
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Contorted graphene nanoribbons from vat dyes: synthesis, properties and charge carrier mobility†
Ali
Darvish
a,
Madison
Mooney
b,
Tiago C.
Gomes
 b,
Félix
Gagnon
a,
Simon
Rondeau-Gagné
b and
Jean-François
Morin
*a
b,
Félix
Gagnon
a,
Simon
Rondeau-Gagné
b and
Jean-François
Morin
*a
aDépartement de Chimie and Centre de Recherche sur les Matériaux Avancés (CERMA), 1045 Ave de la Médecine, Université Laval, Québec, QC G1V 0A6, Canada. E-mail: jean-francois.morin@chm.ulaval.ca
bDepartment of Chemistry & Biochemistry, University of Windsor, Essex-Hall 375-1, Windsor, ON N9B 3P4, Canada
First published on 16th January 2025
Abstract
Vat dyes represent an interesting class of building blocks for organic electronics as they are readily available at low cost and easy to functionalize. Yet, these molecules have never been used to prepare graphene nanoribbons (GNRs). Using the Brønsted acid-catalyzed alkyne benzannulation, two contorted GNRs have been synthesized from vat orange 1 and vat orange 3 in a few synthetic steps. These GNRs absorb light in the visible region with bandgap values around 2.0 eV and have been tested in organic field-effect transistors (OFETs) to measure their charge transport ability. Hole mobility values of up to 1.34 × 10−2 cm2 V−1 s−1 were measured in a bottom-gate top-contact device architecture.
Introduction
Graphene has attracted lots of attention in various areas since its discovery in 2004 due to its remarkable properties such as its exceptional electrical conductivity,1,2 high thermal conductivity,3,4 mechanical strength and quantum Hall effect.5,6 However, its semi-metallic nature (zero-band gap) restricts its application as an active semiconducting component in most traditional electronic devices.7,8 One approach to overcome this issue is to confine the electron delocalization in one dimension by forming graphene nanoribbons (GNRs), which opens a bandgap whose value is correlated with the width and edge configuration of the GNRs.9–14 Over the past decade, several examples of soluble GNRs have been reported using a bottom-up approach to prepare well-defined GNRs with controlled edge structures.15–20 In most cases, these GNRs are synthesized from polymeric precursors containing small aromatic units such as benzene, naphthalene,21 and heterocycles (for doped GNRs)19 bonded together to form the GNR backbone. Consequently, when wider GNRs are desired, monomers containing several aromatic units must be prepared. This process often involves multiple synthetic steps, making the overall synthesis labor-intensive and time-consuming. A potential solution to this challenge is the use of larger aromatic units with multiple fused rings as building blocks for the monomers. In this context, polycyclic aromatic hydrocarbons (PAHs) such as pyrene,22 coronene,23 and perylene24 have been explored, leading to larger structures in fewer synthetic steps. However, examples of GNRs derived from large PAHs through solution-phase synthesis remain limited, highlighting the need for further investigation in this area.Herein, we report the synthesis, characterization and device performances of new contorted GNRs, namely 2,9-dibromodibenzo[b,def]chrysene-7,14-dione (vat orange 1) and 4,10-dibromodibenzo[def,mno]chrysene-6,12-dione (vat orange 3, Fig. 1). We have shown in a previous report that annulation of these acenoacene scaffolds lead to contorted polycyclic aromatic hydrocarbons (PAHs), due the presence of two bulky substituents in the bay region.25 Nonetheless, a significant decrease of the bandgap value was observed upon annulation, while maintaining excellent stability under ambient conditions. GNR1 and GNR2, which can be regarded as polymeric version of these contorted PAHs, were prepared using the Brønsted acid-catalyzed alkyne benzannulation reaction developed by Swager and optimized by Chalifoux and coworkers for polycyclic aromatic hydrocarbons (PAHs) and GNRs.26–28 The structural integrity of the GNRs was verified using various techniques including UV-visible absorption and nuclear magnetic resonance (NMR). The GNRs were also tested in field-effect transistors.
