
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceExploring a solid-state nanopore approach for single-molecule protein detection from single cells†
Zi-Qi
Zhou
a,
Shao-Chuang
Liu
 *a,
Jia
Wang
a,
Ke-Le
Chen
a,
Bao-Kang
Xie
a,
Yi-Lun
Ying
ab and
Yi-Tao
Long
*a
*a,
Jia
Wang
a,
Ke-Le
Chen
a,
Bao-Kang
Xie
a,
Yi-Lun
Ying
ab and
Yi-Tao
Long
*a
aMolecular Sensing and Imaging Center, School of Chemistry and Chemical Engineering, Nanjing University, Nanjing, 210023, P. R. China. E-mail: yitaolong@nju.edu.cn; shaochuangliu@nju.edu.cn
bChemistry and Biomedicine Innovation Center, Nanjing University, Nanjing, 210023, P. R. China
First published on 7th April 2025
Abstract
Direct protein analysis from complex cellular samples is crucial for understanding cellular diversity and disease mechanisms. Here, we explored the potential of SiNx solid-state nanopores for single-molecule protein analysis from complex cellular samples. Using the LOV2 protein as a model, we designed a nanopore electrophoretic driver protein and fused it with LOV2, thereby enhancing the capture efficiency of the target protein. Then, we performed ex situ single-cell protein analysis by directly extracting the contents of individual cells using glass nanopipette-based single-cell extraction and successfully identified and monitored the conformational changes of the LOV2 protein from single-cell extracts using SiNx nanopores. Our results reveal significant differences between proteins measured directly from single cells and those obtained from purified samples. This work demonstrates the potential of solid-state nanopores as a powerful tool for single-cell, single-molecule protein analysis, opening avenues for investigating protein dynamics and interactions at the cellular level.
Introduction
Measuring proteins at the single-cell level is essential for understanding cellular heterogeneity, elucidating protein function, and capturing dynamic interactions that govern biological processes and diseases.1–5 A key barrier in achieving this goal is the absence of protein amplification methods similar to PCR for nucleic acids,6 making it difficult to detect and quantify proteins within individual cells, especially low-abundance proteins. Overcoming this limitation requires the development of techniques capable of analyzing proteins at the single-molecule level while preserving their native state within complex biological environments.7–10 Existing single-cell protein analysis methods, such as mass spectrometry11–13 and single-molecule fluorescence microscopy,14–17 have made significant advances. However, these techniques often require complex sample preparation and large sample populations, and suffer from limitations in throughput. Achieving single-molecule sensitivity to detect target proteins in complex cellular context remains a critical challenge.Solid-state nanopores have emerged as a versatile platform for single-molecule analysis,18,19 offering exceptional sensitivity by detecting ionic current blockage as individual molecules translocate through the pores. These nanopores can be precisely engineered to control their size and surface properties, making them ideal for analyzing molecules with diverse sizes, charges, and surface characteristics. This versatility has enabled solid-state nanopores to be used in studying DNA,20 proteins,21–23 carbohydrates,24–26 and small molecules.27,28 However, most studies were focused on purified or synthetic samples, with limited exploration of their performance in complex biological samples. Unlike standardized purified samples, cellular extracts are highly heterogeneous,29 containing a vast array of biomolecules that can introduce noise and hinder the selective detection of target proteins. The broad range of biomolecule concentrations and complex molecular interactions within cellular samples further complicate the direct application of nanopores for single-cell protein analysis. Despite the promise of nanopore technology, its implementation in single-cell studies remains challenging.
Among the different types of solid-state nanopores, only nanopipettes have been used for single-cell analysis due to their needle-like geometry and ability to extract individual cellular contents.30–32 However, this geometry limits high-throughput capabilities, making it difficult to meet the increasing demands of single-cell protein analysis. In contrast, silicon nitride (SiNx) solid-state nanopores offer enhanced sensitivity, selectivity, and specificity for single-molecule detection,18,33 enabling real-time monitoring of dynamic conformational changes and protein–protein interactions.34–36 Furthermore, advancements in micro- and nanofabrication techniques have allowed for the miniaturization of solid-state nanopores and the development of high-throughput arrays, which could significantly improve the analysis throughput of complex cellular samples. Despite these advancements, the application of SiNx solid-state nanopores for single-cell analysis remains largely unexplored, with few studies directly utilizing them for complex biological samples. This gap limits the broader integration of solid-state nanopore technology into single-cell analysis.
Herein, we demonstrate the feasibility of using SiNx solid-state nanopores for direct single-molecule protein analysis from individual cell extracts. By designing a nanopore electrophoretic driver (NEPD) protein to enhance protein capture efficiency, we successfully detected and monitored conformational changes of a model photoswitchable protein from single cells. Our findings highlight the potential of solid-state nanopores for analyzing proteins in their native cellular environment, paving the way for future applications in single-cell studies.
