and TmPyPb function as the hole transport layer and electron transport layer, respectively. TCTA serves as the electron blocking layer. The detailed EL performance data are summarized in Table 2. The nondoped OLED devices demonstrate remarkable stable EL performance, with their EL spectra remaining almost unchanged as the driving voltage increases from 3 to 4, 5, and 6 V (Fig. 4b). At a driving voltage of 5 V, the EL peaks of TDBA-Ph and TDBA-Pd are 487 nm and 488 nm, respectively, while their Commission Internationale de l’Eclairage (CIE) coordinates are (0.17, 0.36) and (0.18, 0.39). | ||
| Fig. 4 (a) Nondoped OLED device configuration with an energy level diagram, and the molecular structures of the materials used in devices. (b) EL spectra at driving voltages of 3, 4, 5, and 6 V. (c) The current density and luminance versus voltage (J–V–L) characteristics. (d) EQE–luminance curves, the inset shows the CIE coordinates of EL spectra. (e) The power efficiency and current efficiency versus luminance. | ||
| Emitter | λ [nm] | FWHM [nm] | V on [V] | CEmaxc [cd A−1] | PEmaxc [lm W−1] | EQEd [%] | Roll-offe [%] | CIEf [x, y] | |
|---|---|---|---|---|---|---|---|---|---|
|
a Maximum EL wavelength.
b Turn-on voltage recorded at a luminance of 1 cd m−2.
c Maximum current efficiency and power efficiency.
d Maximum EQE and EQE at luminances of 1000, 5000, and 10000 cd m−2.
e Efficiency roll-off at luminances of 1000, 5000, and 10000 cd m−2.
f CIE coordinates of EL spectra recorded at 5 V.
|
|||||||||
| Nondoped device | TDBA-Ph | 487 | 59 | 2.3 | 27.2 | 30.1 | 12.4/12.1/10.6/8.9 | 2.4/14.9/33.0 | (0.17, 0.36) |
| TDBA-Pd | 488 | 66 | 2.4 | 28.1 | 32.9 | 11.7/11.2/9.8/8.7 | 5.1/17.1/30.6 | (0.18, 0.39) | |
| Doped device | TDBA-Ph | 483 | 59 | 2.6 | 45.9 | 42.1 | 23.0/22.8/20.1/17.7 | 0.9/12.6/23.0 | (0.15, 0.31) |
| TDBA-Pd | 474 | 59 | 2.8 | 32.5 | 33.2 | 21.4/19.0/14.9/12.7 | 11.2/30.4/40.7 | (0.14, 0.20) | |
The slight redshift relative to their solid-state fluorescence can be attributed to the formation of exciplexes under electrical excitation.
Nondoped OLED devices typically suffer from inferior EL performance because of the detrimental π-stacking effect. Nevertheless, both emitters in this study show excellent device performance. The current density and luminance versus voltage (J–V–L) characteristics of OLEDs are depicted in Fig. 4c. TDBA-Ph and TDBA-Pd exhibit low turn-on voltages (at a luminance of 1 cd m−2) of 2.3 V and 2.4 V, respectively, highlighting their exceptional carrier injection and transport capabilities. However, both devices exhibit a relatively low EQEmax of 12.4% for TDBA-Ph and 11.7% for TDBA-Pd, respectively, which can be attributed to their low solid-state PLQY (Fig. 4d). The devices show a very slight efficiency roll-off, at high luminance levels of 1000, 5000, and 10000 cd m−2, and the EQEs of TDBA-Ph and TDBA-Pd are still maintained at 12.1%, 10.6%, 8.9% and 11.2%, 9.8%, 8.7%, respectively, indicating efficiency roll-offs of 2.4%, 14.9%, 33.0% and 5.1%, 17.1%, 30.6%. Although the EQE of TADB-Pd is slightly lower than that of TDBA-Ph, its power efficiency (PEmax) and current efficiency (CEmax) are both significantly enhanced, reaching 32.9 lm W−1 and 28.1 cd A−1, respectively, compared to TDBA-Ph's 30.1 lm W−1 and 27.2 cd A−1 (Fig. 4e). Notably, the Lmax increases from 25600 cd m−2 for TDBA-Ph to 42498 cd m−2 for TDBA-Pd. This enhancement can be ascribed to the ability of the pyridyl moiety to adjust the molecular energy levels, thereby facilitating more efficient charge injection and balanced transport. Additionally, molecular conformation analysis reveals dimensions of 22.738 × 15.016 × 12.130 Å for TDBA-Ph and 22.816 × 15.200 × 10.601 Å for TDBA-Pd, with respective mean radii of 6.004 Å and 5.985 Å (Fig. S16). The more planar conformation of TDBA-Pd is conducive to forming compact π-stacking in the solid-state, which in turn contributes to enhanced luminance in OLED devices.