 | ||
| Fig. 1 Structures of GNR1 and GNR2 from vat orange 3 and vat orange 1. | ||
Results and discussion
Recently, we reported the highly regioselective alkyne benzannulation on vat orange 1 and 3 derivatives bearing alkoxy chains at the 7, 14 and 6, 12 positions, respectively (Fig. 2).25 The methanesulfonic acid (MSA) catalyzed cyclization occurs under kinetic control at the more crowded positions, close to the alkoxy chains, causing the PAH scaffold to bend significantly. The cyclization was completed in less than 10 minutes and all the starting material was consumed. No trace of other isomers from cyclization at other possible positions was detected. This high regioselectivity on both vat orange 1 and vat orange 3 derivatives upon alkyne benzannulation indicates that these could be used as building blocks to prepare regioregular GNRs. | ||
| Fig. 2 Brønsted acid-catalyzed alkyne benzannulation on vat dye derivatives showing high regioselectivity towards the most sterically hindered positions. Adapted from ref. 25. | ||
Synthesis of the GNRs
The synthesis of GNR1 and GNR2 is shown in Scheme 1. Starting from vat orange 3, a reduction-alkylation sequence was performed using a procedure previously optimized for vat orange 3.29 A branched alkyl chain was used to ensure optimal solubility of the polymeric precursors and GNRs. Then, a palladium-catalyzed debromination followed by an iridium-catalyzed borylation at the 2 and 8 positions were achieved to yield compound 7. This compound was thus engaged in a Suzuki–Miyaura polymerization reaction with compound 4 (prepared following a reported protocol for a very similar molecule).25 to provide P1 in 85% yield. It is worth noticing that the presence of methyl groups in ortho position relative to the alkyne are not necessary for the alkyne benzannulation to proceed, but it prevents side reactions during the ring closure due to steric effect, ensuring high structural fidelity of the resulting GNR.30 The polymer was thoroughly purified by precipitation in methanol, followed by washing with methanol and hexanes in a Soxhlet apparatus to removed catalyst residue and the low molecular weight fraction. Finally, P1 was treated with methanesulfonic acid (MSA) in dichloromethane using the same conditions previously optimized for vat dyes.25 Upon addition of MSA, the solution rapidly changed color, suggesting a very fast conversion of P1 to GNR1. The color of the solution