
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRetro-forward synthesis design and experimental validation of potent structural analogs of known drugs†
Ahmad
Makkawi‡
a,
Wiktor
Beker‡
b,
Agnieszka
Wołos‡
b,
Sabyasachi
Manna
a,
Rafał
Roszak
b,
Sara
Szymkuć
b,
Martyna
Moskal
b,
Aleksei
Koshevarnikov
ab,
Karol
Molga
b,
Anna
Żądło-Dobrowolska
*a and
Bartosz A.
Grzybowski
 *acd
*acd
aInstitute of Organic Chemistry, Polish Academy of Sciences, Warsaw, Poland. E-mail: anna.zadlo@icho.edu.pl
bAllchemy, Inc., Highland, IN, USA
cCenter for Algorithmic and Robotized Synthesis (CARS), Institute for Basic Science (IBS), Ulsan, 44919, Republic of Korea. E-mail: nanogrzybowski@gmail.com
dDepartment of Chemistry, Ulsan Institute of Science and Technology, UNIST, Ulsan, 44919, Republic of Korea
First published on 19th March 2025
Abstract
Generation of structural analogs to “parent” molecule(s) of interest remains one of the important elements of drug development. Ideally, such analogs should be synthesizable by concise and robust synthetic routes. The current work illustrates how this process can be facilitated by a computational pipeline spanning (i) diversification of the parent via substructure replacements aimed at enhancing biological activity, (ii) retrosynthesis of the thus generated “replicas” to identify substrates, (iii) forward syntheses originating from these substrates (and synthetically versatile “auxiliaries”) and guided “towards” the parent, and (iv) evaluation of the candidates for target binding and other medicinal–chemical properties. This pipeline proposes syntheses of thousands of readily makeable analogs in a matter of minutes, and is deployed here to validate by experiment seven structural analogs of Ketoprofen and six analogs of Donepezil. The concise, computer-designed syntheses are confirmed in 12 out of 13 cases, offering access to several potent inhibitors. While the synthesis-design component is robust, binding affinities are predicted less accurately although still to the order-of-magnitude, which may be valuable in discerning promising from inadequate binders.
1. Introduction
Recent years have brought revolutionary advances1–10 in the use of computers to autonomously plan chemical syntheses of arbitrary targets, all the way up to complex natural products.8–10 One of the prominent areas of application of these algorithms has been in drug discovery where the synthesis design is part of algorithmic pipelines11–15 intended to predict target and off target binding as well as ADME-Tox properties. While the premium is, without doubt, on discovering potent candidates featuring unprecedented scaffolds16 and binding modes, many drugs are derivatives of the old ones and “the best way to discover a new drug is to start with an old one”.17 Accordingly, analogs structurally similar to some desired “parent” molecules continue to be sought in drug screening and lead-optimization campaigns18 and efforts to accelerate this process with the help of computers date back to at least the 1990s with many ingenious approaches undertaken since then.There are two general schools of thought about the analog design problem. In the “classic” approach, a computer program performs in silico reactions to produce virtual molecules. The reactions are typically applied in the “forward” direction (starting from some static collections of starting materials19–25) and generate molecular spaces which, nowadays, can be quite enormous25 (e.g., a few billion virtual molecules in Enamine's REAL space and up to 10 (ref. 20) in Merck KGaA's MASSIV collection). These spaces are subsequently pruned for target similarity or other properties of interest. There are also solutions that use retrosynthetic pathways of the parent as input and generate analogs by replacing the original starting materials by structurally or functionally similar blocks.26 The second family of approaches relies on the burgeoning generative AI models.27–34 These methods often combine target-similarity with concurrent predictions of other properties.35 While the very synthesizability of the generated structures has historically been a challenge,36–39 there has been significant recent progress, as detailed in an excellent recent ref. 40. We observe, however, that irrespective of the approach taken, studies in this area are rarely accompanied by experimental validations of computer-designed syntheses (see ref. 23, 24 and 41) and/or of the predicted potency of the proposed analogs. Such validations are urgently needed as, ultimately, they will decide wider adoption of these methods (which, one may argue, is not guaranteed given the widely publicized, recent setbacks of AI in industrial drug discovery42).
With this in mind, the overriding objective of our current work is not to argue for the advantages or disadvantages of any particular approach (they all have some) but to test by experiment the particular analog-design pipeline we have been developing for several years now. This pipeline is along the abovementioned “classical” lines of in silico synthesis and encompasses substructure replacements within the parent (to diversify the parent scaffold and, hopefully, enhance biological activity), retrosynthetic generation of substrates retaining mutual reactivity, “guided” forward synthesis to produce large numbers of easily synthesizable structural analogs of the parent, and estimation of these candidates’ binding affinity to the desired target. Within this scheme, the two key questions we ask relate to the correctness of the computer-designed analog syntheses and to the accuracy of predicting their binding affinities.
Considering two parent molecules (Ketoprofen and Donepezil), the outcomes of our studies are nuanced. On the positive side, experiments validate concise, computer-designed syntheses of seven analogs of Ketoprofen and five analogs of Donepezil (against one failed route). Six Ketoprofen analogs are μM binders to human cyclooxygenase-2 (COX-2), with one offering slightly better binding than the parent drug (0.61 μM vs. 0.69 μM). For Donepezil, all five analogs show submicromolar binding to acetylcholinesterase, AChE, with one having nanomolar affinity close to that of the parent (36 nM vs. 21 nM). At the same time, binding predictions – which had guided selection of analogs for synthesis validation – by three different docking programs and a neural-network, match the experimental values only to within an order-of-magnitude. These results make us conclude that (i) the synthesis-planning aspects of computerized analog design are nowadays robust, and (ii) common affinity-prediction tools may help select promising binders but cannot discriminate between moderate (μM) vs. high-affinity (nM) ones.
