
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceChatbot-assisted quantum chemistry for explicitly solvated molecules†
Rohit S. K.
Gadde
 a,
Sreelaya
Devaguptam
a,
Fangning
Ren
a,
Rajat
Mittal
b,
Lechen
Dong
a,
Yao
Wang
a and
Fang
Liu
*a
a,
Sreelaya
Devaguptam
a,
Fangning
Ren
a,
Rajat
Mittal
b,
Lechen
Dong
a,
Yao
Wang
a and
Fang
Liu
*a
aDepartment of Chemistry, Emory University, Atlanta, GA 30322, USA. E-mail: fang.liu@emory.edu
bDepartment of Physics and Astronomy, Clemson University, Clemson, SC 29631, USA
First published on 29th January 2025
Abstract
Advanced computational chemistry software packages have transformed chemical research by leveraging quantum chemistry and molecular simulations. Despite their capabilities, the complicated design and the requirement for specialized computing hardware hinder their applications in the broad chemistry community. Here, we introduce AutoSolvateWeb, a chatbot-assisted computational platform that addresses both challenges simultaneously. This platform employs a user-friendly chatbot interface to guide non-experts through a multistep procedure involving various computational packages, enabling them to configure and execute complex quantum mechanical/molecular mechanical (QM/MM) simulations of explicitly solvated molecules. Moreover, this platform operates on cloud infrastructure, allowing researchers to run simulations without hardware configuration challenges. As a proof of concept, AutoSolvateWeb demonstrates that combining virtual agents with cloud computing can democratize access to sophisticated computational research tools.
1. Introduction
Computational chemistry has significantly advanced chemistry research in recent decades, from revealing reaction mechanisms1,2 and interpreting spectroscopy3 to generating training sets for artificial intelligence (AI) assisted design and discovery.4,5 Advances in theoretical methods and software empower researchers to tackle increasingly complex problems, yet learning to use these tools properly becomes increasingly difficult. Computational chemistry packages, whether for electronic structure calculations or molecular simulations, invariably demand the users' familiarity with the underlying theories and package-specific options, alongside sufficient computing resources for executing them. Many chemical processes necessitate the synergistic usage of multiple packages, posing additional challenges for researchers across the broad chemical science community.The past decades have seen a growing trend of developing open-source, automated workflows to address this issue. Quantum mechanical (QM) calculation workflows, such as Material projects,6 QCDB,7 and MolSimplify,8 have significantly enhanced data generation efficiency and software interoperability for data-driven research on solid-state materials and molecules. In addition, the AutoSolvate toolkit9 has streamlined the modeling of explicitly solvated molecules by synergizing QM calculations, force field fitting, and molecular dynamics (MD) simulations, allowing efficient computational investigations of real-life solution phase chemical processes.
However, two major challenges remain. Firstly, crucial simulation parameters must still be set manually, forcing the users to delve into lengthy user manuals. Secondly, the workflows often require high-performance computing resources that are not readily accessible to non-computational researchers. These barriers render the workflows unfriendly to students and experimental chemists. Consequently, experimental researchers frequently struggle to simulate chemical processes in realistic experimental environments, which require synergistic utilization of multiple simulation packages. For example, the solvent environment plays vital roles in synthesis, catalysis, and energy storage, where fast and accurate descriptions of solvents in the simulation are essential. However, available automated computational workflows are usually limited to gas-phase or implicit solvent quantum chemistry calculations. Learning to perform complex simulations that explicitly describe solvents on high-performance computing clusters is time-prohibitive for experimental researchers. Hence, a user-friendly computation platform that requires minimal simulation experience and hardware prerequisites is essential for expanding access to computational chemistry within the broader community.
Here, we introduce AutoSolvateWeb, a chatbot-assisted, cloud-based computational platform for quantum chemistry studies of explicitly solved molecules, as a proof of concept to address the challenges concurrently. Having achieved automation through the AutoSolvate workflow, AutoSolvateWeb further addresses the accessibility challenge by integrating a chatbot using the Google Dialogflow CX framework.10 The chatbot educates users through natural language conversations, guides users to configure parameters for modeling explicitly solvated molecules, triggers calculations in the backend, and retrieves the results after completion. In addition, AutosolvateWeb fulfills hardware requirements by conducting all calculations on accessible cloud-computing resources, providing convenient access for anyone with an internet-accessible device.
This work innovatively employs a chatbot to assist users of scientific software, potentially reshaping how scientists interact with advanced research tools. AI chatbot frameworks such as Google Dialogflow CX,10 Microsoft Azure Bot service, and Amazon Lex find extensive applications in creating virtual assistants for commercial usage, such as online banking11 and customer service.12 Nevertheless, very few AI applications are dedicated to enhancing the user experience of scientific research tools.13–15 Exceptions include ChemVox16 for voice-controlled fixed-type quantum chemistry calculations and Coscientist17 for GPT-4 assisted autonomous chemical experiments. Most scientific software users rely heavily on manuals and expert guidance for multistep calculations. Through the chatbot integration, AutoSolvateWeb enables non-expert users to perform multistep simulations of explicitly solvated molecules for the first time without external aids. This convenience empowers researchers from the broad community to efficiently utilize complex functionalities of scientific computing tools, which marks a significant step forward in integrating AI into scientific research.