remained unchanged even after the addition of an excess amount of the acid. Interestingly, no precipitation was observed upon the formation of GNR1 in dichloromethane at 0 °C. The good solubility of GNR1 can be attributed not solely to the presence of long, branched alkyl chains, but also to the inherent twisted conformation of the GNR structure that makes π-stacking less effective. After 20 minutes, the mixture was neutralized and GNR1 was recovered in 93% yield. The same synthetic sequence was used for the synthesis of GNR2 by starting with compound 8 that has been previously reported by our group.29 As GNR1, GNR2 exhibit excellent solubility in common organic solvents.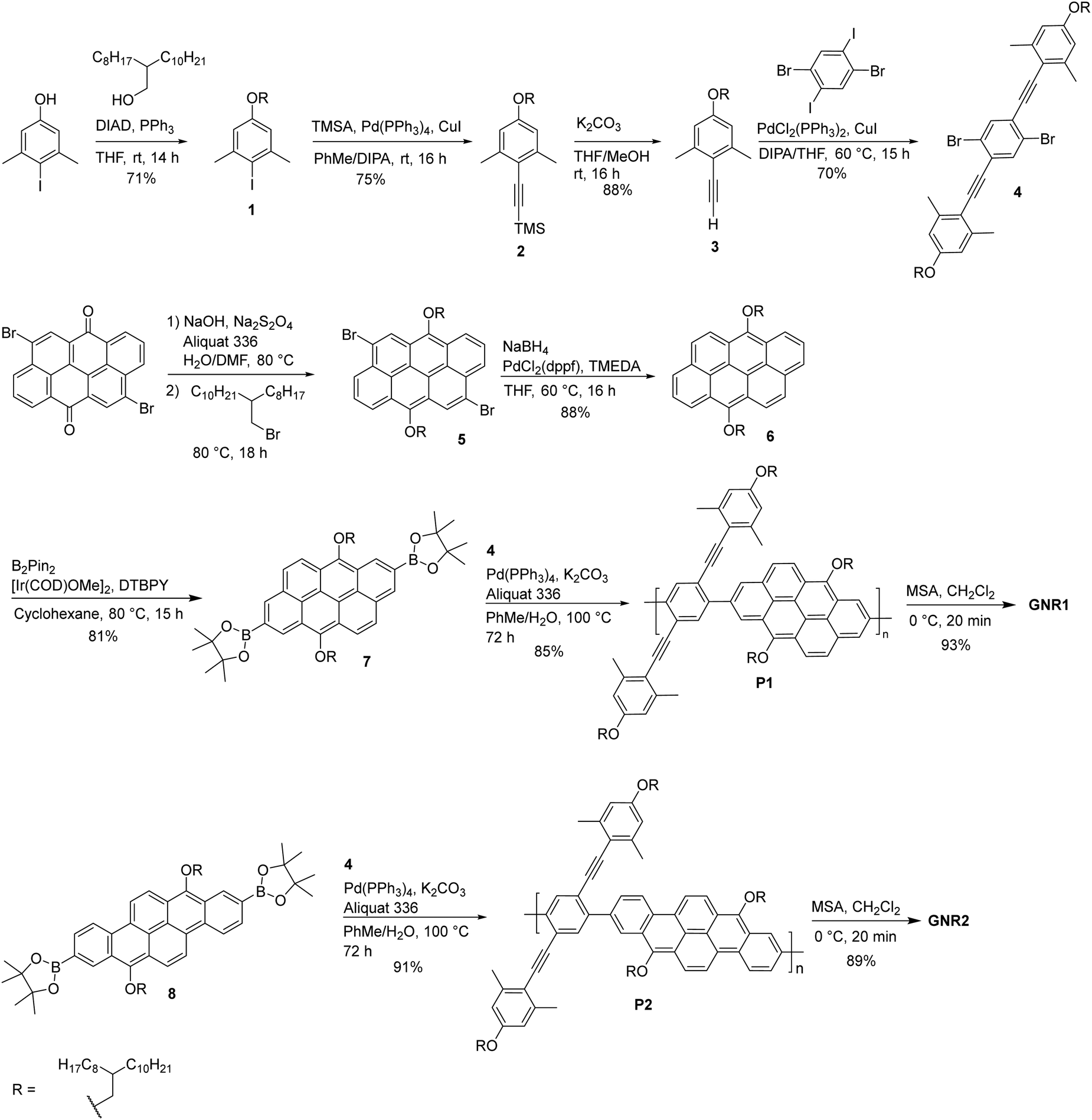 | ||
| Scheme 1 Synthesis of GNR1 and GNR2. | ||
Structural analysis and properties
1H NMR spectra of both GNR1 and GNR2 and their polymeric precursors are shown in Fig. 3. The 1H NMR analysis reveals the same patterns observed for pre- and post-cyclized model compounds (see Fig. 2). For P1, the two aromatic protons of the symmetrical pendant 2,6-dimethyl-4-methoxybenzene appear as one singlet at δ = 6.43 ppm (red dots, Fig. 3a), suggesting a freely rotating phenyl ring. Upon cyclization (Fig. 3b), this singlet splits into two distinct signals at δ = 7.11 and 6.20 ppm due to the constrained rotation of the phenyl facing a long, branched alkoxy chain in the bay region of GNR1. The complete disappearance of the peaks in the aromatic region between δ = 9.20 and 8.60 ppm along with the appearance of a new peak downfield at δ = 9.65 ppm suggest a complete benzannulation reaction. Similar observations can be made for going from P2 to GNR2 (Fig. 3c and d). | ||
| Fig. 3 1H NMR spectra of (a) P1, (b) GNR1, (c) P2 and (d) GNR2 in CDCl3. | ||
Infrared spectroscopy was performed on both GNRs polymers and their polymeric precursors to evaluate the disappearance of the band associated with the C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C bond stretching after benzannulation. The findings are presented in Fig. S17 (ESI†). As expected, the FTIR spectrum of both polymers P1 and P2 displays a peak at 2199 cm−1, whereas this peak almost completely disappears for GNR1 and GNR2, indicating an almost complete benzannulation reaction. However, our attempt to perform Raman spectroscopy was unsuccessful as we were unable to observe the spectra of the polymers and GNRs due to significant fluorescence from both GNRs.
