
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEco-designed ZSM-5 zeolites: biomass-assisted modifications and catalytic evaluation through model reactions
Camille
Longue
 ab,
Anne
Bolmont
b,
Valérie
Ruaux
a,
Aurélie
Vicente
a,
Nourrdine
Chaouati
a,
Marie
Desmurs
a,
Benoît
Louis
*b and
Ludovic
Pinard
*a
ab,
Anne
Bolmont
b,
Valérie
Ruaux
a,
Aurélie
Vicente
a,
Nourrdine
Chaouati
a,
Marie
Desmurs
a,
Benoît
Louis
*b and
Ludovic
Pinard
*a
aUniversité de Caen Normandie, ENSICAEN, CNRS, LCS, 14000 Caen, France. E-mail: ludovic.pinard@ensicaen.fr
bInstitut de Chimie et Procédés pour l'Energie, l'Environnement et la Santé (ICPEES) – UMR 7515, Université de Strasbourg, ECPM, CNRS, 67087 Strasbourg Cedex 2, France. E-mail: blouis@unistra.fr
First published on 24th March 2025
Abstract
This study evaluates the impact of biomass addition on the physicochemical properties of ZSM-5 zeolites. Three families of zeolites were synthesized hydrothermally: a reference zeolite without biomass, one with lignin, and another combining lignin and sugarcane bagasse. Biomass has been shown to modify the zeolite structure by reducing the crystal size, favouring aluminium incorporation within the framework and reducing the number of defects as internal silanols. These modifications are attributed to the chemical interactions between biomass and inorganic precursors present in solution. The catalytic performance of these zeolites was analysed in n-hexane cracking and in the methanol to olefin (MTO) reactions. Zeolites synthesized with biomass demonstrated improved catalytic stability and selectivity towards light olefins, thanks to an enhanced diffusion path. Lignin, in particular, helped minimize structural defects, thus improving the catalyst lifetime. The addition of biomass offers significant advantages for tailoring zeolite properties while using renewable and abundant resources. This innovative approach opens up interesting prospects for the sustainable design of catalytic materials. It also enables agricultural and industrial wastes to be recycled into high value-added applications, strengthening the links between green chemistry and industrial performance.
Sustainability spotlightZeolites are by far the most important catalysts used in refineries, particularly in the fluid catalytic cracking (FCC) process. It is not exaggerating to state that not a single drop of gasoline has been produced by a means other than the FCC process. Unfortunately, with growing fossil fuel utilization and industrial activities, along with a world population increase above 7 billion in 2022, the amount of greenhouse CO2 emission is continuously increasing, leading to severe climate change issues. A new paradigm therefore needs to be set with the emergence of alternative energy resources. In the energy context of the 21st century, these amazing microporous materials are still leaders for performing acid-catalyzed reactions. Biomass residues such as lignin or sugar cane bagasse were used to set a sustainable route to produce ZSM-5 zeolites. In addition, it has been shown that both textural and chemical properties of these “bio-sourced” materials could be tuned (almost) at will to design efficient catalysts, which are themselves applied in green processes to produce either key building blocks of the chemical industry or fuels. Our work emphasizes the importance of the following UN sustainable development goals: affordable and clean energy (SDG 7), industry, innovation, and infrastructure (SDG 9), and climate action (SDG 13). |
1. Introduction
For millennia, nature has designed fascinating mineral structures with remarkable properties, whilst only recently mankind has begun to exploit their potential.1–4 Zeolites are solid microporous crystalline materials composed of silicon and aluminium atoms generating a 3D framework.5 Their unique structure and physico-chemical properties make them essential materials for numerous applications (catalysis, fertilisers, water purification, etc.).6–9 Indeed, under appropriate synthesis conditions, these properties can be customised ad infinitum thanks to the large number of parameters that can be adjusted during the synthesis step making zeolites particularly attractive.10,11 One of them, Zeolite Socony Mobil-5 (ZSM-5), emerged several years ago as a promising material for catalysis in both existing and emerging technologies, thanks to its indisputable shape selectivity.12–14ZSM-5 exhibits a MFI framework, with a Si/Al ratio ranging from 12 to infinity.15 The MFI structure was first synthesized by Argauer and Landolt in 1972.16 They used an organic structure-directing agent (OSDA), tetrapropylammonium (TPA+), with hydroxide and bromide as counterions. ZSM-5 features a ten-membered ring (10-MR) structure. It forms straight channels with medium pore sizes (0.53–0.56 nm), intersecting with sinusoidal channels (0.51–0.55 nm).17,18 ZSM-5 has two types of acid sites: Brønsted and Lewis.5 Brønsted acid sites (BASs) are typically represented by acidic hydroxyl groups (–O–Al–O(H)–Si–O–) within the zeolite, while Lewis acid sites (LASs) function as electron-pair acceptors. The discovery of this first synthesis strategy led to the creation of many others depending on the application: seed-assisted, hierarchical, template-free, metal incorporation, solvent-free, and microwave-assisted syntheses and have been largely described in the literature.11,19,20
In 2010, the Louis group published a new strategy for preparing ZSM-5 zeolites.21 The shape and physico-chemical properties could be modified by adding biomass, in particular sugarcane bagasse (SCB), directly to the synthesis gel.
Though covalent bonding prevails in inorganic porous solids, the zeolite organization at the molecular level involves several species brought in contact by non-covalent interactions.22–25 The formation of the zeolite crystalline structures is therefore governed by electrostatic, van der Waals and hydrophobic forces known as supramolecular interactions. In spite of being considered as ‘hard matter’ themselves, internal voids present in zeolites remain ‘soft matter’ and a parallel can be drawn with phenomena found in Nature.
The rationale behind the use of biomass or its extracted molecules in the zeolite synthesis medium was to perturb the whole system and to impact the inorganic precursor organization. This led us to define the Bio-Sourced Secondary Template (BSST) concept, mainly focusing on MFI zeolite synthesis.22–28
Afterward, other syntheses were developed by our team involving other types of biomass, such as lignin or vanillin, to synthesize other zeolite topologies such as GIS, *BEA or LTA.22,27–30 A mechanism based on biomass acting as a bio-sourced secondary template (BSST) was suggested to explain previously unseen levels of aluminium, resulting in extremely low Si/Al, and yielding ZSM-5 materials.24 In addition, the BSST strategy allowed generating mesoporosity and sometimes led to unconventional crystal morphologies. It would also provide interesting results in acid catalysis. Moreover, lignin and sugarcane bagasse, for example, are abundant resources on Earth, since they are mainly derived from by-products and/or waste from the paper industry and also the manufacture of sugar and biofuels.31–34 They are therefore valuable materials. Flores et al. recently reported a protocol using rice husk as a source of silicon for the synthesis of chabazite-K and merlinoite zeolites.35 Li et al. prepared hierarchical ZSM-5 using natural cellulose as a template with different preparation methods.36 Several syntheses involving biomass are listed in the literature and are based on the recovery of agricultural waste.37–39
In this work, ZSM-5 zeolites have been obtained using lignin and a mixture of lignin and sugarcane bagasse, with higher Si/Al ratios than those commonly observed in the literature for this type of biomass. The resulting zeolites were compared with pristine ZSM-5 (referred to as the reference) synthesized without biomass. The aim is to see whether the syntheses are reproducible at higher Si/Al ratios, approximately 45, and investigate the impact of biomass on the final material properties. What about aluminium incorporation and siting, and crystal formation? The observed effects were highlighted by several characterization techniques and catalytic tests with model reactions such as n-hexane cracking and MTO.