2. Results
2.1 Overview of AutoSolvateWeb functionality
AutoSolvateWeb's primary functionality is to automate explicit solvent simulations for an arbitrary user-specified organic molecule solvated in arbitrary organic solvents. The outputs of AutoSolvateWeb are solvation configurations sampled from molecular dynamics simulations, representing 3D structures of solute molecules immersed in a specific number of explicit solvent molecules. These configurations are valuable to chemists studying solute conformation in solvent environments or exploring solute–solvent interactions (e.g., hydrogen bonding). Additionally, these configurations serve as starting points for quantum chemistry calculations to predict molecular properties (e.g., redox potential), simulate molecular spectra (e.g., UV/Vis and IR spectra), and investigate chemical reaction mechanisms in the solution phase. Table 1 summarizes typical computational studies requiring explicit solvation configurations and, therefore, can benefit from the simulations automated by AutoSolvateWeb.| Properties | Target properties | When is the explicit solvent needed | Further calculations |
|---|---|---|---|
| Solvation configuration | Visualization,21 conformer sampling22,23 | Always | Visualization, MD trajectory analysis, and conformational distribution analysis |
| Ground state interaction | H-bond,24 interaction energy25 | Always | QM calculation, energy decomposition analysis (EDA), weak-interaction analysis, visualization |
| Thermodynamics | Density, solubility, vaporization enthalpy (ΔHvap),26 solvation free energy (ΔGsol)27 | H-bond, non-bonded | QM calculation, thermodynamic integration |
| Spectrum | UV/Vis,28 IR,29 VCD,30 Raman,311H/13C NMR32 | H-bond | QM excited-state calculation (UV/Vis), Hessian (IR, Raman), or chemical shift calculation (NMR) |
| Excited state | Redox potential,33 charge transfer34 | H-bond, polar solvent | QM optimization of the initial and final state, compute Hessian and zero-point vibrational energy (ZPVE), electronic coupling |
| Nonadiabatic process | Charge/exciton transfer rate,35 decay pathway, decoherence36 | Solvent participates in the reaction | Generate initial geometry, then use corresponding nonadiabatic dynamic methods |
| Chemical reaction | Reaction pathway, catalysis mechanism, activation energy37–39 | H-bond, adsorption, dynamic response | ab initio MD (AIMD) or QM/MM, cluster-continuum model, TS-search, meta-dynamics |
AutoSolvateWeb's automation of simulations is achieved through the command-line-based AutoSolvate backend, which generates explicit solvation configurations through three steps: force field and solvent box generation (“Step-1”), MD simulation (“Step-2”), and microsolvated cluster generation (“Step-3”). In Step-1, a solvent box accommodating a user-provided solute molecule surrounded by solvent molecules (water, methanol, acetonitrile, chloroform, or NMA) is constructed, with the missing General Amber Force Field (GAFF)18 parameters determined by quantum chemistry calculations. Then, solvation configurations are sampled in Step-2 using the AMBER19 molecular dynamics package, with optional QM/MM simulations conducted with the GPU-accelerated quantum chemistry package, TeraChem.20 Finally, in Step-3, users have the option to extract microsolvated clusters of customized sizes from MD trajectories as inputs for other quantum chemistry packages. More details about the software design, usage, and applications of the command-line-based AutoSolvate toolkit are available in our previous publication.9
Although the original command-line-based AutoSolvate Toolkit has automated the simulation workflow, it still presents challenges for users without a background in computational chemistry. Each of the three steps requires a command with multiple user-specified keywords, necessitating familiarity with the Linux shell environment and additional time to learn the command syntax. Furthermore, some backend software packages integrated with AutoSolvate require specialized computing hardware (e.g., TeraChem requiring GPUs), which demands the correct configuration of both software and hardware. These challenges may deter many potential users, such as chemistry undergraduates lacking computational training, from effectively utilizing the tool. To address these challenges, we developed AutoSolvateWeb, a web-based interface that leverages cloud computing resources to eliminate software and hardware configuration issues. A built-in chatbot gathers the necessary keywords through natural language conversations with users and automatically prepares the commands for the three steps, freeing users from the need to learn complex command syntax.
2.2 The chatbot design philosophy for AutoSolvateWeb
We designed the chatbot for AutoSolvateWeb with the intention to balance specialization with flexibility, efficiency, and capability. Currently, two types of chatbots are commonly considered for scientific software interfaces: traditional chatbots and large language models (LLMs). Traditional chatbots are rule-based or intent-driven, matching inputs to a limited set of predefined responses, such as, “If the user says X, respond with Y”.40 Consequently, traditional chatbots often struggle with nuanced conversations and may indicate that they cannot understand the query. In contrast, LLMs are built on advanced architectures like Transformers41 (e.g., OpenAI's GPT, Google's BERT) and can encode complex contextual relationships across large amounts of data. Consequently, LLMs exhibit contextual understanding and can generate responses to complex, open-ended queries.42 While LLMs appear more capable than traditional chatbots, our specific goal of assisting users in setting input keywords for a specialized scientific software package poses unique challenges. A disadvantage of LLMs for our task is the complexity and overhead to deploy. Building an LLM from scratch is computationally expensive, requiring substantial resources for training, inference, and deployment.41 Fine-tuning existing LLMs, such as ChatGPT, is also challenging due to the lack of high-quality, domain-specific data. AutoSolvate, being a new computational package, has little readily available data for training, and curating sufficient data would require substantial effort. Additionally, LLMs may generate inconsistent responses depending on the phrasing or context of a query. In our use case, this inconsistency could result in the generation of inconsistent input keywords for the same molecular systems, compromising the reproducibility of resulting simulations.Based on these considerations, we design the chatbot using the Google DialogFlow CX42 framework (Fig. 1), which combines the advantages of lightweight, intent-driven chatbots with the additional flexibility provided by generative AI (LLMs). To ensure that all necessary input keywords are collected consistently, we have predefined a dialog flow where the chatbot proactively asks users questions to gather information about the solvated molecules and select each keyword needed for preparing input commands for the AutoSolvate backend (Fig. 1). Users are generally expected to respond to these questions sequentially. However, experienced users can bypass certain questions and proceed directly to another stage of the simulation by providing instructions such as “Go to Step-3” (more details available in Section 5.2 and ESI Text S1†). This aspect of the chatbot design is intent-based, like traditional chatbots.