C bond stretching after benzannulation. The findings are presented in Fig. S17 (ESI†). As expected, the FTIR spectrum of both polymers P1 and P2 displays a peak at 2199 cm−1, whereas this peak almost completely disappears for GNR1 and GNR2, indicating an almost complete benzannulation reaction. However, our attempt to perform Raman spectroscopy was unsuccessful as we were unable to observe the spectra of the polymers and GNRs due to significant fluorescence from both GNRs.
The molecular weight values (Mn and Mw) of P1, P2, GNR1 and GNR2 were determined using size exclusion chromatography (SEC), employing polystyrene standards and 1,2,4-trichlorochlorobenzene as the eluent at 110 °C. The results are presented in Table 1.
| Polymer | M n (kg mol−1) | M w (kg mol−1) | Đ (Mn/Mw) | X n |
|---|---|---|---|---|
M
n represents the number average molecular weight,  indicate the weight average molecular weight, and Đ denotes the dispersity index.a The molecular weight and Đ values for P1 and P2 were determined by size exclusion chromatography (SEC) at 135 °C. This involved utilizing 1,2,4-trichlorobenzene as the eluent and employing polystyrene as the standard for measurement. indicate the weight average molecular weight, and Đ denotes the dispersity index.a The molecular weight and Đ values for P1 and P2 were determined by size exclusion chromatography (SEC) at 135 °C. This involved utilizing 1,2,4-trichlorobenzene as the eluent and employing polystyrene as the standard for measurement. |
||||
| P1 | 23 | 59 | 2.5 | 13 |
| P2 | 27 | 86 | 3.1 | 15 |
| GNR1 | 24 | 48 | 2.0 | 13 |
| GNR2 | 27 | 81 | 3.0 | 15 |
The degree of polymerization (Xn) for both P1 and P2 are similar at 13 and 15, respectively. Interestingly, upon cyclization, the Mn values of both GNRs remain relatively the same, meaning that the rigidification of the structures did not result in a significant change in their hydrodynamic volume.
The photophysical properties of the GNRs and their polymeric precursors P1 and P2 have been determined by UV-visible and fluorescence spectroscopy and the results are summarized in Table 2 and Fig. 4. Upon benzannulation on P1 to yield GNR1, an important red-shift of 160 nm (from 470 to 630 nm) was observed, indicative of a significant extension of the effective conjugation length. Interestingly, the benzannulation on P2, which has a similar λmax value (470 and 456 nm for P1 and P2 in chloroform, respectively) did not produce such a significant red shift (44 nm). This can be explained in part by the increased lateral conjugation (larger GNR width) for GNR1 compared to GNR2. For both GNR1 and GNR2, the absorption band features a vibronic structure similar to that of their precursor, indicating a well-defined structure. Moreover, the solid-state spectrum of GNR1 and GNR2 are slightly red shift (2 nm for both) compared to the solution state, suggesting that the GNRs structure are rigid and that no significant π–π stacking is taking place in the solid state. Finally, GNR2 exhibits a featureless emission band in the visible region with a λemi value of 585 nm, while GNR1 exhibit an intense peak centered at λemi 643 nm with a shoulder at ca. 700 nm, which can be assigned as a vibronic band. The small Stokes shift value (13 and 34 nm for GNR1 and GNR2, respectively) is another indication of the rigid nature of the conjugated backbone. The photophysical properties of GNR1 and GNR2 were also compared to their model compounds (Fig. 2).25GNR1 and GNR2 are red shifted by 85 and 45 nm compared to MC1 and MC2, respectively.
