
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDeaminase-driven random mutation enables efficient DNA mutagenesis for protein evolution†
Ying
Hao‡
a,
Tong-Tong
Ji‡
a,
Shu-Yi
Gu‡
b,
Shan
Zhang
a,
Yao-Hua
Gu
b,
Xia
Guo
a,
Li
Zeng
a,
Fang-Yin
Gang
b,
Jun
Xiong
b,
Yu-Qi
Feng
 *a,
Neng-Bin
Xie
*b and
Bi-Feng
Yuan
*abcd
*a,
Neng-Bin
Xie
*b and
Bi-Feng
Yuan
*abcd
aCollege of Chemistry and Molecular Sciences, Department of Radiation and Medical Oncology, Zhongnan Hospital of Wuhan University, Wuhan University, Wuhan 430071, China. E-mail: bfyuan@whu.edu.cn; yqfeng@whu.edu.cn
bDepartment of Occupational and Environmental Health, School of Public Health, Research Center of Public Health, Renmin Hospital of Wuhan University, Wuhan University, Wuhan 430071, China. E-mail: nengbinxie@whu.edu.cn
cHubei Provincial Center for Disease Control and Prevention & NHC Specialty Laboratory of Food Safety Risk Assessment and Standard Development, Wuhan 430079, China
dHubei Key Laboratory of Biomass Resource Chemistry and Environmental Biotechnology, Wuhan University, Wuhan 430071, China
First published on 15th April 2025
Abstract
Protein evolution has emerged as a crucial tool for generating proteins with novel characteristics. A key step in protein evolution is the mutagenesis of protein-coding DNA. Error-prone PCR (epPCR) is a frequently used technique, but its low mutation efficiency often requires multiple rounds of mutagenesis, which can be time-consuming. To address this, we developed a novel DNA mutagenesis strategy termed deaminase-driven random mutation (DRM). DRM utilizes the engineered cytidine deaminase A3A-RL and the engineered adenosine deaminase ABE8e to introduce a broad spectrum of mutations, including C-to-T, G-to-A, A-to-G, and T-to-C, in both the protein-coding strand and the complementary strand. This approach enables the generation of a multitude of DNA mutation types within a single round of mutagenesis, resulting in a higher DNA mutagenic capability than epPCR. The results show that the DRM strategy exhibits a 14.6-fold higher DNA mutation frequency and produces a 27.7-fold greater diversity of mutation types compared to epPCR, enabling a more comprehensive exploration of the genetic landscape. This enhanced mutagenic capability increases the chances of discovering novel and useful mutants. With its ability to produce high-quality DNA products and the superior protein mutant generation capacity, DRM is an attractive tool for researchers seeking to engineer new proteins or improve existing ones.
Introduction
Proteins have been extensively utilized in diverse fields, including scientific research and industrial production.1,2 However, the limitations of natural proteins in meeting the demands of these applications have become increasingly apparent. To overcome these constraints, protein evolution has emerged as a vital tool for generating proteins with novel characteristics, thereby expanding their potential uses.3–5 Protein evolution is a complex, dynamic process by which protein sequences, structures, and functions undergo changes over time, driven by genetic variations, mutations, and natural selection.6,7 This evolutionary process enables proteins to adapt to new environments, acquire new functions, and diverge into distinct families with specialized roles.8,9 A crucial step in protein evolution is the mutagenesis of protein-coding DNA, which introduces genetic variations that can lead to changes in protein sequences, structures and functions.10,11 Through this process, proteins can acquire enhanced stability, altered substrate specificity, or improved catalytic activity.12,13Several technologies have been developed to induce DNA mutagenesis, including radiation mutagenesis, chemical mutagenesis, and biological mutagenesis.14,15 Radiation mutagenesis utilizes high-energy radiations to break chemical bonds in DNA, resulting in a range of mutations, such as base substitutions and deletions.16,17 However, this approach has significant drawbacks, including the requirement for expensive equipment and the potential health risks associated with exposure to high-energy radiation.18 Chemical mutagenesis has been employed in microbial strain improvement and plant breeding, leveraging chemical agents like ethyl methanesulfonate (EMS) and acridine orange to generate DNA mutations.19,20 EMS introduces alkyl groups to DNA bases, leading to base substitutions or frame shifts during replication, while acridine orange can intercalate between DNA bases, causing base insertions or deletions.21–23 Additionally, nucleobase analogs can be incorporated into DNA during replication, resulting in mispairing of nucleobases.24,25 Nevertheless, chemical mutagenesis also poses health risks to researchers and suffers from low mutation efficiency, necessitating multiple rounds of treatment to achieve a diverse range of mutations.26 These limitations render chemical mutagenesis inadequate for meeting the demands of DNA mutagenesis in protein evolution.
Error-prone PCR (epPCR) is a widely utilized biological mutagenesis technique for generating DNA mutations during protein evolution.27,28 This method exploits the inherent error-prone nature of Taq DNA polymerase in the presence of manganese ions (Mn2+), which reduces the enzyme's fidelity and leads to base mutations during PCR amplification.29,30 The frequency of base mutations typically increases with rising concentrations of Mn2+.31 However, excessive Mn2+ can significantly impede PCR amplification efficiency, resulting in low yields of PCR products and limiting the effectiveness of the method. Furthermore, the relatively low mutation efficiency of epPCR often necessitates multiple rounds of mutagenesis to achieve a diverse array of mutation types, which can be time-consuming and labor-intensive.32 This limitation underscores the need for a novel strategy that can efficiently introduce a wide range of mutations into target DNA sequences without compromising PCR yields or posing health risks to researchers.
DNA modifying enzymes, particularly deaminases, have emerged as powerful tools for introducing base mutations into DNA.33–36 For instance, the apolipoprotein B mRNA editing enzyme catalytic subunit 3A (A3A) can deaminate cytosine (C) to uracil (U), resulting in C to thymine (T) mutations in DNA.37–43 Recently, an engineered variant of tRNA-specific adenosine deaminase, ABE8e, was developed to efficiently deaminate adenosine (A) to inosine (I) in DNA, which is then interpreted as guanine (G) during PCR amplification, leading to A-to-G mutations.44–46 Building upon this foundation, our research focused on an engineered A3A variant (A3A-RL), which exhibits comparable deamination activities towards cytosines in diverse sequence contexts.47 By combining the use of cytosine deaminase (A3A-RL) and adenosine deaminase (ABE8e), we can introduce C-to-T, G-to-A, A-to-G, and T-to-C mutations in both the protein-coding strand and the complementary strand, thereby expanding the scope of mutagenesis.
Leveraging the characteristics of A3A-RL and ABE8e, we developed a novel DNA mutagenesis strategy termed deaminase-driven random mutation (DRM) that enables the generation of a multitude of DNA mutation types within a single round of mutagenesis. This approach has the potential to dramatically shorten the time required for protein evolution by introducing a wide range of mutations in a single step, thereby accelerating the discovery of novel proteins with improved or entirely new functions. The DRM strategy offers a powerful tool for protein engineering, allowing researchers to efficiently explore the vast sequence space of proteins and unlock their full potential for various applications in biotechnology, medicine, and beyond.