Results and discussion
Principle of single-molecule detection of intracellular proteins by nanopores
Fig. 1a illustrates the principle of single-cell protein analysis using SiNx solid-state nanopores. Unlike conventional methods, the solid-state nanopore approach allows direct addition of cellular samples into the nanopore chamber for single-molecule analysis, eliminating the need for complex sample preprocessing. Under an external electric field, individual molecules translocate through the nanopore, generating distinct current blockages based on their physical and chemical properties. These unique signatures enable the identification of target proteins. Additionally, by tuning nanopore size and surface properties, the sensitivity and specificity of single-cell protein analysis can be optimized.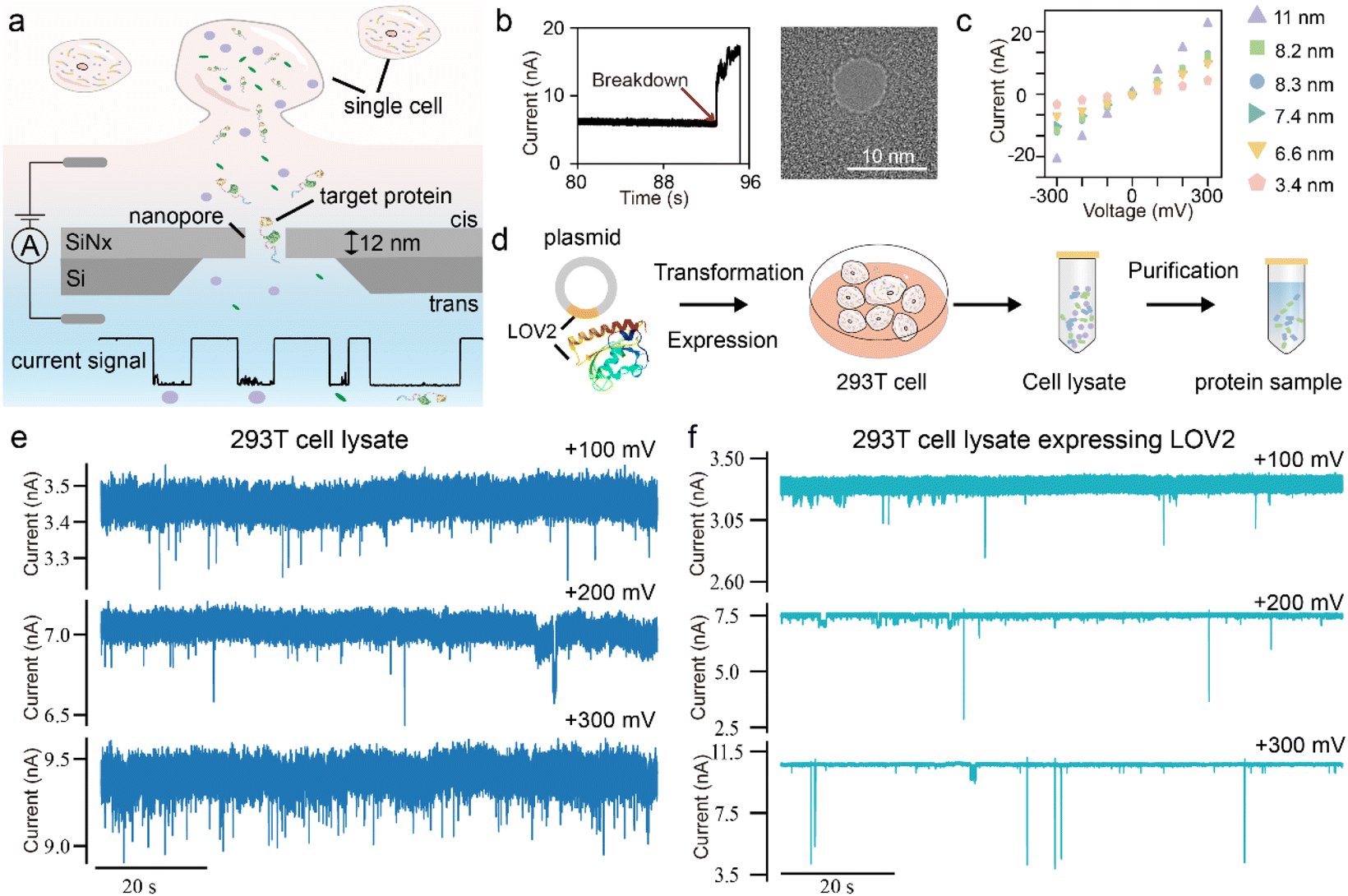 | ||
| Fig. 1 The schematic illustration of the solid-state nanopore for single-cell analysis. (a) The schematic diagram and the experimental set-up of the solid-state nanopore for single-cell analysis. (b) The nanopore fabrication by controlled breakdown and characterization by TEM. (c) The I–V curve characterization of the nanopore fabricated by controlled breakdown. (d) The workflow for protein expression and purification. Two types of samples were prepared: a cell lysate sample containing all cellular components, and a purified protein sample. (e) The representative current trace of the 293T cell lysate, and (f) cell lysate expressing protein LOV2 at +100 mV, +200 mV and +300 mV. The signals were acquired using 8.64 nm and 8.84 nm nanopores, respectively. | ||
The SiNx solid-state nanopores were fabricated via controlled dielectric breakdown (CBD) and transmission electron microscopy (Fig. 1b). The silicon nitride membrane was 12 ± 2 nm thick, and nanopores of varying sizes were prepared for protein measurement (Fig. 1c). These solid-state nanopores exhibit linear and symmetric I–V curves in 1 M KCl, 10 mM Tris–HCl, pH 8.0 buffer, without distinct rectification effects. As shown in Fig. 1a, cell samples were added to the cis chamber of the nanopore flow cell. Ionic current of the nanopore was recorded using an amplifier with a sampling rate of 100 kHz and a 10 kHz low-pass filter.