2. Results
2.1 Components of the computational pipeline (Fig. 1)
 | ||
Fig. 1 Scheme of the algorithmic pipeline for substrate selection and subsequent analog generation. (a) For a given target/“parent” of interest (node colored in red), the algorithm first identifies its substructures that can be altered by replacements likely to result in activity enhancement.49 This creates several (for a typical drug-like parent, ∼10–100) parent replicas (light red). The algorithm then expands retrosynthetic networks to find viable synthetic routes to all of these molecules and retains their commercially available starting materials. Since easily makeable analogs are ultimately sought, retrosynthesis is limited to the depth of five steps and using only 180 reaction classes popular in medicinal chemistry. Retrosynthetic searches stop when reaching commercially available substrates, here, those from Mcule's catalog, https://mcule.com/database/, of ∼2.5 million chemicals. The set of retrosynthetically-derived substrates (nodes colored violet) is further augmented by 23 simple yet synthetically useful chemicals (here, additional nodes colored in light violet). (b) All of the said substrates serve as the zero-th generation, G0, for the guided forward search in which, after each round of reactions, only some W molecules most similar to the parent are retained (dark-blue nodes, here W = 150). In this way, the forward synthesis is gradually “focused” towards parent's structural analogs. We note that because of this truncation, the sizes of the networks do not explode. This also allows the use of larger collections of reaction transforms (here, ∼25![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rules from Allchemy) to achieve more synthetically diverse outcomes, while limiting the times of network propagation, typically to several minutes. 000 rules from Allchemy) to achieve more synthetically diverse outcomes, while limiting the times of network propagation, typically to several minutes. | ||
Given these requirements, the straightforward method of disconnecting the parent into the starting materials by retrosynthesis may be overly simplistic. Retrosynthesis of only the parent molecule generates substrates that, in the forward direction, can be reassembled into the parent itself, into some intermediates en route to this parent, and usually some molecules in which alternative reactions of the starting materials are performed. However, this approach does not generate diverse analogs. The set of starting materials can, of course, be diversified by adding molecules similar to those found by retrosynthesis – we tested this approach early on but many of the “similars” to the starting materials featured functional group patterns that were unsuitable or problematic for the subsequent forward synthesis (see ESI, Section S2†). Mindful of this, we augmented the retrosynthetic protocol in two ways:
First, by performing substructure replacements (intended to enhance biological activity and digitized, in large part, according to Novartis’ tables from ref. 49) within the parent and only then performing retrosyntheses of these “replica” molecules. The replacements are performed at either peripheral groups or internal motifs such as 1,3-disubstituted benzene rings or piperazines (see ESI, Fig. S2†). They increase structural diversity of the starting materials while, simultaneously, retaining functional groups necessary for the “mix-and-match” reactivity between blocks derived from different replicas. Here, in all retrosyntheses, we used the Allchemy algorithm50 although other reliable retrosynthetic engines2–4,51 can be used.
Second, by adding to G0 some simple yet synthetically useful chemicals with which to further modify and/or activate molecules within the forward networks. While various choices are possible, we used a static, “minimal” set of 23 popular chemicals (see ESI, Fig. S4†) chosen for synthetic versatility. For example, N-chlorosuccinimide, N-bromosuccinimide and nitric acid enable electrophilic aromatic substitutions, bis(pinacolato)diboron opens the way to Suzuki couplings, DAST can functionalize molecules with fluoride, mesyl chloride activates alcohols for SN2 reaction, ethyl magnesium bromide can engage in Kulinkovich cyclopropanation while hydrazine, thiourea, azide and trimethyl orthoformate enable formation of various heterocycles, etc. An example of a search with and without these additional chemicals in provided in the ESI, Section Section S3.†
Regarding the synthesis components 1.1 and 1.2, we note that retrosynthesis may be significantly slower than guided network expansion. With Allchemy's full reaction database, retrosynthetic analysis of a typical drug-like molecule takes only 1–5 min, but for the tens of replicas created in the first stage of the pipeline, these times are already in hours – that is, much longer than for the forward synthesis up to 3–4 generations (4–6 min). Consequently, these two elements of the pipeline use different sets of reaction rules: for retrosynthesis, to reduce the size of retrosynthetic search networks, only 180 reaction classes most popular in medicinal chemistry, and for the forward synthesis, all 25307 reaction rules available in Allchemy. In this way, both retrosynthetic and forward searches complete within minutes which, based on the feedback on the pipeline's external users appears to be a practically acceptable limit for this type of application. The entire pipeline, integrated in a form of a user-friendly WebApp is available for academic testing at https://analogs.allchemy.net/ with the user manual provided in the ESI, Section S1.† The source-code for guided network expansion is deposited at https://zenodo.org/records/7371247.
2.3 Application of the pipeline to specific targets
We applied the above approach (1)–(3) to identify readily synthesizable analogs to anti-inflammatory Ketoprofen and to Donepezil, a medication used to treat Alzheimer-type dementia. | ||
| Fig. 2 Search for Ketoprofen's analogs. The panels illustrate the key steps along the analog design pipeline: (a) Ketoprofen and some of its replicas featuring substructure replacements aimed at enhancing biological activity;49 (b) some of the substrates derived by retrosynthesis of molecules from (a) as well as, on the right, examples of the simple reagents with which to functionalize or activate the substrates (for all 23, see ESI, Fig. S4†); (c) Allchemy screenshot of some of the top-ranking analogs generated by guided expansion of a reaction network starting from molecules in (b). The analogs shown here are sorted by similarity to the Donepezil parent and are centered around the 2-(3-benzoylphenyl)acetic acid with modifications introduced mostly in the α-position of carboxylic acid and as substituents on the aromatic ring. Clicking on any of the “tiles” provides synthesis details (these and other options to visualize the synthetic networks are detailed in the ESI, Section S1†). Some of the analogs ultimately committed to synthesis are shown on pink backgrounds. Visible on the left is the panel with medicinal–chemical functionalities by which the analogs can be filtered (e.g., general drug-likeness,79 binding to specific proteins, various ADME-Tox models, PAINS and substructure-based filters, see user manual in the ESI†). | ||
Within this set, we focused on molecules 1–7 that form a small synthetic cluster summarized in Fig. 3a. These analogs are all derivatives of the commercially available 2-(3-benzoylphenyl)acetic acid and were not reported or tested for cyclooxygenase-2, COX-2, binding before (only compound 1 was synthesized in ref. 53 but in the context of hydrocarboxylation methodology with no biological studies). Moreover, the proposed syntheses all appear concise and straightforward, and were successfully carried out in yields indicated next to the arrows (under conditions identical or very similar to those suggested by the software and without thorough optimization).