 | ||
| Fig. 1 AutoSolvateWeb's chatbot design philosophy. The command–line interface at the top is transformed into a chatbot-based interaction. The chatbot combines a predefined dialog flow focusing on collecting each keyword from users, some definition intents to answer users' clarifying questions, and the generative fallback to avoid conversational derailment. | ||
Our chatbot also allows users to ask clarifying questions about terminologies mentioned in its prompts. For example, when the chatbot asks the user to specify the solvent, the user can inquire: “What does solvent mean?” The chatbot first attempts to match such questions with a predefined intent, “UserQuestions”, which responds the user with standard definitions of keywords and terminologies related to AutoSolvate (Fig. 1, ESI Text S1 and Table S1†). This ensures that domain-specific answers are delivered consistently, avoiding potential inconsistencies or hallucinations commonly associated with LLMs.43 If the user's question does not match any predefined intent or any parameter relevant to AutoSolvateWeb, the query is handled by the generative fallback feature of Google's latest generative LLMs. In such cases, the chatbot uses LLMs to generate a response informing the user that their question is out of scope and encourages them to address the chatbot's previous query (Fig. 1). This approach mitigates the risk of conversational derailment, a common issue with LLMs when deployed in open-ended conversational systems. By integrating intent-based design with generative AI capabilities, the chatbot achieves a balance between deterministic and flexible interaction styles, meeting the specific requirements of scientific software assistance. More technical details of the conversation flow are available in Section 5 and ESI Fig. S1.†
2.3 AutoSolvateWeb cloud server structure
With the aforementioned design philosophy, we implement AutoSolvateWeb on the JetStream2 (ref. 44) cloud computing platform. Fig. 2 illustrates the architecture of AutoSolvateWeb, composed of four containerized services: the Node server, the Nginx server, the chatbot server, and the AutoSolvate server, orchestrated via the Docker containerization platform. The Node server hosts the webpage with the chatbot frontend and necessary scripts to initiate chat sessions via API calls to Google's Dialog flow CX API through the chatbot server. Each user prompt on the chatbot server triggers a REST API request for the virtual agent, facilitating calculation setup via natural language conversations. Once all input parameters are validated and confirmed by the user, automated simulations will be triggered on the AutoSolvate server, which executes the command–line-based AutoSolvate toolkit on the CPU/GPU instances of JetStream2 (ref. 44) cloud infrastructure. Finally, the outputs (solvation configuration) are returned to the Node server upon completion. | ||
| Fig. 2 AutosolvateWeb workflow on the JetStream2 cloud computing platform. (1) The user communicates with the chatbot server through the Ngnix server; (2) the chatbot retrieves the required structure from PubChem; (3) the chatbot server makes a REST API request to Google's Dialog flow CX virtual agent; (4) the chatbot server sends the necessary information to the AutoSolvate Server and triggers calculations; (5) the calculation results are returned to the Node Server for visualization and downloading; (6) the user downloads the calculation results through the Ngnix reverse proxy. | ||
2.4 Chatbot-assisted explicit solvent simulation
Once the user confirms to proceed to generate solvation configurations, the chatbot switches to the second stage of conversation and sequentially guides the user through the three steps of running AutoSolvate. Fig. 3 depicts a sample conversation for Step-1 between the user and the chatbot. In the first step, the user provides the solute structure by uploading an XYZ or providing the IUPAC name, prompting the chatbot to download the corresponding structure from PubChem. The chatbot then navigates the user through the parameter setup procedure via natural language conversation. Suggested response buttons are provided in the chat box to simplify this process further. | ||
| Fig. 3 Example conversation for running Step-1. Some dialogue has been omitted for brevity, including (1) the user specifies the charge and spin multiplicity of the solute; (2) the user provides the solvent box size. | ||
For example, specifying the solvent can be done by typing “water”, “the solvent is water”, “use water”, or clicking on the “water” button in the chat box (see ESI Table S2† for all supported solvents). Further, while prompting for a parameter, the chatbot provides a link to a webpage with its definition for further reading. Alternatively, the user can ask the chatbot with the phrase, “What is the definition of …” and receive the answer in the chat box. Once all parameters are set, the chatbot automatically validates them before confirming with the user to initiate the calculation. The validation includes checking the type and range of each parameter (e.g., whether spin multiplicity is a positive integer) and whether the combination of different parameters defines a valid chemical system (e.g., the compatibility of the net charge and spin multiplicity for a given molecule). At this step, the user can either confirm or reset the input parameters. Upon user confirmation, the chatbot sends all required information to the backend via the chatbot server, initiating the AutoSolvate workflow and awaiting calculation completion. A compressed folder containing all essential output files (descriptions available in ESI Table S3†) can be downloaded by clicking the “Download” button on the web page or the “Download Step-1 Button” in the chat box. Clicking the “Show output for Step-1” button will display the image of the generated solvent box with JSmol.45
Typically, Step-1 takes less than 92 seconds for small to medium-sized closed-shell solute molecules with up to 33 heavy atoms (Fig. 4 and ESI Text S2†), meaning the users can get the results almost instantly. For open-shell solute molecules (spin multiplicity greater than 1) with up to 33 heavy atoms, Step-1 takes up to 774 seconds due to the requirement to evaluate the restrained electrostatic potential (RESP)47 atomic partial charges with the GAMESS quantum chemistry package (ESI Table S4†). A built-in queue on the web interface allows users to check their job status (ESI Fig. S3†). The queue will keep the output files for up to 14 days, which allows the user to temporarily step away when waiting for a lengthy job to complete.
 | ||
| Fig. 4 Runtime for Step-1 of AutoSolvateWeb. The test systems comprise the water solvent and selected small to medium-sized solutes, with solute charge 0 and spin multiplicity 1. For each test system, timings for the AM1-BCC46 charge fitting (default option for closed-shell solutes) and RESP47 charge fitting are done separately. Each solute's structure is shown in the inset, along with the number of heavy atoms and basis functions. | ||
The chatbot then prompts the user to proceed to Step-2 by asking, “Do MD simulation?” The user can proceed or redo the previous step by typing in “restart Step-1” or terminate this workflow by saying “goodbye” or its synonyms. If the user chooses to proceed to Step-2, the chatbot will guide them in configuring input parameters for various types of classical mechanics (MM) or QM/MM MD simulations, then automatically generate the simulation input files and execution scripts (Fig. 5). Specifically, the workflow assumes the sequential performance of MM minimization, heating to a target temperature, constant temperature constant volume equilibration (NVT ensemble), constant temperature constant pressure equilibration (NPT ensemble), and constant energy constant volume simulation (NVE ensemble), followed by optional QM/MM simulations (minimization, heating up, NVT, and NVE). After specifying the simulation temperature and pressure, the chatbot asks whether the simulations will be performed with the “dry run” mode, meaning that only the input files will be generated without actual MD simulations running on the cloud. If the “dry run” mode is off, a short simulation will run on the cloud for demonstration purposes only due to resource limitations on JetStream 2. Simulation length is restricted to up to 100 steps for each stage of MM simulations and 10 steps for all QM/MM simulation stages, with output trajectories included in the download zip file upon job completion. If the “dry run” mode is on, the chatbot asks the user to specify the length of each simulation stage. Only the simulation input files will be generated for download, which is useful for users intending to run production simulations on their own computing clusters. Finally, the user can proceed to Step-3 for microsolvated molecule cluster extraction based on the trajectories generated in Step-2. Users can download these files along with the outputs of previous steps by clicking the “Download Step-3 Output” button in the chat window.