To assess the feasibility of solid-state nanopores for protein detection in cellular samples, we first conducted measurements using conventional cell lysates. As a model target, we selected the exogenously expressed light-oxygen-voltage-sensing domain (LOV2 domain) of Avena Sativa phototropin 1 protein due to its light-regulated intramolecular conformational changes, making it ideal for evaluating the performance of nanopore measurements in detecting conformational dynamics. Fig. 1d outlines the protein expression and purification workflow. We analyzed two types of samples: the control group of 293T cell lysate (CL) and cell lysate expressing protein LOV2 (CL-LOV2). As shown in Fig. 1e, under voltages ranging from +100 mV to +300 mV, we observed blockage signals upon adding the lysate samples into the nanopore. This confirms that cellular components in the lysate can be captured by the nanopore and generate detectable signals. Compared to the CL sample, the CL-LOV2 sample exhibited distinct signal characteristics with notable differences in blockage depth, indicating the ability of the SiNx to differentiate target proteins within complex cellular environments. However, the relatively low capture efficiency in direct lysate measurements highlights a challenge in specifically detecting low-abundance proteins in cellular samples.
Design of the nanopore electrophoretic driver protein
While SiNx solid-state nanopores provide high sensitivity for single-molecule measurements, the complex surface charge distribution and diverse shapes of proteins complicate their translocation,35,37,38 making it challenging to achieve stable and consistent single-molecule signals from cell samples. Although previous studies have explored strategies such as those based on supercharged polypeptides,39 biotin binding,40 and molecular carriers41,42 to improve the capture in nanopores, their effectiveness in complex biological samples remains underexplored.To address this challenge, we designed a nanopore electrophoretic driver (NEPD) protein to enhance the capture of target proteins under the electric field of the nanopore. This protein was meticulously designed and optimized using AlphaFold2. As shown in Fig. 2a, the NEPD protein has two main components: an enhanced electrophoretic driver and an enhanced green fluorescent protein (EGFP), linked by a flexible linker. The sequence of the electrophoretic driver is (DE)11(EAAAK)2(GGGGS)(H)6(GGGGS)2. The DE repetitive sequence determines the negative charge of protein, which enhances the electrophoretic properties of the target protein in the nanopore. This region is referred to as the electrophoretic domain (E-domain). EGFP serves solely as a reporter to confirm target protein expression. The His6 tag is used for protein purification. The α-helical EAAAK linker and flexible GGGS linker connect these components. This design ensures that the NEPD protein adopts an extended linear conformation with a uniform negative surface charge distribution, which facilitates its capture by the nanopore. Then, the NEPD protein is co-expressed with the target LOV2 protein within cellular systems via a GS-XTEN-GS linker (SGGSSGGSSGSETPGTSESATPESSGGSSGGS). These enhanced electrophoretic properties of the NEPD protein enable efficient and selective capture of the target LOV2 protein into the nanopore. Subsequently, we expressed both the standalone NEPD protein and the fusion protein (NEPD–LOV2) in 293T cells. Confocal fluorescence imaging and single-molecule TIRF microscopy confirmed successful expression and purification (Fig. 2b and S1†).
 | ||
| Fig. 2 Design and characterization of the nanopore electrophoretic driver protein and the LOV2 target protein. (a) Domain architecture of the fusion protein consisting of the nanopore electrophoretic driver (NEPD) protein and LOV2 target protein. (b) The fluorescence image of 293T cells expressing NEPD–LOV2 and the purified NEPD–LOV2 protein. (c) The representative current trace of the cell lysate expressing NEPD–LOV2 at +300 mV. (d) The representative current trace and individual example events of purified NEPDT and NEPD–LOV2 proteins at +300 mV using a 4.39 nm nanopore. (e) Scatter plots of blockage current versus dwell time (N = 239), and corresponding box with strip plots of blockage current and dwell time for NEPD and NEPD–LOV2 proteins, along with the histogram distribution. (f) Schematic of the AsLOV2 Domain upon light-induced unfolding. (g) The representative current trace and individual example events of the purified NEPD–LOV2 protein under dark conditions and blue light illumination (450 nm). The applied voltage is +300 mV. (h) Scatter plot (N = 3003) and corresponding histogram statistics of blockage current versus dwell time for the NEPD–LOV2 protein under dark conditions and blue light illumination. | ||
To validate the electrophoretic capture, we analyzed the cell lysates of the 293T expressing NEPD–LOV2 fusion protein (CL-NEPD–LOV2). As shown in Fig. 2c and S2,† the CL-NEPD–LOV2 samples exhibited numerous blockage events. These results indicate that our engineered NEPD protein effectively enhances the capture of the target LOV2 protein from complex cellular samples via solid-state nanopores.
To identify target proteins from complex cell samples. We first tested the purified NEPD and NEPD–LOV2 proteins. Fig. 2d shows the representative current trace of the NEPD and NEPD–LOV2 proteins at a voltage of +300 mV using an ∼4.39 nm nanopore. Both proteins produced distinct translocation events characterized by multiple stepwise transitions and significant fluctuations at each step. Previous studies have proved that these current fluctuations correspond to the dynamic conformational changes of the protein, enabling analysis of transient conformations and folding–unfolding processes.43Fig. 2e shows the scatter plots of dwell time versus peak amplitude for both proteins. The translocation of a single protein produced a current blockade (ΔI) of 0.41 nA for NEPD and 0.58 nA for the NEPD–LOV2 protein, with mean dwell times of 0.34 ms and 0.42 ms, respectively. These results demonstrate that the SiNx solid-state nanopores can clearly distinguish between the NEPD protein and the fused target protein.