λ
solmax![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) a [nm] a [nm] |
λ
filmmaxa [nm] |
λ
solonsetb [nm] |
E optical [eV] | λ Emis [nm] | ϕ F [%] | E HOMO [eV] | |
|---|---|---|---|---|---|---|---|
|
a Absorption wavelengths of the lowest energies.
b Estimated from the onset of the UV-vis spectra.
c Calculated at the λonset.
d The λmax of the emission spectra.
e Calculated according to EHOMO = −[(Eoxvs.Ag/Ag+) – (Eferrocenevs.Ag/Ag+1/2) + 4.8] from the electrochemical measurements performed in the thin fil state. All optical measurements in solution have been performed in chloroform.
f The quantum yield have been measured in CH2Cl2 using cresyl violet perchlorate (560 nm) (ϕF = 54 in MeOH) for GNR1 and rhodamine B (510 nm) (ϕF = 70 in EtOH) for GNR2 as the standards.
|
|||||||
| P1 | 470 | N/A | 490 | 2.53 | N/A | — | N/A |
| GNR1 | 630 | 632 | 655 | 1.89 | 643 | 10 | −4.67 |
| P2 | 456 | N/A | 504 | 2.46 | N/A | — | N/A |
| GNR2 | 551 | 553 | 576 | 2.15 | 585 | 33 | −4.74 |
 | ||
| Fig. 4 UV-visible absorption and emission (in chloroform for solution measurements) spectra of (a) P1 and GNR1, (b) P2 and GNR2. | ||
To get insights on the electronic properties of both GNRs, DFT calculations were performed on oligomers (two repeated units) using B3LYP/6-31+G(d,p) using Gaussian31 to determine the frontiers orbitals distribution and energy (Fig. S29, ESI†). The calculated bandgap of GNR1 (2.10 eV) is smaller than that of GNR2 one (2.34 eV), which agrees with the experimental results suggesting a higher bandgap for GNR2 than for GNR1.
Differential scanning calorimetry (DSC) and thermogravimetric analysis (TGA) were utilized to measure the thermal properties, and the corresponding results can be found in Fig. S18 and S19 (ESI†), respectively. The DSC analysis did not indicate a distinct glass transition temperature (Tg) nor a fusion transition. Consequently, it can be inferred that the GNRs are amorphous. TGA analyses were conducted under a nitrogen atmosphere at a heating rate of 10 °C per minute. The decomposition temperature (Td) values, determined at 5% weight loss, are 310 °C and 270 °C for GNR1 and GNR2, respectively.
GNRs for OFET application
GNR1 and GNR2 were used as active layers in organic field-effect transistors (OFETs) to evaluate their electronic properties. Bottom-gate top-contact devices were fabricated using solution deposition via spin-coating of GNR1 and GNR2 in toluene and 2-methyl tetrahydrofuran (2-MeTHF), with the experimental procedure detailed in the OFET device section in the ESI.† Both solvents are explored for specific reasons – toluene is a commonly used high-boiling point solvent for OFET fabrication, while 2-MeTHF offers a greener alternative with lower toxicity and environmental impact. Briefly, the polymers were dissolved in the selected solvent at a concentration of 5 mg mL−1 and stirred at 70 °C for 1 hour before spin-coating at 1000 rpm onto OTS-modified doped Si/SiO2 substrates.32 The coated substrates were then annealed at 150 °C for 30 minutes in an N2 glovebox. Gold electrodes were deposited through physical vapor deposition. Transfer and output curves were measured in a N2 glovebox for all devices, with representative curves shown in Fig. S25 and S26 (ESI†). The charge carrier mobility was extracted from the transfer curves through linear fitting of IDS1/2vs. VGS in the saturation regime using IDS(sat) = (WC/2L)μsat(VG − Vth)2. Since some of the transfer curves exhibited slight non-linearity, a slope of IDS1/2vs. VGS from approximately VGS = −15 V to −55 V was used to avoid over- or underestimation of mobilities (shown as dotted lines in Fig. S25, ESI†). The results obtained from the fabricated OFETs are summarized in Table 3.| GNR | Solvent | μ aveh/μmaxh [cm2 V−1 s−1] | I ON/IaveOFF | V aveth [V] |