2. Experimental
2.1 ZSM-5 synthesis
ZSM-5 zeolites were synthesized by the hydrothermal method using the following chemicals without further purification or treatment: sodium aluminate (NaAlO2, Al2O3 50–56%, Sigma-Aldrich), sodium chloride (NaCl, 99%, Sigma-Aldrich), tetrapropylammonium hydroxide (TPAOH, 40%, Sigma-Aldrich), tetraethylorthosilicate (TEOS, 99%, Sigma-Aldrich), ammonium nitrate (NH4NO3, 98%, Sigma-Aldrich), kraft lignin (Borregaard, Norway) and sugarcane bagasse in natura (Brazil). The initial synthesis gel composition was 1.0 SiO2: 0.01 Al2O3: 0.37 NaCl: 0.30 TPAOH: 62 H2O. From this gel, three samples were synthesized: (i) the designated reference (R) or by adding either (ii) 600 mg of kraft lignin (L) or (iii) an equimassic mixture of lignin/sugarcane bagasse (300 mg/300 mg) (L + S). NaAlO2 (0.094 g), NaCl (1.16 g), TPAOH (8.18 g) and distilled water were mixed together until a clear solution was obtained. Then, TEOS (11.2 g) and/or L (600 mg) or L + S (300 mg/300 mg) were added to the previous solution. The gel was aged for 3 h under vigorous stirring at room temperature. The final pH was 12. The gel was placed inside a 100 mL Teflon-lined autoclave and the synthesis was performed at 443 K for 24 h. After crystallization, the solid was recovered by filtration, washed with distilled water until a neutral pH was reached and dried overnight at 373 K. The obtained crystals in a sodium form were then calcined under air with a temperature ramp of 2 K min−1 at 823 K for 5 h to remove organics.ZSM-5 zeolites were ion-exchanged to yield the acidic form. Two successive exchange steps were carried out using an aqueous solution of 2 M NH4NO3 (1 g of zeolite per 50 mL) for 1 h at 353 K followed by filtration and drying each time. Then, calcination under air was performed with a temperature ramp of 2 K min−1 at 723 K for 4 h to remove ammonia and yield the H form.
The synthesis yield, based on silicon, was calculated using the following formula:
 | (1) |
2.2 Characterization
CHN analysis was carried out using a Thermo Fisher Scientific Flash 2000 for biomass nature, coke nature and total carbon percentage.Scanning electron microscopy (SEM) images were taken with a Zeiss Gemini SEM 500 microscope working at 9 kV accelerating voltage and equipped with an energy dispersive X-ray spectrometer (EDX) for bulk elemental analyses. Prior to the observation, the samples were placed on aluminium brackets with carbon tape and metallized with gold. The images were used to access particle size with the help of Image J software.
In situ ATR-FTIR measurements were performed for biomass analysis. Infrared spectra were recorded on a Bruker IFS 66v/S FTIR spectrometer equipped with a deuterated triglycine sulfate (DTGS) detector (4 cm−1 resolution).
Powder X-ray diffraction (XRD) patterns were recorded on a Bruker D8 Advance diffractometer operated at 40 kV and 20 mA and equipped with an energy dispersive detector using a monochromatic CuKα radiation source (λ = 1.5418 Å). The patterns were collected in the 2θ = 5–55° range using a step size of 0.02° and an acquisition time of 0.5 s. The relative crystallinity (RC) was estimated from the peak area in the 2θ range from 22 to 25°.40 The relative crystallinity of the L + S zeolite was arbitrarily set to 100%.
N2 adsorption–desorption isotherms were measured using a Micromeritics ASAP 2420 apparatus at 77 K. Prior to the analysis, the samples were degassed under vacuum (10 mHg) for 1 h at 363 K and then for 4 h at 623 K. The total pore volume (Vtot) was calculated at P/P0 = 0.98 while the external surface area (Sext) and the micropore volume (Vmicro) were estimated by the t-plot method. The Harkins–Jura equation and the thickness range between 5 and 6 Å were used. The mesopore volume (Vmeso) was calculated by subtracting Vmicro from Vtot. Density functional theory (DFT) calculations were used to draw the pore size distribution.41
Inductively coupled plasma mass spectrometry (ICP-MS) elemental analysis was carried out using an Agilent 7900 ICP-MS. Prior to the analysis, all samples (50 mg) were dissolved in an acid medium composed of a mixture of hydrofluoric acid and aqua regia (6![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 vol. ratio) for 1 h at 383 K followed by neutralization with boric acid (2 g) and Milli-Q water (10 mL) and stirred overnight. Then, the samples were diluted up to 100 mL and filtered.
:1 vol. ratio) for 1 h at 383 K followed by neutralization with boric acid (2 g) and Milli-Q water (10 mL) and stirred overnight. Then, the samples were diluted up to 100 mL and filtered.
Solid state magic-angle spinning nuclear magnetic resonance (MAS NMR) spectra of 27Al and 29Si nuclei and {1H}29Si cross-polarisation (CP) were recorded with a single pulse on a Bruker Avance 500 MHz (11.7 T) spectrometer using 4 mm outer diameter zirconia rotors and a spinning frequency of 12 kHz. Tetramethylsilane (TMS) was used as a reference for the 29Si chemical shift and aluminium nitrate solution (Al(NO3)3, 0.1 M) was used as a reference for the 27Al chemical shift. The Si/Al ratio of the framework (Si/AlF) was calculated using the peak areas obtained after decomposition of the spectra and using the DMfit software. The following formula was used:
 | (2) |
An in situ home-made FT-IR cell called “Pelicaen” recently developed in the LCS laboratory for adsorption experiments was used. The spectroscopic analyses were performed with pyridine (Py) as a base probe molecule using a self-supported pellet (∼20 mg, 2 cm2) of the zeolite sample. FT-IR spectra were collected on a ThermoScientific Nicolet Magna FTIR iS50 spectrometer equipped with a mercury, cadmium, and tellurium (MCT) detector (4 cm−1 resolution). The samples were first activated by heating at 623 K for 4 h under high vacuum and the background spectra were collected. Pyridine vapor was next introduced after cooling the cell at 423 K for 15 min adsorption. Then, 15 min desorption took place. All spectra were recorded at room temperature and normalized to the area and mass of the pellet. The concentration of Brønsted (PyH+) and Lewis (PyL) acid sites was calculated from the band areas of adsorbed Py at 1545 cm−1 and 1455 cm−1, respectively, using the Beer–Lambert–Bouguer law. Molar extinction coefficients of 1.13 cm μmol−1 and 1.28 cm μmol−1 for Brønsted and Lewis acid sites were used, respectively.43
2.3 Catalytic tests
The n-hexane cracking catalytic tests were performed in four parallel fixed-bed reactors under atmospheric pressure at 813 K. Different amounts of the same catalyst, with a particle size range of 0.2–0.4 mm, were loaded into each reactor (20, 40, 60, and 80 mg) and subjected to a pre-treatment at 813 K under a nitrogen flow for 12 h. n-Hexane (99.99%, Sigma-Aldrich) was then diluted in a nitrogen flow and introduced into the reactors at the reaction temperature, with a N2/C6H14 molar ratio of 11. The activity of each zeolite was evaluated by determining the conversion while adjusting the weight hourly space velocity (WHSV). The reaction products were analyzed online using a GC450 gas chromatography system equipped with a Cp-Al2O3/Na2SO4 capillary column (50 m, 10 μm) coupled with an FID detector. The turnover frequency (TOF) was estimated as follows: | (3) |
The MTO catalytic tests were performed in a fixed bed reactor under atmospheric pressure at 723 K. First, each catalyst was shaped into particles with size in the range of 0.2–0.4 mm and 0.1 g was packed in a tubular quartz reactor (7 mm internal diameter) between two quartz wool plugs. Then, the catalyst was pre-treated with a heating ramp of 5 K min−1 at 823 K under a nitrogen flow of 55 mL min−1 for 1 h. It was cooled next to the reaction temperature. The MTO reaction was carried out at a WHSV of 2.1 gMeOH gcat−1 h−1 (STP) with liquid methanol (0.0044 mL min−1), instantaneously vaporised at the reactor inlet and diluted in a continuous flow of nitrogen (50 mL min−1). The N2/MeOH molar ratio was 21. The gas composition at the reactor outlet was analyzed using an online gas chromatograph (Chromatec, Crystal 9000) equipped with an FID detector (PONA column). Conversion (X) and selectivity (S) have been calculated according to (4) and (5), respectively. Dimethylether (DME) is a condensation product of methanol (MeOH) and is thus considered a reactant.