Further, to assess reproducibility, we conducted parallel tests using nanopores with diameters ranging from 4 nm to 8 nm (Fig. S3–S7†). The data revealed that even slight variations in nanopore size led to significant differences in blockage currents and dwell times, indicating the high sensitivity of protein translocation to pore dimensions and the challenge of achieving consistent data across different nanopores.
Next, we investigated the light-induced conformational changes of the NEPD–LOV2 protein using a nanopore with a pore diameter of ∼9.34 nm. As shown in Fig. 2f, the LOV domain, functioning as a photo switch, undergoes a photochemical reaction under blue light (450 nm) that induces α-helical structural alterations. This process is reversible, with a few seconds of excitation time under blue light followed by tens of seconds of recovery in the dark. Fig. 2g shows the representative current traces and individual events under both dark and blue light conditions. Under blue light, increased background noise was observed, reducing the signal-to-noise ratio. Statistical analysis indicated that upon blue light illumination, the ΔI of the NEPD–LOV2 protein increased from 0.34 nA to 0.55 pA, and the dwell time increased from 0.27 ms to 0.34 ms (Fig. 2h). These trends were consistent across the voltage range from 150 to 300 mV (Fig. S8†). We attribute these changes to the light-induced unfolding of the LOV2 domain, which increases both the effective excluded volume and the translocation length of the protein. Above all, these results demonstrate that fusing the target protein with the NEPD protein significantly enhances its capture by SiNx nanopores, enabling effective identification and characterization of its dynamic conformational changes.
Single cell analysis using solid-state nanopores
The cell lysis process disrupts intracellular components, potentially altering the native characteristics of proteins and complicating accurate analysis. Compared to bulk cell lysate analysis, single-cell protein detection enables a more precise assessment of cellular heterogeneity. However, with only approximately 200 pg of total protein per cell,44 extracting single-cell contents efficiently while maintaining molecular integrity remains a significant challenge. In addition, some biomarkers are very low in some biological samples, requiring high sensitivity and resolution.45,46 In this study, we employed nanopipettes to extract the contents of individual 293T cells. As shown in Fig. 3a, nanopipettes were fabricated using a laser-based pipette puller, producing tip diameters of approximately 1 μm. Mounted on a micromanipulator for precise control, these nanopipettes were guided into single cells under real-time fluorescence microscopy (Fig. 3b and c). Negative pressure was applied to extract cellular contents, which were subsequently transferred into the cis chamber of the SiNx nanopore flow cell for single-molecule analysis. Within 10–15 minutes after adding the cell sample, we effectively collected signals originating from single-cell extracts. This ex situ single-cell approach eliminates the need for complete cell lysis and purification, enabling the direct analysis of intracellular proteins while maintaining conditions that closely resemble their native cellular environment. | ||
| Fig. 3 (a) The schematic illustration of the extraction and measurement of samples from single cells. (b) The fluorescence image of 293T cells expressing NEPD–LOV2. (c) The bright field image of a nanopipette inserting into a single cell. (d) The representative current trace and individual example events for detecting the samples of a single 293T cell. (e) Scatter plots of blockage current versus dwell time, and the corresponding blockage current histogram distribution of the sample extracted from a single 293T cell at +300 mV (N = 305) and +400 mV (N = 381). (f) The representative current trace and individual example events for detecting the samples of a single 293T cell expressing NEPD–LOV2 and (g) after adding the purified NEPD–LOV2 protein, at +300 mV, +400 mV and +500 mV. Additional raw current trace is shown in Fig. S15 and S16.† (h) Scatter plot with the marginal histogram statistics of blockage current versus dwell time for the single 293T cell samples expressing NEPD–LOV2 and the signals after the additional purified NEPD–LOV2 added at +300 mV, +400 mV (i) and + 500 mV (j), respectively. | ||
Fig. 3d presents representative current traces and individual events from a single original 293T cell using an ∼9.28 nm nanopore. Statistical analysis at +300 mV and +400 mV revealed average current blockades of 0.38 nA and 0.57 nA, respectively (Fig. 3e). Obviously, these signals originate from the single-molecule substances within individual cells as they pass through the nanopore; however, direct identification of the specific proteins remains challenging.
Subsequently, we analyzed signal 293T cells expressing the NEPD–LOV2 fusion protein under dark conditions. Fig. 3f shows numerous blockage events recorded at different voltages. To further compare the signals obtained from single-cell samples with purified proteins, additional purified NEPD–LOV2 proteins were introduced into the same nanopore (Fig. 3g). Statistical analysis revealed significant differences between single-cell measurements of unmodified 293T cells (Fig. 3e) and those expressing NEPD–LOV2 (Fig. 3h–j). These results indicate that NEPD–LOV2 proteins were preferentially captured from single-cell samples, confirming the feasibility of our approach for detecting target proteins from individual cells.