To further enhance EL performance, doped devices were fabricated, where the EML employs DBFPO as the host and the emitters as the guest at doping concentration of 20 wt% (Fig. 5a). These doped OLED devices also exhibit stable EL spectra at driving voltages ranging from 3 to 6 V (Fig. 5b), presenting emission peaks and CIE coordinates of 487 nm/(0.15, 0.31) for TDBA-Ph, and 474 nm/(0.14, 0.20) for TDBA-Pd, respectively. However, TDBA-Ph-based device demonstrates superior performance in terms of CEmax and PEmax with values of 45.9 cd A−1/42.1 lm W−1 for TDBA-Ph, and 32.5 cd A−1/33.2 lm W−1 for TDBA-Pd (Fig. 5c). Consequently, the EQEmax of TDBA-Ph (23.0%) is marginally higher than that of TDBA-PD (21.4%) (Fig. 5d). Both devices also exhibit alleviated efficiency roll-off. At high luminance levels of 1000, 5000, and 10000 cd m−2, the EQEs of TDBA-Ph remain at 22.8%, 20.1%, and 17.7%, corresponding to roll-offs of 0.9%, 12.6%, and 23.0%, respectively; for TDBA-Pd, the EQEs are 19.0%, 14.9%, and 12.7%, with roll-offs of 11.2%, 30.4%, and 40.7%, respectively. The significant mitigation of efficiency roll-off can be attributed to the emitters’ distorted molecular conformations, and short τd in the sub-microsecond range. The OLED devices successfully exhibit blueshifted emission while maintaining comparable device performance, which undoubtedly validates the feasibility of our design strategy.
 | ||
| Fig. 5 (a) Doped OLED device configuration with an energy level diagram. (b) EL spectra at driving voltages of 3, 4, 5, and 6 V. (c) The current density and luminance versus voltage (J–V–L) characteristics. (d) EQE–luminance curves, the inset shows the CIE coordinates of EL spectra. | ||
Conclusions
In summary, two blue TADF emitters of TDBA-Ph and TDBA-Pd were designed and synthesized by substituting the phenyl group with a pyridyl group on the TAT donor. The introduction of the pyridyl group facilitates the formation of intramolecular C–H⋯N hydrogen bonds, thereby suppressing molecular distortion and reducing electron density. Consequently, the diminished electron-donating ability of the donor unit and lowered HOMO energy result in a blueshifted emission for TDBA-Pd. When doped into the DBFPO film at a concentration of 20 wt%, TDBA-Ph and TDBA-Pd exhibit high PLQYs of 94% and 88%, accompanied by rapid RISC rates of 1.85 × 106 s−1 and 1.57 × 106 s−1. The non-doped OLED devices based on these emitters exhibit remarkable performance: TDBA-Ph exhibits a Lmax of 25600 cd m−2 and an EQEmax of 12.4%, while those of TDBA-Pd reach 42498 cd m−2 and 11.7%, respectively. Additionally, both devices show exceptionally low efficiency roll-off: at luminance of 1000/5000/10000 cd m−2, the roll-off values are 2.4%/14.9%/33.0% for TDBA-Ph and 5.1%/17.1%/30.6% for TDBA-Pd. In the doped OLED device, TDBA-Ph and TDBA-Pd exhibit significantly enhanced EQEmax of 23.0% and 21.4%, respectively. They also exhibit alleviated efficiency roll-off, with values of 0.9%/12.6%/23.0% for TDBA-Ph and 11.2%/30.4%/40.7% for TDBA-Pd at luminance of 1000/5000/10000 cd m−2. This work validates the critical role of the sp2-hybridized nitrogen atom in regulating the photophysical properties and offers a viable strategy for the rational design of blue TADF emitters.
