
Blue-emitting iridium(III) phosphors with functional imidazo[4,5-b]pyridin-2-ylidene cyclometalates: designs aimed at greater steric hindrance†
Chengcheng
Wu‡
a,
Yixin
Wu‡
b,
Kai-Ning
Tong‡
a,
Martin
Kuhn
 c,
Shek-Man
Yiu
b,
Yu-Cheng
Kung
d,
Wen-Yi
Hung
d,
Jie
Yan
*b,
Xiuwen
Zhou
*ce,
Guodan
Wei
*a and
Yun
Chi
*b
c,
Shek-Man
Yiu
b,
Yu-Cheng
Kung
d,
Wen-Yi
Hung
d,
Jie
Yan
*b,
Xiuwen
Zhou
*ce,
Guodan
Wei
*a and
Yun
Chi
*b
aInstitute of Materials Research, Tsinghua University, Shenzhen 518055, China. E-mail: weiguodan@sz.tsinghua.edu.cn
bDepartment of Chemistry, Department of Materials Science and Engineering, and Center of Super-Diamond and Advanced Films (COSDAF), City University of Hong Kong, Hong Kong, SAR. E-mail: jyanae@connect.ust.hk; yunchi@cityu.edu.hk
cSchool of Mathematics and Physics, The University of Queensland, Brisbane, Queensland 4072, Australia
dDepartment of Optoelectronics and Materials Technology, National Taiwan Ocean University, Keelung 20224, Taiwan
eCentre of the Materials Science, School of Chemistry and Physics, Queensland University of Technology, Brisbane, Queensland 4000, Australia. E-mail: xiuwen.zhou@qut.edu.au
First published on 8th May 2025
Abstract
Ir(III) carbene complexes are a class of promising phosphors used in constructing high-performance blue OLEDs. In this work, two series of efficient blue-emitting Ir(III) carbene complexes, namely, f-ct3ax/bx and f-ct9ax/bx, were designed, synthesized and characterized. These Ir(III) carbene complexes possessed bulky substituents (both 2-xylenyl and tert-butyl group) at both cyclometalating and non-coordinated aryl sites and a dislocated 6- or 5-cyano substituent on the imidazo[4,5-b]pyridin-2-ylidene fragment, achieving blue phosphorescence. These Ir(III) emitters showed excellent photophysical properties in degassed toluene solution with high PLQYs (72–98%) and short radiative lifetimes (0.91–3.08 μs), making them excellent candidates for the fabrication of blue PhOLEDs. As a result, the blue PhOLEDs based on f-ct9ax and f-ct9bx with a 5-cyano substituent exhibited maximum EQEs of 24.2% and 23.3%, respectively, which were higher than those of f-ct3ax and f-ct3bx with a 6-cyano substituent (EQEmax = 22.1% and 20.0%, respectively). Moreover, the hyper-OLEDs featuring the f-ct9ax sensitizer and v-DABNA terminal emitter delivered a narrowband blue emission with a peak maximum at 468 nm and CIEx,y coordinates of (0.123, 0.095). This champion device also achieved a highest maximum EQE of 31.9% and an EQE of 20.9% at a practical luminance of 1000 cd m−2. These results demonstrate the potential of these Ir(III) carbene complexes in constructing blue OLEDs with high efficiency and color purity for future applications.
Introduction
Luminescent transition metal complexes have been extensively studied as labels, sensors and probes for biological systems,1 as sensitizers for photosynthesis and solar cells,2 and as emitters for organic light-emitting diode (OLED) applications.3 In particular, Ir(III)-based metal phosphors are prominent in serving as OLED emitters owing to their ability in effectively harnessing singlet and triplet excitons, giving unitary internal quantum efficiency.4 The majority of these Ir(III) complexes possess traditional hetero-aromatic chelates such as 1-phenyl-1H-pyrazole, 2-phenylpyridine, 1-phenylisoquinoline, and their functional derivatives, exhibiting tuned emission wavelength across the whole visible spectral region.5 Their unique features include high photophysical and chemical stabilities and excellent emission efficiency with shortened radiative lifetimes. Additionally, there is a growing interest in substituting relevant N-donor chelates with carbene-based (C-donor) chelates in these Ir(III) phosphors for the achievement of higher emission energy and better efficiency.6As demonstrated in literature, cyclometalating carbene chelates can provide an easy access to the Ir(III)-based blue OLED emitters owing to their destabilized LUMO energy level and stronger dative bonding interaction from carbene-to-iridium metal atom. Particularly, the Ir(III) complexes featuring cyclometalating carbenes such as benzo[d]imidazol-2-ylidene (pmb),7 imidazo[4,5-b]pyridin-2-ylidene (pmp),8 and imidazo[4,5-b]pyrazin-2-ylidene (pmpz)9 have shown efficient ultraviolet to sky-blue emission. Their structures are depicted in Scheme 1. Similar to the electronegative skeletal N-atom, CF3 and cyano groups in the chelates tpb18b and mfcp10 can lower the LUMO energy of the carbene entity, giving the desired blue emission comparable to that of pmpz. These carbene chelates with a single N-alkyl substituent are known to afford many efficient Ir(III) emitters; however, they show relatively inferior chemical stability compared with their counterparts with dual N-aryl substituents, such as cb9a and tpz.9b However, one major problem encountered for the dual N-aryl groups is the possible formation of multiple products induced by competitive cyclometalation, particularly for chelate cb. Fortunately, functional benzo[d]imidazol-2-ylidene with either the greatly asymmetrical N-aryl appendages or bulky peri-substituent on the benzo subunit rather than the traditional N-mesityl pendant brilliantly solved this synthetic challenge and afforded a new family of Ir(III) blue OLED emitters with better product selectivity and promising device performances. These newly invented blue carbene chelates with dual N-aryl substituents (namely: (ct1),11 (ct4),12 (ct8)13 and (CN1)14) are also depicted in Scheme 1 as models for scrutiny.