Interestingly, statistical analysis also revealed significant differences in the blockage currents between signals from single-cell samples and purified proteins. As shown in Fig. 3h, the signals from the single-cell sample exhibited a larger blockage depth with an average blockage current of 4.98 nA at +300 mV, compared to 0.28 nA for the purified NEPD–LOV2 protein. Additionally, at +400 mV, the signal distribution of the single-cell samples expressing NEPD–LOV2 displayed two peaks (ΔI of 2.05 nA and 6.21 nA), whereas the purified protein signals shifted to ΔI of 1.48 nA and 5.20 nA (Fig. 3i). At +500 mV, single-cell samples expressing the NEPD–LOV2 protein exhibited a single broad peak at ΔI of 6.44 nA, while the purified protein signals showed two peaks at ΔI of 1.70 nA and 6.67 nA (Fig. 3j).
These results suggest that proteins obtained directly from single cells exhibit distinct characteristics compared to their purified counterparts through cell lysis. Notably, each signal from single-cell samples displayed unique blockage patterns (Fig. 3f and g), including larger blockage depths and prolonged dwell times. This observation suggests that proteins in their native cellular environment may interact with other biomolecules, forming complexes that alter their translocation profiles compared to purified proteins.
We also investigated light-induced conformational changes of the NEPD–LOV2 protein in the single-cell extracts. Fig. 4a shows the representative current traces and individual events from a single 293T cell expressing NEPD–LOV2 under blue light, while Fig. 4b presents the corresponding signals after adding purified NEPD–LOV2 proteins (Fig. S17†). As shown in Fig. 4c–e, the blockage current signals from both single-cell samples and purified proteins were distributed over a broader range under blue light compared to dark conditions. This suggests that in its open state, the LOV2 domain may interact more readily with various cellular components, leading to alterations in the composition and conformation of the NEPD–LOV2 complex. These interactions could contribute to the observed variability in translocation signals.
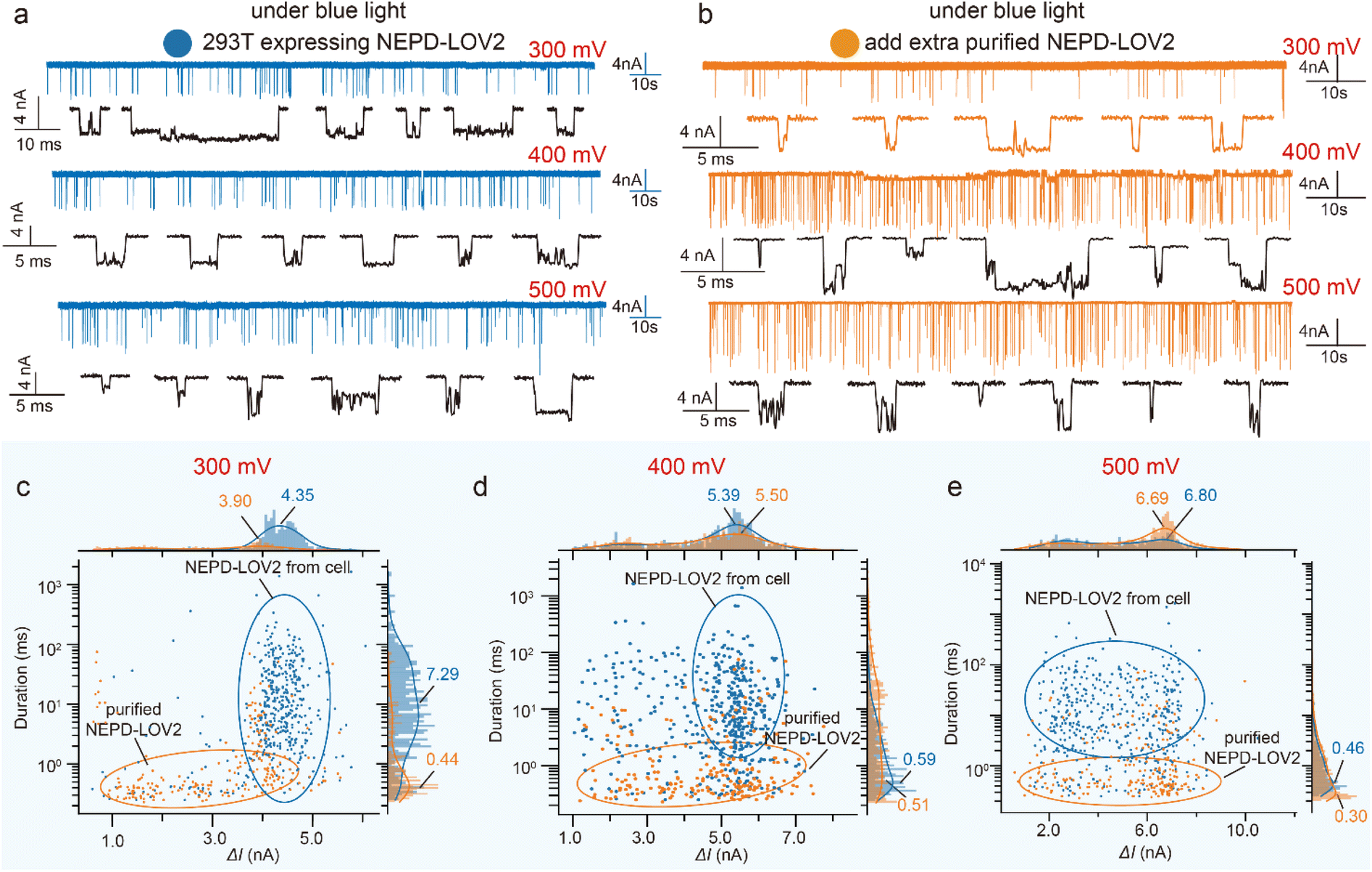 | ||
| Fig. 4 (a) The representative current trace and individual example events for detecting the samples of a single 293T cell expressing NEPD–LOV2 under blue light and (b) after adding the purified NEPD–LOV2 protein, at +300 mV, +400 mV and +500 mV. (c) Scatter plot with the marginal histogram statistics of blockage current versus dwell time under blue light for the single 293T cell samples expressing NEPD–LOV2 and the signals after the additional purified NEPD–LOV2 added at +300 mV, +400 mV (d) and +500 mV (e), respectively. | ||
Conclusion
In this study, we developed a SiNx solid-state nanopore-based approach for the direct analysis of proteins from complex cellular samples. By engineering an electrophoretic driver protein, we enhanced the capture efficiency, enabling the selective detection of target proteins within cellular extracts. Our findings demonstrate that SiNx solid-state nanopores offer a powerful tool for the detection of proteins directly from single cells and provide insights into their dynamic behavior and different conformational states. Importantly, we observed significant differences between proteins measured directly from single cells and their purified counterparts, highlighting the advantage of ex situ single-cell protein analysis in preserving native conformations and molecular interactions.While solid-state nanopores hold great promise, further optimization is required to address challenges such as nanopore size sensitivity and protein capture efficiency. Future developments may involve enhancing nanopore arrays and integrating complementary techniques