Author contributions
B. Z. Tang, Z. Li and Y. Xie conceived and supervised the project. Y. Xie and X. Li designed the experiments, synthesized, and characterized the emitters, and wrote the manuscript. D. Xie and D. Ma conducted OLED fabrication and measurement. X. Song helped in theoretical calculations and provided suggestions on experiments. Y. Xie and Z. Li supervised, reviewed and revised the manuscript.Conflicts of interest
There are no conflicts to declare.Data availability
The data supporting this article, including synthesis and characterization, general information, 1H/13C NMR spectra, TGA and DSC curves, electroluminescent properties, photophysical properties, have been included as part of the Supplementary information (SI). See DOI: https://doi.org/10.1039/d5tc02857d.Acknowledgements
We acknowledge financial support from the Open Fund of the State Key Laboratory of Luminescent Materials and Devices (South China University of Technology) (No. 2024-skllmd-04) and the National Natural Science Foundation of China (No. W2411008).References
- T. Yu, L. Liu, Z. Xie and Y. Ma, Sci. China: Chem., 2015, 58, 907–915 CrossRef
.
- C. W. Tang and S. A. VanSlyke, Appl. Phys. Lett., 1987, 51, 913–915 CrossRef
- C. Adachi, M. A. Baldo, M. E. Thompson and S. R. Forrest, J. Appl. Phys., 2001, 90, 5048–5051 CrossRef
- Z. Zhang, R. Xia, K. Wang, Y. Wu, P. Zang, X. Gan, Z. Liao, B. Wei, P. Wu, S. Bräse and Z. Wang, Aggregate, 2024, 5, e588 CrossRef
- W. Liu, C. Zhang, R. Alessandri, B. T. Diroll, Y. Li, H. Liang, X. Fan, K. Wang, H. Cho, Y. Liu, Y. Dai, Q. Su, N. Li, S. Li, S. Wai, Q. Li, S. Shao, L. Wang, J. Xu, X. Zhang, D. V. Talapin, J. J. de Pablo and S. Wang, Nat. Mater., 2023, 22, 737–745 CrossRef PubMed
- S.-W. Liu, C.-C. Lee, C.-H. Wang, J.-H. Lee, C.-T. Chen and J.-K. Wang, Chem. Phys. Lett., 2009, 474, 207–211 CrossRef
- Z. Ruan, Y. Shan, Y. Gong, C. Wang, F. Ye, Y. Qiu, Z. Liang and Z. Li, J. Mater. Chem. C, 2018, 6, 773–780 RSC
- B. Zhang, B. Li, H. Zhang, B. Ma, J. Lou, X. Dong, D. Yang, B. Z. Tang and Z. Wang,, Aggregate, 2024, 6, e726 CrossRef
- K. Wang, X. Ou, X. Niu, Z. Wang, F. Song, X. Dong, W. Guo, H.-Q. Peng, Z. Zhao, J. W. Y. Lam, J. Sun, H. Wu, S.-Y. Yu, F. Li and B. Z. Tang, Aggregate, 2024, 6, e667 CrossRef
- Y. Gao, J. Lu, Q. Liao, S. Li, Q. Li and Z. Li, Natl. Sci. Rev., 2023, 10, nwad239 CrossRef PubMed
- X. Xiao, Z.-Z. Huo, B. Yang, Z.-J. Chen, L. Yuan, C.-H. Li and Y.-X. Zheng, Sci. China: Chem., 2025, 68, 4224–4233 CrossRef