Experimental methods
Materials and reagents
2′-Deoxynucleoside triphosphates (dATP, dGTP, dCTP, and TTP) were obtained from Sangon Biotech (Shanghai, China). Accurate Taq DNA polymerase was purchased from Accurate Biology (Wuhan, China). Q5 High-Fidelity master mix was purchased from New England Biolabs (Ipswich, MA, USA). Tween-20, manganese(II) chloride (MnCl2), magnesium chloride (MgCl2), ethylene diamine tetraacetic acid (EDTA) and Tri (hydroxymethyl) aminomethane hydrochloride (Tris–HCl) were purchased from Sinopharm Chemical Reagent Co., Ltd (Shanghai, China). All the solvents and chemical reagents were of analytical grade.Preparation of DNA template for epPCR and DRM
A 321-bp double-stranded DNA (MT-1) was used as a template to evaluate the DNA mutagenic capabilities of epPCR and DRM. To synthesize MT-1, 1 ng of pUC19 plasmid was used as a template for PCR amplification. The PCR reaction was performed in a 50 μL solution containing 25 μL of Q5 high-fidelity master mix (New England Biolabs) and 2 μL each of MT-F and MT-R primers (10 μM). The sequences of MT-1 and the primers are listed in Table S1.† The PCR program consisted of an initial denaturation at 95 °C for 3 min, followed by 30 cycles of 95 °C for 30 s, 65 °C for 30 s, and 68 °C for 30 s, with a final extension at 68 °C for 10 min. The PCR products were separated by agarose gel electrophoresis and then purified using an agarose gel extraction kit (Zymo Research).Expression and purification of A3A-RL and ABE8e proteins
To produce A3A-RL proteins, the coding sequence of A3A-RL was cloned into the pET-41a (+) plasmid between the XbaI and XhoI restriction sites, with a human rhinovirus 3C protease (HRV 3C) digestion site inserted between the glutathione S-transferase (GST) tag and the A3A-RL protein (Fig. S1†). The resulting plasmid (pET-41a-A3A-RL) was transformed into E. coli BL21(DE3) pLysS cells for protein expression. The coding sequence and amino acid composition of A3A-RL are listed in Tables S2 and S3,† respectively. Protein expression, purification, and culturing of transformed E. coli cells were performed as previously described.48–51 The detailed procedures are provided in the ESI.† Purified A3A-RL proteins were stored at −80 °C in a solution containing 50 mM NaCl, 50 mM Tris–HCl (pH 7.5), 0.5 mM dithiothreitol, 0.01 mM EDTA, and 0.01% Tween-20.For ABE8e protein production, the coding sequence of ABE8e was cloned into the pET-49b plasmid between the MluI and XhoI restriction sites, with an HRV 3C digestion site inserted between the GST tag and the ABE8e protein (Fig. S2†). The resulting plasmid (pET-49b-ABE8e) was transformed into E. coli BL21(DE3) pLysS cells for protein expression. The coding sequence and amino acid composition of ABE8e are listed in Tables S2 and S3,† respectively. Protein expression, purification, and culturing of transformed E. coli cells were performed as previously described.52,53 The detailed procedures are provided in the ESI.† Purified ABE8e proteins were stored at −80 °C in a solution containing 50 mM Tris–HCl (pH 7.5), 40% glycerol, and 0.5 mM dithiothreitol. The purified proteins were analyzed by SDS-PAGE (Fig. S2 and S3†) and their concentrations were determined using a BCA protein assay kit (Beyotime, Shanghai, China).
Optimization of the concentrations of Mn2+ in epPCR
To determine the optimal concentration of Mn2+ for epPCR, six sets of experiments were conducted under identical conditions, with the exception of varying Mn2+ concentrations. The PCR amplification was performed in a 50 μL reaction mixture containing 5 μL of 10× buffer, 0.2 mM dNTPs, 0.2 mM each of dATP and dGTP, 1 unit of Accurate Taq DNA polymerase, 0.4 μM MT-F primer, and 0.4 μM MT-R primer. The primer sequences are provided in Table S1.† The reaction mixture also included 0.5 mM MgCl2 and varying amounts of MnCl2, resulting in final Mn2+ concentrations ranging from 0 to 0.9 mM. The PCR amplification protocol consisted of an initial denaturation step at 95 °C for 10 min, followed by 30 cycles of denaturation at 95 °C for 30 s, annealing at 65 °C for 30 s, and extension at 72 °C for 30 s. A final extension step was performed at 72 °C for 10 min. The resulting PCR products were analyzed by agarose gel electrophoresis.Optimization of the deamination reaction in DRM using A3A-RL or ABE8e
To achieve an optimal mutation rate and generate diverse mutation types in DRM, we optimized the deaminase concentration and deamination reaction time. Typically, 40 ng of MT-1 double-stranded DNA (dsDNA) was first denatured to single-stranded DNA (ssDNA) by heating to 95 °C for 10 min in a 20% dimethylsulfoxide (DMSO) solution and then chilled in ice water. The deamination reaction using different concentrations of A3A-RL was then carried out at 37 °C for various times in a 20 μL solution containing 20 mM 2-morpholinoethanesulfonate (MES) (pH 6.5), 2 μL of DMSO, and 0.1% Triton X-100. The deamination reaction was terminated by heating at 95 °C for 10 min. Subsequently, 5 ng of deaminase-treated DNA was used as the template for PCR amplification. PCR amplification was performed in a 50 μL solution containing 10 μL of 5× reaction buffer, 1 unit of Accurate Taq DNA polymerase, 0.2 mM dNTP, 0.4 μM MT-F primer, and 0.4 μM MT-R primer (Table S1†). The PCR reaction consisted of an initial denaturation at 95 °C for 5 min, 30 cycles of 95 °C for 30 s, 65 °C for 30 s, 72 °C for 30 s, and an additional 10 min of elongation at 72 °C.Similarly, 40 ng of denatured MT-1 dsDNA was used as a template in the deamination reaction using ABE8e. Briefly, the ABE8e deamination reaction was performed in a 10 μL solution containing 50 mM Tris–HCl (pH 7.5), 10 mM DTT, 20% DMSO, and different concentrations of ABE8e. The mixtures were incubated at 37 °C for various times and quenched by heating at 95 °C for 10 min. Then, 5 ng of ABE8e-treated DNA was amplified using PCR, as described above.
The resulting PCR products were subjected to colony sequencing to determine the mutation rate and mutation types in DRM. Colony sequencing was performed according to previous studies.54,55 Briefly, PCR products were ligated into the pClone007 Versatile Simple vector (Tsingke, Beijing, China). The resulting vectors were then transformed into E. coli DH5α cells. Individual clones were randomly picked, lysed in TE buffer, amplified by PCR using MT-F and MT-R primers (Table S1†), and the PCR products were then sequenced using an ABI3700 (Applied Biosystems, Inc.). Ten positive clones from each sample were picked and subjected to sequencing.
High-throughput sequencing library construction for epPCR and DRM
For the epPCR method, a 50 μL reaction mixture was prepared, containing 40 ng of MT-1 dsDNA, 5 μL of 10× buffer, 0.5 mM MgCl2, 0.7 mM MnCl2, 0.4 mM dATP and dGTP, 0.2 mM dCTP and dTTP, 1 unit of Accurate Taq DNA polymerase, 0.4 μM MT-F primer, and 0.4 μM MT-R primer (Table S1†). The PCR amplification program consisted of an initial denaturation at 95 °C for 10 min, followed by 30 cycles of 95 °C for 30 s, 65 °C for 30 s, 72 °C for 30 s, and a final elongation at 72 °C for 10 min.For the DRM method, 40 ng of MT-1 dsDNA was denatured to ssDNA by heating to 95 °C for 10 min in a 20% DMSO solution and then chilled in ice water. The deamination reaction was carried out using 2 μM A3A-RL at 37 °C for 3 h in a 20 μL solution containing 20 mM MES (pH 6.5), 2 μL of DMSO, and 0.1% Triton X-100. The deamination reaction was terminated by incubating at 95 °C for 10 min. Subsequently, 5 ng of deaminase-treated DNA was used as the template for PCR amplification. Next, 40 ng of PCR products (amplified from A3A-RL-treated MT-1 DNA) were used for the ABE8e deamination reaction, which was carried out in a 10 μL solution containing 50 mM Tris–HCl (pH 7.5), 10 mM DTT, 20% DMSO, and 8 μM ABE8e. The reaction mixture was incubated at 37 °C for 3 h and then quenched by heating at 95 °C for 10 min. Finally, 5 ng of ABE8e-treated DNA was amplified using PCR.