|---|---|---|---|---|
| GNR1 | Toluene | 0.013 ± 0.001/0.015 | 105 | −3.9 |
| GNR1 | 2-MeTHF | 0.007 ± 0.001/0.008 | 105 | −2.6 |
| GNR2 | Toluene | 0.012 ± 0.003/0.015 | 105 | −5.7 |
| GNR2 | 2-MeTHF | 0.008 ± 0.001/0.010 | 104 | −7.2 |
The devices fabricated from GNR1 exhibited good hole mobilities, with a maximum average of 1.34 × 10−2 cm2 V−1 s−1 when processed in toluene. The polymer also demonstrated high ION/OFF (105) values and low threshold voltages. Although the average mobilities for GNR2 were slightly lower (1.22 × 10−2 cm2 V−1 s−1 in toluene) and the devices showed slightly higher threshold voltages, the performance was still comparable to that of GNR1. Interestingly, both nanoribbons showed improved performance when processed from toluene compared to 2-MeTHF, which can be attributed to the combination of a slightly lower solubility in this solvent and increased surface roughness in thin films. Nonetheless, GNR1 and GNR2 both demonstrated good solubility and film formation when processed from toluene and 2-MeTHF. This is particularly noteworthy given that graphene nanoribbons are often insoluble due to strong π–π interactions and typically require additional chemical functionalization to access solution deposition methods which can disrupt their charge transport properties.33 Given that GNRs can often only be processed in devices from chemical vapor deposition or on-surface synthesis (both costly and not easily scalable methods), this feature is particularly promising for printed electronics. It is also important to note that the favorable device characteristics obtained from thin film formation in a non-halogenated solvent like 2-MeTHF (more eco-friendly alternatives to common halogenated solvents) open new avenues toward a scalable and sustainable fabrication manufacturing of GNR-based OFETs.34,35
To gain deeper insights into the properties of GNR1 and GNR2 in thin film, atomic force microscopy (AFM) was used to evaluate the nanoscale morphology in the solid state. This technique was used not only to examine the influence of chemical design on thin film nanostructure, but also to assess the impact of the processing solvent (toluene or 2-MeTHF) on thin film properties and OFET performance. As shown in Fig. S27 and S28 (ESI†), films of both GNRs processed from toluene exhibited smooth surfaces, without large aggregates. An RMS roughness of 1.73 and 1.16 nm was measured for GNR1 and GNR2, respectively, when processed from toluene. In contrast, thin film of GNR1 prepared using 2-MeTHF displayed significantly higher roughness (RMS = 14.9 nm) and the presence of large aggregated was observed by AFM. This result can be attributed to the higher polarity and lower boiling point of 2-MeTHF, which can promote aggregation upon solution deposition. Similar observations have been reported in previous studies utilizing polar solvents for semiconducting polymers thin film formation.36,37 For GNR2, the impact of processing in 2-MeTHF on aggregation is significantly less pronounced compared to GNR1, with only a minimal increase in surface roughness. This difference can be attributed to the superior solubility of GNR2 in 2-MeTHF relative to GNR1. Overall, the increased surface roughness provides a partial explanation for the reduced OFET performance observed for both GNRs when processed from 2-MeTHF.