 | (4) |
 | (5) |
3. Results and discussion
3.1 Biomass characterization
The chemical composition and particle size of lignin and sugarcane bagasse are presented in Table 1. The H/C ratios are not the same for the two types of biomass. Lignin has a lower H/C ratio because it is a highly branched carbon-rich biopolymer (aromatic, phenolic and hydrocarbon groups).31 Indeed, it is composed of three main types of monomers: p-coumaryl alcohol (H unit), coniferyl alcohol (G unit) and sinapyl alcohol (S unit), with an average H/C ratio of 1.19. In contrast, SCB has a higher H/C ratio, as it is mainly composed of cellulose and hemicellulose polysaccharides.34 In general, this fibrous biomass contains 35–50% cellulose, 20–25% hemicellulose and 15–25% lignin.44 The expected H/C ratio is therefore often lower than that expected from sugars (H/C ∼ 2), as this biomass is not solely composed of sugars. In terms of elemental analysis, carbon and oxygen are present in majority, regardless of the type of biomass. However, lignin shows traces of sodium and sulfur in compliance with standards, as it is a kraft lignin from the sulfate pulping process.45The morphological properties of the biomass are shown in Fig. 1. Particle sizes were measured for lignin, which is defined by smooth and hollow spheres (Fig. 1(a)). The average internal diameters of these spheres measure 7 μm (Table 1 and Fig. 1(b)). SCB is instead defined by an irregular and fractured surface (Fig. 1(d)). In addition, randomly distributed holes on the biomass surface are visible in the SEM images. Their average sizes are indicated as 0.1 μm for lignin and 0.8 μm for SCB (Table 1 and Fig. 1(c, e)).
 | ||
| Fig. 1 SEM images of lignin (a) and sugarcane bagasse (d) with corresponding particle size distribution (b) and distribution of hole sizes (c) in lignin and (e) in SCB. | ||
The chemical and structural differences in the biomass are shown in Fig. 2, which illustrates the complexity of the biomass and the contributions of lignin, cellulose and hemicellulose for each sample. The two ATR-FTIR spectra in Fig. 2(a and b) show a dominant peak in the 3400 cm−1 region, corresponding to valence vibrations of hydroxyl groups, typical of polysaccharides such as cellulose in SCB, phenolic groups in lignin or physically adsorbed water.44,46 However, the peak observed in lignin at 3396 cm−1 is less intense than that in SCB, at 3334 cm−1, suggesting a greater abundance of hydroxyl groups in SCB, probably due to its high cellulose and hemicellulose content. Both spectra also show bands around 2900 cm−1 linked to vibrations of aliphatic methyl groups.47 However, lignin shows an additional band at 2836 cm−1, attributed to the vibrations of methyl functions involved in methoxy bonds, characteristic of the G and S units of its structure.47 This peak is not observed in SCB, reflecting the difference in chemical composition between these two types of biomass: SCB is rich in cellulose and hemicellulose, whereas lignin contains a high proportion of phenolic and methoxy units. Bands around 1605 cm−1 and 1510 cm−1 in both spectra are attributed to aromatic ring vibrations, but are more pronounced in lignin, consistent with its aromatic-rich structure.48 In SCB, these bands are present but exhibit a lower intensity, reflecting the lower lignin content in this biomass. A prominent band at 1725 cm−1 is observed in the SCB spectrum, corresponding to the vibrations of carbonyl groups, potentially associated with esters or acetyl groups in hemicellulose.48 This band is absent or very weak in lignin, indicating that carbonyl groups are less abundant in its structure. In the regions of 1455 cm−1 and 1420 cm−1, both types of biomass exhibit bands associated with methyl group deformations, although their intensity is slightly stronger in lignin.48 In contrast, SCB shows an additional peak around 1367 cm−1, which is linked to another methyl group deformation, more specific to the polysaccharide compounds cellulose and hemicellulose.48 A distinct peak at 1320 cm−1 is observed in both spectra, but is attributed to different functional groups in each biomass. In lignin, this peak may be linked to the vibration of a C–O group in a S unit, while in SCB it is associated with a C–H vibration in cellulose.48 In addition, a peak at 1240 cm−1 can be seen in SCB (Fig. 2(b)), associated with S unit ring vibrations of lignin and the stretching of carbonyl groups in lignin and hemicellulose.46 A peak at 1259 cm−1 is observed in lignin in Fig. 2(a), corresponding to the C–O stretching in lignin and to the C–O bonds in the aromatic G units of the methoxy groups.46 Finally, both types of biomass show significant bands around 1160 cm−1 and 1030 cm−1. The first may correspond to C–O–C vibrations in cellulose and hemicellulose, as well as to in-plane C–H deformation of a lignin G unit, and may also be associated with secondary alcohols.46 The second band can be attributed to in-plane C–H deformation of aromatic compounds and C–O deformation of primary alcohols.46 However, these bands are more pronounced in SCB, confirming its higher cellulose and hemicellulose contents with respect to lignin.
 | ||
| Fig. 2 ATR-FTIR spectra of lignin (a) and SCB (b). | ||
Biomass was characterized by several analytical techniques in an attempt to better define its role in the zeolite syntheses.
3.2 Zeolite characterization
XRD patterns of as-prepared R, L and L + S zeolites are presented in Fig. 3. All three zeolites exhibit characteristic patterns of a MFI type structure, confirming the formation of ZSM-5.49 | ||
| Fig. 3 XRD patterns of R, L and L + S zeolites. | ||
Table 2 shows the physico-chemical properties of the three synthesized zeolites. The synthesis yields range between 75 and 79%, representing high yields frequently reported in the literature.50,51 Furthermore, the relative crystallinities show a high crystallization rate, over 90%. These results can also be corroborated by the micropore volumes of the samples, which vary between 0.16 and 0.17 cm3 g−1, confirming that these are well-crystallized MFI-type zeolites.52 The external surfaces of all samples remain relatively small due to the large size of the crystals, while the mesoporous volumes are almost negligible (≤0.07 cm3 g−1). Indeed, reference R exhibits crystals of significant size, measuring around 11.3 μm, whilst samples L and L + S show smaller crystals, with average dimensions of 6.1 μm and 6.3 μm, respectively.
| Units | R | L | L + S | |
|---|---|---|---|---|
| a Based-on Si. b Calculated according to ref. 40. c Estimated by the t-plot method. d V meso = Vtot − Vmicro (Vtot: adsorbed volume at P/P0 = 0.98). e Obtained from SEM images. f Determined by ICP-MS analysis. g Calculated by 29Si MAS NMR. h Estimated by 27Al MAS NMR. i Concentration of Brønsted [H+] and Lewis [L] acid sites probed by thermodesorption of pyridine at 423 K. j Extracted from n-hexane cracking catalytic tests. | ||||
| Synthesis yielda | % | 76 | 75 | 79 |
| RCb | % | 92.6 | 92.0 | 100 |
| S ext | m2 g−1 | 74 | 56 | 53 |
| V micro | cm3 g−1 | 0.16 | 0.16 | 0.17 |
| V meso | cm3 g−1 | 0.07 | 0.07 | 0.06 |
| Particle sizee | μm | 11.3 | 6.1 | 6.3 |
| Si/Alf | — | 45 | 41 | 46 |
| Si/AlFg | — | 31.2 | 53.8 | 35.1 |
| FAl (δ = 54 ppm)h | % | 95.2 | 95.8 | 98.0 |
| EFAl (δ = 0 ppm)h | % | 4.8 | 4.2 | 2.0 |
| [PyH+]i | μmol gcat−1 | 257 | 251 | 272 |
| [PyL]i | μmol gcat−1 | 26 | 16 | 13 |
| [Al]theof | μmol gcat−1 | 287 | 307 | 272 |
| TOFj | h−1 | 250 | 264 | 204 |
| A 0 | mmol h−1 gcat−1 | 64 | 66 | 56 |
| P/Oj | mol mol−1 | 0.54 | 0.52 | 0.61 |
Bernardon et al.53 showed that ZSM-5 crystals obtained with SCB led to nearly the same Si/Al ratio of 47. However, larger spherical crystals of 7–8 μm were formed by an aggregation of 300–400 nm rectangular building blocks.53 It is worth mentioning that a correlation could neither be established between the size of the SCB particles nor the cellulose/hemi-cellulose solvated species.
In contrast, A link can be made between the average internal diameter of the lignin spheres of 7 μm (Table 1) and the average sizes of the formed crystals for zeolites L and L + S (Table 2).