to improve the precision and throughput of protein analysis, ultimately making solid-state nanopores a more robust tool for studying proteins in their native cellular contexts. Moreover, the ionic current signals from nanopores lack molecular specificity based on chemical composition, primarily relying on surface properties for identification. For specific substances such as DNA, amino acids, and polysaccharides, accurate identification models can be developed through comprehensive measurements and modelling of standard samples. However, in complex protein samples, different proteins can exhibit significant signal degeneracy. One potential solution is to monitor dynamic conformational changes and molecular interactions, using dynamic fingerprint information for more precise single-molecule identification. This ability to detect dynamic conformational changes is a key advantage of solid-state nanopores over other techniques. Although, our previous work demonstrated the capture of transient conformations during peptide folding and unfolding,34 constructing an analytical model based on the dynamic changes of proteins requires a substantial amount of nanopore data from single-molecule proteins. Furthermore, by combining detection techniques like Raman spectroscopy with the efficient and controllable single-molecule capture capabilities of solid-state nanopores, more accurate protein identification can be achieved in single-cell analysis. In summary, our study underscores the potential of SiNx solid-state nanopores for protein detection and characterization in complex biological contexts.
Data availability
All data are available in the main text or ESI.†Author contributions
S.-C. L., Z.-Q. Z., and Y.-T. L. designed the research; Z.-Q. Z. conducted the experiment; Z.-Q. Z. and S.-C. L. analysed data; J. W. and B.-K. X. helped in nanopore fabrication. K.-L. C. helped in cell culture. S.-C. L. and Z.-Q. Z. wrote the original draft of the paper; Y.-T. L., Y.-L. Y. contributed to funding acquisition. All authors reviewed and approved the paper. S.-C. L., and Y.-T. L. supervised the project.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (22204073) and the National Key Research and Development Program of China (2022YFB3205601).Notes and references
- J. Lim, C. Park, M. Kim, H. Kim, J. Kim and D.-S. Lee, Exp. Mol. Med., 2024, 56, 515–526 CrossRef CAS PubMed.
- F. Sun, H. Li, D. Sun, S. Fu, L. Gu, X. Shao, Q. Wang, X. Dong, B. Duan, F. Xing, J. Wu, M. Xiao, F. Zhao, J.-D. J. Han, Q. Liu, X. Fan, C. Li, C. Wang and T. Shi, Sci. China:Life Sci., 2025, 68 DOI:10.1007/s11427-023-2561-0.
- L. Heumos, A. C. Schaar, C. Lance, A. Litinetskaya, F. Drost, L. Zappia, M. D. Lücken, D. C. Strobl, J. Henao, F. Curion, H. B. Schiller and F. J. Theis, Nat. Rev. Genet., 2023, 24, 550–572 CrossRef CAS PubMed.
- Z. Jehan, in Single-Cell Omics, ed. D. Barh and V. Azevedo, Academic Press, 2019, pp. 3–19 Search PubMed.
- G. Misevic, J. Biomed. Res., 2021, 35, 264–276 CrossRef PubMed.
- Single-cell proteomics: challenges and prospects, Nat. Methods, 2023, 20, 317–318 Search PubMed.
- H. M. Bennett, W. Stephenson, C. M. Rose and S. Darmanis, Nat. Methods, 2023, 20, 363–374 CrossRef CAS PubMed.
- J. A. Alfaro, P. Bohländer, M. Dai, M. Filius, C. J. Howard, X. F. van Kooten, S. Ohayon, A. Pomorski, S. Schmid, A. Aksimentiev, E. V. Anslyn, G. Bedran, C. Cao, M. Chinappi, E. Coyaud, C. Dekker, G. Dittmar, N. Drachman, R. Eelkema, D. Goodlett, S. Hentz, U. Kalathiya, N. L. Kelleher, R. T. Kelly, Z. Kelman, S. H. Kim, B. Kuster, D. Rodriguez-Larrea, S. Lindsay, G. Maglia, E. M. Marcotte, J. P. Marino, C. Masselon, M. Mayer, P. Samaras, K. Sarthak, L. Sepiashvili, D. Stein, M. Wanunu, M. Wilhelm, P. Yin, A. Meller and C. Joo, Nat. Methods, 2021, 18, 604–617 CrossRef CAS PubMed.