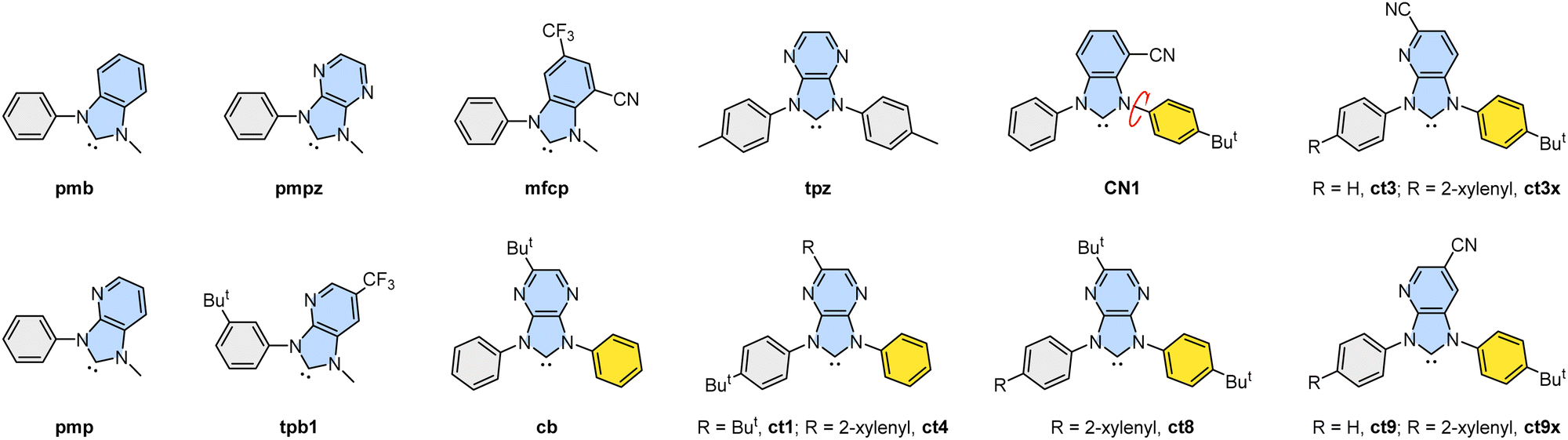 | ||
| Scheme 1 Drawings of carbene chelates mentioned in this study. N-Aryl groups marked with grey color indicate the preferred reaction sites for the synthesis of Ir(III) metal complexes. | ||
Now, we turned to investigate the photophysical properties by the addition of the steric encumbering 2-xylenyl (or 2,6-dimethylphenyl) group,15 which is a close analogue of mesityl and other bulky aromatics, to the cyano functionalized imidazo[4,5-b]pyridin-2-ylidene (ct3) and (ct9),16 and the resulting chelates are depicted as (ct3x) and (ct9x). Their design principles are conceptually motived by literature precedents; for example, the mesityl group on the Ir(III) emitters is known to enhance their emission efficiencies by reducing triplet–triplet quenching without affecting blue color purity,17 and the mesityl groups increase the solubility and reduce concentration quenching in devices; thus, the blue OLED devices obtained from mesitylated emitters are more efficient than those from their parent emitters.18 Similarly, the mesitylation of Pt(II) emitters is a facile strategy to hinder aggregations or Pt⋯Pt interactions, allowing fabrication of efficient blue OLED devices,19 or even drastically enhancing OLED efficiencies.20 Equally important, radicals with multiple mesityl substituents showed strongly improved photoluminescence, allowing fabrication of deep red or even near infrared OLED devices with a maximum external quantum efficiency (EQE) of ∼28% at 689![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) nm.21 Lastly, the mesityl group can offer better stability and performance of boron–nitrogen based thermally activated delayed fluorescent (TADF) emitters by locking the mesityl group at the selective locations.22
nm.21 Lastly, the mesityl group can offer better stability and performance of boron–nitrogen based thermally activated delayed fluorescent (TADF) emitters by locking the mesityl group at the selective locations.22
With chelates (ct3x)H2+ and (ct9x)H2+ in hand, we further attempted the synthesis and characterization of the associated homoleptic Ir(III) carbene complexes: f-ct3xa, f-ct3xb, and f-ct9xa, f-ct9xb, respectively. The density functional theory (DFT) and respective time-dependent (TD) DFT calculations were conducted to make a direct comparison between the theoretical results and those obtained by experimental means. As for the potential applications, we executed the fabrication of both the phosphorescent and hyperphosphorescent blue OLED devices,23 and the latter employed a Förster resonance energy transfer (FRET) process from the Ir(III) dopant sensitizers to the multiple resonance (MR) TADF terminal emitter v-DABNA. It seemed that all these Ir(III) carbene emitters could deliver device performances better than the majority of blue OLED devices documented in the literature.
Results and discussion
Synthesis and characterizations
The synthesis of carbene pro-chelates (ct3x)H2+ and (ct9x)H2+, demanded a custom-made reagent 4-(2,6-dimethylphenyl)aniline (or 4-(2-xylenyl)aniline)24 and two functional 3-nitropyridine starting materials (namely, 2,6-dibromo-3-nitropyridine and 2,5-dibromo-3-nitropyridine) following multi-step procedures depicted in Scheme S1 of the ESI.† Next, the treatment of these pro-chelates with mer-IrCl3(tht)3, using sodium acetate as promoter in refluxing o-dichlorobenzene solution afforded two distinctive classes of Ir(III) phosphors: f-ct3ax and f-ct3bx, and f-ct9ax and f-ct9bx; all of them can be separated and purified by silica gel column chromatography, followed by recrystallization at RT. Scheme 2 indicated the schematic drawings of these Ir(III) carbene complexes, showing the presence of three 4-(2-xylenyl)phenyl cyclometalates for isomer a and only two relevant aryl cyclometalates for isomer b, while the remaining (i.e., the third) aryl cyclometalate of isomer b carried the 4-t-butyl phenyl cyclometalate. Other products (the isomers c and d)11,25 that can be expected by random permutation of aryl cyclometalates have not been observed, despite that their close analogue ct9H2+ yielding the third isomer f-ct9c in ∼4% during initial synthesis.16a This situation is probably attributed to the steric hindrance imposed by the 2-xylenyl groups, which disallowed more than two bulky 4-(2-xylenyl)phenyl groups from the N-aryl appendages to reside within the triangular region of the octahedral coordination framework. | ||
| Scheme 2 Chemical drawings of Ir(III) carbene complexes f-ct3ax/bx and f-ct9ax/bx. | ||
The 1H NMR spectroscopy and high-resolution mass spectrometry can be employed to provide the initial structural assignment. In particular, the 1H NMR spectra of both a isomers showed only one kind of carbene cyclometalate, which unambiguously ruled out the formation of meridional geometry or asymmetrically arranged carbene cyclometalates, i.e., the expected pattern of corresponding isomer b.26 Subsequently, single crystal X-ray structural analyses were executed on all obtained Ir(III) derivatives to decipher this ambiguity, while their molecular structures and essential metric parameters were depicted in Fig. S1–S4 (ESI†) and corresponding captions. The gross metric parameters were consistent with the anticipated structures, but with minor distortions that can be attributed to the inter-chelate repulsions, inter-molecular packing within the crystal lattices, or both factors. Moreover, the corresponding b isomers seemed to always be in higher yields in reference to a isomers in both systems. Apparently, this minute change of product ratio may originate from the electronic effect of relocating the cyano group at either the 5- or 6-position on the imidazo[4,5-b]pyridin-2-ylidene as the only variation introduced.
Photophysical, thermal and electrochemical characterization
The UV-Vis absorption and emission spectra were recorded in toluene to probe their photophysical properties. As shown in Fig. 1, these Ir(III) complexes display very similar patterns of absorption bands: in particular, the pair of Ir(III) complexes f-ct3ax and f-ct3bx exhibit a more red-shifted (i.e., lower-energy) absorption onset (∼445 nm) than the other pair of Ir(III) complexes f-ct9ax and f-ct9bx, in which the onset occurred at ∼435 nm. Moreover, the lowest energy absorption peak max. occurred at ∼397 nm for f-ct3ax/bx and ∼389 nm for f-ct9ax/bx, as summarized in Table 1. These absorption bands are assigned to the metal-to-ligand charge transfer (MLCT) transition and are akin to other reported fac-arranged homoleptic Ir(III) carbene complexes.27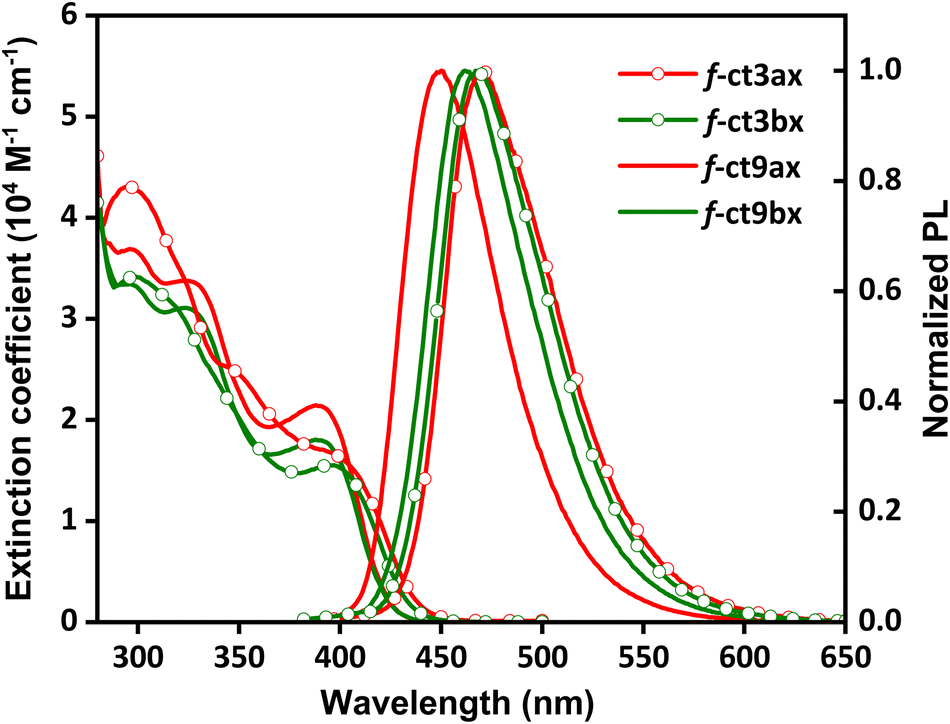 | ||