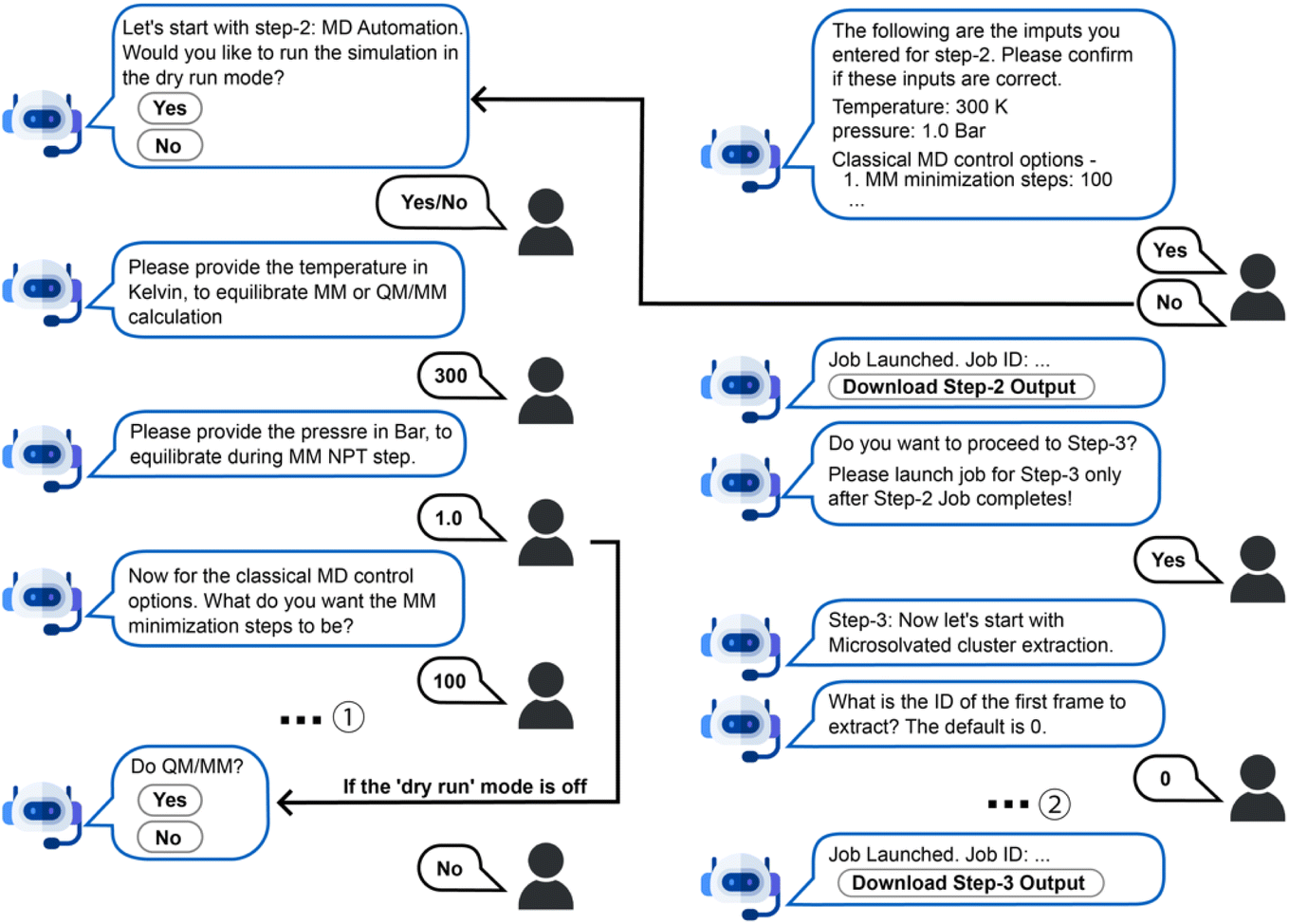 | ||
| Fig. 5 An example conversation for running Step-2 and Step-3. Some dialogue has been omitted for brevity, including (1) the user specifies the length of the MM heat up, NVT, NPT and NVE step; (2) the user provides the extraction interval and shell thickness for Step-3. | ||
2.5 Example: innovative laboratory course for solvatochromism
AutoSolvateWeb allows chemists, including those without a computational background, to conduct complex calculations, thus making computational analysis more accessible for undergraduate laboratory courses. Here, we present an example of how AutoSolvateWeb can be used to design innovative laboratory courses centered around the solvatochromic effect.Solvatochromism is a photophysical phenomenon where the color (wavelength) of light absorbed or emitted by a molecule shifts in response to changes in the solvent's polarity and other intermolecular forces.48 This effect is particularly useful in photochemistry for investigating solvent environments, as the shifts can indicate the nature of solute–solvent interactions, such as hydrogen bonding and dipole–dipole interactions. Solvatochromic shifts are complex and influenced by a range of solvent effects, including polarization, structural modifications, and dynamic reorientation of the solvent around the solute in excited states.
Solvatochromism introduces students to the complex solvation concept and can be used to teach dye synthesis, UV/Vis spectroscopy, solvent polarity, and molecular dipole moment. Lab projects centered on solvatochromism have been adapted to undergraduate lab courses in organic chemistry,49,50 analytical chemistry, and physical chemistry.51 However, most existing lab projects only provide qualitative explanations of solvatochromism, where the effect is described as the differential stabilization of the HOMO and LUMO by polar solvents. A quantitative computational investigation of the frontier orbitals' energy changes in various solvents is not integrated into the lab project, likely because it requires a complex computational protocol involving the generation of solvation configurations. For students with limited or no computational chemistry training, completing these tasks within the lab timeframe would be impractical, creating a barrier to directly illustrate how the solvent environment impacts the solute's electronic structure.
Here, we demonstrate the potential use of AutoSolvateWeb to introduce a computational component in the solvatochromism lab. We take the lab project proposed by González-Arjona et al. as an example, where UV/Vis spectra of Reichardt's dye (Fig. 6), also known as Betaine 30, are recorded in solvents of different polarities and hydrogen bond effects.51 The experimental UV/Vis spectrum peak of Reichardt's dye significantly blue shifts from 620 nm to 520 nm when the solvent changes from acetonitrile to methanol, despite their similar relative dielectric constants (36.65 and 33.00, respectively). Since implicit solvent models like the polarizable continuum model (PCM)52 mostly account for the solvent effects through the solvent’ dielectric constant, they cannot distinguish the different impacts of acetonitrile to methanol. The differences in computed HOMO–LUMO gaps between the two solvents, together with the excitation energy computed by time-dependent DFT (TDDFT)53 with both linear-response54 and state-specific55 PCM, were insignificant, failing to reproduce the experimental spectrum shift (ESI Text S3†). Hence, students need to explore the explicit solvation configurations to understand the origin of this blue shift. The students will use AutoSolvateWeb to obtain solvation configurations of Reichardt's dye in acetonitrile and methanol and perform quantum chemistry following our instructions below.