The resulting PCR products from either epPCR or DRM were end-repaired and adenylated using a Hieff NGS Ultima Endprep Mix kit (Yeasen Biotechnology Co., Ltd, Shanghai). The pre-T5 and pre-T7 (Table S1†) was then ligated to both ends of the repaired DNA using a Hieff NGS Ultima DNA Ligation Module kit (Yeasen), and the resulting DNA was purified using 0.9× KAPA Pure beads. The DNA products were then amplified by PCR with 10 cycles using P5-index primer and P7-index primer (Table S1†). The PCR amplification was carried out in a 50 μL solution containing 25 μL of Q5 high-fidelity master mix (New England Biolabs), 2 μL each of P5-index primer and P7-index primer (10 μM). The PCR amplification program consisted of an initial denaturation at 95 °C for 10 min, followed by 10 cycles of 95 °C for 30 s, 65 °C for 30 s, and 68 °C for 30 s, and a final elongation at 68 °C for 10 min. The PCR products were purified with 0.8× KAPA Pure beads and examined using 1.5% agarose gel electrophoresis. Library quality was assessed on an Agilent Bioanalyzer 2100 system. Finally, the library was sequenced on an Illumina NovaSeq 6000 platform (Gentlegen gene Co., Ltd, Jiangsu, China). The schematic diagram of library preparation for epPCR and DRM are shown in Fig. S3.†
High-throughput sequencing data analysis
High-throughput sequencing data analysis was conducted in a Linux environment utilizing Miniconda for package and environment management. FastQC (v0.12.1) was employed to assess the quality of the raw sequencing data, providing metrics such as base quality, GC content, and sequence duplication rates. Low-quality bases and adapter sequences were trimmed using Trim Galore (v0.6.10). The cleaned reads were then aligned to the reference sequence with BWA MEM (v0.7.18). Sequencing depth was calculated using the depth function of Samtools (v1.16.1). Following sequence merging with FLASH (v1.2.11), the proportions of mutation bases and analyses of base distribution and variant frequencies were performed in R (v4.3.2).Random mutation of the coding DNA of EGFP
To introduce mutations into the coding DNA of EGFP, both epPCR and DRM were employed, and 40 ng of EGFP DNA was used as template. For epPCR, PCR amplification was performed in a 50 μL reaction mixture containing 5 μL of 10× buffer, 0.5 mM MgCl2, 0.7 mM MnCl2, 0.4 mM dATP and dGTP, 0.2 mM dCTP and dTTP, 1 unit of Accurate Taq DNA polymerase, 0.4 μM of EGFP-F primer, and EGFP-R primer (Table S1†). The PCR program consisted of an initial denaturation at 95 °C for 10 min, followed by 30 cycles of 95 °C for 30 s, 65 °C for 30 s, and 72 °C for 75 s, with a final elongation at 72 °C for 10 min. The DRM protocol for EGFP DNA mutagenesis was identical to that used for MT-1 DNA mutagenesis.The resulting PCR products were purified using 0.6× KAPA Pure beads and then cloned into the pETDuet-1 plasmid between the SpeI and BamHI restriction enzyme digestion sites (Fig. S4†). The recombinant plasmid was transformed into E. coli DH5α strains, which were then cultured on LB agar plates supplemented with ampicillin at 37 °C for 12 h. A total of 400 clones were randomly selected and further cultured in LB medium containing 10 g per L tryptone, 5 g per L yeast extract, and 10 g per L NaCl. The cultures were grown at 37 °C with shaking at 180 rpm in the presence of 10 μg per mL ampicillin. When the optical density at 600 nm (OD600) reached 0.5, 1 mM IPTG was added to induce EGFP protein expression. The induction was carried out at 20 °C for 16 h. The E. coli cells were harvested by centrifugation at 5000 g for 5 min and washed twice with PBS buffer. Equal amounts of harvested cells were resuspended in PBS buffer and transferred to 96-well plates. The green fluorescence intensity was measured using a MD SpectraMax i3x system (Molecular Devices).
Results and discussion
Principle of the DRM method
epPCR has been frequently used to introduce random mutations into protein-coding DNA for protein evolution. However, the low mutation rate of epPCR often requires multiple rounds of mutation to generate diverse mutation types. Therefore, there is a need for a more efficient mutation strategy. Here, we propose a deaminase-driven random mutation (DRM) strategy that combines the deaminase activities of A3A-RL and ABE8e to generate diverse mutation types in protein evolution. In DRM approach, A3A-RL randomly deaminates cytosines in both the protein-coding strand and its complementary strand, resulting in C-to-U transition in both strands (Fig. 1A). After PCR amplification, these mutations give rise to C-to-T and G-to-A mutations in the protein-coding strand (Fig. 1B). Simultaneously, ABE8e randomly deaminates adenines in both strands, resulting in A-to-I transition in both strands (Fig. 1A). After PCR amplification, these mutations give rise to A-to-G and T-to-C mutations in the protein-coding strand (Fig. 1B). Therefore, the combined use of A3A-RL and ABE8e enables the generation of C-to-T, G-to-A, A-to-G, and T-to-C mutations in the protein-coding strand, providing a versatile tool for introducing diverse DNA mutation types. | ||
| Fig. 1 Principle of DRM method. (A) A3A-RL catalyzes the deamination of C to U, which is paired with A during DNA replication, resulting in C-to-T mutation. ABE8e catalyzes the deamination of A to I, which is paired with C, leading to A-to-G mutation. (B) Schematic illustration of the DRM method. In the DRM process, dsDNA is first denatured at 95 °C, resulting in the generation of ssDNA. The ssDNA is then subjected to successive deamination by A3A-RL and ABE8e, which catalyze the conversion of C to U and A to I, respectively. This leads to the formation of U and I in both strands of the ssDNA, ultimately resulting in C-to-T, G-to-A, A-to-G, and T-to-C mutations during subsequent DNA replication. | ||
Development of DRM
To achieve random mutation using the DRM strategy, it is essential to optimize the deamination reaction to avoid excessive deamination of cytosines and adenines. To this end, we investigated the impact of different concentrations of A3A-RL and ABE8e, as well as varying reaction durations, on the deamination of cytosines and adenines within the MT-1 DNA sequence.For A3A-RL, 40 ng of MT-1 DNA was treated with different concentrations of the enzyme for various durations at 37 °C. The deaminase-treated DNAs were amplified and subjected to colony sequencing (Fig. 2A). The results showed that the average number of deaminated cytosines increased with the increased A3A-RL concentration and reaction durations (Fig. 2B and C). Specifically, treatment with 2 μM A3A-RL resulted in an average of 2.6, 4.3, 10.9, and 19.6 deaminated cytosines after 1 h, 2 h, 3 h, and 4 h treatment, respectively (Fig. 2B). When treated with different concentrations of A3A-RL for 3 h, the average number of deaminated cytosines was 3.1, 4.6, 14.9, and 30 for 0.02 μM, 0.2 μM, 2 μM, and 20 μM A3A-RL, respectively (Fig. 2C). For ABE8e, 40 ng of MT-1 DNA was treated with different concentrations of the enzyme for various durations at 37 °C (Fig. 2A). The results showed that the average number of deaminated adenines increased with the increased ABE8e concentration and reaction durations (Fig. 2D and E). Specifically, treatment with 8 μM ABE8e resulted in an average of 1.4, 3.0, 13.6, and 20.7 deaminated adenines after 1 h, 2 h, 3 h, and 4 h treatment, respectively (Fig. 2D). When treated with different concentrations of ABE8e for 3 h, the average number of deaminated adenines was 1.7, 2.3, 4.7, and 13.2 for 0.008 μM, 0.08 μM, 0.8 μM, and 8 μM ABE8e, respectively (Fig. 2E).