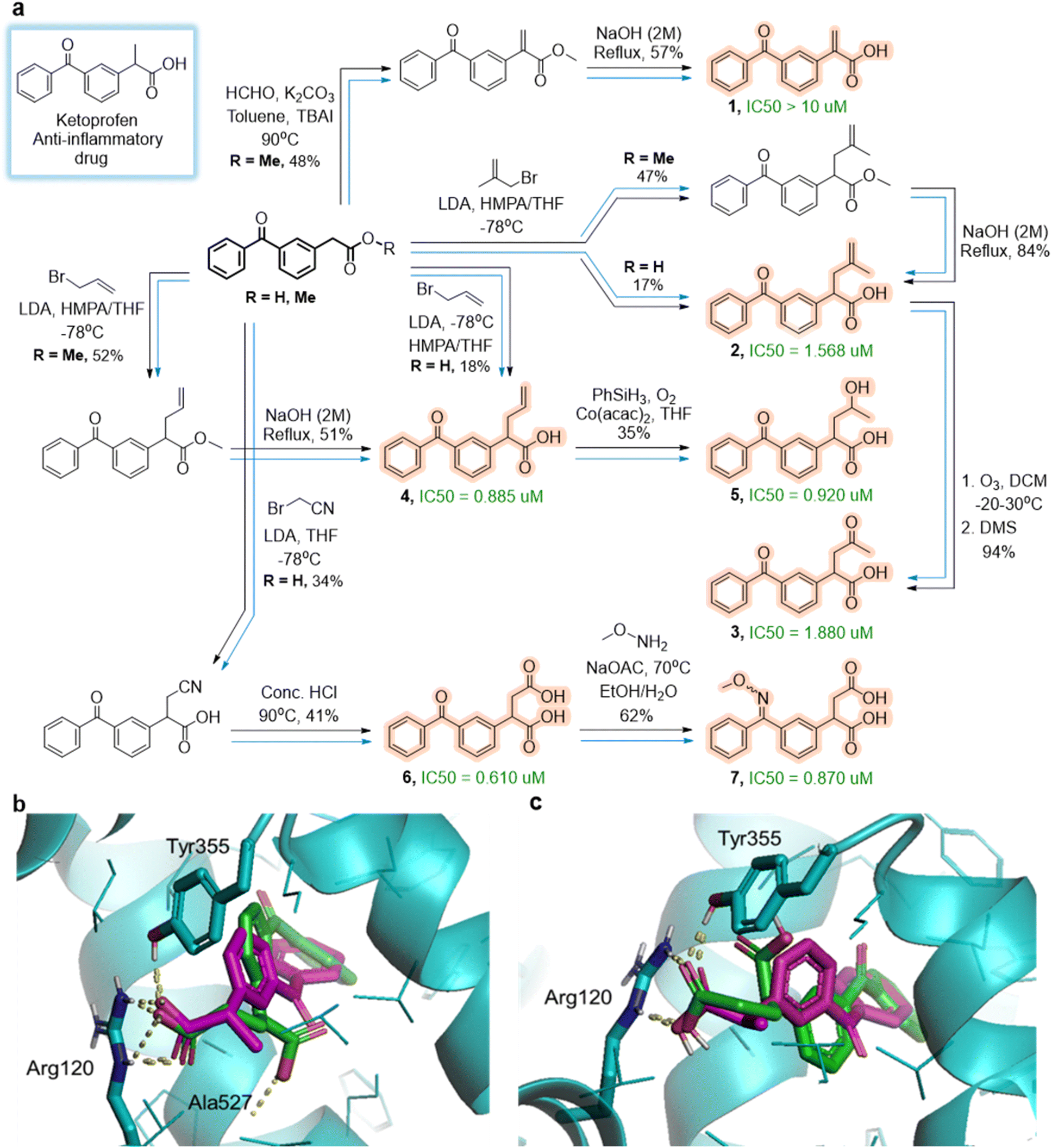 | ||
| Fig. 3 Ketoprofen's analogs committed to synthesis. (a) A small network of algorithm-planned syntheses of analogs 1 through 7 starting from 2-(3-benzoylphenyl)acetic acid and/or ester derivatives (R = H or R = Me). Experimental pathways (tracked by black arrows) follow the algorithm's original suggestions (blue arrows). Conditions and isolated yields are given next to reaction arrows. IC50 values are given next to the analogs. (b) and (c) All analogs were docked into COX-2 protein. The alignment of docking poses from AutoDock 4 for (b) (S)-Ketoprofen and its analog (S)-6, and (c) (R)-Ketoprofen and its analog (R)-6. Key, protein-ligand hydrogen bonds are depicted as yellow dotted lines. The carboxylic acid moiety of (S)-Ketoprofen (colored in magenta) forms two hydrogen bonds with Arg120 and one with Tyr355. Its analog (S)-6 (b, colored in green) is predicted to be similarly positioned inside the active site and to form three hydrogen bonds: with Arg120, with Tyr355 and, for the second carboxylic acid moiety, with Ala527. For (R)-Ketoprofen (colored in magenta in panel (c)) and its analog (R)-6 (colored in green), the carboxylic acid moieties in both compounds form two hydrogen bonds (with Arg120 and with Tyr355). Raw files with these and all other docking experiments using AutoDock 4, AutoDock Vina, and Dock 6 (ref. 54–57) are deposited at https://zenodo.org/records/14571461. | ||
In selecting these particular structures, we were also guided by predictions of their binding affinities to COX-2. To this end, we used three popular docking programs, AutoDock 4, AutoDock Vina, Dock 6.54–57 These programs used the 5IKR PDB structure and predicted the binding scores of the analogs 1–7 (averaged over possible stereoisomers of a given racemic analog) to be comparable or more favorable than those of Ketoprofen, which we also docked for reference (top portion of Table 1). Inspection of the docking poses (see examples in Fig. 3b and c and remaining analogs deposited at https://zenodo.org/records/14571461) revealed that these molecules should, indeed, be able to engage in numerous favorable interactions with the COX-2 active site. In parallel, we sought binding strength estimates using Allchemy's neural network, NN, trained on 1752921 protein assays from the ChEMBL 29 database58 and spanning activity values for 3843 one-hot-encoded targets and 863471 ligands represented as concatenation of Morgan fingerprints with radius 3 (using AllChem.GetMorganFingerprint function in RDKit59), Xfp pharmacophore fingeprints60 and selection of MACCS keys.61 The architecture of this feedforward multitasking NN is similar to the one used in ref. 62 and 63, where it was shown to provide the best performance for ChEMBL activity prediction. The main difference is that we used batch normalization, and assigned different activity thresholds to different protein families (according to the values defined in ref. 64). These alterations slightly improved performance to ROC AUC = 0.87. Here, this network predicted that the affinities of the analogs we synthesized should be on-the-order-of micromolar.