Fig. 4 presents a tentative model for ZSM-5 crystal growth inside lignin spheres. These spheres may act as micro-autoclaves and can limit and control the size of the crystals. The strong alkaline medium induces the formation of negatively charged aromatic molecules, yielding hydrophobic interactions. The aromatic species released in solution may be stabilized via π-stacking interactions, thereby preventing Ostwald ripening.24
 | ||
| Fig. 4 Proposed zeolite crystal growth model based on a self-assembly mechanism inside lignin spheres. | ||
Fig. 5 shows the textural analysis of the synthesized zeolites. Fig. 5(a) shows typical type I isotherms for microporous materials, with a slight presence of mesopores of around 3 nm for all samples (Fig. 5(b)). These small mesopores can be attributed to the intercrystalline pores between crystallites.
 | ||
| Fig. 5 N2 physisorption isotherms (a) and micropore size distributions derived from the adsorption branch (b) of R, L and L + S zeolites. | ||
Fig. 6 shows the morphology and particle size distribution of R, L and L + S zeolites. The R reference is in the form of non-regular, rough spherical particles (Fig. 6(a)), whereas L and L + S zeolites are in a more regular spherical form that appears smoother. Furthermore, the histograms of particle size distribution show a more homogeneous size distribution in the case of zeolites prepared with biomass (Fig. 6(e and f)) than in the case of the reference (Fig. 6(d)). Biomass could therefore be a tool for controlling particle size.
 | ||
| Fig. 6 SEM images of (a) R, (b) L and (c) L + S zeolites with associated particle size distribution in (d–f) histograms. | ||
ICP-MS and NMR analyses were used to identify the composition and chemical environment of the as-prepared zeolites (Table 2). To complete these data, 27Al, 29Si and 1H–29Si cross-polarization MAS NMR spectra are grouped together in Fig. 7. The total Si/Al ratios ranged from 41 to 46 and were as expected since they correspond approximately to the Si/Al ratios initially present in the synthesis gel for the three zeolites (Table 2). In addition, the framework Si/Al ratios (Si/AlF) could be calculated from the signal intensities in the 29Si MAS NMR spectra shown in Fig. 7(b). Several signals of varying intensity were detected between −100 and −120 ppm. The signals positioned between −100 and −108 ppm correspond to Q4(1Al) and Q3 sites, i.e. Si(3Si, 1Al) and Si(3Si, 1OH) species, respectively, with similar resonance positions.54 Moreover, the presence of silanol species in the zeolites could be assessed using the non-quantitative {1H}29Si CP MAS NMR analysis shown in Fig. 7(c). The other signals located between −108 and −120 ppm are linked to Q4(0Al) sites, i.e. Si(0Al) species exhibiting the same environment but with different tetrahedral T positions in the zeolite structure.55 The three Si/AlF ratios obtained are not the same for the three zeolites. L has a higher ratio of 53.8 than the result from ICP-MS analysis, while R and L + S have lower ratios of 31.2 and 35.1 to the total Si/Al ratio. In the case of L, the Si/AlF ratio may be greater because 4.2% of the aluminium atoms are in the form of extra-framework aluminium (EFAl) species. The EFAl species are visible in the 27Al MAS NMR spectrum in Fig. 7(a) at 0 ppm and could be quantified by an additional integration of the signal at about 54 ppm representing the framework aluminium (FAl) atoms linked to the structure in the tetrahedral position. For the other two zeolites, R and L + S, the Si/AlF values may have been overestimated since the EFAl contents were 4.8% and 2.0%, respectively, equivalent to that of L. This may be explained by the fact that the peaks of the Q4Si(1Al) and Q3 sites overlap and may result in an overestimation of this signal.56 This hypothesis is even more likely in the case of R, whose {1H}29Si CP MAS NMR analysis, shown in Fig. 7(c), revealed the existence of Q3 silanol groups, i.e. defects such as terminal silanols.57 According to these results, the biomass addition during the synthesis appears to slightly reduce the formation of EFAl and silanol defects.
 | ||
| Fig. 7 27Al (a), 29Si (b) and {1H}29Si CP MAS NMR spectra (c) of R, L and L + S synthesized zeolites. | ||
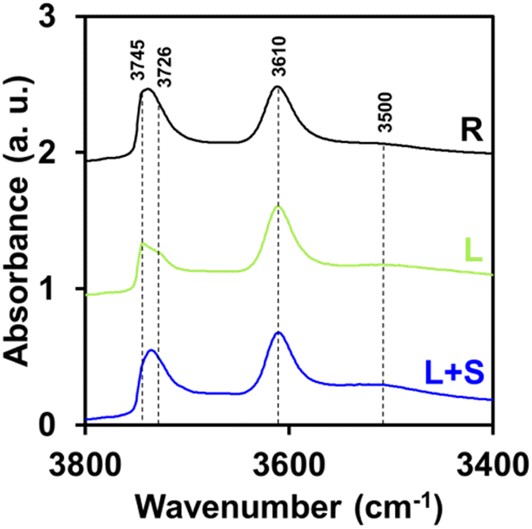
Fig. 8 shows the infrared spectra of the three zeolites R, L and L + S in the region of stretching vibrations of hydroxyl groups. All three materials exhibit characteristic bands of MFI zeolites. From left to right, the bands at 3745 and 3726 cm−1 correspond to isolated silanols present on the outer surface and isolated terminal silanols internal to the zeolite structure, respectively.58 The band at 3610 cm−1 corresponds to bridged hydroxyls, i.e. BAS.58 The weak band at around 3500 cm−1 corresponds to internal silanol nest groups.58 No signal was detected around 3660 cm−1 corresponding to EFAl-OH. This means that the EFAl species detected in the 27Al MAS NMR spectrum in Fig. 7(a) may exist in another form, i.e. a neutral or cationic form.59 For silanols located between 3745 and 3726 cm−1, R and L + S zeolites have stronger signals than the L zeolite. Moreover, the L zeolite appears to have fewer internal defects than the other two zeolites. Barbera et al. showed that in general the proportion of internal silanols was uniformly distributed in a zeolite, whilst the proportion of silanols on the external surface varies proportionally with the size of the external surface and therefore the crystal size.60 In this case, L and L + S have half the crystal size of R but L + S still appears to have band intensities close to those of R. The syntheses were carried out in a strongly alkaline environment (11 < pH < 13). The pH of the synthesis medium plays a key role in the formation of silanols within the zeolites.61 In a highly alkaline environment (pH > 10), the partial dissolution of silicate precursors favours the creation of silanol nests, as well as an increase in internal and external silanol groups, due to increased disruption of the crystal lattice. Conversely, under more neutral or slightly acidic conditions, crystallization occurs in a more orderly process, limiting not only the formation of silanol nests, but also that of internal and external silanols. The slight variations in pH in a strongly alkaline environment during synthesis could therefore explain the differences observed in the content and distribution of silanols in the materials.61
 | ||
| Fig. 8 FT-IR spectra of R, L and L + S zeolites in the OH-stretching region after activation at 623 K. | ||
Pyridine adsorption measurements followed by FT-IR were then performed to investigate the acidic properties of the three ZSM-5 zeolites. As shown in Table 2, the three zeolites obtained with or without biomass exhibit similar BAS concentrations, ranging from 251 to 272 μmol gcat−1. The same applies to LAS concentrations, which again range from 13 to 26 μmol gcat−1 for all three zeolites. The low quantity of LAS suggests that a low quantity of EFAl species exists in the materials, consistent with 27Al MAS NMR data. In addition, the total aluminium amounts calculated by ICP-MS, with values close to the acidities calculated by Py-FT-IR, reveal that the majority of the aluminium has been well integrated within the framework for all ZSM-5. Altogether, these values are in line with the same acidic properties exhibited by the three materials, despite the different synthesis routes undertaken.
3.3 Model reactions
The as-prepared zeolites were then tested in two model reactions: n-hexane cracking and MTO.The R and L zeolites yielded TOFs close to 250 and 264 molecules converted per BAS per hour, unlike the L + S zeolite, which exhibited a lower value of 204 molecules converted per site per hour. The same trend is observed for measured activities. These values are slightly lower than those obtained from the slope of the linear relationship between initial activity and concentration of BAS in a series of commercial ZSM-5 zeolites.64 This difference may result from the large size of zeolite crystals which can induce diffusional limitations. The n-hexane cracking reaction further provides evidence of the similarity of the acidic properties of the three catalysts.