| Fig. 1 UV-Vis absorption and emission spectra of the studied Ir(III) complexes recorded in toluene at RT. | ||
| abs λmaxa (nm) | em λmaxb (nm) | FWHMc (nm) | PLQYd (%) | τ obs (μs) | τ rad (μs) | k r (105 s−1) | k nr (105 s−1) | |
|---|---|---|---|---|---|---|---|---|
| a Extinction coefficient (ε) is given in parentheses with a unit of 104 M−1 cm−1. b Recorded in degassed toluene at a conc. of 10−5 M at RT. c Full width at half maximum. d Coumarin 102 (C102) in methanol (PLQY = 87% and λmax = 480 nm) was employed as standard. | ||||||||
| f-ct3ax | 296 (4.3), 397 (1.7) | 470 | 63 | 72 | 0.99 | 1.38 | 7.27 | 2.83 |
| f-ct3bx | 298 (3.4), 398 (1.5) | 467 | 63 | 82 | 2.46 | 3.08 | 3.25 | 0.81 |
| f-ct9ax | 326 (3.4), 389 (2.1) | 450 | 56 | 98 | 0.89 | 0.91 | 11.0 | 0.22 |
| f-ct9bx | 324 (3.1), 389 (1.8) | 462 | 60 | 94 | 1.15 | 1.22 | 8.17 | 0.52 |
Next, the photoluminescence was recorded in degassed toluene at RT. As expected, all spectra exhibited a structureless spectral pattern, which is consistent with the common MLCT assignment. Notably, their emission peak max. follows the trend of 470 nm (f-ct3ax) and 467 nm (f-ct3bx) > 450 nm (f-ct9ax) and 462 nm (f-ct9bx). Hence, this indicated that, for those with identical N-aryl cyclometalates around the Ir(III) metal center, the imidazo[4,5-b]pyridin-2-ylidene with 5-cyano substituent (i.e., f-ct9ax/bx) would induce lesser bathochromic shift than those with the 6-substituted cyano group (i.e., f-ct3ax/bx), leading to significant differences in emission peaks (i.e., 450 nm of f-ct9axvs. 470 nm of f-ct3ax). Additionally, the shallower LUMO energy level was recorded for f-ct9ax (vide infra), affirming the distinguished electron-withdrawing ability of the 5-cyano substituent. Third, the difference in emission energy was smaller for their second isomers f-ct3bx (467 nm) and f-ct9bx (462 nm), which is obviously caused by the relocation of one 4-(2-xylenyl)phenyl entity to the uncoordinated pendent site. Finally, all these Ir(III) complexes exhibited high photoluminescence quantum yield (PLQY) between 72% and 98% and observed lifetimes between 0.89 μs and 2.46 μs. In contrast to the majority of TADF emitters which typically exhibit long lifetimes and relatively slow radiative decay rate constants, these complexes demonstrate much shorter radiative lifetimes and fast radiative rates. These characteristics effectively alleviate the accumulation of triplet excitons in the EML of OLED devices, thereby reducing efficiency roll-offs at high current densities.28 As revealed in Table 1, their radiative rate constant (kr) calculated using the equation
| kr = PLQY/τobs |
Cyclic voltammetry (CV) was executed to collect their electrochemical data (Fig. S6, ESI†). In general, the reversible oxidation process occurred at the Ir(III) metal center, while their recorded onsets spanned a narrow range: 0.68–0.72 eV. Notably, both b isomers, i.e., f-ct3bx and f-ct9bx, exhibited a less positive electrochemical potential compared with their a counterparts, which may originate from the incorporation of one more electron rich 4-t-butylphenyl cyclometalate at the Ir(III) center, as previously documented.9c,11 Subsequently, their HOMO and LUMO energy levels were calculated using these onset potentials and the optical energy gaps were estimated from the photophysical measurements and compiled in Table 2. Furthermore, decomposition temperatures (Td, 5% weight loss) of 461, 462, 439 and 451 °C were recorded for f-ct3ax/bx and f-ct9ax/bx, respectively. (Fig. S7, ESI†) Despite having lowered Td data than their parent complexes (457–483 °C) and procession of three bulky xylenyl groups, sublimation can still be executed without notable decomposition.
| Complex |
E
onsetox
(eV) |
E HOMO (eV) |
E
optg
(eV) |
E LUMO (eV) | T d,5% (°C) |
|---|---|---|---|---|---|
| a Electrochemical potentials were measured in an acetonitrile solution of TBAPF6 at 0.1 M. Eonsetox is the onset potential of the oxidation wave. b HOMO = −(Eonsetox + 4.8). c Energy gap = 1240/[PLonset (nm)]. d LUMO = HOMO + energy gap. e TGA is recorded under N2 flow. | |||||
| f-ct3ax | 0.70 | −5.50 | 2.86 | −2.64 | 461 |
| f-ct3bx | 0.69 | −5.49 | 2.87 | −2.62 | 462 |
| f-ct9ax | 0.72 | −5.52 | 2.99 | −2.53 | 439 |
| f-ct9bx | 0.68 | −5.48 | 2.91 | −2.57 | 451 |
Theoretical investigation
A time-dependent density functional theory (TD-DFT)29 based method with the B3LYP functional30 was employed to acquire theoretical insights into the photophysical properties of Ir(III) complexes f-ct3ax/bx and f-ct9ax/bx. Low-energy electronic excited states, in particular the first singlet S1 and the first triplet T1, of these Ir(III) complexes in the toluene solvent were investigated to understand the observed photophysical properties and the micro-mechanism of how chelate modifications affect these properties. Further computational details are provided in the Experimental section of the ESI.†Computed excitation energies from the S0 to S1 state were obtained as 432 nm and 430 nm for f-ct3ax and f-ct3bx, respectively and 409 nm and 418 nm for f-ct9ax and f-ct9bx, respectively (cf.Table 3), corresponding to the tail area of the experimental absorption curves at ∼420–440 nm (cf.Fig. 1). For S0 → T1 transitions, the excitation energies were found to be 458 nm, 457 nm, 436 nm, and 446 nm for f-ct3ax, f-ct3bx, f-ct9ax, and f-ct9bx, respectively (cf.Table 3). This closely mirrors the observed experimental trends for emission energies (470 nm, 467 nm, 450 nm, and 462 nm for f-ct3ax, f-ct3bx, f-ct9ax, and f-ct9bx, respectively) (cf.Fig. 1 and Table 1). This indicates that (i) the isomerization of f-ct3ax/3bx has negligible effects on the emission wavelength, and (ii) the emission energy is blue-shifted by the structural changes from f-ct3ax/bx to f-ct9ax/bx (i.e., relocating the cyano group from the 6- to 5-position). A small deviation is found between the calculated and measured excitation energies (within 0.09 eV). Such a difference is common for this class of Ir(III) complexes and can be mainly ascribed to the uncertainties of the employed level of theoretical methods and the approximated molecular geometry of the emissive state.