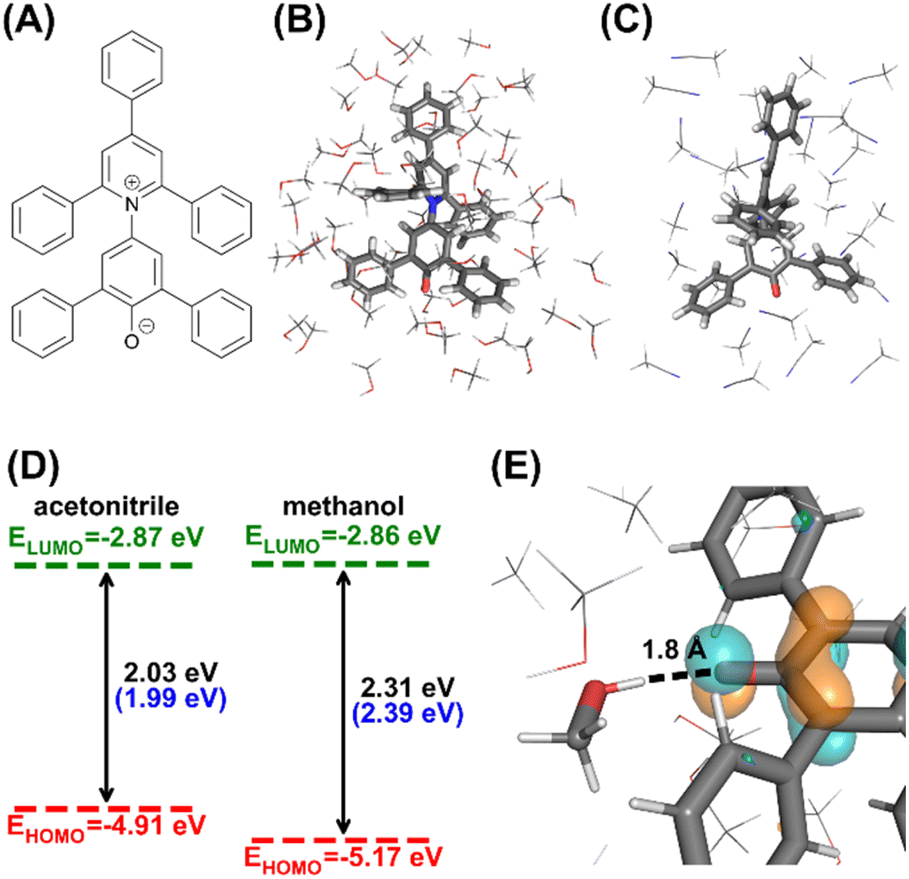 | ||
| Fig. 6 Expected computational results for the solvatochromism lab. (A) 2D structure of the Reichardt's dye. (B) Microsolvated cluster extracted in methanol. (C) Microsolvated cluster extracted in acetonitrile. (D) Average computed frontier orbital energies and HOMO–LUMO gaps for 10 acetonitrile and methanol-solvated clusters. Values in blue brackets are experimental absorption maxima in corresponding solvents.51 (E) Close-up look of the HOMO orbital of a methanol solvated cluster (isovalue 0.05 a.u.), focusing on the oxygen atom on the Reichardt's dye. The solute and the methanol molecule forming hydrogen bonds are drawn in sticks. | ||
The first part of the computational component is generating solvation configurations of Reichardt's dye in acetonitrile and methanol through conversations with the AutoSolvateWeb chatbot, with a complete record of the conversations detailed in ESI Text S4.† In Step-1, the name of the solute (“Reichardt's dye”) and solvents are provided to the chatbot, which then generates a 45 × 45 × 45 Å solvent box of a Reichardt's dye in 1193 methanols and another box of a Reichardt's dye in 1075 acetonitriles, together with classical force field parameters. In Step-2, MD samplings are performed for each solvent box, resulting in a 1 ns long NPT production trajectory at 298 K and 1 bar. In Step-3, students would extract microsolvated clusters with a 4.0 Å thick solvent shell from the MD trajectories and obtain 10 solvation configurations with an interval of 100 ps. Example clusters are shown in Fig. 6.
The second part is QM calculations on the solvated dye to understand the solvatochromism shift from acetonitrile to methanol. The students will perform DFT (PBE0 (ref. 56 and 57)/6-31G* (ref. 58 and 59)) calculations of the solvation configurations, with an additional PCM model applied to the cluster to account for the electrostatic effects of bulk solvents. The calculations can reproduce the experimentally observed absorption difference of 0.40 eV between methanol and acetonitrile, which can be mainly attributed to the 0.26 eV decrease in the HOMO energy. This is because the HOMO is localized around the solute's oxygen atom (Fig. 6). The oxygen atom forms hydrogen bonds with protic solvents like methanol, changing the local electrostatic environment and lowering the HOMO energy, thus affecting the absorption shift. With the above computational experiment with AutoSolvateWeb, students without computational chemistry backgrounds can perform multistep calculations and intuitively understand the solvatochromism through the computed results. The example computation results are included in the ESI 3.†
This proposed computational component is appropriate for an organic chemistry lab where students have learned basic concepts like HOMO and LUMO but are not trained in electronic structure theory. For computational chemistry lab or advanced physical chemistry lab, the proposed lab can be extended by using excited-state electronic structure methods [e.g., TDDFT or complete active space self-consistent field60 (CASSCF)] for the quantum chemistry calculation part to obtain calculated shifts that agree with the experiment even better. Regardless, the explicit solvation configurations are still the foundation for the QM calculations.
3. Discussion
AutoSolvateWeb pioneers the integration of chatbots to streamline complex computational workflows in a user-friendly manner, enabling cloud-based, easily operated solution-phase chemistry data generation. Through interactions with AutoSolvateWeb's chatbot, users without prior experience in MD and QC software can perform multiple-step explicit solvent simulations without delving into lengthy documentation, flattening the learning curve for new computational chemistry software. Moreover, AutosolvateWeb enables researchers to participate in data generation and sharing without specialized computing hardware, democratizing data-driven research in related fields.As a proof-of-concept tool, AutosolvateWeb still has ample room for improvement. This section highlights some current limitations and discusses plans for future development.