P calculated by group contribution method80 as well as predictions of Allchemy's machine-learning models for hERG cardiotoxicity (on a 0–1 scale), degree of human plasma protein binding, hPPB, related to drug absorption and efficacy (0–100%), and degree of blood–brain barrier, BBB, penetration (0–1). Although none of the models used Ketoprofen or Donepezil molecules for training/testing, the key predictions match experimental data. For instance, the hERG model correctly predicts Donepezil to be cardiotoxic (values close to 1), which is in line with experimental studies evidencing that Donepezil binds to hERG81 with IC50 = 1.3 μm and can cause QTc prolongation.82 By contrast, the model predicts low values of hERG inhibition for Ketoprofen (and its analogs), which agrees with experiments confirming its low cardiotoxicity.83 In turn, the hPPB model predicts high, >90% bound fractions for both Ketoprofen and Donepezil and their analogs – while lower hPPB values are, in principle, desired in drug design, these predicted values agree with experimental measurements for Ketoprofen (99%84) and Donepezil (95.6%85). In turn, the blood–brain barrier, BBB, penetration model predicts high values for Donepezil, which is expected for a CNS drug and experimentally confirmed.86 Ketoprofen also crosses the BBB87 but we note that some of the analogs, more polar ones, are predicted to have significantly lower values
| Compound | K i AutoDock Vina (μM) | K i AutoDock (μM) | Dock 6 (score) | logP |
hERG (0–1) | hPPB (0–100%) | BBB (0–1) |
|---|---|---|---|---|---|---|---|
| Ketoprofen | 0.613 | 0.284 | −33.03 | 3.106 | 0.04 | 98.40 ± 1.20 | 0.91 |
| 1 | 0.413 | 0.202 | −34.85 | 3.015 | 0.03 | 98.50 ± 1.00 | 0.84 |
| 2 | 0.814 | 0.268 | −34.62 | 4.052 | 0.1 | 98.80 ± 1.30 | 0.64 |
| 3 | 0.579 | 0.161 | −33.40 | 3.065 | 0.01 | 97.00 ± 1.30 | 0.42 |
| 4 | 1.138 | 0.224 | −34.82 | 3.662 | 0.09 | 98.60 ± 1.20 | 0.69 |
| 5 | 0.538 | 0.348 | −36.74 | 2.857 | 0.01 | 94.30 ± 1.20 | 0.46 |
| 6 | 0.451 | 0.154 | −33.19 | 2.561 | 0 | 96.50 ± 2.70 | 0.37 |
| 7 | 0.534 | 0.264 | −39.15 | 2.728 | 0 | 95.80 ± 3.60 | 0.29 |
|
|||||||
| Donepezil | 0.046 | 0.037 | −43.78 | 4.361 | 0.96 | 91.70 ± 1.30 | 0.92 |
| 11 | 0.086 | 0.061 | −41.76 | 4.357 | 0.98 | 90.80 ± 1.10 | 0.9 |
| 12 | 0.064 | 0.069 | −42.54 | 4.671 | 0.95 | 95.80 ± 1.20 | 0.75 |
| 13 | 0.204 | 0.097 | −43.06 | 3.722 | 0.92 | 88.90 ± 1.10 | 0.66 |
| 14 | 0.037 | 0.053 | −42.55 | 3.930 | 0.92 | 93.90 ± 4.70 | 0.77 |
| 16 | 0.027 | 0.011 | −46.98 | 3.833 | 0.97 | 89.60 ± 1.20 | 0.75 |
To verify these predictions, we ran spectrofluorometric COX-2 human inhibition assay65,66 and quantified the IC50 values. These values are marked in green next to the specific analogs in Fig. 3a. As seen, one analog, 1, is binding poorly, >10 μm, but the remaining six are micromolar binders with one, 6, exhibiting slightly better affinity than the parent Ketoprofen, 0.61 μM vs. 0.69 μM.
Retrosyntheses of the parent and all 18 replicas took only 3 min and generated 44 substrates which were combined with 23 synthetically useful, simple chemicals. Three molecules overlapped between these two sets so that the number of unique starting materials in G0 was 64. The forward search up to G3 generated, within 5 min, a network of 3619 products of which 2222 (∼61%) had similarity to parent above 0.7.
Fig. 4a shows six molecules that were committed to synthesis: the replica 11 of the parent drug and analogs 12–15 forming a small cluster entailing only six reactions based on the aforementioned aldehyde 9, ester 10 as well as ketone 17. Additionally, we included the synthesis of analog 16 that had been found by an earlier version of the algorithm, and used 8 and 18 as starting materials. All these reactions were executed under computer-suggested conditions, save for the Swern rather than Dess–Martin oxidation of 13 to 14. In five out of six cases, the reactions gave the expected products although in poor to moderate yields (no condition optimization was attempted). In one case, the fluorinated derivative 15 was not obtained because of elimination to 12. While there are literature examples of fluorination proceeding on β-hydroxy ketones, Allchemy's pKa model67 predicts that the cH position within our cyclic aryl ketone is more acidic, hence promoting elimination vs. substitution.