The P/O ratio, calculated from initial reaction rates, is a very straightforward way of quantifying the contributions of primary and secondary cracking reactions.65 When the P/O ratio is equal to 1, only primary cracking occurs (α-scission of carbonium ion), whereas when it is less than 1, secondary cracking reactions take place (β-scission of carbenium ion).65 Values of P/O are 0.54, 0.52 and 0.61 for R, L and L + S samples, respectively (Table 2), thus indicating an important contribution of secondary cracking. The L + S sample displays a P/O ratio higher than the other two zeolites, demonstrating lower selectivity for secondary cracking of reaction products. The increase in proximity between BASs leads to an increase in secondary reactions.62 The studied zeolites present approximately the same number of BAS, then observed values of P/O may indicate a heterogeneous distribution of BASs. Indeed, the presence of Al-rich zones in the crystal where acid sites are closer to each other can promote the secondary cracking of olefins to the detriment of their desorption. This behaviour seems to be more pronounced in the R and L samples, demonstrating a more heterogeneous distribution of BASs in their crystallites.
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) 2), propene (C3), butenes (C4), butane (C4), pentenes plus pentane (C5) and aliphatics plus aromatics (C6+) at different MeOH and DME conversion levels and various reaction durations are presented in Fig. 9(b–d). Fig. 9(b) shows the catalytic data acquired at 100% conversion at the initial reaction time. The data collected in Fig. 9(c) were calculated at 47–59% iso-conversion during deactivation, whilst Fig. 9(d) presents results acquired at 21–29% iso-conversion in the deactivation regime.
2), propene (C3), butenes (C4), butane (C4), pentenes plus pentane (C5) and aliphatics plus aromatics (C6+) at different MeOH and DME conversion levels and various reaction durations are presented in Fig. 9(b–d). Fig. 9(b) shows the catalytic data acquired at 100% conversion at the initial reaction time. The data collected in Fig. 9(c) were calculated at 47–59% iso-conversion during deactivation, whilst Fig. 9(d) presents results acquired at 21–29% iso-conversion in the deactivation regime.
 | ||
| Fig. 9 (a) MeOH and DME conversion over the TOS, cumulative product selectivity and the ethene/propene ratio at (b) 100% conversion of MeOH and DME at the initial time, (c) 47–59% iso-conversion during deactivation and, (d) 21–29% iso-conversion under deactivation conditions. Black crosses correspond to conversions in product selectivity. | ||
The pore structure (cage size and shape, pore openings) and acid site characteristics (strength, density) of zeolites are among the parameters that influence the lifetime, stability and selectivity in the MTO reaction.11Fig. 9 shows significant differences in stability between the three catalysts and some differences in product selectivity during the course of the reaction. All catalysts led to an initial conversion of 100%. After only 10 h of reaction, a sudden deactivation appears for R and continues slowly. A more abrupt deactivation then appears for L + S a few hours on stream after R. The more stable L catalyst suddenly deactivates after more than 30 h of reaction. As far as product selectivity is concerned, a high olefin selectivity dominates at the start of the reaction for all catalysts. When methanol conversion drops and deactivation takes place, the selectivity towards C6+ and methane increases as deactivation progresses.
The MTO process is based indirectly on a hydrocarbon pool mechanism (HCP) which occurs within the pores of the zeolites, producing light olefins, alkanes and aromatics.67 Several descriptors such as methane formation or the distribution of products such as ethene or propene can be used to improve the understanding of the reaction mechanism(s).68 Methane formation is a descriptor of deactivation since it is linked to the development of hydrogen-poor coke molecules. In this case, methane represents the hydrogen-rich co-product and is formed via a dehydrogenation reaction of coke molecules by methanol.69 The HCP mechanism in MTO with ZSM-5 zeolites is based on a dual cycle mechanism. The first cycle forms predominantly ethene via polymethylbenzenes (PMB) in successive methylation and dealkylation reactions, known as the aromatic-based cycle. The second cycle predominantly produces propene via long alkenes through methylation and oligomerization reactions followed by β-scission, the so-called alkene-based cycle. The ethene/propene ratio can then be used to rationalize product distribution and study the relative contribution of each cycle during the reaction.
As explained previously, the MTO reaction is sensitive to the type of zeolites tested and all the parameters that characterize them. First, compared with the R catalyst synthesized without biomass, the L + S and L catalysts obtained with biomass demonstrate enhanced stability over time (e.g. 100% of methanol conversion). Stability and selectivity towards products are descriptors that can be linked to the crystal size. Indeed, the R catalyst has crystal sizes almost twice as large as those of the L and L + S catalysts (11.3 μm vs. 6.1 and 6.3 μm, respectively). At the initial reaction time, the ethene/propene ratio is higher in the case of R, at almost 0.4, unlike L and L + S whose ratio is around 0.3 (Fig. 9(b)). In addition, R already exhibits a selectivity of around 20% towards C6+, unlike the other two catalysts whose selectivities are around 10%. Next, the deactivation patterns shown in Fig. 9(c and d) at different conversion levels and reaction times show that R retains this lead in C6+ production compared with L and L + S. The size of the crystals plays a crucial role in the diffusion path of the molecules and their transformation. Large crystals favour the formation of aromatic molecules and smaller crystals favour the production of light olefins thanks to shorter diffusion paths. When the dominant cycle is the aromatic-based cycle, the catalyst's lifetime may be reduced due to limited diffusion within the zeolite pores of the species generated. The density of acid sites and their accessibility therefore vary with the size of the crystals, making the dual-cycle mechanism dependent on this parameter, as demonstrated in several studies.70–72 Khare et al. introduced a MTO descriptor called NH+ that correlates crystal size and the density of acid sites on ethene formation, i.e. the relative propagation of the aromatic-based cycle.73
Catalytic performance can also be affected by the structure of the catalysts. The performance of the L catalyst compared with that of the R and L + S catalysts could be explained by the more discrete presence of structural defects, i.e. internal silanols. It could also be due to a slightly larger pore opening size as shown in Fig. 5(b). The IR spectrum presented in Fig. 8 showed that L exhibited fewer internal silanols than the other two catalysts. Internal defects may be one factor responsible for faster deactivation of the catalysts in the MTO reaction.60,74 In fact, internal defects can play a crucial role in the diffusion of molecules by modifying the reaction pathways and favouring the accumulation of carbon deposits. The distribution of acid sites could also be modified. In addition, R shows a catalytic behaviour in which the mechanism is mainly controlled by the aromatic-based cycle. In this case, internal silanols could hinder the diffusion of PMBs and lead to faster deactivation due to the coverage of the active sites or pore blocking. As a result, silanols can act as anchoring sites for coke precursor molecules that subsequently undergo polymerisation processes.
In addition, the P/O ratios presented in Table 2 and derived from the n-hexane cracking reaction revealed a more or less homogeneous distribution of aluminium (i.e. BAS) in the samples. The location of these atoms in the structure and their concentration can impact strongly the catalytic performance depending on the sites in which they are positioned.11 MFI structures in which the straight and sinusoidal channels are enriched in BASs will tend to favour the propagation of the olefin-based cycle, while those in which the intersections are enriched in BASs will favour the propagation of the aromatic-based cycle.55 This parameter can also have an influence on the lifetime of the catalyst.75 It is possible in the case of R that the intersections of its structure are slightly more concentrated in BASs, unlike L whose straight and sinusoidal channels could be enriched in BASs. This could explain a slightly higher ethene/propene ratio at the start of the reaction for R and greater stability for L (Fig. 9(a and b)). In fact, the channels with BASs contrary to intersections allow better stabilisation of small transition states thanks to their linear geometry and their confinement effect by maximising the electrostatic interactions between the reactive species and the acid sites.76 This allows the rapid diffusion of light products (i.e. light olefins), limiting the creation of bulky intermediates and delaying the formation of coke. The addition of biomass during synthesis would optimize the positioning of the aluminium atoms to improve catalyst stability, especially lignin.