- A. Endo, K. Sato, K. Yoshimura, T. Kai, A. Kawada, H. Miyazaki and C. Adachi, Appl. Phys. Lett., 2011, 98, 083302 CrossRef
- H. Uoyama, K. Goushi, K. Shizu, H. Nomura and C. Adachi, Nature, 2012, 492, 234–238 CrossRef PubMed
- M. Y. Wong and E. Zysman-Colman, Adv. Mater., 2017, 29, 1605444 CrossRef PubMed
- X. Cai and S.-J. Su, Adv. Funct. Mater., 2018, 28, 1802558 CrossRef
- Y. Zou, S. Gong, G. Xie and C. Yang, Adv. Opt. Mater., 2018, 6, 1800568 CrossRef
- Q. Zhang, B. Li, S. Huang, H. Nomura, H. Tanaka and C. Adachi, Nat. Photonics, 2014, 8, 326–332 CrossRef CAS
- X. Yu, D. Cui, M. Wang, Z. Wang, M. Wang, D. Tu, V. Bregadze, C. Lu, Q. Zhao, R. Chen and H. Yan, Chin. Chem. Lett., 2025, 36, 110520 CrossRef CAS
- J. Liu, Z. Feng, C. Peng, Y. Yu, S. Yang, Z. Jiang and L. Liao, Chin. Chem. Lett., 2023, 34, 107634 CrossRef CAS
- M. Lu, S. Liao, J. Li, Z. Yu, N. Zhao, Z. Xie, S. Chen, L. Dang and M.-D. Li, Chin. Chem. Lett., 2025, 36, 110066 CrossRef CAS
- L. Yuan, Y.-P. Zhang and Y.-X. Zheng, Sci. China: Chem., 2024, 67, 1097–1116 CrossRef CAS
- Y.-C. Wu, G.-T. Huang, M.-Y. Lian, R. Liang, H.-L. Deng, F. Gan, Y.-F. Zhang, N.-B. Yi, L.-Y. Tian, C.-P. Ma and Y. Wei, Chin. J. Polym. Sci., 2023, 41, 1609–1616 CrossRef CAS
- Y. Wang, Z. Ma, J. Pu, D. Guo, G. Li, Z. Chen, S.-J. Su, H. Deng, J. Zhao and Z. Chi, Aggregate, 2024, 5, e585 CrossRef CAS
- J. Wang, H. Hafeez, S. Tang, T. Matulaitis, L. Edman, I. D. W. Samuel and E. Zysman-Colman, Aggregate, 2024, 5, e571 CrossRef CAS
- Y.-C. Wu, G.-T. Huang, M.-Y. Lian, R. Liang, H.-L. Deng, F. Gan, Y.-F. Zhang, N.-B. Yi, L.-Y. Tian, C.-P. Ma and Y. Wei, Chin. J. Polym. Sci., 2023, 41, 1609–1616 CrossRef CAS
- X. Tang, L. S. Cui, H. C. Li, A. J. Gillett, F. Auras, Y. K. Qu, C. Zhong, S. T. E. Jones, Z. Q. Jiang, R. H. Friend and L. S. Liao, Nat. Mater., 2020, 19, 1332–1338 CrossRef CAS PubMed
- T.-L. Wu, M.-J. Huang, C.-C. Lin, P.-Y. Huang, T.-Y. Chou, R.-W. Chen-Cheng, H.-W. Lin, R.-S. Liu and C.-H. Cheng, Nat. Photonics, 2018, 12, 235–240 CrossRef CAS
- X. Song, X. Li, X. Ye, Y. Xie, Y. Zhang, L. Duan and Z. Li, Adv. Opt. Mater., 2025, 13, 2500515 CrossRef CAS
- C. Qu, Y. Zhu, L. Liang, K. Ye, Y. Zhang, H. Zhang, Z. Zhang, L. Duan and Y. Wang, Adv. Opt. Mater., 2023, 11, 2203030 CrossRef CAS
- T. Huang, Q. Wang, G. Meng, L. Duan and D. Zhang, Angew. Chem., Int. Ed., 2022, 61, e202200059 CrossRef CAS PubMed
- T. Huang, Q. Wang, S. Xiao, D. Zhang, Y. Zhang, C. Yin, D. Yang, D. Ma, Z. Wang and L. Duan, Angew. Chem., Int. Ed., 2021, 60, 23771–23776 CrossRef CAS
- S. Hirata, Y. Sakai, K. Masui, H. Tanaka, S.