 | ||
| Fig. 4 Donepezil's analogs committed to synthesis. (a) A small network of algorithm-planned syntheses of analogs 11 through 16. Blue arrows highlight Allchemy's suggestions, black arrows highlight the experimentally executed pathways. Conditions and isolated yields are given above reaction arrows. IC50 values are given next to the analogs. Note: As synthesis of 9 and 10 from simpler starting materials was previously described, we followed the literature procedure (10 was made in two steps and aldehyde 9 from ester 10 in another 2 steps with Allchemy-proposed alcohol as an intermediate; ester 10 and alcohol are commercially available but relatively expensive). All analogs were docked into acetylcholinesterase from Electrophorus electricus. Docking poses from AutoDock Vina are aligned for (b) (R)-donepezil and its analog (S)-14, and (c) (R)-donepezil and its analog (1R,2S)-13. Key, protein-ligand hydrogen bond interactions are traced by yellow dotted lines. The carbonyl oxygen of (R)-Donepezil's (colored in magenta) indanone ring forms a hydrogen bond with Phe295 –NH whereas nitrogen from piperidine ring hydrogen-bonds with Tyr124 –OH. Its analog (S)-14 (b, colored in green) forms hydrogen bonds with Phe295 –NH, but instead of carbonyl oxygen from indanone ring, carbonyl oxygen from 2-(1-benzylpiperidin-4-yl)acetyl side chain is involved. Analog (1R,2S)-13 (c, colored in green) forms two hydrogen bonds between Phe295 –NH and carbonyl oxygen of indanone ring and hydroxyl oxygen of analog's side chain as well as and one hydrogen bond between carbonyl oxygen of indanone ring and Arg296 –NH2. Raw files with these and all other docking experiments using (AutoDock 4, AutoDock Vina, Dock 6)54–57 are deposited at https://zenodo.org/records/14571461. | ||
As in the case of Ketoprofen, these analogs were chosen because most were not reported before (compound 11 is known as an intermediate in the synthesis of acetylcholinesterase, AChE, inhibitors but its activity was not evaluated68) and because their predicted binding scores (by AutoDock 4, AutoDock Vina, Dock 6)54–57 were comparable or better than those of the Donepezil parent, which we also docked for reference (see bottom portion of Table 1). Allchemy's neural network also predicted micromolar-level binding.
These predictions were verified in spectrophotometric AChE inhibition assay,65,69 which quantified the IC50 values given next to the specific analogs in Fig. 4a. The replica 11 had IC50 = 144 nM and two analogs were submicromolar, 991 nM for 16 and 362 nM for 13. However, the two remaining analogs were significantly more potent, 88 nM for 14 and 36 nM for 12. This last value rivals the potency measured for Donepezil itself, 21 nM.
3. Discussion and conclusions
The above results substantiate three major conclusions. First, the quality of synthetic predictions appears satisfactory with 12 out of 13 analogs made according to the computer-designed routes. This is perhaps not unexpected given that predictions of programs such as Allchemy or Chematica/Synthia had previously been validated on targets more challenging than our analogs.3,8,10,44 In this light, one can argue that all these simple syntheses could have been designed without much effort by a human expert – while this is true, the computer may be viewed as a useful “calculator” accelerating the straightforward but otherwise tedious steps of the design process, from the creation of replicas via substructure replacements, through the selection of commercially available substrates, to forward synthesis.Second, whereas the synthesis part is robust, the prediction of analog's properties remains challenging. This is illustrated in Fig. 5 which plots the experimentally measured IC50's against the predictions of the docking programs – as seen, the correlations are quite poor and even discounting the outlier 1, are limited to ∼0.45 for AutoDock Vina (green markers in the inset to the upper plot). We observe that such values are in line with comparative studies of docking methods70 where similar, low correlations were reported. In parallel, Allchemy's internal neural network trained on 1.75 million of protein assays from ChEMBL estimated the potency of our analogs to be micromolar, which is true for most but not all of them (e.g., not for 1 which is >10 μM and not for 12 and 14 for which IC50 values are in tens of nM). Taken together, these results reinforce the view that neither the docking nor NN models can currently predict the affinity accurately – though they can perhaps be accurate to the order-of-magnitude, which can still be useful in distinguishing very poor from decent binders.
 | ||
| Fig. 5 The relationship between the experimental IC50 (μM) and Ki (μM) values (from AutoDock 4, blue, and AutoDock Vina, green) or the docking scores (from Dock 6, orange). Insets zoom on the regions which, in the full plots, are shaded in gray and do not include the poorly binding analog. | ||
Third and last, it should be remembered that in developing potential drugs, binding affinity is but one of the important metrics and one should also evaluate ADME-Tox properties. There are nowadays multiple machine-learning models to evaluate these properties (e.g., ref. 71–73 for hERG models, ref. 74 for plasma protein binding, PPB, or ref. 75–78 for blood–brain barrier, BBB, penetration) and some have also been implemented in Allchemy's pipeline (see user manual in ESI, Section S1†), offering respectable accuracies ∼0.8–0.9 and realistic predictions (see Table 1 and its caption). Thus, one could also think of using these metrics as part of a multiobjective scoring function35 to guide the synthesis of molecules that, ultimately, meet several desirable criteria at once. The multiobjective approach is, arguably, more conceptually elegant than the synthesize-and-then-evaluate pipeline we pursued here. On the other hand, it should be noted that ADME-Tox models suffer from the scarcity of publically available data (typically, few thousand molecules per model) and are largely untested (and likely less reliable) in out-of-box predictions. Using such metrics to make decisions about which molecules to synthesize can eliminate some interesting candidates from consideration. Our thinking when developing the “synthesis-first” pipeline was that synthesis planning – being the most robust component – should be unhindered by additional constraints, especially that it yields large collections of candidate molecules in very short times. These molecules can then be evaluated by other models/filters, with a human expert making choices which metrics to prioritize (or trust). This said, we recognize that if much larger synthetic spaces are to be explored (with much bigger substrate sets or higher beam width, W, see ref. 44 and 45), then multiobjective scoring should be considered to narrow down and accelerate this exploration.
Data availability
All docking poses are deposited at https://zenodo.org/records/14571461. The source-code for guided network expansion is deposited at https://zenodo.org/records/7371247. Sample of the database of reaction templates is deposited at https://zenodo.org/records/15001486. All synthesis results are deposited under the “saved results” tab of the WebApp at https://analogs.allchemy.net/(see user manual in the ESI†). To test the WebApp under two-week, free academic access, please send an email from an academic address to E-mail: admin@allchemy.net for access credentials.Author contributions
A. M. and S. M. performed syntheses. A. M. and A. Ż.-D. Performed the binding assays. W. B., A. W., R. R., S. S., M. M, A. K. and K. M. developed the algorithms and the WebApp. A. Ż.-D. performed docking studies and supervised the syntheses. B. A. G. conceived and supervised the project.Conflicts of interest
W. B., A. W., R. R., S. S., M. M., A. K., K. M. and B. A. G. are consultants and/or stakeholders of Allchemy, Inc. Allchemy software is property of Allchemy Inc.Acknowledgements
Development of the Analogs module within the Allchemy platform (by W. B., A. W., R. R., S. S., M. M., A. K. and K. M.) was been supported by internal funds of Allchemy, Inc. Syntheses performed by A. M. and analyses performed by A. K. were supported by the National Science Center, Poland (grant Maestro, # 2018/30/A/ST5/00529 to B. A. G.). Synthesis, binding assays and docking studies by A. Ż.-D. and S. M. were supported by the National Science Centre, Poland (grant SONATA 2020/39/D/ST4/01890 to A. Ż.-D.). Analysis of results and writing of the paper by B. A. G. was supported by the Institute for Basic Science, Korea (project code IBS-R020-D1). The authors thank the Mcule team for providing access to their catalog and standardizing the prices information therein to per-gram.References
- S. Szymkuć, E. P. Gajewska, T. Klucznik, K. Molga, P. Dittwald, M. Startek, M. Bajczyk and B. A. Grzybowski, Angew. Chem., Int. Ed., 2016, 55, 5904–5937 Search PubMed.