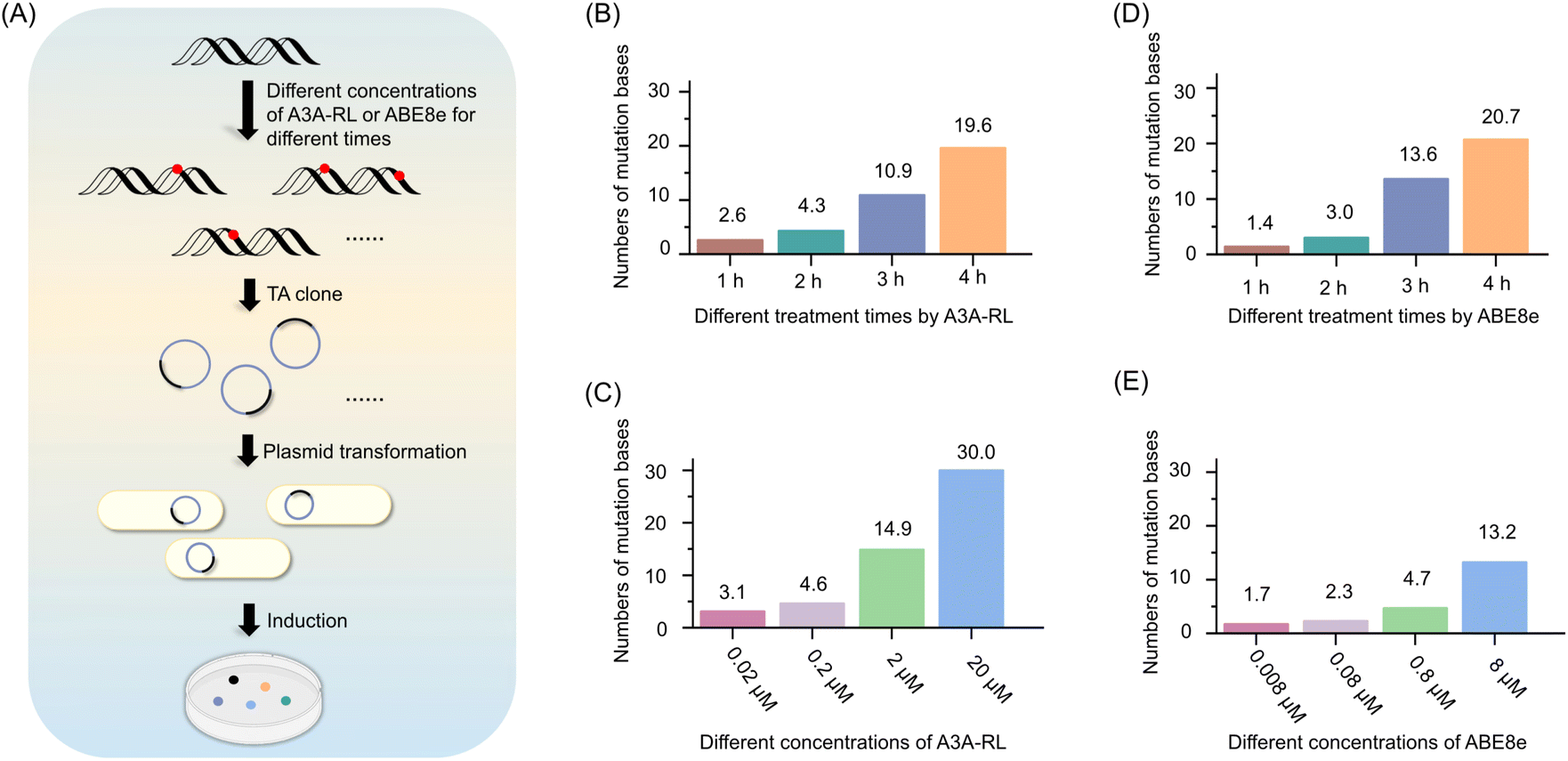 | ||
| Fig. 2 Assessing the impact of A3A-RL and ABE8e concentrations and reaction times on cytosine and adenine deamination by colony sequencing. (A) Schematic illustration for the assessment by colony sequencing. (B) The number of mutations with A3A-RL treatment for different durations. (C) The number of mutations with A3A-RL treatment for different concentrations. (D) The number of mutations with ABE8e treatment for different durations. (E) The number of mutations with ABE8e treatment for different concentrations. MT-1 DNA was used for the assessment. | ||
To avoid excessive deamination of cytosines and adenines in the DRM strategy, we conducted the deamination reactions with 2 μM of A3A-RL and 8 μM of ABE8e for 3 h at 37 °C. These conditions were chosen to balance the level of deamination and minimize the risk of excessive mutations. Moreover, it is worth noting that the mutation rate can be controlled by adjusting either the deaminase concentration or the deamination reaction time, highlighting the flexibility of the DRM strategy as an approach for DNA mutagenesis.
Optimization of Mn2+ concentration in epPCR
In epPCR, Mn2+ is added to reduce the fidelity of Taq DNA polymerase. Mn2+ can affect the size of the polymerase's catalytic center and destabilize the interaction between the polymerase and DNA, resulting in more frequent base mismatches.56,57 Traditionally, the base mutation rate increases with higher concentrations of Mn2+.56,57 However, excessive Mn2+ can inhibit PCR amplification in epPCR. To maximize mutation frequency while ensuring adequate PCR product yield, we optimized the Mn2+ concentration used in epPCR. A series of PCR reactions were conducted under identical conditions with varying Mn2+ concentrations (Fig. 3A). We also utilized A3A-RL and ABE8e to conduct the DRM under optimized conditions (Fig. 3B). The results indicated a progressive decrease in PCR products as Mn2+ concentration increased (Fig. 3C). Notably, no PCR product was detected at 0.9 mM Mn2+ (Fig. 3C). Consequently, 0.7 mM Mn2+ was selected as the optimal concentration, balancing mutation rate and detectable PCR product yield. In contrast, the deamination reactions in the DRM method did not adversely affect PCR amplification, producing more PCR products than epPCR using 0.7 mM Mn2+ (Fig. 3C and D).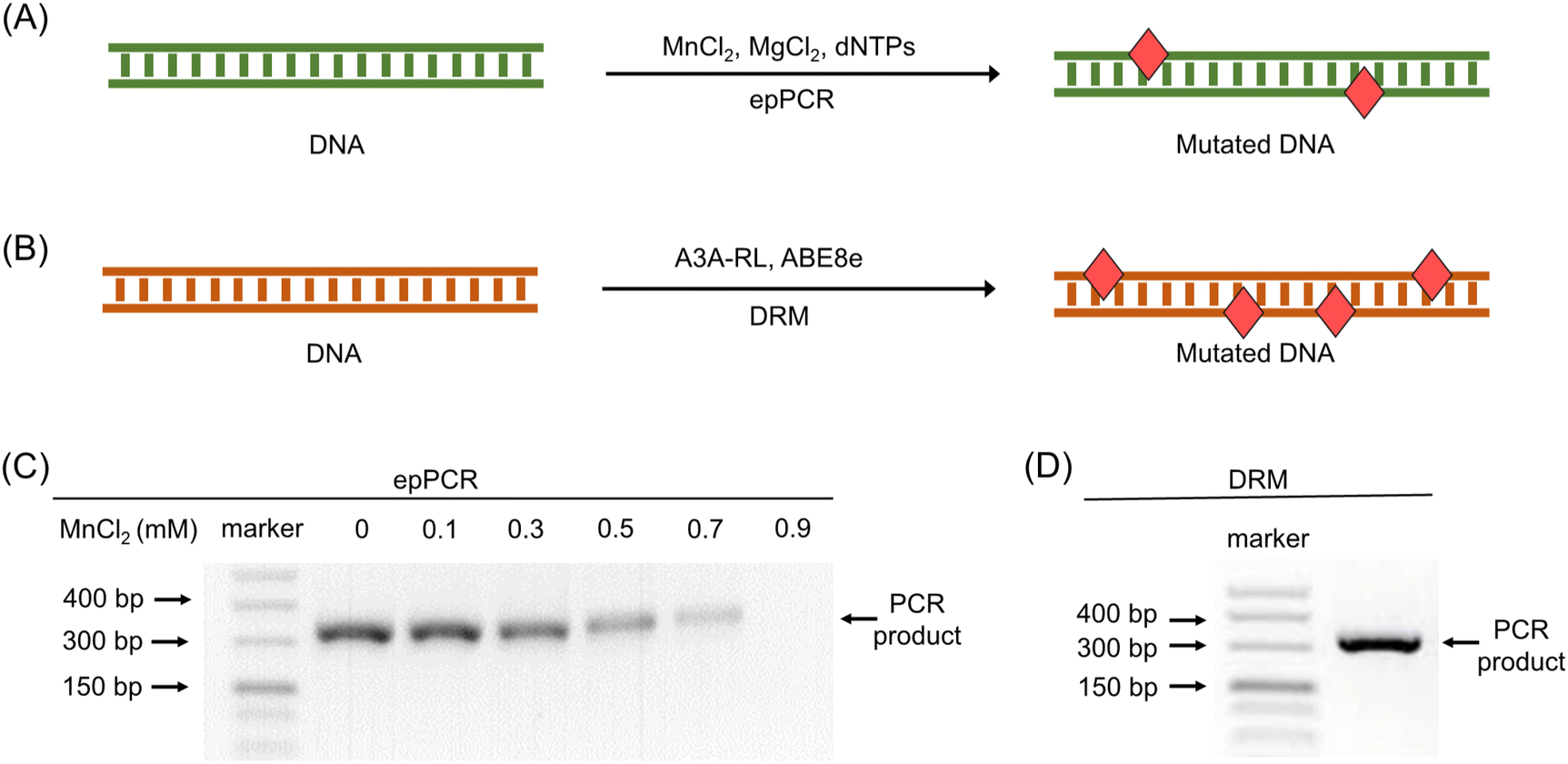 | ||
| Fig. 3 Optimization of Mn2+ concentration in epPCR. (A) Schematic illustration for the reaction of epPCR. (B) Schematic illustration for the reaction of DRM. (C) Evaluation of the PCR products in epPCR using different concentrations of Mn2+ in the PCR reaction. (D) Evaluation of the PCR products in DRM. | ||
Mutation frequencies produced by epPCR and DRM
To assess the DNA mutagenic capabilities of epPCR and DRM, we subjected 40 ng of MT-1 DNA to both methods (Fig. 4A). The resulting PCR products were used for library construction and analyzed via high-throughput sequencing (Fig. S3†). Both methods yielded comparable sequencing depths, with approximately 2 million clean reads each (Fig. S5†). | ||
| Fig. 4 Comparison of the mutation frequencies produced by epPCR and DRM through high-throughput sequencing analysis. (A) Schematic illustration of the DNA mutation frequencies produced in epPCR and DRM. (B) The mutation frequencies generated by epPCR and DRM. (C–E) The mutation frequencies generated by epPCR from three parallel experiments. (F–H) The mutation frequencies generated by DRM from three parallel experiments. | ||
The mutation frequency, defined as the proportion of reads containing mutated bases relative to the total number of reads, was used to compare the DNA mutagenic capabilities of epPCR and DRM. The epPCR method yielded a relatively low mutation frequency, with approximately 5.0% of PCR products harboring mutated bases, corresponding to around 0.1 million mutants (Fig. 4B). Three parallel experiments using epPCR resulted in mutation frequencies of 4.95%, 4.29%, and 5.76% (Fig. 4C–E). In contrast, the DRM method exhibited a significantly higher mutation frequency, with 73.12% of PCR products containing mutated bases, equivalent to approximately 1.46 million mutants (Fig. 4B). Three parallel experiments using DRM yielded mutation frequencies of 72.55%, 74.20%, and 72.60% (Fig. 4F–H). These results demonstrate that DRM generates a substantially larger proportion of PCR products with base mutations compared to epPCR.
Mutation types produced by epPCR and DRM