| State | ε (nm eV−1) | f | Orbital contribution (>20%) | Assignmentb | |
|---|---|---|---|---|---|
| a Calculated by TD-DFT using B3LYP functional and PCM for modelling the toluene solvent (see ESI for further details). b Rate of the MLCT character of the T1 state was obtained as the product of the difference in the metal contribution (from occupied to virtual NTOs) and the eigenvalue of the corresponding NTO pair (cf.Fig. 2). | |||||
| f-ct3ax | T1 | 458/2.71 | 0 | HOMO → LUMO (64%) | MLCT (22.8%), ILCT, LC, LLCT |
| S1 | 432/2.87 | 0.04 | HOMO → LUMO+1 (78%) | ||
| f-ct3bx | T1 | 457/2.72 | 0 | HOMO → LUMO (44%) | MLCT (21.8%), ILCT, LC, LLCT |
| HOMO → LUMO+1 (28%) | |||||
| S1 | 430/2.88 | 0.05 | HOMO → LUMO (85%) | ||
| f-ct9ax | T1 | 436/2.84 | 0 | HOMO → LUMO (78%) | MLCT (23.1%), ILCT, LC, LLCT |
| S1 | 409/3.03 | 0.11 | HOMO → LUMO+1 (92%) | ||
| f-ct9bx | T1 | 446/2.78 | 0 | HOMO → LUMO+1 (50%) | MLCT (19.6%), ILCT, LC, LLCT |
| HOMO → LUMO (27%) | |||||
| S1 | 418/2.97 | 0.04 | HOMO → LUMO (62%) | ||
| HOMO → LUMO+1 (30%) | |||||
In the next step, the calculated T1 excited states were further investigated to analyze the electron behaviors during transition and its connection to the observed radiative rate. The primary molecular orbitals (MOs) involved in S0 → T1 (and S0 → S1) electronic excitations are described in Table 3. Multiple MO pairs contribute to each T1 excited state. To achieve an understanding of the electronic character of the excited state transition, a predominant pair of natural transition orbitals (NTOs)31 was constructed to express each T1 excited state. This would allow us to gain a clear and concise orbital assignment. The resulting main NTO pairs of the T1 excited states are shown in Fig. 2. It reveals that the occupied NTOs of all four complexes are delocalized at both the metal atom and aryl cyclometalates, while the virtual NTOs are mainly localized on the carbene skeletons including the cyano group. This suggests that metal-to-ligand charge transfer (MLCT), intra-ligand charge transfer (ILCT), ligand-centered (LC) transitions, and ligand-to-ligand charge transfer (LLCT) characters are involved in the T1 excited state. Additionally, f-ct9bx was localized on one of three chelates, leading to a more localized nature and a larger orbital overlap than the other three complexes.
 | ||
| Fig. 2 Natural transition orbital (NTO) pairs representing the T1 excited states of the investigated Ir(III) complexes at their geometrically optimized ground state, including the contribution of the Ir center to the NTOs. The NTO eigenvalues are 0.85 and 0.83 for f-ct3ax and f-ct3bx, and 0.83 and 0.99 for f-ct9ax and f-ct9bx, respectively. Overlap of orbitals between occupied and virtual NTOs is 0.51 (a.u.) for both f-ct3ax and f-ct3bx, 0.48 (a.u.) for f-ct9ax, and 0.52 (a.u.) for f-ct9bx. | ||
Following this, we quantified the MLCT percentage for the T1 transition (cf.Table 3) and the orbital overlap of electronic orbitals characterizing the T1 state (cf.Fig. 2), as these quantities were identified as important factors of the radiative rate constant in our previous work.11 The MLCT percentage is an indicator of the ratio of heavy metal involvement in the T1 state, where greater MLCT induces more efficient phosphorescence due to the improvement of intersystem crossing. Furthermore, orbital overlap is related to the electronic transition dipole moment, where a larger orbital overlap in the orbital pair representing the T1 state also leads to an enhanced radiative rate. For the four Ir(III) investigated here, higher MLCT percentages are found for the more symmetrical complexes f-ct3ax (22.8%) and f-ct9ax (23.1%) compared with their asymmetric counterparts, f-ct3bx (21.8%) and f-ct9bx (19.6%). Orbital overlap is similar for f-ct3ax and f-ct3bx (0.51 a.u., cf.Fig. 2) but it is slightly larger for f-ct9bx (0.52 a.u.) than that for f-ct9ax (0.48 a.u.). Overall, f-ct9ax has the greatest MLCT percentage and achieves the fastest radiative rate (11.0 × 105 s−1) among all four complexes.
Electroluminescence
Encouraged by their promising photophysical properties, thermal stability and volatility, these Ir(III) carbene complexes were employed as both dopants and sensitizers in the fabrication of vacuum-deposited OLED devices. First, the electroluminescence (EL) of these Ir(III) complexes was evaluated as dopants using the following device architecture: ITO/1,4,5,8,9,11-hexaazatriphenylenehexacarbonitrile (HATCN, 10 nm)/4,4′,4′′-tris(N-carbazolyl)-triphenylamine (TAPC, 40 nm)/4,4′-bis(N-carbazolyl)-1,1′-biphenyl (TCTA, 5 nm)/9,9′-spirobifluorene-2,2′-diphenylamine (CzSi, 5 nm)/x wt% emitter in CzSi (20 nm)/2,7-diphenyl-9,9′-spirobifluorene (DPEPO, 5 nm)/4,4′-bis(2,2′-diphenylvinyl)-1,1′-biphenyl (TSPO1, 4 nm)/lithium quinolate (Liq, 2 nm)/Al (120 nm). In this structure, HATCN and Liq functioned as hole-injecting and electron-injecting layers, TAPC and TCTA served as the corresponding hole-transporting layers, while TSPO1 and DPEPO acted as the electron-transporting and hole-blocking layers, respectively. Additionally, CzSi was employed as both the electron-blocking layer and host material of the emission layer (EML) for its high triplet energy gap. Energy level diagram and molecular structures of organic functional materials are illustrated in Fig. 3(a) and Fig. S8 (ESI†), respectively.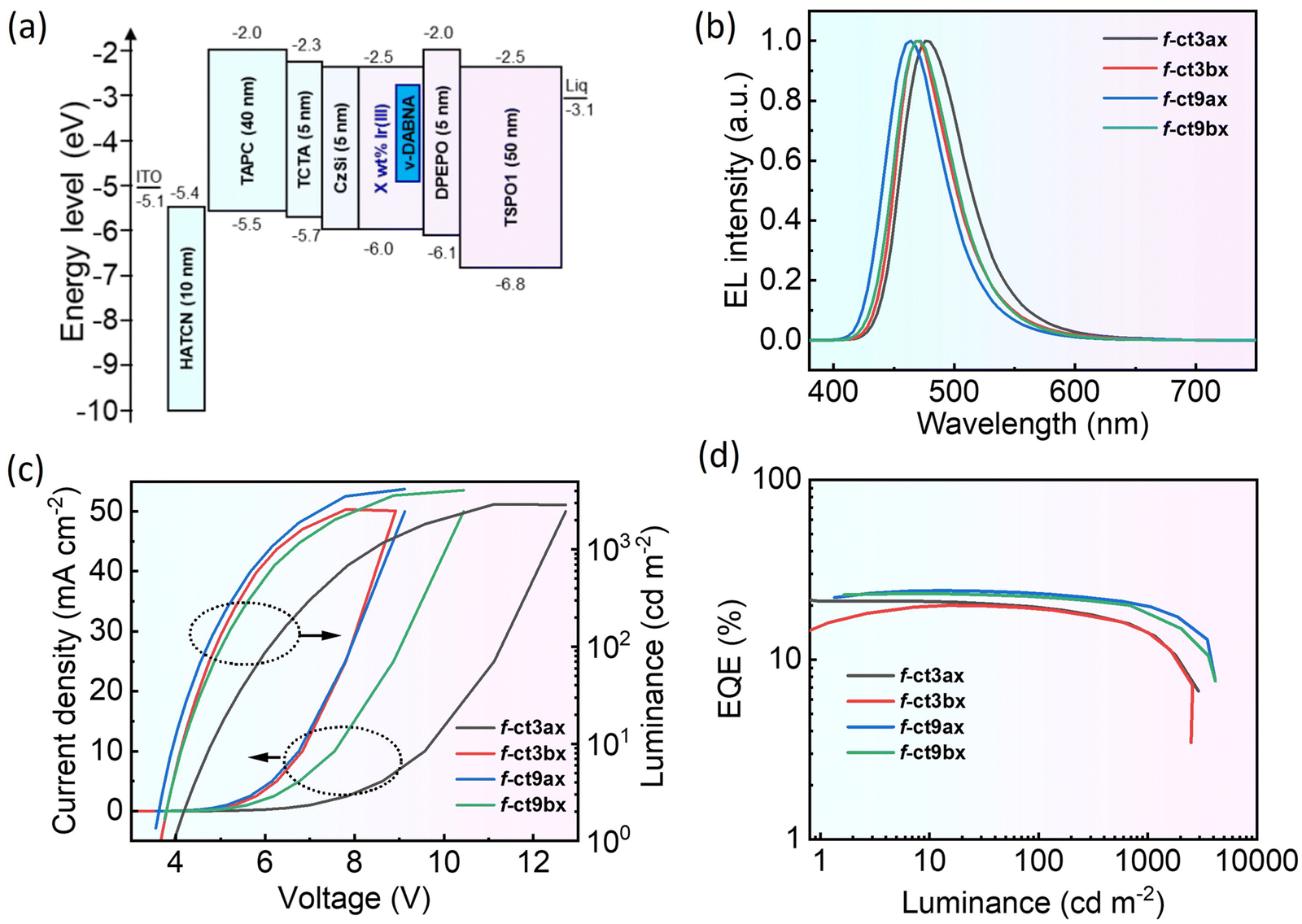 | ||
| Fig. 3 Performances of OLED devices with Ir(III) phosphors at 15 wt% in the CzSi host. (a) Device architecture and energy level diagram of the materials employed. (b) EL spectra measured at a current density of 10 mA cm−2. (c) Corresponding current density–voltage–luminance (J–V–L) characteristics. (d) Plot of EQE versus luminance of devices. | ||
The devices were first examined using f-ct9ax with a varied concentration from 7.5 wt% to 15 wt% to optimize the performance. As shown in Table S3 and Fig. S9 (ESI†), the higher doping ratio could lower the turn-on voltage and concurrently improve device performance. Such phenomenon can be attributed to enhanced carrier mobility or charge balance, which resulted in significant improvement in current density (CE) and power efficiency (PE), together with higher maximum luminance (Lm). Notably, a bathochromic shift and larger CIEy coordinate were also observed at the elevated doping ratio, which is likely caused by the intermolecular stacking interactions between dopant and host molecules. These findings resemble the reports on homoleptic Ir(III) emitters bearing parent cyano functionalized imidazo[4,5-b]pyridin-2-ylidene cyclometalates.16 Upon further increasing the dopant concentration to 17.5 wt%, we observed a slight decline in maximum brightness, a gradual red shifting in emission, and a minute decrease in maximum CE, PE and EQE, respectively. Overall, the best result was achieved at 15 wt%, which was considered as the optimal doping ratio for phosphors, and then applied in the fabrication of all blue emissive PhOLED devices. Their electroluminescence performances are depicted in Fig. 3 and Table 4 for scrutiny.