- C. W. Coley, L. Rogers, W. H. Green and K. F. Jensen, ACS Cent. Sci., 2017, 3, 1237–1245 CrossRef CAS PubMed.
- T. Klucznik, B. Mikulak-Klucznik, M. P. McCormack, H. Lima, S. Szymkuć, M. Bhowmick, K. Molga, Y. Zhou, L. Rickershauser, E. P. Gajewska, A. Toutchkine, P. Dittwald, M. P. Startek, G. J. Kirkovits, R. Roszak, A. Adamski, B. Sieredzińska, M. Mrksich, S. L. J. Trice and B. A. Grzybowski, Chem, 2018, 4, 522–532 CAS.
- M. H. S. Segler, M. Preuss and M. P. Waller, Nature, 2018, 555, 604–610 CrossRef CAS PubMed.
- C. W. Coley, D. A. Thomas, J. Lummiss, J. N. Jaworski, C. P. Breen, V. Schultz, T. Hart, J. S. Fishman, L. Rogers, H. Gao, R. W. Hicklin, P. P. Plehiers, J. Byington, J. S Piotti, W. H Green, A. J. Hart, T. F. Jamison and K. F. Jensen, Science, 2019, 365, eaax1566 CrossRef CAS PubMed.
- A. F. de Almeida, R. Moreira and T. Rodrigues, Nat. Rev. Chem., 2019, 3, 589–604 CrossRef.
- P. Schwaller, A. C. Vaucher, R. Laplaza, C. Bunne, A. Krause, C. Corminboeuf and T. Laino, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2022, 12, e1604 Search PubMed.
- B. Mikulak-Klucznik, P. Gołębiowska, A. A. Bayly, O. Popik, T. Klucznik, S. Szymkuć, E. P. Gajewska, P. Dittwald, O. Staszewska-Krajewska, W. Beker, T. Badowski, K. A. Scheidt, K. Molga, J. Mlynarski, M. Mrksich and B. A. Grzybowski, Nature, 2020, 588, 83–88 CrossRef CAS.
- Y. Lin, R. Zhang, D. Wang and T. Cernak, Science, 2023, 379, 453–457 CrossRef CAS.
- T. Klucznik, L. D. Syntrivanis, S. Baś, B. Mikulak-Klucznik, M. Moskal, S. Szymkuć, J. Mlynarski, L. Gadina, W. Beker, M. D. Burke, K. Tiefenbacher and B. A. Grzybowski, Nature, 2024, 625, 508–515 CAS.
- P. Jia, J. Pei, G. Wang, X. Pan, Y. Zhu, Y. Wu and L. Ouyang, Green Synth. Catal., 2022, 3, 11–24 CrossRef CAS.
- A. Zhavoronkov, Y. A. Ivanenkov, A. Aliper, M. S. Veselov, V. A. Aladinskiy, A. V. Aladinskaya, V. A. Terentiev, D. A. Polykovskiy, M. D. Kuznetsov, A. Asadulaev, Y. Volkov, A. Zholus, R. R. Shayakhmetov, A. Zhebrak, L. I. Minaeva, B. A. Zagribelnyy, L. H. Lee, R. Soll, D. Madge, L. Xing, T. Guo and A. Aspuru-Guzik, Nat. Biotechnol., 2019, 37, 1038–1040 CrossRef CAS.
- D. E. Graff, E. I. Shakhnovich and C. W. Coley, Chem. Sci., 2021, 12, 7866–7881 RSC.
- T. J. Struble, J. C. Alvarez, S. P. Brown, M. Chytil, J. Cisar, R. L. DesJarlais, O. Engkvist, S. A. Frank, D. R. Greve, D. J. Griffin, X. Hou, J. W. Johannes, C. Kreatsoulas, B. Lahue, M. Mathea, G. Mogk, C. A. Nicolaou, A. D. Palmer, D. J. Price, R. I. Robinson, S. Salentin, L. Xing, T. Jaakkola, W. H. Green, R. Barzilay, C. W. Coley and K. F. Jensen, J. Med. Chem., 2020, 63, 8667–8682 CrossRef CAS.
- A. V. Sadybekov and V. Katritch, Nature, 2023, 616, 673–685 CrossRef CAS.
- R. Roszak, L. Gadina, A. Wołos, A. Makkawi, B. Mikulak-Klucznik, Y. Bilgi, K. Molga, P. Gołębiowska, O. Popik, T. Klucznik, S. Szymkuć, M. Moskal, S. Baś, R. Frydrych, J. Mlynarski, O. Vakuliuk, D. T. Gryko and B. A. Grzybowski, Nat. Commun., 2024, 15, 10285 CrossRef CAS.
- T. N. K. Raju, Lancet, 2000, 356, 436 CrossRef.
- J. Fischer and C. R. Ganellin, Analogue-Based Drug Discovery II, Wiley-VCH, 2010, DOI:10.1002/9783527630035.
- M. Hartenfeller and G. Schneider, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2011, 1, 742–759 CAS.
- G. Schneider and U. Fechner, Nat. Rev. Drug Discovery, 2005, 4, 649–663 CrossRef CAS PubMed.
- G. Schneider, M. L. Lee, M. Stahl and P. Schneider, J. Comput. Aided Mol. Des., 2000, 14, 487–494 CrossRef CAS PubMed.
- X. Q. Lewell, D. B. Judd, S. P. Watson and M. M. Hann, J. Chem. Inf. Comput. Sci., 1998, 38, 511–522 CrossRef CAS PubMed.