3.1 Supported solvation systems
The current version of AutoSolvateWeb supports single organic molecules as the solute. These solutes must contain only elements compatible with GAFF, including hydrogen (H), carbon (C), nitrogen (N), oxygen (O), phosphorus (P), sulfur (S), and halogens fluorine (F), chlorine (Cl), bromine (Br), and iodine (I). Metals, other main-group elements, and noble gases are not yet supported. Additionally, specific solutes with unique bonding patterns not defined in GAFF are incompatible. For example, the diphenyliodonium cation is unsupported because its iodine atom forms two single bonds with two phenyl groups, which deviates from the bonding patterns for iodine defined in GAFF.To expand the range of chemical systems that can be simulated, we have already implemented new functionalities in the development branches of the AutoSolvate command–line interface. These updates support arbitrary organic solvents, an arbitrary number of solvent/solute species, and transition metal complexes as solutes. These functionalities will be integrated into AutoSolvateWeb in the near future.
3.2 Accuracy of the explicit solvent simulations
The solvation configurations generated by the AutoSolvate backend have been used to calculate various molecular properties. For instance, the thermodynamic integration approach was employed to calculate the redox potentials of a diverse set of 165 organic redox couples using solvation configurations generated by AutoSolvate.61 These calculations achieved a mean absolute error (MAE) of 0.64 V compared to experimental measurements, which was significantly reduced to 0.19 V when a machine learning-based error correction algorithm was applied to mitigate systematic errors.61 Notably, only 3 out of the 165 explicit solvent calculations resulted in large-error outliers (errors >1 V compared to experiments), a significant improvement over the corresponding implicit solvent calculations, which yielded 11 outliers.61 Additionally, other research groups have applied AutoSolvate to investigate solvation conformations of natural products21,62,63 and to simulate Raman spectra.64However, users should be aware of potential limitations in accuracy arising from the method or computing resources. The AutoSolvate backend relies on non-polarizable force fields, such as GAFF and Amber force fields, for classical MD and QM/MM simulations. While these force fields are computationally efficient, they fail to accurately describe certain solvated systems because they cannot account for dynamic polarization effects.65,66 Complex solvation phenomena, such as ion pairing in electrolyte solutions or solute-induced solvent structure perturbations, are inadequately represented without polarization effects.67 In such cases, users can employ AutoSolvateWeb to generate the initial solvated structures, but further sampling with polarizable force fields, machine learning potentials, semi-empirical methods, or even ab initio molecular dynamics is necessary to incorporate dynamic polarization effects and obtain higher-quality solvation configurations. We are also extending the AutoSolvate backend's workflow to include additional sampling steps using semi-empirical or higher-level methods.
Additionally, sufficiently long simulation trajectories are required for publication-quality sampling. For instance, our previous study indicates that 600 ps of NPT sampling is necessary for calculating the redox potential of solvated small organic redox couples.61 Due to resource limitations on the JetStream2 cloud computing platform, AutoSolvateWeb allows only a limited number of steps for MM and QM/MM sampling when executed directly through the platform, as mentioned in the Result section. The web interface primarily serves to demonstrate functionality and educate users about simulation concepts. Therefore, users aiming to generate publication-quality MD trajectories are encouraged to utilize AutoSolvateWeb's “dry run” mode, which generates force field parameters and simulation input files without executing the simulations on the cloud. These input files can then be used to run long simulations (at the nanosecond time scale) on local computing resources. We also plan to apply for additional computing resources on JetStream2 to support AutoSolvateWeb, enabling longer simulation steps for each user session.
3.3 Cyberinfrastructure
We have implemented several strategies to ensure the web interface can handle high user traffic and manage computational workloads effectively. To evaluate the chatbot's performance under heavy traffic, we conducted stress tests simulating thousands of concurrent users accessing the website (ESI Tables S5–S8†). The results demonstrated that the website successfully loaded with 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 concurrent users and maintained a very low probability (0.4%) of loading errors even with 25000 concurrent users (ESI Table S8†). However, the chatbot could handle only up to 120 simultaneous users due to a limitation in the DialogFlow service, which allows a maximum of 1200 text queries per minute (ESI Tables S6 and S7†). Beyond this threshold, some users may experience chatbot failures in responding to their queries. In such cases, users can retry sending their query, and the chatbot will resume functioning once the traffic peak subsides. Moreover, traffic exceeding 120 concurrent users is unlikely during the initial launch of this specialized chemistry software website. Therefore, we expect users to experience smooth interactions with the chatbot under typical conditions.
000 concurrent users and maintained a very low probability (0.4%) of loading errors even with 25000 concurrent users (ESI Table S8†). However, the chatbot could handle only up to 120 simultaneous users due to a limitation in the DialogFlow service, which allows a maximum of 1200 text queries per minute (ESI Tables S6 and S7†). Beyond this threshold, some users may experience chatbot failures in responding to their queries. In such cases, users can retry sending their query, and the chatbot will resume functioning once the traffic peak subsides. Moreover, traffic exceeding 120 concurrent users is unlikely during the initial launch of this specialized chemistry software website. Therefore, we expect users to experience smooth interactions with the chatbot under typical conditions.
To manage the hardware resources for backend molecular dynamics (MD) simulations, we implemented job queuing on the Node server to prevent excessive server strain, as detailed in the Section 5.4 and ESI Text S5.† Currently, the system allows a maximum of four CPU jobs and one GPU job at any given time, regardless of the number of active users. To ensure a single user's lengthy simulation does not monopolize the queue, we imposed limits on the number of simulation steps, as discussed in the previous subsection. In future updates, we aim to secure additional cloud computing resources, which will enable a higher number of simultaneous jobs and reduce restrictions on simulation steps.
To utilize cloud computing resources more efficiently, AutoSolvateWeb can implement data storage and reuse functionalities to ensure compliance with the FAIR data principles.68 The AutosolvateWeb chatbot can now clarify terminologies, guide parameters setup, perform fact-checking, and initiate MD and QM calculations. Nevertheless, due to chatbot architecture limitations, users must adhere to a specific sequence of conversations, as explained in the Result section. Despite offering some flexibility in language input, chatbots may still misinterpret user requests if their phrasing deviates from predefined patterns. Large language models (LLMs), featured by the OpenAI's GPT, have consistently demonstrated remarkable versatility in recent years. While training an LLM for field-specific tasks remains data-demanding and computationally expensive, we expect that with advancements in AI, field-specific LLM will be more accessible in chatbot frontend design. Users will be able to perform highly complex calculations using natural language, bypassing predefined steps and patterns.