Y. Lee, H. Nomura, N. Nakamura, M. Yasumatsu, H. Nakanotani, Q. Zhang, K. Shizu, H. Miyazaki and C. Adachi, Nat. Mater., 2015, 14, 330–336 CrossRef CAS
- D. H. Ahn, S. W. Kim, H. Lee, I. J. Ko, D. Karthik, J. Y. Lee and J. H. Kwon, Nat. Photonics, 2019, 13, 540–546 CrossRef CAS
- K. Kawasumi, T. Wu, T. Zhu, H. S. Chae, T. V. Voorhis, M. A. Baldo and T. M. Swager, J. Am. Chem. Soc., 2015, 137, 11908–11911 CrossRef CAS PubMed
- L. Tu, Y. Xie, Z. Li and B. Z. Tang, SmartMat, 2021, 2, 326–346 CrossRef CAS
- W. Che, Y. Xie and Z. Li, Asian J. Org. Chem., 2020, 9, 1262–1276 CrossRef CAS
- M. C. Gather, A. Kohnen and K. Meerholz, Adv. Mater., 2011, 23, 233–248 CrossRef CAS PubMed
- Y. Im, S. Y. Byun, J. H. Kim, D. R. Lee, C. S. Oh, K. S. Yook and J. Y. Lee, Adv. Funct. Mater., 2017, 27, 1603007 CrossRef
- X. Li, S. Fu, Y. Xie and Z. Li, Rep. Prog. Phys., 2023, 86, 096501 CrossRef CAS PubMed
- T. Hua, X. Cao, J. Miao, X. Yin, Z. Chen, Z. Huang and C. Yang, Nat. Photonics, 2024, 18, 1161–1169 CrossRef CAS
- X. He, J. Lou, B. Li, X. Dong, F. Zhong, W. Liu, X. Feng, D. Yang, D. Ma, Z. Zhao, Z. Wang and B. Z. Tang, Adv. Mater., 2024, 36, e2310417 CrossRef PubMed
- X. Li, Y. Xie and Z. Li, Chem. – Asian J., 2021, 16, 2817–2829 CrossRef CAS
- T. Kim, G. Shin, T. Park and M. Kim, Adv. Funct. Mater., 2024, 35, 2412267 CrossRef
- Y. Liu, C. Li, Z. Ren, S. Yan and M. R. Bryce, Nat. Rev. Mater., 2018, 3, 18020 CrossRef CAS
- U. Deori, G. P. Nanda, C. Murawski and P. Rajamalli, Chem. Sci., 2024, 15, 17739–17759 RSC
- S. Y. Yang, Y. K. Qu, L. S. Liao, Z. Q. Jiang and S. T. Lee, Adv. Mater., 2021, 34, 2104125 CrossRef
- Q. Zhang, J. Li, K. Shizu, S. Huang, S. Hirata, H. Miyazaki and C. Adachi, J. Am. Chem. Soc., 2012, 134, 14706–14709 CrossRef CAS PubMed
- L. Chen, J.-H. Cai, Y.-J. Yu, Y.-K. Qu, S.-Y. Yang, S.-N. Zou, R.-H. Liu, D.-Y. Zhou, L.-S. Liao and Z.-Q. Jiang, Sci. China: Chem., 2023, 67, 351–359 CrossRef
- Y. Xu, C. Li, Z. Li, J. Wang, J. Xue, Q. Wang, X. Cai and Y. Wang, CCS Chem., 2021, 4, 2077–2091 Search PubMed
- P. Rajamalli, V. Thangaraji, N. Senthilkumar, C.-C. Ren-Wu, H.-W. Lin and C.-H. Cheng, J. Mater. Chem. C, 2017, 5, 2919–2926 RSC
- P. Rajamalli, N. Senthilkumar, P. Y. Huang, C. C. Ren-Wu, H. W. Lin and C. H. Cheng, J. Am. Chem. Soc., 2017, 139, 10948–10951 CrossRef CAS PubMed
- F. Ma, G. Zhao, Y. Zheng, F. He, K. Hasrat and Z. Qi, ACS Appl. Mater. Interfaces, 2020, 12, 1179–1189 CrossRef CAS
- X. Cai, Y. Pan, C. Li, L. Li, Y. Pu, Y. Wu and Y. Wang, Angew. Chem., Int. Ed., 2024, 63, e202408522 CrossRef CAS PubMed
- X. Cao, D. Zhang, S. Zhang, Y. Tao and W. Huang, J. Mater. Chem. C, 2017, 5, 7699–7714 RSC
- T. Lu and F. Chen, J. Comput. Chem., 2012, 33, 580–592 CrossRef CAS
- T. Lu, J. Chem. Phys., 2024, 161, 082503 CrossRef CAS
- L. Tu, Y. Fan, C. Bi, L. Xiao, Y. Li, A. Li, W. Che, Y. Xie, Y. Zhang, S. Xu, W. Xu, Q. Li and Z. Li, Sci. China: Chem., 2023, 66, 816–825 CAS
- C. Ruiz, E. M. García-Frutos, D. A. da Silva Filho, J. T. López Navarrete, M. C. Ruiz Delgado and B. Gómez-Lor, J. Phys. Chem. C, 2014, 118, 5470–5477 CrossRef CAS
Footnote |
| † X. Li and D. Xie contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2025 |