- H. M. Vinkers, M. R. de Jonge, F. F. D. Daeyaert, J. Heeres, L. M. H. Koymans, J. H. van Lenthe, P. J. Lewi, H. Timmerman, K. Van Aken and P. A. J. Janssen, J. Med. Chem., 2003, 46, 2765–2773 CrossRef CAS PubMed.
- M. Hartenfeller, H. Zettl, M. Walter, M. Rupp, F. Reisen, E. Proschak, S. Weggen, H. Stark and G. Schneider, PLoS Comput. Biol., 2012, 8, e1002380 CrossRef CAS.
- T. Hoffmann and M. Gastreich, Drug Discovery Today, 2019, 24, 1148–1156 CrossRef CAS.
- U. Dolfus, H. Briem and M. Rarey, J. Chem. Inf. Model., 2022, 62, 3565–3576 CrossRef CAS PubMed.
- B. Sanchez-Lengeling and A. Aspuru-Guzik, Science, 2018, 361, 360–365 CrossRef CAS.
- M. H. S. Segler, T. Kogej, C. Tyrchan and M. P. Waller, ACS Cent. Sci., 2018, 4, 120–131 CrossRef CAS.
- I. Goodfellow, J. Pouget-Abadie, M. Mirza, B. Xu, D. Warde-Farley, S. Ozair, A. Courville and B. Yoshua, in Advances in Neural Information Processing Systems 27 (NIPS 2014), 2014, pp. 2672–2680 Search PubMed.
- D. C. Elton, Z. Boukouvalas, M. D. Fuge and P. W. Chung, Mol. Syst. Des. Eng., 2019, 4, 828–849 RSC.
- J. Bradshaw, B. Paige, M. J. Kusner, M. H. S. Segler and J. M. Hernández-Lobato, arXiv, 2019, preprint, arXiv:1906.05221, DOI:10.48550/arXiv.1906.05221.
- T. Blaschke, J. Arús-Pous, H. Chen, C. Margreitter, C. Tyrchan, O. Engkvist, K. Papadopoulos and A. Patronov, J. Chem. Inf. Model., 2020, 60, 5918–5922 Search PubMed.
- J. Bradshaw, B. Paige, M. J. Kusner, M. H. S. Segler and J. M. Hernández-Lobato, arXiv, 2020, preprint, arXiv:2012.11522, DOI:10.48550/arXiv.2012.11522.
- S. K. Gottipati, B. Sattarov, S. Niu, Y. Pathak, H. Wei, S. Liu, K. M. J. Thomas, S. Blackburn, C. W. Coley, J. Tang, S. Chandar and Y. Bengio, arXiv, 2020, preprint, arXiv:2004.12485, DOI:10.48550/arXiv.2004.12485.
- J. C. Fromer, D. E. Graff and C. W. Coley, Digital Discovery, 2024, 3, 467–481 Search PubMed.
- W. Gao and C. W. Coley, J. Chem. Inf. Model., 2020, 60, 5714–5723 CrossRef CAS.
- P. Renz, D. Van Rompaey, J. K. Wegner, S. Hochreiter and G. Klambauer, Drug Discovery Today: Technol., 2019, 32, 55–63 CrossRef.
- W. P. Walters, R. Barzilay and E. Opin, Drug. Discovery, 2021, 16, 937–947 CAS.
- M. Stanley and M. Segler, Curr. Opin. Struct. Biol., 2023, 82, 102658 CrossRef CAS.
- W. Gao, S. Luo and C. W. Coley, arXiv, 2024, preprint, arXiv:2410.03494, DOI:10.48550/arXiv.2410.03494.
- S. K. Singh, K. King, C. Gannett, C. Chuong, S. Y. Joshi, C. Plate, P. Farzeen, E. M. Webb, L. Kumar-Kunche, J. Weger-Lucarelli, A. N. Lowell, A. M. Brown and S. A. Deshmukh, J. Phys. Chem. Lett., 2023, 14, 9490–9499 CrossRef CAS PubMed.
- D. Lowe, AI Does Not Make It Easy (In the pipeline), Science, 2024, https://www.science.org/content/blog-post/ai-does-not-make-it-easy Search PubMed.
- A. Wołos, R. Roszak, A. Żądło-Dobrowolska, W. Beker, B. Mikulak-Klucznik, G. Spólnik, M. Dygas, S. Szymkuć and B. A. Grzybowski, Science, 2020, 369, eaaw1955 CrossRef.
- A. Wołos, D. Koszelewski, R. Roszak, S. Szymkuć, M. Moskal, R. Ostaszewski, B. T. Herrera, J. M. Maier, G. Brezicki, J. Samuel, J. A. M. Lummiss, D. T. McQuade, L. Rogers and B. A. Grzybowski, Nature, 2022, 604, 668–676 CrossRef PubMed.
- R. Roszak, A. Wołos, M. Benke, Ł. Gleń, J. Konka, P. Jensen, P. Burgchardt, A. Żądło-Dobrowolska, P. Janiuk, S. Szymkuć and B. A. Grzybowski, Chem, 2024, 10, 952–970 CAS.
- K. Molga, E. P. Gajewska, S. Szymkuć and B. A. Grzybowski, React. Chem. Eng., 2019, 4, 1506–1521 RSC.
- F. Strieth-Kalthoff, S. Szymkuć, K. Molga, A. Aspuru-Guzik, F. Glorius and B. A. Grzybowski, J. Am. Chem. Soc., 2024, 146, 11005–11017 CAS.
- B. A. Grzybowski, T. Badowski, K. Molga and S. Szymkuć, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2023, 13, e1630 Search PubMed.
- P. Ertl, E. Altmann, S. Racine and R. Lewis, Eur. J. Med. Chem., 2022, 238, 114483 CrossRef CAS PubMed.
- B. Mikulak-Klucznik, T. Klucznik, W. Beker, M. Moskal and B. A. Grzybowski, Chem, 2024, 10, 1319–1326 CAS.
- S. Genheden, A. Thakkar, V. Chadimová, J.-L. Reymond, O. Engkvist and E. Bjerrum, J. Cheminform., 2020, 12, 70 CrossRef PubMed.
- D. Rogers and M. Hahn, J. Chem. Inf. Model., 2010, 50, 742–754 CrossRef CAS.
- D. Liu, T. Ru, Z. Deng, L. Zhang, Y. Ning and F. E. Chen, ACS Catal., 2023, 13, 12868–12876 CrossRef.