| Dopants | V on | ELa | CEb | PEb | EQEb | L m | CIEa | FWHMa |
|---|---|---|---|---|---|---|---|---|
| (V) | (nm) | (cd A−1) | (lm W−1) | (%) | (cd m−2) | (x, y) | (nm) | |
| a Recorded at a current density of 10 mA cm−2. b Correspond to the maximum value and data recorded at both 100 and 1000 cd m−2. | ||||||||
| f-ct3ax | 3.9 | 476 | 38.2/34.5/23.4 | 35.7/18.1/8.5 | 22.1/19.7/14.2 | 2914 | 0.143, 0.261 | 61 |
| f-ct3bx | 3.7 | 468 | 29.3/27.1/20.8 | 22.3/17.0/10.1 | 20.0/18.9/14.2 | 2578 | 0.141, 0.193 | 53 |
| f-ct9ax | 3.6 | 464 | 28.9/26.7/21.5 | 24.2/17.5/11.0 | 24.2/23.3/19.8 | 4172 | 0.141, 0.137 | 57 |
| f-ct9bx | 3.6 | 470 | 33.1/30.8/23.5 | 27.8/19.1/10.9 | 23.3/22.2/18.1 | 4066 | 0.138, 0.182 | 57 |
As shown in Table 4, all their EL showed red-shifted peak max. compared with their PL recorded in toluene due to the higher polarity of the CzSi. More specifically, the EL peak max. occurred at 476 nm for f-ct3ax, 468 nm for f-ct3bx, 464 nm for f-ct9ax, and 470 nm for f-ct9bx. In particular, f-ct9ax exhibited the largest bathochromic shift among all fabricated devices, i.e., from 450 nm in toluene to 464 nm in CzSi. Also, PhOLEDs based on f-ct9ax and f-ct9bx exhibited higher maximum external quantum efficiencies (max. EQEs) of 24.2% and 23.3%, together with higher Lm of 4172 and 4066 cd m−2. These discrepancies are primarily due to the higher photoluminescence quantum yields (ΦPL) and shorter radiative lifetime (τrad) of f-ct9ax and f-ct9bx (ΦPL = 94–98% and τrad = 0.91–1.22 μs) compared with f-ct3ax and f-ct3bx (ΦPL = 72–82% and τrad = 1.38–3.08 μs). Similarly, the J–V–L characteristics shown in Fig. 3 affirmed the lower turn-on voltages of 3.6 V for f-ct9ax and f-ct9bx, compared with 3.9 V for f-ct3ax and 3.7 V for f-ct3bx. Overall, these findings suggest that f-ct9ax and f-ct9bx, i.e., those with 5-cyano-imidazo[4,5-b]pyridin-2-ylidene, show better OLED performances compared with 6-cyano-imidazo[4,5-b]pyridin-2-ylidene fragments as observed in f-ct3ax and f-ct3bx.
Recently, hyperfluorescent OLEDs (i.e., hyper-OLEDs) have received a great deal of attention due to their efficient Förster energy transfer process (FRET) from sensitizer to the terminal emitter and great improvement in emission efficiency.32 In the meantime, v-DABNA has also established itself as a superb terminal emitter of hyper-OLEDs due to its nearly 100% internal quantum efficiency, and its narrow-band blue emission by the multi-resonance effect.33 Hence, we employed our newly prepared Ir(III) phosphors as the sensitizers and v-DABNA as the terminal emitter to make the hyper-OLEDs,34 or the so-called hyperphosphorescent OLEDs to strongly emphasize the inherent advantages of phosphors. By leveraging the excellent spectral overlap between the absorption of v-DABNA and emission of Ir(III) sensitizers in CzSi thin films (Fig. 4(b)), we confirmed the efficient energy transfer. As depicted in Fig. 4(c) and Table 5, all the fabricated hyper-OLEDs exhibited sharp emission peak max. at 468 nm, with full-width at half-maximum (FWHM) values of approximately 21–23 nm and a CIEy coordinate of ∼0.10. Notably, by capitalizing on the superior OLED performances and efficient FRET, the f-ct9ax based hyper-OLED achieved a max. EQE of up to 31.9%, together with CIEx,y coordinates of (0.123, 0.095) and a high EQE of 20.9% at a practical luminance of 1000 cd m−2. These results represent one of the best hyper-OLED devices to date.35 Overall, these findings highlight the potential of Ir(III) phosphors in serving as sensitizers to produce blue emissive hyper-OLEDs, offering both improved device efficiency and color purity.
 | ||
| Fig. 4 Performances of hyper-OLED devices with 15 wt% of Ir(III) sensitizers and 1 wt% of v-DABNA terminal emitter. (a) Structural drawing of v-DABNA. (b) Spectral overlap between the absorption of v-DABNA and emission of Ir(III) sensitizers in CzSi thin film. (c) EL spectra at a current density of 10 mA cm−2. (d) Corresponding CIEx,y coordinates. (e) Current density–voltage–luminance (J–V–L) characteristics and (f) plot of EQE versus luminance of OLED devices. | ||
| Dopants | V on (V) | ELa (nm) | CEb (cd A−1) | PEb (lm W−1) | EQEb (%) | L m (cd m−2) | CIEa (x, y) | FWHMa (nm) |
|---|---|---|---|---|---|---|---|---|
| a Recorded at a current density of 10 mA cm−2. b Correspond to the maximum value and data recorded at both 100 and 1000 cd m−2. | ||||||||
| f-ct3ax/v-DABNA | 3.8 | 468 | 27.7/21.6/17.2 | 27.9/13.1/8.3 | 28.1/21.6/16.3 | 7550 | 0.129, 0.131 | 23 |
| f-ct3bx/v-DABNA | 4.0 | 468 | 20.3/12.4/9.3 | 20.8/6.42/3.74 | 26.1/15.3/10.6 | 4335 | 0.125, 0.106 | 22 |
| f-ct9ax/v-DABNA | 3.9 | 468 | 25.1/21.3/17.0 | 20.1/11.1/6.63 | 31.9/27.0/20.9 | 6053 | 0.123, 0.095 | 21 |
| f-ct9bx/v-DABNA | 3.9 | 468 | 24.7/21.3/15.6 | 18.7/10.4/5.19 | 29.7/25.0/19.4 | 5540 | 0.124, 0.103 | 21 |
Both the EQE1000 (i.e., EQE recorded at 1000 cd m−2) and J90 (current density recorded at 90% of max. EQE) were estimated for these OLEDs in giving an estimation of overall performance via a conceptual index of Figure of Merit (FOM).36 Table S4 (ESI†) depicted both the EQE1000 and J90 of several recently reported Ir(III) carbene complexes, and the data derived from the closely related analogues f-ct3a/b to f-ct9a/b without the mesityl groups and using the common unipolar and high triplet energy gap host material. Remarkably, these Ir(III)-based OLED devices showed competitive performances as demonstrated by their adequate EQE1000 and J90 data compared with the record-setting TADF OLED devices with blue emission, as depicted in Fig. S10 (ESI†). We attributed these to the more effective MLCT contribution that has reduced the concentration of high-energy triplet excitons within the EML of OLEDs in reference to the contribution from LLCT and even TSCT (through space charge transfer) processes that have occurred in the Ir(III) carbene complexes37 as well as typical TADF emitters.