We further analyzed the DNA mutation types generated by epPCR and DRM to gain a deeper understanding of the mutagenic capabilities of these methods. The mutation type is defined as the number of reads with unique base mutation patterns, providing insight into the diversity of mutations introduced by each method (Fig. 5A). The epPCR method generated a limited number of mutation types, with a total of 631 different mutation types (Fig. 5B). We performed three parallel experiments, which yielded 348, 970, and 574 mutation types, respectively (Fig. 5C). Although there was some variation in the number of mutation types between these experiments, the overall number of mutation types remained relatively low. In contrast, the DRM method exhibited a significantly higher capacity for generating diverse mutation types. Approximately 17![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 211 different mutation types were identified, indicating a much broader range of mutagenic outcomes compared to epPCR (Fig. 5B). We also conducted three parallel experiments using the DRM method, which resulted in 13510, 18752, and 19370 mutation types, respectively (Fig. 5D). The consistency of these results across multiple experiments underscores the robustness of the DRM method in introducing a wide array of mutation types.
211 different mutation types were identified, indicating a much broader range of mutagenic outcomes compared to epPCR (Fig. 5B). We also conducted three parallel experiments using the DRM method, which resulted in 13510, 18752, and 19370 mutation types, respectively (Fig. 5D). The consistency of these results across multiple experiments underscores the robustness of the DRM method in introducing a wide array of mutation types.
 | ||
| Fig. 5 Comparison of the mutation types produced by epPCR and DRM through high-throughput sequencing analysis. (A) Schematic illustration of the DNA mutation types produced in epPCR and DRM. (B) The numbers of mutation types generated by epPCR and DRM. (C) The numbers of mutation types generated by epPCR from three parallel experiments. (D) The numbers of mutation types generated by DRM from three parallel experiments. | ||
These results collectively demonstrate that DRM not only generates a higher frequency of mutations but also introduces a significantly more diverse range of mutation types compared to epPCR. This enhanced mutagenic capability makes DRM a more versatile tool for applications requiring the introduction of a broad spectrum of genetic variations.
Mutation distribution by epPCR and DRM
We conducted a comprehensive examination of the distribution of base mutations introduced by both the epPCR and DRM methods. The results showed clear differences in the mutational profiles generated by these two approaches. The epPCR method was found to primarily introduce mutants carrying a relatively small number of base mutations, with the majority of mutants harboring between 1 and 9 base mutations (Fig. 6A). Specifically, the percentage of mutants containing between 1 and 3 base mutations accounts for approximately 78.56% of the total number of mutants. This suggests that epPCR tends to introduce mutations in a more targeted and limited manner. In contrast, the DRM method introduced a much broader range of base mutations, with mutants carrying from 1 to 75 base mutations (Fig. 6B). This extensive range of mutational outcomes indicates that DRM is capable of introducing a wide array of genetic changes, from subtle single-base mutations to more profound multi-base mutations. The higher number of base mutations observed in DRM is likely a key contributor to its greater diversity of mutations compared to epPCR, as it allows for a more comprehensive exploration of the genetic landscape. To validate the consistency of our results, we conducted three parallel experiments, which yielded similar results, thereby confirming the reproducibility of our observations (Fig. S6 and S7†). | ||
| Fig. 6 Comparison of the mutation distributions by epPCR and DRM. (A) The distribution of mutants with various mutated bases generated by epPCR. The epPCR method was found to primarily introduce mutants carrying a relatively small number of base mutations, with the majority of mutants harboring between 1 and 9 base mutations. (B) The distribution of mutants with various mutated bases generated by DRM. The DRM method introduced a much broader range of base mutations, with mutants carrying from 1 to 75 base mutations. | ||
Overall, the results demonstrate that while both epPCR and DRM can introduce mutations, DRM generates a significantly broader and more diverse spectrum of mutations due to its ability to introduce a wider range of base mutations. This highlights the potential of DRM as a powerful tool for exploring the vast genetic landscape and uncovering novel genetic variants with potential applications in various fields.