Conclusion
In conclusion, two twisted graphene nanoribbons (GNRs) were synthesized in a few synthetic steps from readily available, low-cost vat orange 1 and vat orange 3, utilizing a Brønsted acid-catalyzed alkyne benzannulation reaction. These highly soluble GNRs, with bandgap values around 2.0 eV, were incorporated as active layers in organic field-effect transistors (OFETs) to assess their electronic properties. Achieving hole mobilities of up to 1.34 × 10−2 cm2 V−1 s−1, these GNRs demonstrate promising potential for printed electronics. Notably, they can be processed from eco-friendly 2-MeTHF and exhibit favorable charge transport properties, highlighting their suitability for sustainable electronic device fabrication.Data availability
Additional data supporting this article have been uploaded as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
J.-F. Morin and S. R.-G. thank the Natural Sciences and Engineering Research Council of Canada (NSERC) for support through a Discovery Grants (RGPIN-2019-04215 and RGPIN-2022-04428). M. M. and F. G. thank NSERC for financial support through a Canada Postgraduate Scholarship – Doctoral (PGS-D).References
- A. K. Geim, Science, 2009, 324, 1530–1534 CrossRef.
- A. K. Geim and K. S. Novoselov, Nat. Mater., 2007, 6, 183–191 CrossRef PubMed.
- A. A. Balandin, Nat. Mater., 2011, 10, 569–581 CrossRef PubMed.
- Y. Xu, Z. Li and W. Duan, Small, 2014, 10, 2182–2199 CrossRef PubMed.
- K. S. Novoselov, Z. F. Jiang, Y. S. Zhang, S. V. Morozov, H. L. Stormer, U. Zeitler, J. C. Maan, G. S. Boebinger, P. Kim and A. K. Geim, Science, 2007, 315, 1379 CrossRef PubMed.
- Y. Zhang, Y. W. Tan, H. L. Stormer and P. Kim, Nature, 2005, 438, 201–204 CrossRef PubMed.
- L. Zhang, Y. Cao, N. S. Colella, Y. Liang, J. L. Brédas, K. N. Houk and A. L. Briseno, Acc. Chem. Res., 2015, 48, 500–509 CrossRef PubMed.
- J. Mei, Y. Diao, A. L. Appleton, L. Fang and Z. Bao, J. Am. Chem. Soc., 2013, 135, 6724–6746 CrossRef PubMed.
- D. Moran, F. Stahl, H. F. Bettinger, H. F. Schaefer and P. V. R. Schleyer, J. Am. Chem. Soc., 2003, 125, 6746–6752 CrossRef PubMed.
- K. Nakada, M. Fujita, G. Dresselhaus and M. S. Dresselhaus, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 17954 CrossRef PubMed.
- M. R. Philpott and Y. Kawazoe, J. Chem. Phys., 2011, 134, 124706 CrossRef PubMed.
- S. Fujii and T. Enoki, Acc. Chem. Res., 2013, 46, 2202–2210 CrossRef PubMed.
- T. Enoki, Y. Kobayashi and K. I. Fukui, Int. Rev. Phys. Chem., 2007, 26, 609–645 Search PubMed.
- M. Kastler, J. Schmidt, W. Pisula, D. Sebastiani and K. Müllen, J. Am. Chem. Soc., 2006, 128, 9526–9534 CrossRef PubMed.
- W. Yang, A. Lucotti, M. Tommasini and W. A. Chalifoux, J. Am. Chem. Soc., 2016, 138, 9137–9144 CrossRef PubMed.
- Y. L. Lee, F. Zhao, T. Cao, J. Ihm and S. G. Louie, Nano Lett., 2018, 18, 7247–7253 CrossRef PubMed.
- J. Liu, B. W. Li, Y. Z. Tan, A. Giannakopoulos, C. Sanchez-Sanchez, D. Beljonne, P. Ruffieux, R. Fasel, X. Feng and K. Müllen, J. Am. Chem. Soc., 2015, 137, 6097–6103 CrossRef PubMed.
- M. Daigle, D. Miao, A. Lucotti, M. Tommasini and J.-F. Morin, Angew. Chem., Int. Ed., 2017, 56, 6213–6217 CrossRef PubMed.
- D. Miao, M. Daigle, A. Lucotti, J. Boismenu-Lavoie, M. Tommasini and J.-F. Morin, Angew. Chem., Int. Ed., 2018, 57, 3588–3592 CrossRef.