Conclusion
We designed and synthesized two distinctive series of Ir(III) carbene complexes (f-ct3ax/bx and f-ct9ax/bx) featuring both the dislocated 6- and 5-cyano substituent on imidazo[4,5-b]pyridin-2-ylidene cyclometalates and bulky 4-(2-xylenyl)phenyl group located at one of the N-aryl appendages. They exhibited excellent photophysical properties in degassed toluene solution at RT, particularly with blue emission located in the region of 450–470 nm and high PLQY in the range of 72–98%. Hence, all of them are employed in the fabrication of OLED devices, giving superior EL performances with max. EQEs of 24.2% and 23.3% for PhOLEDs based on f-ct9ax and f-ct9bx, respectively. Furthermore, the hyper-OLED device based on f-ct9ax and v-DABNA exhibited a narrow-band blue emission peak at 468 nm with a FWHM of 21 nm, together with max. EQE of up to 31.9% with a CIEx,y coordinate of (0.123, 0.095). These results demonstrate the potential pros and cons of the introduction of bulky groups in the Ir(III) metal phosphors and provide valuable insights into the development of next-generation blue OLED phosphors with improved performance and color purity.Experimental section
General information and materials
All reactions were conducted under a N2 atmosphere. Commercially available reagents were used without further purification and solvents were dried prior to use. 1H NMR spectra were measured with a Bruker Avance III 400 MHz NMR instrument. The high-resolution mass spectra were obtained on a Sciex X500R Q-TOF with acetonitrile applied as the solvent. UV-Vis spectra were recorded on a HITACHI UH-4150 spectrophotometer. The steady-state emission spectrum was measured with an Edinburgh FLS980 spectrofluorometer. The lifetime studies were performed by a time-correlated single photon counting system (TCSPC). All sample solutions were degassed using at least three freeze–pump–thaw cycles and the quantum yields were calculated using the standard sample which has a known quantum yield. Cyclic voltammetry was conducted on a CHI621A electrochemical analyzer. The oxidation and reduction potentials were measured using a glassy carbon working electrode with 0.1 M of NBu4PF6 in CH3CN. The potentials were referenced externally to the ferrocenium/ferrocene (Fc+/Fc) couple.Synthesis of iridium(III) complexes f-ct3ax and f-ct3bx
To a 50 mL flask, the following components were added: G4 (0.9 g, 1.5 mmol), sodium acetate (0.41 g, 5 mmol), mer-IrCl3(tht)3 (0.28 g, 0.5 mmol) and o-dichlorobenzene (25 mL). The mixture was refluxed for 24 hours with vigorous stirring. After removal of the solvent under vacuum, the residue was dissolved into CH2Cl2 and washed with deionized water. The organic phase was dried over anhydrous Na2SO4, filtered, concentrated to dryness and the residue was purified by column chromatography with a mixture of n-hexane and ethyl acetate (5/2, v/v) to afford f-ct3ax (Rf = 0.33) and f-ct3bx (Rf = 0.38) according to their elution sequence. Further recrystallization from a layered solution of CH2Cl2 and methanol gave a yellow solid of f-ct3ax (60 mg, 8%) and a yellow solid of f-ct3bx (187 mg, 24%), respectively.Spectroscopic data of f-ct3ax: HRMS (ESI) for C93H82IrN12 [M + H]+: calcd 1559.6409, found 1559.6335; 1H NMR (400 MHz, CDCl3) δ 8.78 (d, J = 8.0 Hz, 3H), 7.41 (d, J = 8.0 Hz, 3H), 7.39 (d, J = 7.6 Hz, 3H), 6.96 (t, J = 7.6 Hz, 3H), 6.92–6.79 (m, 12H), 6.62 (d, J = 7.6 Hz, 3H), 6.44 (d, J = 8.4 Hz, 6H), 6.35 (s, 3H), 1.58 (s, 6H), 1.56 (s, 6H), 1.50 (s, 6H), 1.01 (s, 27H).
Selected crystal data of f-ct3ax: CCDC deposition number: 2371311. C96H84IrN12Cl9; M = 1917.00; monoclinic; space group = P21/c; a = 25.9520(10) Å, b = 20.3652(9) Å, c = 18.3252(7) Å; β = 100.632(2); V = 9518.9(7) Å3; Z = 4; ρCalcd = 1.338 g cm−3; μ = 5.434 mm−1; F(000) = 3896.0, λ(Cu-Kα) = 1.54178 Å; T = 213 (2) K; index range: −32 ≤ h ≤ 32, −25 ≤ k ≤ 25, −21 ≤ l ≤ 22; 120657 reflections collected, 19436 independent reflections (Rint = 0.0766), max./min. transmission ratio: 0.372, data/restraints/parameters = 19436/919/1245, GOF = 1.050, final R1[I > 2σ(I)] = 0.0545 and wR2(all data) = 0.1657.
Spectroscopic data of f-ct3bx: HRMS (ESI) for C93H82IrN12 [M + H]+: calcd 1559.6409, found 1559.6424; 1H NMR (400 MHz, CDCl3) δ 8.83 (d, J = 8.0 Hz, 1H), 8.78 (d, J = 8.0 Hz, 1H), 8.38 (d, J = 8.4 Hz, 1H), 7.67 (d, J = 8.4 Hz, 1H), 7.61 (d, J = 8.4 Hz, 1H), 7.45 (dd, J = 8.4, 1.6 Hz, 1H), 7.38 (t, J = 7.6, 2H), 7.34 (dd, J = 8.4, 1.6 Hz, 1H), 7.11 (t, J = 7.6 Hz, 1H), 7.06–6.92 (m, 8H), 6.90–6.81 (m, 8H), 6.75 (td, J = 8.4, 2.0 Hz, 2H), 6.64 (d, J = 8.4 Hz, 2H), 6.50 (d, J = 8.0 Hz, 2H), 6.44 (dd, J = 8.4, 2.0 Hz, 1H), 6.33 (dd, J = 7.6, 1.6 Hz, 2H), 6.26 (dd, J = 8.4, 2.0 Hz, 1H), 1.99 (s, 3H), 1.65 (s, 3H), 1.61–1.53 (m, 9H), 1.29 (s, 3H), 1.04 (s, 9H), 1.02 (s, 9H), 0.95 (s, 9H).
Selected crystal data of f-ct3bx: CCDC deposition number: 2371313. C93H81IrN12; M = 1558.89; monoclinic; space group = P21/c; a = 15.6795(4) Å, b = 21.1239(5) Å, c = 26.1636(6) Å; β = 92.2430(10)°; V = 8659.1(4) Å3; Z = 4; ρCalcd = 1.196 g cm−3; μ = 3.367 mm−1; F(000) = 3200.0, λ(Cu-Kα) = 1.54178 Å; T = 193 (2) K; index range: −19 ≤ h ≤ 18, −25 ≤ k ≤ 26, −32 ≤ l ≤ 32; 76411 reflections collected, 17652 independent reflections (Rint = 0.0372), max./min. transmission ratio: 0.808, data/restraints/parameters = 17652/0/970, GOF = 1.020, final R1[I > 2σ(I)] = 0.0269 and wR2(all data) = 0.0763.