EGFP mutants produced by epPCR and DRM
To assess the efficiency of epPCR and DRM in generating protein mutants, we randomly mutated the EGFP coding DNA using both methods and compared the fluorescence intensity of E. coli cells expressing EGFP mutants to that of cells expressing wild-type EGFP protein (Fig. 7A). Mutants with fluorescence differing by more than 10% from the wild-type EGFP protein were considered true mutants.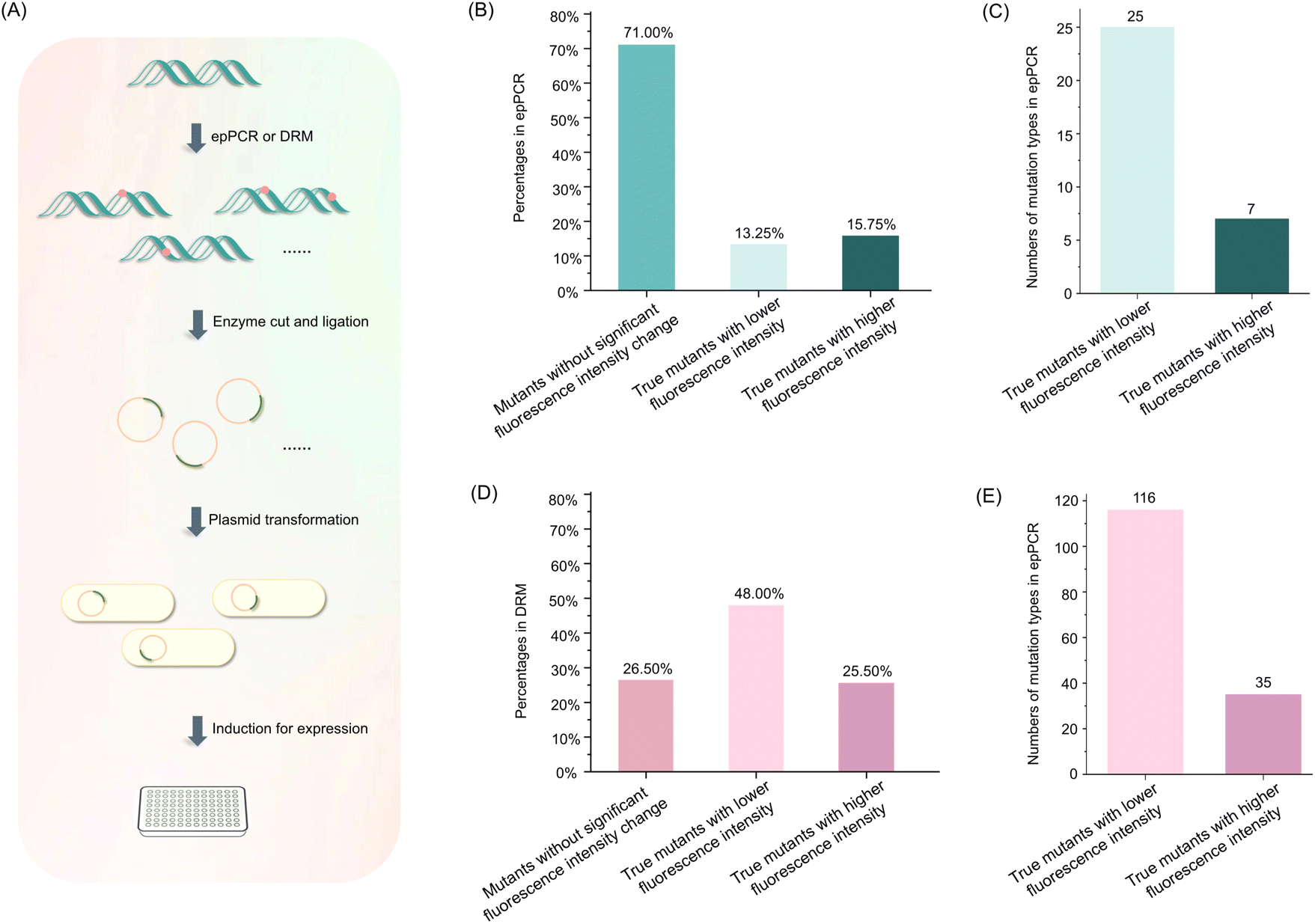 | ||
| Fig. 7 Evaluation of the EGFP mutants produced by epPCR and DRM. (A) The Schematic illustration for evaluation of the EGFP mutants produced by epPCR and DRM. (B) Percentages of true EGFP mutants produced by epPCR method. (C) Numbers of mutation types of true mutants produced by epPCR method. (D) Percentages of true EGFP mutants produced by DRM method. Mutants with fluorescence differing by more than 10% from the wild-type EGFP protein were considered true mutants. (E) Numbers of mutation types of true mutants produced by DRM method. | ||
The results revealed significant differences in the efficiency of epPCR and DRM in generating true protein mutants. Among the 400 mutants analyzed, 13.25% of EGFP mutants generated by epPCR exhibited lower fluorescence, while 15.75% displayed higher fluorescence, resulting in a total of 116 true mutants and 32 mutation types (Fig. 7B and C). The 32 mutation types include 25 mutants with lower fluorescence intensity and 7 with higher fluorescence intensity (Fig. 7C). In contrast, the DRM method yielded a substantially higher number of mutants, with 48% of EGFP mutants showing lower fluorescence and 25.5% exhibiting higher fluorescence, resulting in 294 true mutants and 151 mutation types (Fig. 7D and E). The 151 mutation types include 116 mutants with lower fluorescence intensity and 35 with higher fluorescence intensity (Fig. 7E). The fluorescence intensity of each EGFP mutant is depicted in Fig. S8.† The sequences of different mutation types are listed in Table S4.† Among the 400 clones analyzed, EGFP exhibited a mutation frequency of 32.25% with epPCR, while a significantly higher mutation frequency of 77.75% was observed with DRM (Fig. S9†). Notably, all EGFP mutants, whether exhibiting lower or higher fluorescence than wild-type EGFP from both epPCR and DRM methods, demonstrated a 100% mutation frequency (Fig. S9†). In addition, we did not observe that the cultures expressing EGFP mutants exhibited fluorescence colors other than green. This clear difference in the number of true mutants and mutation types generated by each method underscores the superior efficiency of DRM in producing protein mutants.
Besides epPCR, other DNA mutagenesis methods like DNA shuffling and Sequence saturation mutagenesis (SeSaM) have been developed (Fig. S10†). The DNA shuffling method involves fragmenting DNA with DNase I, followed by reassembly through self-priming PCR, which can introduce frame shifts, insertions, or deletions.58 However, DNase I shows sequence bias, preferring cleavage near pyrimidine bases, which limits the diversity of recombined products.59 The SeSaM approach fragments DNA into random-length pieces and then adds deoxyinosine to the 3′-termini of the fragments.60 During PCR, the deoxyinosine is replaced by one of the four standard nucleotides, generating base mutations. However, this method also exhibits bias, as most mutations occur at adenine sites, reducing its effectiveness in generating mutations at other base positions.
Compared to other methods, DRM offers several advantages in DNA mutagenesis. Firstly, DRM can generate sufficient PCR products without the interference of Mn2+, which can hinder PCR amplification in epPCR and lead to lower yields. Secondly, DRM exhibits a much higher DNA mutagenic capability than epPCR, resulting in an increased mutation frequency and a greater diversity of DNA mutation types. This enhanced mutagenic capability allows for a more comprehensive exploration of the genetic landscape, increasing the chances of discovering novel and useful mutants. Thirdly, DRM's protein mutant generation capability is superior to that of epPCR, reducing the time required for protein evolution and enabling the rapid development of new proteins with desired properties. Fourthly, the DRM method allows for the modulation of the DNA mutation rate by adjusting the concentrations and reaction times of deaminases, offering a flexible approach to DNA mutagenesis. It should be noted that the DRM method requires two deaminases of A3A-RL and ABE8e, necessitating their expression and purification. Additionally, since A3A-RL and ABE8e treatments need be performed sequentially, further efforts are needed to enable the deamination reaction to be completed in a single step, which would simplify the overall procedure.