To illustrate deactivation, the product distributions obtained at iso-conversion during the MTO reaction in Fig. 9(c and d) are shown. Catalyst deactivation is a complex mechanism. It can be strongly affected by coke deposition, the number of acid sites available, their spacing due to the sporadic disappearance of BASs and the availability of reactants.77,78 Coke is responsible for deactivating the catalysts, but it is nonetheless a reaction product. The nature of the coke is strongly linked to the topology of the catalysts, ZSM-5 in this case, and is a very good trace of the deactivation process.79,80Table 3 shows the mass and chemical composition of the coke formed for all catalysts with their remaining micropore volume. At the end of the reaction, R has a carbon content of 6.4%, unlike L and L + S, which have carbon contents of 9.0 and 9.2%, respectively. This means that the L and L + S catalysts demonstrate a higher resistance to coke. During the reaction, the biomass-based catalysts have an increasingly high selectivity towards methane, increasing from 0% at the start of the reaction to 5% at the end, unlike R, where the selectivity towards methane remains low throughout the reaction (below 1%). As mentioned previously, this may be due to the methanol dehydrogenation reaction of coke molecules trapped in the zeolites. In particular, the methane production observed may be lower in the case of R because the final carbon content obtained is lower than for the other two catalysts, in which more molecules were certainly retained. As far as the nature of the coke is concerned, R has a slightly higher H/C ratio of 0.86 than L and L + S, whose ratios are around 0.70. The longer the reaction duration, the greater the mass of carbon due to the growth of the coke molecules also influenced by the high reaction temperature. R underwent faster deactivation, so the coke may have slower formation kinetics. In addition, these coke levels may correspond to aromatic molecules such as alkylpyrenes or naphthalene derivatives typical of internal coke corresponding to the structure of ZSM-5 zeolites.80 Lastly, the final micropore volumes listed in Table 3 confirm the presence of internal coke with a loss of at least 65% of the starting micropore volume for all three catalysts.
These catalytic tests revealed a beneficial effect of the addition of biomass, and more specifically lignin, on catalyst performance: both in terms of stability and selectivity towards light olefins.
4. Conclusion
In this work, a series of three ZSM-5 zeolites were synthesized using a bottom-up approach, including lignin, a mixture of lignin and sugarcane bagasse, and a reference zeolite (without biomass). The physico-chemical properties of the two types of biomass and of the zeolites obtained were characterized using several techniques. The model reactions for the cracking of n-hexane and MTO were further used to characterize the catalytic response of each catalyst during the tests.It appears that the biomass-prepared zeolites are well crystallized, exhibiting similar morphologies and analogous acidic properties to the pristine R zeolite. The main differences that were highlighted following the addition of biomass were a reduction in crystal size, an improvement in the incorporation of aluminium into the lattice and a reduction in the formation of internal silanol defects. Crystal growth could have been guided by the lignin spheres that would have defined their final size. The lignin could have acted as a micro-autoclave during the synthesis. With regard to the incorporation of aluminium and the reduction of defects, it is possible that the addition of biomass led to better dissolution of the species, yielding more interactions between the species in solution during the alkaline hydrolysis of the functional groups in the biomass. The synthesis based on the biomass mixture resulted in a very low level of EFAl, while the lignin-based synthesis resulted in a lower level of internal silanol defects.
The n-hexane cracking reaction revealed similar acidic strengths with slight differences in the distribution of aluminium in the samples. However, MTO revealed dissimilar results in response to differences in crystal size, structure and Al siting. The lignin-based synthesis stood out, showing the positive impact of its addition during synthesis on catalytic performance, with a reduction in crystal size, fewer defects and optimized positioning of aluminium atoms.
This synthesis route offers a sustainable solution for the challenging design of new zeolites, opening up infinite potential thanks to the diversity and abundance of available biomass.
Data availability
Neither software nor code has been included in this study. The data supporting this article have been included in the core of the manuscript.Conflicts of interest
The authors declare no competing interests.Acknowledgements
The financial support from the Normandy Region and SATT Conectus Alsace is acknowledged by the authors.References
- P. Rocher, Mémento roches et minéraux industriels Zéolites naturelles, BRGM, 45000 Orléans, France, 1995 Search PubMed.
- R. L. Hay, Geologic occurrence of zeolites, in Natural Zeolites: Occurrence, Properties, Use, ed. L. B. Sand and F. A. Mumpton, Pergamon Press, New York, USA, 1978, pp 135–143 Search PubMed.
- J. Weitkamp, Zeolites and catalysis, Solid State Ionics, 2000, 131, 175–188 CAS.
- R. M. Barrer, Zeolites and their synthesis, Zeolites, 1981, 1, 130–140 CAS.
- E. Roland and P. Kleinschmit, Zeolites, in Ullmann's Encyclopedia of Industrial Chemistry, John Wiley & Sons, Ltd, 2000 Search PubMed.
- M. Guisnet and J.-P. Gilson, Zeolites for Cleaner Technologies, Catalytic Science Series, ed. G. J. Hutchings, Imperial College Press, 2002 Search PubMed.
- R. Jarosz, J. Szerement, K. Gondek and M. Mierzwa-Hersztek, The use of zeolites as an addition to fertilisers – A review, Catena, 2022, 213, 106125 CAS.
- C. G. Flores, H. Schneider, N. Marcilio, L. Ferret and J. C. Oliveira, Potassic zeolites from Brazilian coal ash for use as a fertilizer in agriculture, Waste Manage., 2017, 70, 263–271 CAS.
- R. M. Far, B. van der Bruggen, A. Verliefde and E. Cornelissen, A review of zeolite materials used in membranes for water purification: history, applications, challenges and future trends, J. Chem. Technol. Biotechnol., 2022, 97, 575–596 CrossRef.
- J. D. Rimer, M. Kumar, R. Li, A. I. Lupulescu and M. D. Oleksiak, Tailoring the physicochemical properties of zeolite catalysts, Catal. Sci. Technol., 2014, 4, 3762–3771 RSC.
- H. S. Kamaluddin, X. Gong, P. Ma, K. Narasimharao, A. D. Chowdhury and M. Mokhtar, Influence of zeolite ZSM-5 synthesis protocols and physicochemical properties in the methanol-to-olefin process, Mater. Today Chem., 2022, 26, 101061 Search PubMed.
- E. G. Derouane, New Aspects of Molecular Shape-Selectivity: Catalysis by Zeolite ZSM-5, in Studies in Surface Science and Catalysis, ed. B. Imelik, C. Naccache, Y. B. Taarit, J. C. Vedrine, G. Coudurier and H. Praliaud, Elsevier, 1980, pp. 5–18 Search PubMed.
- E. G. Derouane and Z. Gabelica, A novel effect of shape selectivity: Molecular traffic control in zeolite ZSM-5, J. Catal., 1980, 65, 486–489 CAS.
- T. Cordero-Lanzac, A. G. Gayubo, A. T. Aguayo and J. Bilbao, The MTO and DTO processes as greener alternatives to produce olefins: A review of kinetic models and reactor design, Chem. Eng. J., 2024, 494, 152906 CAS.
- P. Payra and P. K. Dutta, Zeolites: A Primer, in Handbook of Zeolite Science and Technology, CRC Press, 2003 Search PubMed.
- R. J. Argauer and G. R. Landolt, Crystalline zeolite ZSM-5 and method of preparing the same, US3702886A, 1972.
- G. T. Kokotailo, S. L. Lawton, D. H. Olson and W. M. Meier, Structure of synthetic zeolite ZSM-5, Nature, 1978, 272, 437–438 CAS.
- D. H. Olson, G. T. Kokotailo, S. L. Lawton and W. M. Meier, Crystal structure and structure-related properties of ZSM-5, J. Phys. Chem., 1981, 85, 2238–2243 CAS.
- A. Deneyer, Q. Ke, J. Devos and M. Dusselier, Zeolite Synthesis under Non-conventional Conditions: Reagents, Reactors, and Modi Operandi, Chem. Mater., 2020, 32, 4884–4919 CAS.
- R. Jain, A. J. Mallette and J. D. Rimer, Controlling Nucleation Pathways in Zeolite Crystallization: Seeding Conceptual Methodologies for Advanced Materials Design, J. Am. Chem. Soc., 2021, 143, 21446–21460 CAS.