Synthesis of iridium(III) complexes f-ct9ax and f-ct9bx
To a 50 mL flask, the following substances were added: H4 (0.9 g, 1.5 mmol), sodium acetate (0.41 g, 5 mmol), mer-IrCl3(tht)3 (0.28 mg, 0.5 mmol) and o-dichlorobenzene (25 mL). The mixture was refluxed for 24 hours with vigorous stirring. After removal of solvent under vacuum, the residue was dissolved into CH2Cl2 and washed with deionized water three times. The organic phase was dried over anhydrous Na2SO4, filtered, concentrated to dryness and the residue was purified by column chromatography with a mixture of n-hexane and ethyl acetate (3/1, v/v) to afford f-ct9ax (Rf = 0.38) and f-ct9bx (Rf = 0.43) according to their elution sequence. Further recrystallization from a layered solution of CH2Cl2 and methanol gave a yellow solid of f-ct9ax (187 mg, 24%) and a yellow solid of f-ct9bx (300 mg, 38%), respectively.Spectroscopic data of f-ct9ax: HRMS (ESI) for C93H82IrN12 [M + H]+: calcd 1559.6409, found 1559.6305; 1H NMR (400 MHz, CDCl3) δ 8.80 (d, J = 8.0 Hz, 3H), 8.66 (d, J = 1.6 Hz, 3H), 7.45 (d, J = 8.0 Hz, 3H), 6.99 (d, J = 1.6 Hz, 3H), 6.96 (t, J = 7.2 Hz, 3H), 6.89 (d, J = 7.2 Hz, 3H), 6.85 (d, J = 7.2 Hz, 3H), 6.83 (dd, J = 8.0, 1.6 Hz, 3H), 6.58 (d, J = 8.0 Hz, 3H), 6.43 (d, J = 6.8 Hz, 3H), 6.41 (d, J = 6.8 Hz, 3H), 6.37 (d, J = 1.6 Hz, 3H), 1.57 (s, 9H), 1.51 (s, 9H), 1.06 (s, 27H).
Selected crystal data of f-ct9ax: CCDC deposition number: 2407904. C96H87IrN12Cl6; M = 1813.67; triclinic; space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) ; a = 13.015(3) Å, b = 15.040(5) Å, c = 24.206(9) Å; α = 99.607(14)°, β = 99.685(11)°, γ = 106.715(10)°; V = 4355(2) Å3; Z = 2; ρCalcd = 1.383 g cm−3; μ = 5.079 mm−1; F(000) = 1852.0, λ(Cu-Kα) = 1.54178 Å; T = 213 (2) K; crystal size: 0.35 × 0.18 × 0.04 mm; 59316 reflections collected, 17783 independent reflections (Rint = 0.0412), data/restraints/parameters = 17783/288/1117, GOF = 1.032, final R1[I > 2σ(I)] = 0.0321 and wR2(all data) = 0.0853.
; a = 13.015(3) Å, b = 15.040(5) Å, c = 24.206(9) Å; α = 99.607(14)°, β = 99.685(11)°, γ = 106.715(10)°; V = 4355(2) Å3; Z = 2; ρCalcd = 1.383 g cm−3; μ = 5.079 mm−1; F(000) = 1852.0, λ(Cu-Kα) = 1.54178 Å; T = 213 (2) K; crystal size: 0.35 × 0.18 × 0.04 mm; 59316 reflections collected, 17783 independent reflections (Rint = 0.0412), data/restraints/parameters = 17783/288/1117, GOF = 1.032, final R1[I > 2σ(I)] = 0.0321 and wR2(all data) = 0.0853.
Spectroscopic data of f-ct9bx: HRMS (ESI) for C93H82IrN12 [M + H]+: calcd 1559.6409, found 1559.6292; 1H NMR (400 MHz, CDCl3) δ 8.85 (d, J = 8.0 Hz, 1H), 8.75 (d, J = 8.0 Hz, 1H), 8.64 (d, J = 1.6 Hz, 1H), 8.57 (d, J = 1.6 Hz, 1H), 8.54 (d, J = 1.6 Hz, 1H), 8.38 (d, J = 1.6 Hz, 1H), 7.56 (d, J = 8.4 Hz, 1H), 7.45 (dd, J = 8.4, 2.0 Hz, 1H), 7.39 (dd, J = 8.4, 2.0 Hz, 1H), 7.13 (t, J = 7.6 Hz, 1H), 7.05–6.93 (m, 10H), 6.90–6.87 (m, 4H), 6.83 (d, J = 2.0 Hz, 1H), 6.80 (dd, J = 8.0, 2.0 Hz, 1H), 6.72 (dd, J = 8.4, 2.0 Hz, 2H), 6.64 (d, J = 8.4 Hz, 2H), 6.60–6.53 (m, 2H), 6.43 (dd, J = 8.4, 2.0 Hz, 1H), 6.36 (d, J = 1.6 Hz, 1H), 6.34 (d, J = 1.6 Hz, 1H), 6.26 (dd, J = 8.4, 2.0 Hz, 1H), 1.98 (s, 3H), 1.68–1.51 (m, 12H), 1.33 (s, 3H), 1.05 (s, 9H), 1.03 (s, 9H), 0.95 (s, 9H).
Selected crystal data of f-ct9bx: CCDC deposition number: 2407906. C94.5H84IrN12Cl3; M = 1686.28; monoclinic; space group P21/c; a = 15.419(4) Å, b = 40.541(10) Å, c = 16.158(4) Å; β = 99.742(8) °; V = 9955(4) Å3; Z = 4; ρCalcd = 1.125 g cm−3; μ = 3.678 mm−1; F(000) = 3452.0, λ(Cu-Kα) = 1.54178 Å; T = 193 (2) K; crystal size: 0.29 × 0.22 × 0.03 mm; 130544 reflections collected, 20249 independent reflections (Rint = 0.0564), data/restraints/parameters = 20249/184/1063, GOF = 1.031, final R1[I > 2σ(I)] = 0.0466 and wR2(all data) = 0.1386.