4. Conclusions
In conclusion, AutosolvateWeb innovatively integrates a chatbot frontend and cloud computing with automated workflow, significantly reducing the knowledge and hardware barriers in using computational chemistry packages and democratizing data-driven research across the chemical science community. We anticipate the adoption of analogous strategies to incorporate AI across various domains of basic science, transforming the utilization of advanced scientific tools.5. Methods
AutoSolvateWeb consists of four containerized applications: a web application, a reverse proxy application, a virtual agent proxy service, and an AutoSolvate cloud server application. Due to robust containerization, all applications can be horizontally scaled seamlessly on both traditional and elastic clusters. All the containerized applications are currently deployed on a single cloud instance (4 cores with NVIDIA A100 GPU) on Jetstream2.5.1 Web application and reverse proxy application
The web application engages with users to authenticate identity, define input parameters and files, and visualize molecular geometry. This service also manages user-specific file storage and facilitates inter-server calls to launch computation jobs. Interactive visualization of input and output molecular geometry files is enabled by the JSMol viewer.45 SSL ensures the security of all communication between the web application and the user's browser. In addition, strategically positioned CAPTCHA verifications from hCAPTCHA69 have been integrated throughout the web application workflow to prevent abuse by spam bots. Since all four applications are currently deployed on a single cloud instance, a reverse proxy application using Nginx70 redirects requests to appropriate applications. Additionally, since a reverse proxy also acts as a load balancer, any future scalability (i.e., deploying to multiple cloud instances) is secured.5.2 Virtual agent
We have employed Google's DialogFlow CX to develop the virtual agent because of the extensive documentation, ease of annotation, and flexibility in designing agent responses. All the interactions between the virtual agent and users rely on a virtual agent proxy service. Apart from handling server–server authentication with Google Cloud Platform (GCP), this proxy service also acts as a gateway to restructure the virtual agent's responses to deliver complex end–user interaction experiences (such as loading visualizations on job completion and uploading files as part of the conversation) on the webpage.Conversation in DialogFlow CX is a collection of flows embedded in a finite state machine (ESI Fig. S1†). Each flow may be made up of multiple pages. Each page may have an entry fulfillment (parameters to be filled in by the user during the conversation) and a route that decides the transition to the next page. Each route is either associated with an intent or some condition (Boolean equation) based on the parameters filled by the user or both. The virtual agent in AutoSolvateWeb has a single flow of twelve pages and six intents. Each of the pages represents a state in the conversation (ESI Table S9†), allowing the user to (1) choose between uploading a solute file or downloading from PubChem API; (2) specify the solute name; (3) upload a geometry file; (4) choose the recommended parameters for the solute in Step-1 if the solute is downloaded from PubChem API; (5) set all the solute and solvent parameters manually in Step-1; (6) confirm the inputs of Step-1; (7) choose if Step-2 is to be run in the “dry run” mode; (8) set parameters for MM minimization in Step-2; (8) choose to add QM/MM to Step-2; (9) set parameters for QM/MM in Step-2; (10) confirm inputs for Step-2; (11) set parameters for Step-3; (12) confirm inputs for Step-3. Pages 2, 6, 10, and 12 post webhooks to the Autosolvate web server as part of their fulfillment. Currently, the six intents are to: (1) recognize a successful task; (2) recognize a failed task; (3) recognize a Boolean True input from the user; (4) recognize a Boolean False input from the user; (5) recognize if the user wants to upload the solute file; (6) recognize if the user wants to download the solute file from PubChem. Additionally, there are three more intents corresponding to restarting each of the simulation steps, which are triggered in response to a phrase fuzzy matching the following: (1) “Run Step-1” or “Restart Step-1”; (2) “Run Step-2 [in dry run mode][ with QM]” or “Run Step-2 [in normal mode] [with QM]”; (3) “Run Step-3” or “Restart Step-3”. It is worth noting that user prompts need not match the exact phrase of an intent. DialogFlow CX can map a similar phrase to their respective intents. More detailed explanation of the intent-based conversations is available in ESI Text S1.†
Also, all the responses from the virtual agent have a “recommended response” for the user – except for prompts that ask the user to set input parameters. The “recommended response” aims to familiarize new users with the chatbot workflow. The users might also choose to input any of the above-discussed intents to steer the conversation.
5.3 AutoSolvate cloud server
Finally, the Autosolvate cloud server application executes the computation job only at the request of the web application or the DialogFlow virtual agent through an authenticated webhook. The containerized image of this application has an Autosolvate Conda environment, all the third-party software (AMBER,19 GAMESS71 and TeraChem20) orchestrated by the Autosolvate framework and distributed computing frameworks such as OpenMPI.72 Hence, this container may be deployed seamlessly on any traditional cluster or a cloud computing instance. The server application has four workers to handle job requests, which are processed synchronously. Each job request spawns a new process to execute the job. Further, the GPU drivers are exposed only to this application.5.4 Job queue
When a job is requested by the user, it is queued and will be executed when a computing resource is available. The job status and outputs are accessible from the web interface. A user can have up to two jobs in the queue at any time: one job of either Step-1 or Step-3 and one job of Step-2. The implementation of the job queue is discussed in ESI Text S5 and Fig. S3.†5.5 Computational details
For Step-1 force field fitting, the partial charges are determined by GAMESS at the HF/6-31G* level of theory if the RESP47 charge method is selected, or by AmberTools' Antechamber module to obtain the semi-empirical AM1-BCC46 charges otherwise. For Step-2, the classical molecular dynamics simulations use the GAFF force field for the solute and well-established force fields for the five supporting solvents summarized in ESI Table S2.† The QM/MM simulation treats the solute as the QM region at HF or B3LYP level of theory based on the user's selection, using either the 6-31G* basis set or LANL2DZ effective core potentials for the transition metals, Iodine, and Bromine.Data availability
AuotSolvateWeb (https://autosolvate.che230059.projects.jetstream-cloud.org) is available as a web server and can be directly used without installation (ESI Text S6 and Fig. S2†). The source code of the AutoSolvate backend is available as open source on GitHub (https://www.github.com/Liu-group/AutoSolvate). A video tutorial for interacting with the chatbot on AutoSolvateWeb is available on YouTube (https://youtu.be/kBhugQ6cbc0). Instructions about reproducing the demo shown in the video tutorial are also available in ESI Text S6 and S7 and ESI 1.† All output files of an example AutoSolvateWeb job (Supplementary_Data1), the Cartesian coordinates of solute molecules used for scaling analysis (Supplementary_Data2), and the example results for the innovative course design (Supplementary_Data3). See https://figshare.com/s/2bcfa0860bbecf8467f6.Author contributions
F. L. conceived this project. R. S. K. G. designed the cloud computing orchestration architecture. R. S. K. G. and R. M. implemented the frontend web server, supervised by Y. W. and F. L. jointly. R. S. K. G. and S. D. designed and implemented the chatbot. L. D. and F. L. contributed to the code improvement of the backend. F. R., R. S. K. G. and F. L. wrote the first draft of the manuscript, and all authors commented on and revised the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
F. L. was supported by a DOE Office of Science Early Career Research Program Award, managed by the DOE BES CPIMS program under award number DE-SC0025345. F. R. was supported by the Cottrell Scholar Award #CS-CSA-2024-099, sponsored by the Research Corporation for Science Advancement. R. S. K. G., S. D., R. M., and Y. W. acknowledge support from the U.S. Department of Energy, Office of Science, Basic Energy Sciences, under Early Career Award No. DE-SC0024524. This work used JetStream2 at Indiana University through allocation CHE230134 from the Advanced Cyberinfrastructure Coordination Ecosystem: Services & Support (ACCESS) program, which is supported by National Science Foundation grants #2138259, #2138286, #2138307, #2137603, and #2138296. The chatbot using Google DialogFlow CX is based upon work supported by the Google Cloud Research Credits program with the award GCP19980904.Notes and references
- G.-J. Cheng, X. Zhang, L. W. Chung, L. Xu and Y.-D. Wu, Computational Organic Chemistry: Bridging Theory and Experiment in Establishing the Mechanisms of Chemical Reactions, J. Am. Chem. Soc., 2015, 137, 1706–1725 Search PubMed.