- C. Tao, L. Jiao, O. V. Yazyev, Y. C. Chen, J. Feng, X. Zhang, R. B. Capaz, J. M. Tour, A. Zettl, S. G. Louie, H. Dai and M. F. Crommie, Nat. Phys., 2011, 7, 616–620 Search PubMed.
- A. Keerthi, C. Sánchez-Sánchez, O. Deniz, P. Ruffieux, D. Schollmeyer, X. Feng, A. Narita, R. Fasel and K. Müllen, Chem. – Asian J., 2020, 15, 3807–3811 CrossRef PubMed.
- S. Obermann, W. Zheng, J. Melidonie, S. Böckmann, S. Osella, N. Arisnabarreta, L. A. Guerrero-León, F. Hennersdorf, D. Beljonne, J. J. Weigand and M. Bonn, Chem. Sci., 2023, 14, 8607–8614 RSC.
- Y. Kinno, H. Omachi and H. Shinohara, Appl. Phys. Express, 2019, 13, 015002 CrossRef.
- S. R. Peurifoy, Q. Xu, R. May, N. A. Gadjieva, T. J. Sisto, Z. Jin, L. E. Marbella and C. Nuckolls, Chem. Sci., 2020, 11, 9978–9982 RSC.
- A. Darvish, F. Lirette, I. Fernández and J.-F. Morin, Chem. – Eur. J., 2024, e202403456 Search PubMed.
- W. Yang and W. A. Chalifoux, Synlett, 2017, 625–632 CAS.
- M. B. Goldfinger and T. M. Swager, J. Am. Chem. Soc., 1994, 116, 7895–7896 CrossRef CAS.
- M. B. Goldfinger, K. B. Crawford and T. M. Swager, J. Am. Chem. Soc., 1997, 119, 4578–4593 CrossRef CAS.
- F. Gagnon, V. Tremblay, A. Soldera, M. U. Ocheje, S. Rondeau-Gagné, M. Leclerc and J.-F. Morin, Mater. Adv., 2022, 3, 599–603 RSC.
- W. Zheng, T. Ikai and E. Yashima, Angew. Chem., Int. Ed., 2021, 133, 11394–11399 CrossRef.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, L. Williams, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 09, Revision D.01, Gaussian Inc., Wallingford, CT, 2016 Search PubMed.
- Y. Ito, A. A. Virkar, S. Mannsfeld, H. O. Joon, M. Toney, J. Locklin and Z. Bao, J. Am. Chem. Soc., 2009, 131, 9396–9404 CrossRef CAS.
- B. Genorio and A. Znidarsic, J. Phys. D: Appl. Phys., 2014, 47, 094012 CrossRef CAS.
- C. M. Alder, J. D. Hayler, R. K. Henderson, A. M. Redman, L. Shukla, L. E. Shuster and H. F. Sneddon, Green Chem., 2016, 18, 3879–3890 RSC.
- C. S. Slater, M. J. Savelski, D. Hitchcock and E. J. Cavanagh, J. Environ. Sci. Health, Part A, 2016, 51, 487–494 CrossRef CAS.
- M. Mooney, Y. Wang, A. Nyayachavadi, S. Zhang, X. Gu and S. Rondeau-Gagné, ACS App. Mater. Interfaces., 2021, 13, 25175–25185 CrossRef CAS PubMed.
- M. Mooney, A. Nyayachavadi, A. Awada, E. Iakovidis, Y. Wang, M. N. Chen, Y. Liu, J. Xu, Y. C. Chiu, X. Gu and S. Rondeau-Gagné, Polym. Chem., 2023, 14, 562–572 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Details of experimental procedures, 1H NMR, 13C NMR, UV-visible spectroscopy, conditions for each parameter of dispersion, IR spectroscopy, AFM, and DFT calculations (PDF). See DOI: https://doi.org/10.1039/d4tc05043f |
| This journal is © The Royal Society of Chemistry 2025 |