- J. Swaminathan, A. A. Boulgakov, E. T. Hernandez, A. M. Bardo, J. L. Bachman, J. Marotta, A. M. Johnson, E. V. Anslyn and E. M. Marcotte, Nat. Biotechnol., 2018, 36, 1076–1082 CrossRef CAS PubMed.
- V. Marx, Nat. Methods, 2023, 20, 350–354 CrossRef CAS PubMed.
- S.-E. Ong and M. Mann, Nat. Chem. Biol., 2005, 1, 252–262 CrossRef CAS PubMed.
- C. Lombard-Banek, S. A. Moody and P. Nemes, Angew. Chem., Int. Ed., 2016, 55, 2454–2458 CrossRef CAS PubMed.
- T. K. Cheung, C.-Y. Lee, F. P. Bayer, A. McCoy, B. Kuster and C. M. Rose, Nat. Methods, 2021, 18, 76–83 CrossRef CAS PubMed.
- B. D. Reed, M. J. Meyer, V. Abramzon, O. Ad, O. Ad, P. Adcock, F. R. Ahmad, G. Alppay, J. A. Ball, J. Beach, D. Belhachemi, A. Bellofiore, M. Bellos, J. F. Beltrán, A. Betts, M. W. Bhuiya, K. Blacklock, R. Boer, D. Boisvert, N. D. Brault, A. Buxbaum, S. Caprio, C. Choi, T. D. Christian, R. Clancy, J. Clark, T. Connolly, K. F. Croce, R. Cullen, M. Davey, J. Davidson, M. M. Elshenawy, M. Ferrigno, D. Frier, S. Gudipati, S. Hamill, Z. He, S. Hosali, H. Huang, L. Huang, A. Kabiri, G. Kriger, B. Lathrop, A. Li, P. Lim, S. Liu, F. Luo, C. Lv, X. Ma, E. McCormack, M. Millham, R. Nani, M. Pandey, J. Parillo, G. Patel, D. H. Pike, K. Preston, A. Pichard-Kostuch, K. Rearick, T. Rearick, M. Ribezzi-Crivellari, G. Schmid, J. Schultz, X. Shi, B. Singh, N. Srivastava, S. F. Stewman, T. Thurston, T. R. Thurston, P. Trioli, J. Tullman, X. Wang, Y.-C. Wang, E. A. G. Webster, Z. Zhang, J. Zuniga, S. S. Patel, A. D. Griffiths, A. M. van Oijen, M. McKenna, M. D. Dyer and J. M. Rothberg, Science, 2022, 378, 186–192 CrossRef CAS PubMed.
- J. Swaminathan, A. A. Boulgakov, E. T. Hernandez, A. M. Bardo, J. L. Bachman, J. Marotta, A. M. Johnson, E. V. Anslyn and E. M. Marcotte, Nat. Biotechnol., 2018, 36, 1076–1082 CrossRef CAS PubMed.
- J. A. Alfaro, P. Bohländer, M. Dai, M. Filius, C. J. Howard, X. F. van Kooten, S. Ohayon, A. Pomorski, S. Schmid, A. Aksimentiev, E. V. Anslyn, G. Bedran, C. Cao, M. Chinappi, E. Coyaud, C. Dekker, G. Dittmar, N. Drachman, R. Eelkema, D. Goodlett, S. Hentz, U. Kalathiya, N. L. Kelleher, R. T. Kelly, Z. Kelman, S. H. Kim, B. Kuster, D. Rodriguez-Larrea, S. Lindsay, G. Maglia, E. M. Marcotte, J. P. Marino, C. Masselon, M. Mayer, P. Samaras, K. Sarthak, L. Sepiashvili, D. Stein, M. Wanunu, M. Wilhelm, P. Yin, A. Meller and C. Joo, Nat. Methods, 2021, 18, 604–617 CrossRef CAS PubMed.
- K. Okamoto, M. Hiroshima and Y. Sako, Biophys. Rev., 2018, 10, 317–326 CrossRef CAS PubMed.
- C. Dekker, Nat. Nanotechnol., 2007, 2, 209–215 CrossRef CAS PubMed.
- K. Lee, K.-B. Park, H.-J. Kim, J.-S. Yu, H. Chae, H.-M. Kim and K.-B. Kim, Adv. Mater., 2018, 30, 1704680 CrossRef PubMed.
- J. Li, M. Gershow, D. Stein, E. Brandin and J. A. Golovchenko, Nat. Mater., 2003, 2, 611–615 Search PubMed.
- E. C. Yusko, B. R. Bruhn, O. M. Eggenberger, J. Houghtaling, R. C. Rollings, N. C. Walsh, S. Nandivada, M. Pindrus, A. R. Hall, D. Sept, J. Li, D. S. Kalonia and M. Mayer, Nat. Nanotechnol., 2017, 12, 360–367 Search PubMed.
- J. Houghtaling, C. Ying, O. M. Eggenberger, A. Fennouri, S. Nandivada, M. Acharjee, J. Li, A. R. Hall and M. Mayer, ACS Nano, 2019, 13, 5231–5242 Search PubMed.