- F. Ocampo, J. A. Cunha, M. R. de Lima Santos, J. P. Tessonnier, M. M. Pereira and B. Louis, Synthesis of zeolite crystals with unusual morphology: Application in acid catalysis, Appl. Catal., A, 2010, 390, 102–109 CAS.
- E. S. Gomes, G. Lutzweiler, P. Losch, A. V. Silva, C. Bernardon, K. Parkhomenko, M. M. Pereira and B. Louis, Strategy to design zeolite catalysts in the presence of biomass, Microporous Mesoporous Mater., 2017, 254, 28–36 CAS.
- E. S. Gomes, D. A. G. Aranda, M. M. Pereira and B. Louis, ZSM-5 synthesis by the assistance of biomass and biomass-derivate compounds, Microporous Mesoporous Mater., 2018, 263, 251–256 CAS.
- M. M. Pereira, E. S. Gomes, A. V. Silva, A. B. Pinar, M.-G. Willinger, S. Shanmugam, C. Chizallet, G. Laugel, P. Losch and B. Louis, Biomass-mediated ZSM-5 zeolite synthesis: when self-assembly allows to cross the Si/Al lower limit, Chem. Sci., 2018, 9, 6532–6539 CAS.
- B. Louis, E. Gomes, P. Losch, T. Coelho, A. Faro Jr, J. Pinto and M. Pereira, Biomass-assisted Zeolite Syntheses as a Tool for Designing New Acid Catalysts, ChemCatChem, 2017, 9, 2065–2079 CAS.
- Y. Zhang and B. Louis, Biochar-mediated synthesis of ZSM-5 zeolite for the methanol-to-olefins reaction, Next Mater., 2024, 5, 100259 Search PubMed.
- R. Bingre, C. M. Sayago, P. Losch, L. Huang, Q. Wang, M. M. Pereira and B. Louis, Recent progress in the biomass-mediated synthesis of porous materials, Inorg. Chim. Acta, 2019, 487, 379–386 CAS.
- Q. Zheng and B. Louis, Conversion of Methanol into Hydrocarbons over Biomass-Assisted ZSM-5 Zeolites: Effect of the Biomass Nature, ChemCatChem, 2023, 15, e202300210 CAS.
- Y. Zhang and B. Louis, Tailoring structure and acidity of ZSM-5 zeolite by algae carbon modification: Promoting effect in the MTO reaction, Microporous Mesoporous Mater., 2023, 350, 112431 CAS.
- C. G. Flores, H. Schneider and B. Louis, Lignin as a Bio-Sourced Secondary Template for ZSM-5 Zeolite Synthesis, Catalysts, 2022, 12, 368 CAS.
- A. Tribot, G. Amer, M. Abdou Alio, H. de Baynast, C. Delattre, A. Pons, J.-D. Mathias, J.-M. Callois, C. Vial, P. Michaud and C.-G. Dussap, Wood-lignin: Supply, extraction processes and use as bio-based material, Eur. Polym. J., 2019, 112, 228–240 CrossRef CAS.
- C. Libretti, L. S. Correa and M. A. R. Meier, From waste to resource: advancements in sustainable lignin modification, Green Chem., 2024, 26, 4358–4386 RSC.
- C. T. Hiranobe, A. S. Gomes, F. F. G. Paiva, G. R. Tolosa, L. L. Paim, G. Dognani, G. P. Cardim, H. P. Cardim, R. J. dos Santos and F. C. Cabrera, Sugarcane Bagasse: Challenges and Opportunities for Waste Recycling, Clean Technol., 2024, 6, 662–699 CrossRef.
- S. Pan, H. M. Zabed, Y. Wei and X. Qi, Technoeconomic and environmental perspectives of biofuel production from sugarcane bagasse: Current status, challenges and future outlook, Ind. Crops Prod., 2022, 188, 115684 CAS.
- C. G. Flores, H. Schneider, J. S. Dornelles, L. B. Gomes, N. R. Marcilio and P. J. Melo, Synthesis of potassium zeolite from rice husk ash as a silicon source, Clean. Eng. Technol., 2021, 4, 100201 CrossRef.
- X. Li, H. Fu, S. Shao and Y. Cai, Synthesis of hierarchical HZSM-5 utilizing natural cellulose as templates for promoted production of aromatic hydrocarbons in the catalytic pyrolysis of biomass, Fuel Process. Technol., 2023, 248, 107815 CAS.
- D.-K. Nguyen, V.-P. Dinh, H. Q. Nguyen and N. T. Hung, Zeolite ZSM-5 synthesized from natural silica sources and its applications: a critical review, J. Chem. Technol. Biotechnol., 2023, 98, 1339–1355 CrossRef CAS.
- N. S. Samanta, P. P. Das, P. Mondal, M. Changmai and M. K. Purkait, Critical review on the synthesis and advancement of industrial and biomass waste-based zeolites and their applications in gas adsorption and biomedical studies, J. Indian Chem. Soc., 2022, 99, 100761 CrossRef CAS.
- S. Liu, Z. Cheng, B. Li, H. Zeng, W. Liang, Y. Luo, Y. Bai, H. Gao, X. Pan and X. Shu, Recent advances in biomass-assisted synthesis of hierarchical porous zeolite, Mater. Today Sustainability, 2024, 27, 100917 CrossRef.
- K.-J. Chao, T. Chang Tasi, M.-S. Chen and I. Wang, Kinetic studies on the formation of zeolite ZSM-5, J. Chem. Soc., Faraday Trans. 1, 1981, 77, 547–555 RSC.
- R. Bardestani, G. S. Patience and S. Kaliaguine, Experimental methods in chemical engineering: specific surface area and pore size distribution measurements—BET, BJH, and DFT, Can. J. Chem. Eng., 2019, 97, 2781–2791 CrossRef CAS.
- G. Engelhardt and D. Michel, High Resolution Solid State NMR of Silicates and Zeolites, Berichte Bunsenges, Z. Phys. Chem., 1987, 92, 1059 Search PubMed.
- C. Miranda, J. Urresta, H. Cruchade, A. Tran, M. Benghalem, A. Astafan, P. Gaudin, T. J. Daou, A. Ramírez, Y. Pouilloux, A. Sachse and L. Pinard, Exploring the impact of zeolite porous voids in liquid phase reactions: The case of glycerol etherification by tert-butyl alcohol, J. Catal., 2018, 365, 249–260 CrossRef CAS.
- J. T. Kolawole, A. J. Babafemi, E. Fanijo, S. Chandra Paul and R. Combrinck, State-of-the-art review on the use of sugarcane bagasse ash in cementitious materials, Cem. Concr. Compos., 2021, 118, 103975 CrossRef CAS.
- A. Vishtal and A. Kraslawski, Challenges in industrial applications of technical lignins, Bioresources, 2011, 6, 3547–3568 Search PubMed.
- E. Melro, A. Filipe, D. Sousa, B. Medronho and A. Romano, Revisiting lignin: a tour through its structural features, characterization methods and applications, New J. Chem., 2021, 45, 6986–7013 RSC.
- R. Corrales, F. Mendes, C. Perrone, C. Sant’anna, W. Souza, Y. Abud, E. Bon and V. Ferreira-Leitao, Structural Evaluation of Sugarcane Bagasse Steam Pretreated in the Presence of CO2 and SO2, Biotechnol. Biofuels, 2012, 5, 36 CAS.
- N. Wittner, J. Slezsák, W. Broos, J. Geerts, S. Gergely, S. E. Vlaeminck and I. Cornet, Rapid lignin quantification for fungal wood pretreatment by ATR-FTIR spectroscopy, Spectrochim. Acta, Part A, 2023, 285, 121912 CAS.
- S. Mintova, Verified Syntheses of Zeolitic Materials, Synthesis Commission of the International Zeolite Association, 3rd edn, 2016 Search PubMed.
- Z. Sun, Q. Shu, Q. Zhang, S. Li, G. Zhu, C. Wang, J. Zhang, H. Li and Z. Huang, A Hydrothermal Synthesis Process of ZSM-5 Zeolite for VOCs Adsorption Using Desilication Solution, Separations, 2024, 11, 39 CAS.