Overall, the advantages of DRM make it a more effective method for generating protein mutants. The ability to produce high-quality PCR products, coupled with its enhanced mutagenic capability and superior protein mutant generation capacity, renders DRM an attractive tool for researchers seeking to engineer new proteins or improve existing ones. By leveraging the strengths of DRM, researchers can accelerate the pace of protein evolution, driving innovation in fields such as biotechnology, medicine, and synthetic biology.
Conclusion
In summary, we developed a novel method for random DNA mutagenesis, termed deaminase-driven random mutation (DRM), which leverages the deamination activities of A3A-RL and ABE8e. The combined use of these two enzymes enables the introduction of C-to-T, G-to-A, A-to-G, and T-to-C mutations, allowing all four bases (A, G, C, and T) to be mutated. Compared to other DNA mutagenesis methods, DRM exhibits a significantly higher DNA mutagenic capability, resulting in an increased mutation frequency and a greater diversity of DNA mutation types. Furthermore, DRM generates a broader spectrum of mutations, enabling the generation of a multitude of DNA mutation types within a single round of mutagenesis. This presents a versatile and efficient strategy for DNA mutagenesis in protein evolution. By harnessing the power of DRM, researchers can accelerate protein evolution, driving innovation and advancements in fields such as biotechnology, medicine, and synthetic biology.Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
Bi-Feng Yuan: writing – review & editing, investigation, supervision, conceptualization, funding acquisition. Neng-Bin Xie: writing – review & editing, investigation, supervision, conceptualization. Yu-Qi Feng: investigation, supervision, conceptualization. Ying Hao: writing – original draft, methodology, formal analysis, data curation, conceptualization. Tong-Tong Ji: methodology, formal analysis, data curation, conceptualization. Shu-Yi Gu: formal analysis, data curation, conceptualization. Shan Zhang: methodology, data curation. Yao-Hua Gu: data curation. Xia Guo: data curation. Li Zeng: data curation. Fang-Yin Gang: data curation. Jun Xiong: data curation.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
The work is supported by the National Natural Science Foundation of China (22277093, 22207090), the Fundamental Research Funds for the Central Universities (No. 2042024kf0022), and the Key Research and Development Project of Hubei Province (2023BCB094).References
- A. R. Bergeson and H. S. Alper, Trends Biochem. Sci., 2024, 49, 955–968 CrossRef CAS.
- Q. Fu, J. Zhao, S. Rong, Y. Han, F. Liu, Q. Chu, S. Wang and S. Chen, J. Agric. Food Chem., 2023, 71, 15429–15444 CrossRef CAS PubMed.
- R. Buller, S. Lutz, R. J. Kazlauskas, R. Snajdrova, J. C. Moore and U. T. Bornscheuer, Science, 2023, 382, eadh8615 CrossRef CAS.
- Y. Wang, P. Xue, M. Cao, T. Yu, S. T. Lane and H. Zhao, Chem. Rev., 2021, 121, 12384–12444 CrossRef CAS PubMed.
- S. Zhang, J. Zhu, S. Fan, W. Xie, Z. Yang and T. Si, Chem. Sci., 2022, 13, 7581–7586 RSC.
- E. A. DeBenedictis, E. J. Chory, D. W. Gretton, B. Wang, S. Golas and K. M. Esvelt, Nat. Methods, 2022, 19, 55–64 CrossRef CAS PubMed.
- J. Yang, F. Z. Li and F. H. Arnold, ACS Cent. Sci., 2024, 10, 226–241 CrossRef CAS.
- P. Notin, N. Rollins, Y. Gal, C. Sander and D. Marks, Nat. Biotechnol., 2024, 42, 216–228 CrossRef CAS.
- Z. Liu, S. Chen and J. Wu, Trends Biotechnol., 2023, 41, 1168–1181 CrossRef CAS PubMed.
- M. L. Romero Romero, C. Landerer, J. Poehls and A. Toth-Petroczy, Protein Sci., 2022, 31, e4397 CrossRef CAS.
- M. S. Packer and D. R. Liu, Nat. Rev. Genet., 2015, 16, 379–394 CrossRef CAS PubMed.
- J. Yang, R. G. Lal, J. C. Bowden, R. Astudillo, M. A. Hameedi, S. Kaur, M. Hill, Y. Yue and F. H. Arnold, Nat. Commun., 2025, 16, 714 CrossRef CAS.
- R. J. McLure, S. E. Radford and D. J. Brockwell, Trends Chem., 2022, 4, 378–391 CrossRef CAS.
- A. Zimmermann, J. E. Prieto-Vivas, K. Voordeckers, C. Bi and K. J. Verstrepen, Trends Microbiol., 2024, 32, 884–901 CrossRef CAS PubMed.
- J. Luan, C. Song, Y. Liu, R. He, R. Guo, Q. Cui, C. Jiang, X. Li, K. Hao, A. F. Stewart, J. Fu, Y. Zhang and H. Wang, Nat. Protoc., 2024, 19, 3360–3388 CrossRef CAS.
- S. Shinohara, Y. Fitriana, K. Satoh, I. Narumi and T. Saito, FEMS Microbiol. Lett., 2013, 349, 54–60 CAS.
- E. Sage and N. Shikazono, Free Radic. Biol. Med., 2017, 107, 125–135 CrossRef CAS.
- M. V. Purankar, A. A. Nikam, R. M. Devarumath and S. Penna, Int. J. Radiat. Biol., 2022, 98, 1261–1276 CrossRef CAS PubMed.
- S. Jeyachandran, P. Vibhute, D. Kumar and C. Ragavendran, Mol. Biol. Rep., 2023, 51, 19 CrossRef PubMed.
- X. Sun, X. Li, Y. Lu, S. Wang, X. Zhang, K. Zhang, X. Su, M. Liu, D. Feng, S. Luo, A. Gu, Y. Fu, X. Chen, S. Xuan, Y. Wang, D. Xu, S. Chen, W. Ma, S. Shen, F. Cheng and J. Zhao, Mol. Plant, 2022, 15, 913–924 CrossRef CAS PubMed.
- P. S. Hartman, J. Barry, W. Finstad, N. Khan, M. Tanaka, K. Yasuda and N. Ishii, Mutat. Res., 2014, 766–767, 44–48 CrossRef CAS.
- R. Khanam and R. G. Prasuna, J. Environ. Sci. Eng., 2013, 55, 388–396 CAS.
- M. Sayed, B. Krishnamurthy and H. Pal, Phys. Chem. Chem. Phys., 2016, 18, 24642–24653 RSC.
- N. S. Alavijeh, A. Serrano, M. S. Peters, C. Wolper and T. Schrader, Chem.–Asian J., 2023, 18, e202300637 CrossRef CAS PubMed.
- T. G. Miranda, N. N. Ciribelli, M. F. R. Bihain, A. K. D. Pereira, G. S. Cavallini and D. H. Pereira, Comput. Biol. Chem., 2024, 109 Search PubMed.
- N. A. Haelterman, L. Jiang, Y. Li, V. Bayat, H. Sandoval, B. Ugur, K. L. Tan, K. Zhang, D. Bei, B. Xiong, W. L. Charng, T. Busby, A. Jawaid, G. David, M. Jaiswal, K. J. Venken, S. Yamamoto, R. Chen and H. J. Bellen, Genome Res., 2014, 24, 1707–1718 CrossRef CAS PubMed.
- S. O. Lee and S. D. Fried, Anal. Biochem., 2021, 628, 114266 CrossRef CAS PubMed.
- N. Langreder, D. Schackermann, T. Unkauf, M. Schubert, A. Frenzel, F. Bertoglio and M. Hust, Methods Mol. Biol., 2023, 2702, 395–410 CrossRef CAS PubMed.