- G. M. Morris, R. Huey, W. Lindstrom, M. F. Sanner, R. K. Belew, D. S. Goodsell and A. J. Olson, J. Comput. Chem., 2009, 16, 2785–2791 CrossRef PubMed.
- O. Trott and A. J. Olson, J. Comput. Chem., 2010, 31, 455–461 CrossRef PubMed.
- J. Eberhardt, D. Santos-Martins, A. F. Tillack and S. Forli, J. Chem. Inf. Model., 2021, 61, 3891–3898 CrossRef PubMed.
- W. J. Allen, T. E. Balius, S. Mukherjee, S. R. Brozell, D. T. Moustakas, P. T. Lang, D. A. Case, I. D. Kuntz and R. C. Rizzo, J. Comput. Chem., 2015, 36, 1132–1156 CrossRef.
- ChEMBLdb, http://ftp.ebi.ac.uk/pub/databases/chembl/ChEMBLdb/releases/chembl_29/, accessed February 28, 2025 Search PubMed.
- G. Landrum, RDKit, http://www.rdkit.org, accessed February 28, 2025 Search PubMed.
- M. Awale and J.-L. Reymond, J. Chem. Inf. Model., 2014, 54, 1892–1907 CrossRef PubMed.
- J. L. Durant, B. A. Leland, D. R. Henry and J. G. Nourse, J. Chem. Inf. Comput. Sci., 2002, 42, 1273–1280 CrossRef PubMed.
- A. Mayr, G. Klambauer, T. Unterthiner, M. Steijaert, J. K. Wegner, H. Ceulemans, D.-A. Clevert and S. Hochreiter, Chem. Sci., 2018, 9, 5441–5451 RSC.
- N. Bosc, F. Atkinson, E. Félix, A. Gaulton, A. Hersey and A. R. Leach, J. Cheminform., 2019, 11, 64 CrossRef.
- K. Yang, K. Swanson, W. Jin, C. Coley, P. Eiden, H. Gao, A. Guzman-Perez, T. Hopper, B. Kelley, M. Mathea, A. Palmer, V. Settels, T. Jaakkola, K. Jensen and R. Barzilay, J. Chem. Inf. Model., 2019, 59, 3370–3388 CrossRef.
- S. Asghar, N. Mushtaq, A. Ahmed, L. Anwar, R. Munawar and S. Akhtar, Molecules, 2024, 29, 490 CrossRef.
- SigmaAldrich, Cyclooxygenase 2 human ≥70% (SDS-PAGE), 0858UN-UN1000UN, https://www.sigmaaldrich.com/PL/en/product/sigma/c0858, accessed 8 April 2025.
- R. Roszak, W. Beker, K. Molga and B. A. Grzybowski, J. Am. Chem. Soc., 2019, 141, 17142–17149 CrossRef.
- H. Sugimoto, Y. Iimura, Y. Yamanishi and K. Yamatsu, J. Med. Chem., 1995, 38, 4821–4829 CrossRef CAS.
- SigmaAldrich, Acetylcholinesterase Inhibitor Screening Kit, 324KT-KT1KT, https://www.sigmaaldrich.com/PL/en/product/sigma/mak324, accessed 8 April 2025 Search PubMed.
- A. Pecina, J. Fanfrlík, M. Lepšík and J. Řezáč, Nat. Commun., 2024, 15, 1127 CrossRef CAS PubMed.
- K. Ogura, T. Sato, H. Yuki and T. Honma, Sci. Rep., 2019, 9, 12220 CrossRef PubMed.
- H. Kim and H. Nam, Comput. Biol. Chem., 2020, 87, 107286 CrossRef CAS PubMed.
- X. Zhang, J. Mao, M. Wei, Y. Qi and J. Z. H. Zhang, J. Chem. Inf. Model., 2022, 62, 1830–1839 CrossRef CAS.
- Z. Han, Z. Xia, J. Xia, I. V. Tetko and S. Wu, Eur. J. Pharm. Sci., 2025, 204, 106946 CrossRef CAS PubMed.
- F. Meng, Y. Xi, J. Huang and P. W. Ayers, Sci. Data, 2021, 8, 289 CrossRef CAS PubMed.
- V. Kumar, A. Banerjee and K. Roy, J. Chem. Inf. Model., 2024, 64, 4298–4309 CrossRef CAS.
- S. Cherian Parakkal, R. Datta and D. Das, Mol. Inf., 2022, 41, 2100315 CrossRef CAS.
- B. Mazumdar, P. K. Deva Sarma, H. J. Mahanta and G. N. Sastry, Comput. Biol. Med., 2023, 160, 106984 CrossRef PubMed.
- W. Beker, A. Wołos, S. Szymkuć and B. A. Grzybowski, Nat. Mach. Intell., 2020, 2, 457–465 CrossRef.
- S. A. Wildman and G. M. Crippen, J. Chem. Inf. Comput. Sci., 1999, 39, 868–873 CrossRef CAS.
- Y. J. Chae, H. J. Lee, J. H. Jeon, I.-B. Kim, J.-S. Choi, K.-W. Sung and S. J. Hahn, Brain Res., 2015, 1597, 77–85 CrossRef CAS PubMed.
- S. M. Vogel, L. M. Mican and T. L. Smith, Ment. Health Clin., 2019, 9, 128–132 CrossRef PubMed.
- L. Brandolini, A. Antonosante, C. Giorgio, M. Begnasco, M. d'Angelo, V. Castelli, E. Benedetti, A. Cimini and M. Allegretti, Sci. Rep., 2020, 10, 18337 CrossRef CAS.
- P. J. Hayball, R. L. Nation, F. Bochner, J. L. Newton, R. A. Massy-Westropp and D. P. G. Hamon, Chirality, 1991, 3, 460–466 CrossRef CAS.
- S. L. Rogers, N. M. Cooper, R. Sukovaty, J. E. Pederson, J. N. Lee and L. T. Friedhoff, Br. J. Clin. Pharmacol., 1998, 46, 7–12 CrossRef CAS PubMed.
- W. A. Banks, Adv. Drug. Del. Rev., 2012, 64, 629–639 CrossRef CAS.
- H. Kokki, Pediatric Drugs, 2010, 12, 313–329 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5sc00070j |
| ‡ Authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2025 |