- L. Xu, Y. Yuan, Q. Han, L. Dong, L. Chen, X. Zhang and L. Xu, High yield synthesis of nanoscale high-silica ZSM-5 zeolites via interzeolite transformation with a new strategy, Catal. Sci. Technol., 2020, 10, 7904–7913 CAS.
- I. Batonneau-Gener and A. Sachse, Determination of the Exact Microporous Volume and BET Surface Area in Hierarchical ZSM-5, J. Phys. Chem. C, 2019, 123, 4235–4242 CAS.
- C. Bernardon, B. Louis, V. Beneteau and P. Pale, Diels-Alder reaction between isoprene and methyl acrylate over different zeolites: influence of pore topology and acidity, ChemPlusChem, 2013, 78, 1134–1141 CAS.
- Zeolite Chemistry and Catalysis, ed. A. W. Chester and E. G. Derouane, Springer Netherlands, Dordrecht, 2009, pp. 78–91 Search PubMed.
- R. Feng, B. Liu, P. Zhou, X. Yan, X. Hu, M. Zhou and Z. Yan, Influence of framework Al distribution in HZSM-5 channels on catalytic performance in the methanol to propylene reaction, Appl. Catal., A, 2022, 629, 118422 CAS.
- A. Palčić and V. Valtchev, Analysis and control of acid sites in zeolites, Appl. Catal., A, 2020, 606, 117795 Search PubMed.
- J. Grand, S. N. Talapaneni, A. Vicente, C. Fernandez, E. Dib, H. A. Aleksandrov, G. N. Vayssilov, R. Retoux, P. Boullay, J.-P. Gilson, V. Valtchev and S. Mintova, One-pot synthesis of silanol-free nanosized MFI zeolite, Nat. Mater., 2017, 16, 1010–1015 CAS.
- S. Bordiga, C. Lamberti, F. Bonino, A. Travert and F. Thibault-Starzyk, Probing zeolites by vibrational spectroscopies, Chem. Soc. Rev., 2015, 44, 7262–7341 CAS.
- M. Zheng, Y. Chu, Q. Wang, Y. Wang, J. Xu and F. Deng, Advanced solid-state NMR spectroscopy and its applications in zeolite chemistry, Prog. Nucl. Magn. Reson. Spectrosc., 2024, 140–141, 1–41 CAS.
- K. Barbera, F. Bonino, S. Bordiga, T. V. W. Janssens and P. Beato, Structure–deactivation relationship for ZSM-5 catalysts governed by framework defects, J. Catal., 2011, 280, 196–205 CAS.
- H. Li, J. Yu, K. Du, W. Li, L. Ding, W. Chen, S. Xie, Y. Zhang and Y. Tang, Synthesis of ZSM-5 Zeolite Nanosheets with Tunable Silanol Nest Contents across an Ultra-wide pH Range and Their Catalytic Validation, Angew. Chem., Int. Ed., 2024, 63, e202405092 CAS.
- M. Guisnet and L. Pinard, Characterization of acid-base catalysts through model reactions, Catal. Rev., 2018, 60, 1–100 Search PubMed.
- B. Louis, M. M. Pereira, F. M. Santos, P. M. Esteves and J. Sommer, Alkane activation over acidic zeolites : The first step, Chem.–Eur. J., 2010, 16, 573–576 CrossRef CAS PubMed.
- Y. Shen, S. Zhang, Z. Qin, A. Beuque, L. Pinard, S. Asahina, N. Asano, R. Zhang, J. Zhao, Y. Fan, X. Liu, Z. Yan and S. Mintova, Effect of Mixing Order of Si and Al Sources on the Inner Architecture and Catalytic Performance of ZSM-5 Zeolites, ACS Catal., 2024, 14, 3766–3777 CrossRef CAS.
- T. F. Narbeshuber, H. Vinek and J. A. Lercher, Monomolecular Conversion of Light Alkanes over H-ZSM-5, J. Catal., 1995, 157, 388–395 CrossRef CAS.
- N.-L. Michels, S. Mitchell and J. Pérez-Ramírez, Effects of Binders on the Performance of Shaped Hierarchical MFI Zeolites in Methanol-to-Hydrocarbons, ACS Catal., 2014, 4, 2409–2417 CrossRef CAS.
- S. Ilias and A. Bhan, Mechanism of the Catalytic Conversion of Methanol to Hydrocarbons, ACS Catal., 2013, 3, 18–31 CrossRef CAS.
- S. Ilias, R. Khare, A. Malek and A. Bhan, A descriptor for the relative propagation of the aromatic- and olefin-based cycles in methanol-to-hydrocarbons conversion on H-ZSM-5, J. Catal., 2013, 303, 135–140 CrossRef CAS.
- H. Schulz, About the Mechanism of Methanol Conversion on Zeolites, Catal. Lett., 2018, 148, 1263–1280 CrossRef CAS.
- K. Liu, T. Shoinkhorova, X. You, X. Gong, X. Zhang, S.-H. Chung, J. Ruiz-Martínez, J. Gascon and A. D. Chowdhury, The synergistic interplay of hierarchy, crystal size, and Ga-promotion in the methanol-to-aromatics process over ZSM-5 zeolites, Dalton Trans., 2024, 53, 11344–11353 CAS.
- X. Wang, C. Wang, Y. Chu, Y. Liu, M. Hu, F. Deng, J. Xu and J. Yu, Deciphering the Link between Zeolite Crystal Size, Brønsted Acid Site Distribution, and Dual-Cycle Selectivity in Methanol-to-Olefins over Zeolite, ACS Catal., 2024, 14, 15609–15621 CrossRef CAS.
- D. Dittmann, E. Kaya, D. Strassheim and M. Dyballa, Influence of ZSM-5 Crystal Size on Methanol-to-Olefin (MTO) vs. Ethanol-to-Aromatics (ETA) Conversion, Molecules, 2023, 28, 8046 CrossRef CAS PubMed.
- R. Khare, D. Millar and A. Bhan, A mechanistic basis for the effects of crystallite size on light olefin selectivity in methanol-to-hydrocarbons conversion on MFI, J. Catal., 2015, 321, 23–31 CrossRef CAS.
- K. Lee, S. Lee, Y. Jun and M. Choi, Cooperative effects of zeolite mesoporosity and defect sites on the amount and location of coke formation and its consequence in deactivation, J. Catal., 2017, 347, 222–230 CrossRef CAS.
- T. Liang, J. Chen, Z. Qin, J. Li, P. Wang, S. Wang, G. Wang, M. Dong, W. Fan and J. Wang, Conversion of Methanol to Olefins over H-ZSM-5 Zeolite: Reaction Pathway Is Related to the Framework Aluminum Siting, ACS Catal., 2016, 6, 7311–7325 CrossRef CAS.
- A. Ghorbanpour, J. D. Rimer and L. C. Grabow, Computational Assessment of the Dominant Factors Governing the Mechanism of Methanol Dehydration over H-ZSM-5 with Heterogeneous Aluminum Distribution, ACS Catal., 2016, 6, 2287–2298 CrossRef CAS.
- D. Chen, H. P. Rebo, K. Moljord and A. Holmen, Influence of Coke Deposition on Selectivity in Zeolite Catalysis, Ind. Eng. Chem. Res., 1997, 36, 3473–3479 CAS.
- D. Mores, J. Kornatowski, U. Olsbye and B. M. Weckhuysen, Coke Formation during the Methanol-to-Olefin Conversion: In Situ Microspectroscopy on Individual H-ZSM-5 Crystals with Different Brønsted Acidity, Chem.–Eur. J., 2011, 17, 2874–2884 CAS.
- D. Rojo-Gama, M. Signorile, F. Bonino, S. Bordiga, U. Olsbye, K. P. Lillerud, P. Beato and S. Svelle, Structure–deactivation relationships in zeolites during the methanol–to-hydrocarbons reaction: Complementary assessments of the coke content, J. Catal., 2017, 351, 33–48 CrossRef CAS.
- K. Ben Tayeb, S. Hamieh, C. Canaff, H. Nguyen, H. Vezin and L. Pinard, The radical internal coke structure as a fingerprint of the zeolite framework, Microporous Mesoporous Mater., 2019, 289, 109617 CAS.
| This journal is © The Royal Society of Chemistry 2025 |