- J. N. Copp, P. Hanson-Manful, D. F. Ackerley and W. M. Patrick, Methods Mol. Biol., 2014, 1179, 3–22 CrossRef.
- S. Ebrahimi Fana, A. Fazaeli and M. Aminian, Biotechnol. Lett., 2023, 45, 1159–1167 CrossRef CAS.
- W. Shao, K. Ma, Y. Le, H. Wang and C. Sha, Methods Mol. Biol., 2017, 1498, 497–506 CrossRef CAS.
- J. Yang, A. J. Ruff, M. Arlt and U. Schwaneberg, Biotechnol. Bioeng., 2017, 114, 1921–1927 CrossRef CAS.
- J. Huang, Q. Lin, H. Fei, Z. He, H. Xu, Y. Li, K. Qu, P. Han, Q. Gao, B. Li, G. Liu, L. Zhang, J. Hu, R. Zhang, E. Zuo, Y. Luo, Y. Ran, J. L. Qiu, K. T. Zhao and C. Gao, Cell, 2024, 187, 4426–4428 CrossRef CAS.
- L. Villiger, J. Joung, L. Koblan, J. Weissman, O. O. Abudayyeh and J. S. Gootenberg, Nat. Rev. Mol. Cell Biol., 2024, 25, 464–487 CrossRef CAS.
- K. Seem, S. Kaur, S. Kumar and T. Mohapatra, Crit. Rev. Biochem. Mol. Biol., 2024, 59, 69–98 CrossRef CAS.
- W. Hao, W. Cui, F. Suo, L. Han, Z. Cheng and Z. Zhou, Chem. Sci., 2022, 13, 14395–14409 RSC.
- N. B. Xie, M. Wang, W. Chen, T. T. Ji, X. Guo, F. Y. Gang, Y. F. Wang, Y. Q. Feng, Y. Liang, W. Ci and B. F. Yuan, ACS Cent. Sci., 2023, 9, 2315–2325 CrossRef CAS PubMed.
- J. Xiong, P. Wang, W. X. Shao, G. J. Li, J. H. Ding, N. B. Xie, M. Wang, Q. Y. Cheng, C. H. Xie, Y. Q. Feng, W. M. Ci and B. F. Yuan, Chem. Sci., 2022, 13, 9960–9972 RSC.
- S. Zhang, Y. Liang, T. Feng, X. Guo, M. Wang, T. T. Ji, J. Xiong, X. Xiao, Y. Liu, Y. Liu, W. Ci, N. B. Xie and B. F. Yuan, CCS Chem., 2025 DOI:10.31635/ccschem.025.202405023.
- M. Wang, N. B. Xie, F. Y. Gang, S. Zhang, L. Zeng, T. T. Ji, J. Xiong, X. Guo, Y. Hao, Y. Liu and B. F. Yuan, Sci. China Life Sci., 2025 DOI:10.1007/s11427-024-2702-8.
- X. Guo, J. Wu, T. T. Ji, M. Wang, S. Zhang, J. Xiong, F. Y. Gang, W. Liu, Y. H. Gu, Y. Liu, N. B. Xie and B. F. Yuan, Chem. Sci., 2025, 16, 3953–3963 RSC.
- J. H. Ding, G. Li, J. Xiong, F. L. Liu, N. B. Xie, T. T. Ji, M. Wang, X. Guo, Y. Q. Feng, W. Ci and B. F. Yuan, Anal. Chem., 2024, 96, 4726–4735 CrossRef CAS.
- J. Xiong, K. K. Chen, N. B. Xie, T. T. Ji, S. Y. Yu, F. Tang, C. Xie, Y. Q. Feng and B. F. Yuan, Anal. Chem., 2022, 94, 15489–15498 CrossRef CAS PubMed.
- A. Lapinaite, G. J. Knott, C. M. Palumbo, E. Lin-Shiao, M. F. Richter, K. T. Zhao, P. A. Beal, D. R. Liu and J. A. Doudna, Science, 2020, 369, 566–571 CrossRef CAS PubMed.
- L. Chen, B. Zhu, G. Ru, H. Meng, Y. Yan, M. Hong, D. Zhang, C. Luan, S. Zhang, H. Wu, H. Gao, S. Bai, C. Li, R. Ding, N. Xue, Z. Lei, Y. Chen, Y. Guan, S. Siwko, Y. Cheng, G. Song, L. Wang, C. Yi, M. Liu and D. Li, Nat. Biotechnol., 2023, 41, 663–672 CrossRef CAS.
- C. J. Ma, G. Li, W. X. Shao, Y. H. Min, P. Wang, J. H. Ding, N. B. Xie, M. Wang, F. Tang, Y. Q. Feng, W. Ci, Y. Wang and B. F. Yuan, ACS Cent. Sci., 2023, 9, 1799–1809 CrossRef CAS PubMed.
- E. C. Logue, N. Bloch, E. Dhuey, R. Zhang, P. Cao, C. Herate, L. Chauveau, S. R. Hubbard and N. R. Landau, PLoS One, 2014, 9, e97062 CrossRef PubMed.
- N. B. Xie, M. Wang, T. T. Ji, X. Guo, J. H. Ding, B. F. Yuan and Y. Q. Feng, Chem. Sci., 2022, 13, 7046–7056 RSC.
- N. B. Xie, M. Wang, T. T. Ji, X. Guo, F. Y. Gang, Y. Hao, L. Zeng, Y. F. Wang, Y. Q. Feng and B. F. Yuan, Chem. Sci., 2024, 15, 10073–10083 RSC.
- J. Xiong, K. K. Chen, N. B. Xie, W. Chen, W. X. Shao, T. T. Ji, S. Y. Yu, Y. Q. Feng and B. F. Yuan, Chin. Chem. Lett., 2024, 35, 108953 CrossRef CAS.
- M. Wang, N. B. Xie, K. K. Chen, T. T. Ji, J. Xiong, X. Guo, S. Y. Yu, F. Tang, C. Xie, Y. Q. Feng and B. F. Yuan, Anal. Chem., 2023, 95, 1556–1565 CAS.
- W. X. Shao, Y. H. Min, W. Chen, J. Xiong, X. Guo, N. B. Xie, S. Zhang, S. Y. Yu, C. Xie, Y. Q. Feng and B. F. Yuan, Anal. Chem., 2023, 95, 10588–10594 CrossRef CAS PubMed.
- Y. H. Min, W. X. Shao, Q. S. Hu, N. B. Xie, S. Zhang, Y. Q. Feng, X. W. Xing and B. F. Yuan, Anal. Chem., 2024, 96, 8730–8739 CrossRef CAS PubMed.
- F. Tang, S. Liu, Q. Y. Li, J. Yuan, L. Li, Y. Wang, B. F. Yuan and Y. Q. Feng, Chem. Sci., 2019, 10, 4272–4281 RSC.
- F. Y. Gang, N. B. Xie, M. Wang, S. Zhang, T. T. Ji, W. Liu, X. Guo, S. Y. Gu and B. F. Yuan, Anal. Chem., 2024, 96, 20559–20567 CrossRef CAS PubMed.
- R. C. Cadwell and G. F. Joyce, PCR Methods Appl., 1992, 2, 28–33 CrossRef CAS PubMed.
- R. A. Beckman, A. S. Mildvan and L. A. Loeb, Biochemistry, 1985, 24, 5810–5817 CrossRef CAS PubMed.
- W. P. Stemmer, Nature, 1994, 370, 389–391 CrossRef CAS PubMed.
- J. M. Joern, P. Meinhold and F. H. Arnold, J. Mol. Biol., 2002, 316, 643–656 CrossRef CAS PubMed.
- T. S. Wong, K. L. Tee, B. Hauer and U. Schwaneberg, Nucleic Acids Res., 2004, 32, e26 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Expression and purification of A3A-RL and ABE8e proteins; Tables S1–S4 and Fig. S1–S10. See DOI: https://doi.org/10.1039/d5sc00861a |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2025 |