Single X-ray structural determination
The single crystals suitable for X-ray diffraction study were obtained by the recrystallization indicated in the experimental section. Single crystal X-ray diffraction data were recorded on a Bruker D8 Venture Photon II diffractometer with microfocus X-ray sources using phi and omega scan mode (APEX3) at 233 K.Author contributions
Chengcheng Wu, Yixin Wu, Kai-Ning Tong: investigation, data curation, formal analysis, writing – original draft, review & editing. Martin Kuhn, Shek-Man Yiu, Yu-Cheng Kung: investigation. Wen-Yi Hung: methodology, supervision. Jie Yan: formal analysis, supervision, writing – review & editing. Xiuwen Zhou, Guodan Wei: methodology, supervision, writing – original draft, review & editing, funding acquisition. Yun Chi: conceptualization, methodology, project administration, supervision, writing – original draft, review & editing, funding acquisition.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
Y. C. received his financial support from University Grants Council (CityU 11304221 and CityU 11312722) and City University of Hong Kong, Hong Kong SAR. G. W. obtained her research funding from Shenzhen Science and Technology Innovation Committee (grant no. GJHZ20210705143204013) and Shenzhen Science and Technology Innovation Committee (JCYJ20200109144614514). X. Z. is supported by Discovery Early Career Researcher Award (ARC DECRA DE190100144) from the Australian Research Council and a computing grant from National Computational Infrastructure of Australia.References
-
(a) H. Shi, Y. Wang, S. Lin, J. Lou and Q. Zhang, Dalton Trans., 2021, 50, 6410–6417 RSC
; (b) B. H. Jhun, D. Song, S. Y. Park and Y. You, Top. Curr. Chem., 2022, 380, 35 CrossRef CAS PubMed
-
(a) A. Lin, S. Lee and R. R. Knowles, Acc. Chem. Res., 2024, 57, 1827–1838 CrossRef CAS
-
(a) Y. Chi and P.-T. Chou, Chem. Soc. Rev., 2010, 39, 638–655 RSC
-
(a) H. Yersin, A. F. Rausch, R. Czerwieniec, T. Hofbeck and T. Fischer, Coord. Chem. Rev., 2011, 255, 2622–2652 CrossRef CAS
-
(a) L. Xiao, Z. Chen, B. Qu, J. Luo, S. Kong, Q. Gong and J. Kido, Adv. Mater., 2011, 23, 926–952 CrossRef CAS PubMed
-
(a) Y. Chi, T.-K. Chang, P. Ganesan and P. Rajakannu, Coord. Chem. Rev., 2017, 346, 91–100 CrossRef CAS
-
(a) T. Sajoto, P. I. Djurovich, A. B. Tamayo, J. Oxgaard, W. A. Goddard and M. E. Thompson, J. Am. Chem. Soc., 2009, 131, 9813–9822 CrossRef CAS
-
(a) J. Lee, H.-F. Chen, T. Batagoda, C. Coburn, P. I. Djurovich, M. E. Thompson and S. R. Forrest, Nat. Mater., 2016, 15, 92–98 CrossRef CAS PubMed
-
(a) A. Maheshwaran, V. G. Sree, H.-Y. Park, H. Kim, S. H. Han, J. Y. Lee and S.-H. Jin, Adv. Funct. Mater., 2018, 28, 1802945 CrossRef
- J. Yan, S. F. Wang, C.-H. Hsu, E. H.-C. Shi, C.-C. Wu, P.-T. Chou, S.-M. Yiu, Y. Chi, C. You, I. C. Peng and W.-Y. Hung, ACS Appl. Mater. Interfaces, 2023, 15, 21333–21343 CrossRef CAS
- J. Yan, D.-Y. Zhou, L.-S. Liao, M. Kuhn, X. Zhou, S.-M. Yiu and Y. Chi, Nat. Commun., 2023, 14, 6419 CrossRef CAS
- J. Yan, Y. Wu, I.-C. Peng, Y. Pan, S.-M. Yiu, K.-T. Wong, W.-Y. Hung, Y. Chi and K.-C. Lau, J. Mater. Chem. C, 2023, 11, 12270–12279 RSC
- J. Yan, C. Wu, S.-M. Yiu, M. Kuhn, M. Huang, Y. Zhang, X. Zhou, C. Yang, G. Wei and Y. Chi, Adv. Opt. Mater., 2025, 13, 2402332 CrossRef CAS
- J. Yan, T. Nakamura, X. Tan, S.-M. Yiu, R. Mimura, K. Hoshi, X. Zhou, Y. Chi, H. Sasabe and J. Kido, Chem.
Eng. J., 2024, 488, 150791 CrossRef CAS
- F. Zhou, Y. Pan, W.-Y. Hung, C.-F. Chen, K.-M. Chen, J.-L. Li, S.-M. Yiu, Y.-M. Liu, P.-T. Chou, Y. Chi and K.-C. Lau, Chem. – Eur. J., 2024, 30, e202402636 CrossRef CAS
-
(a) Y. Wu, Y. Xin, Y. Pan, S.-M. Yiu, J. Yan, K. C. Lau, L. Duan and Y. Chi, Adv. Sci., 2024, 11, 2309389 CrossRef CAS PubMed
-
(a) K. Udagawa, H. Sasabe, C. Cai and J. Kido, Adv. Mater., 2014, 26, 5062–5066 CrossRef CAS
-
(a) A. F. Henwood, A. K. Bansal, D. B. Cordes, A. M. Z. Slawin, I. D. W. Samuel and E. Zysman-Colman, J. Mater. Chem. C, 2016, 4, 3726–3737 Search PubMed
- A. Colombo, G. De Soricellis, C. Dragonetti, F. Fagnani, D. Roberto, B. Carboni, V. Guerchais, T. Roisnel, M. Cocchi, S. Fantacci, E. Radicchi and D. Marinotto, J. Mater. Chem. C, 2024, 12, 9702–9715 RSC
-
(a) L.-M. Huang, G.-M. Tu, Y. Chi, W.-Y. Hung, Y.-C. Song, M.-R. Tseng, P.-T. Chou, G.-H. Lee, K.-T. Wong, S.-H. Cheng and W.-S. Tsai, J. Mater. Chem. C, 2013, 1, 7582–7592 RSC
- P. Murto, R. Chowdhury, S. Gorgon, E. Guo, W. Zeng, B. Li, Y. Sun, H. Francis, R. H. Friend and H. Bronstein, Nat. Commun., 2023, 14, 4147 CrossRef CAS PubMed
-
(a) P. Ganesan, D.-G. Chen, W.-C. Chen, P. Gnanasekaran, J.-A. Lin, C.-Y. Huang, M.-C. Chen, C.-S. Lee, P.-T. Chou and Y. Chi, J. Mater. Chem. C, 2020, 8, 4780–4788 RSC
-
(a) D. S. M. Ravinson and M. E. Thompson, Mater. Horiz., 2020, 7, 1210–1217 Search PubMed
- J. Dosso, J. Tasseroul, F. Fasano, D. Marinelli, N. Biot, A. Fermi and D. Bonifazi, Angew. Chem., Int. Ed., 2017, 56, 4483–4487 Search PubMed
- J. Yan, Y. Pan, Z.-H. Qu, Z. Xu, K.-C. Law, D.-Y. Zhou, L.-S. Liao, Y. Chi and K.-C. Lau, Adv. Photon. Res., 2025, 6, 2400151 Search PubMed
-
(a) J. Yan, C. Wu, K.-N. Tong, F. Zhou, Y. Chen, Y. Pan, G. Xie, Y. Chi, K.-C. Lau and G. Wei, Small Methods, 2024, 8, 2301555 CrossRef CAS
-
(a) J. Jin, Z. Zhu, J. Yan, X. Zhou, C. Cao, P.-T. Chou, Y.-X. Zhang, Z. Zheng, C.-S. Lee and Y. Chi, Adv. Photon. Res., 2022, 3, 2100381 CrossRef CAS
-
(a) Z. Yang, N. Qiu, X. Dong, J. Li, P. Zou, D. Yang, D. Ma, B. Z. Tang and Z. Zhao, Adv. Opt. Mater., 2024, 12, 2302677 CrossRef CAS
-
(a) M. E. Casida, J. Mol. Struct., 2009, 914, 3–18 CrossRef CAS
-
(a) P. J. Stephens, F. J. Devlin, C. F. Chabalowski and M. J. Frisch, J. Phys. Chem., 1994, 98, 11623–11627 CrossRef CAS
-
(a) R. L. Martin, J. Chem. Phys., 2003, 118, 4775–4777 CrossRef CAS
-
(a) L. Zhan, A. Ying, Y. Qi, K. Wu, Y. Tang, Y. Tan, Y. Zou, G. Xie, S. Gong and C. Yang, Adv. Funct. Mater., 2021, 31, 2106345 CrossRef CAS
-
(a) T. Hatakeyama, K. Shiren, K. Nakajima, S. Nomura, S. Nakatsuka, K. Kinoshita, J. Ni, Y. Ono and T. Ikuta, Adv. Mater., 2016, 28, 2777–2781 Search PubMed
- C. Wu, K.-N. Tong, K. Shi, Z. Jin, Y. Wu, Y. Mu, Y. Huo, M.-C. Tang, C. Yang, H. Meng, F. Kang and G. Wei, Adv. Sci., 2023, 10, 2301112 CrossRef CAS
-
(a) J. Choi, K. Cheong, S. Han and J. Y. Lee, Adv.
Opt. Mater., 2024, 12, 2401451 CrossRef CAS
-
(a) S. Diesing, L. Zhang, E. Zysman-Colman and I. D. W. Samuel, Nature, 2024, 627, 747–753 CrossRef CAS PubMed
- J. Yan, Y. Wu, M. Huang, L. Cheng, Y. Pan, C.-C. Wu, C.-H. Yeh, J.-L. Li, Y.-D. Lin, Y. Chi, C. Yang, P.-T. Chou and K. C. Lau, Angew. Chem., Int. Ed., 2025, 64, e202424694 CrossRef CAS PubMed
Footnotes |
| † Electronic supplementary information (ESI) available: Synthetic procedures and spectroscopic data of chelates and associated intermediates, TD-DFT investigations, selected analytical data, and non-essential OLED data of the studied Ir(III) emitters. CCDC 2371311, 2371313, 2407904 and 2407906. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d5tc01314c |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2025 |