- D. Fologea, B. Ledden, D. S. McNabb and J. Li, Appl. Phys. Lett., 2007, 91, 053901 Search PubMed.
- Y. Cai, L. Liang, S. Wang, D. Wang and H. Cui, in 2019 IEEE International Conference on Manipulation, Manufacturing and Measurement on the Nanoscale (3M-NANO), 2019, pp. 257–260 Search PubMed.
- Y. Cai, B. Zhang, L. Liang, S. Wang, L. Zhang, L. Wang, H.-L. Cui, Y. Zhou and D. Wang, Plant Commun., 2021, 2 DOI:10.1016/j.xplc.2020.100106.
- J. Im, S. Lindsay, X. Wang and P. Zhang, ACS Nano, 2019, 13, 6308–6318 CrossRef CAS PubMed.
- Q. Zhang, Y. Cheng, P. Cao and Z. Gu, Chin. Chem. Lett., 2019, 30, 1607–1617 CrossRef CAS.
- D.-K. Kwak, H. Chae, M.-K. Lee, J.-H. Ha, G. Goyal, M. J. Kim, K.-B. Kim and S.-W. Chi, Angew. Chem., Int. Ed., 2016, 55, 5713–5717 CrossRef CAS PubMed.
- S. Rajaram, L. E. Heinrich, J. D. Gordan, J. Avva, K. M. Bonness, A. K. Witkiewicz, J. S. Malter, C. E. Atreya, R. S. Warren, L. F. Wu and S. J. Altschuler, Nat. Methods, 2017, 14, 967–970 CrossRef CAS PubMed.
- Y. Zhang, J. Clausmeyer, B. Babakinejad, A. López Córdoba, T. Ali, A. Shevchuk, Y. Takahashi, P. Novak, C. Edwards, M. Lab, S. Gopal, C. Chiappini, U. Anand, L. Magnani, R. C. Coombes, J. Gorelik, T. Matsue, W. Schuhmann, D. Klenerman, E. V. Sviderskaya and Y. Korchev, ACS Nano, 2016, 10, 3214–3221 CrossRef CAS PubMed.
- Y.-L. Ying, Y.-X. Hu, R. Gao, R.-J. Yu, Z. Gu, L. P. Lee and Y.-T. Long, J. Am. Chem. Soc., 2018, 140, 5385–5392 CrossRef CAS PubMed.
- J. Lv, R.-C. Qian, Y.-X. Hu, S.-C. Liu, Y. Cao, Y.-J. Zheng and Y.-T. Long, Chem. Commun., 2016, 52, 13909–13911 Search PubMed.
- J. Li, D. Stein, C. McMullan, D. Branton, M. J. Aziz and J. A. Golovchenko, Nature, 2001, 412, 166–169 CrossRef CAS PubMed.
- S.-C. Liu, Y.-L. Ying, W.-H. Li, Y.-J. Wan and Y.-T. Long, Chem. Sci., 2021, 12, 3282–3289 RSC.
- D. S. Talaga and J. Li, J. Am. Chem. Soc., 2009, 131, 9287–9297 CrossRef CAS PubMed.
- H. Chae, D.-K. Kwak, M.-K. Lee, S.-W. Chi and K.-B. Kim, Nanoscale, 2018, 10, 17227–17235 RSC.
- J. Houghtaling, C. Ying, O. M. Eggenberger, A. Fennouri, S. Nandivada, M. Acharjee, J. Li, A. R. Hall and M. Mayer, ACS Nano, 2019, 13, 5231–5242 CrossRef CAS PubMed.
- W. Si and A. Aksimentiev, ACS Nano, 2017, 11, 7091–7100 CrossRef CAS PubMed.
- X. Wang, T.-M. Thomas, R. Ren, Y. Zhou, P. Zhang, J. Li, S. Cai, K. Liu, A. P. Ivanov, A. Herrmann and J. B. Edel, J. Am. Chem. Soc., 2023, 145, 6371–6382 CrossRef CAS PubMed.
- S. Abu Jalboush, I. D. Wadsworth, K. Sethi, L. C. Rogers, T. Hollis and A. R. Hall, ACS Sens., 2024, 9, 1602–1610 CrossRef CAS PubMed.
- H. Brinkerhoff, A. S. W. Kang, J. Liu, A. Aksimentiev and C. Dekker, Science, 2021, 374, 1509–1513 CrossRef CAS PubMed.
- J. Y. Y. Sze, A. P. Ivanov, A. E. G. Cass and J. B. Edel, Nat. Commun., 2017, 8, 1552 CrossRef PubMed.
- S.-C. Liu, Y.-L. Ying, W.-H. Li, Y.-J. Wan and Y.-T. Long, Chem. Sci., 2021, 12, 3282–3289 RSC.
- N. A. Kulak, G. Pichler, I. Paron, N. Nagaraj and M. Mann, Nat. Methods, 2014, 11, 319–324 CrossRef CAS PubMed.
- Q. Liu, Y. Ouyang, Y. Wang, S. Zhou, Y. Zhan and L. Wang, Adv. Healthcare Mater., 2025, 2405058 CrossRef PubMed.
- L. Zhao, Y. Deng, Y. Wang, S. Zhou, B. Yin, Y. Chen, Y. Wang, J. Li, L. Wang, Y. Lin and L. Wang, Mater. Today Phys., 2024, 46, 101479 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5sc01764e |
| This journal is © The Royal Society of Chemistry 2025 |