- T. Kato, T. Kusakizako, C. Jin, X. Zhou, R. Ohgaki, L. Quan, M. Xu, S. Okuda, K. Kobayashi, K. Yamashita, T. Nishizawa, Y. Kanai and O. Nureki, Structural insights into inhibitory mechanism of human excitatory amino acid transporter EAAT2, Nat. Commun., 2022, 13, 4714 CrossRef CAS PubMed.
- V. Barone, S. Alessandrini, M. Biczysko, J. R. Cheeseman, D. C. Clary, A. B. McCoy, R. J. DiRisio, F. Neese, M. Melosso and C. Puzzarini, Computational molecular spectroscopy, Nat. Rev. Methods Primers, 2021, 1, 38 CrossRef CAS.
- R. Ramakrishnan, P. O. Dral, M. Rupp and O. A. von Lilienfeld, Quantum chemistry structures and properties of 134 kilo molecules, Sci. Data, 2014, 1, 140022 Search PubMed.
- J. S. Smith, O. Isayev and A. E. Roitberg, ANI-1, A data set of 20 million calculated off-equilibrium conformations for organic molecules, Sci. Data, 2017, 4, 170193 CrossRef CAS PubMed.
- A. Jain, S. P. Ong, G. Hautier, W. Chen, W. D. Richards, S. Dacek, S. Cholia, D. Gunter, D. Skinner, G. Ceder and K. A. Persson, Commentary: The Materials Project: A materials genome approach to accelerating materials innovation, APL Mater., 2013, 1, 011002 CrossRef.
- D. G. A. Smith, A. T. Lolinco, Z. L. Glick, J. Lee, A. Alenaizan, T. A. Barnes, C. H. Borca, R. Di Remigio, D. L. Dotson, S. Ehlert, A. G. Heide, M. F. Herbst, J. Hermann, C. B. Hicks, J. T. Horton, A. G. Hurtado, P. Kraus, H. Kruse, S. J. R. Lee, J. P. Misiewicz, L. N. Naden, F. Ramezanghorbani, M. Scheurer, J. B. Schriber, A. C. Simmonett, J. Steinmetzer, J. R. Wagner, L. Ward, M. Welborn, D. Altarawy, J. Anwar, J. D. Chodera, A. Dreuw, H. J. Kulik, F. Liu, T. J. Martínez, D. A. Matthews, H. F. Schaefer III, J. Šponer, J. M. Turney, L.-P. Wang, N. De Silva, R. A. King, J. F. Stanton, M. S. Gordon, T. L. Windus, C. D. Sherrill and L. A. Burns, Quantum Chemistry Common Driver and Databases (QCDB) and Quantum Chemistry Engine (QCEngine): Automation and interoperability among computational chemistry programs, J. Chem. Phys., 2021, 155, 204801 CrossRef CAS PubMed.
- E. I. Ioannidis, T. Z. H. Gani and H. J. Kulik, molSimplify: A toolkit for automating discovery in inorganic chemistry, J. Comput. Chem., 2016, 37, 2106–2117 CrossRef CAS PubMed.
- E. Hruska, A. Gale, X. Huang and F. Liu, AutoSolvate: A toolkit for automating quantum chemistry design and discovery of solvated molecules, J. Chem. Phys., 2022, 156, 124801 CrossRef CAS PubMed.
- Google Inc., Conversational Agents (Dialogflow CX) Documentation, accessed 12 October, 2024 Search PubMed.
- S. F. Suhel, V. K. Shukla, S. Vyas and V. P. Mishra, 2020 8th International Conference on Reliability, Infocom Technologies and Optimization (Trends and Future Directions) (ICRITO), Conversation to Automation in Banking Through Chatbot Using Artificial Machine Intelligence Language, 2020, pp. 611–618 Search PubMed.
- A. Xu, Z. Liu, Y. Guo, V. Sinha and R. Akkiraju, Presented in part at the Proceedings of the 2017 CHI Conference on Human Factors in Computing Systems, Denver, Colorado, USA, 2017 Search PubMed.
- J. Jumper, R. Evans, A. Pritzel, T. Green, M. Figurnov, O. Ronneberger, K. Tunyasuvunakool, R. Bates, A. Žídek, A. Potapenko, A. Bridgland, C. Meyer, S. A. A. Kohl, A. J. Ballard, A. Cowie, B. Romera-Paredes, S. Nikolov, R. Jain, J. Adler, T. Back, S. Petersen, D. Reiman, E. Clancy, M. Zielinski, M. Steinegger, M. Pacholska, T. Berghammer, S. Bodenstein, D. Silver, O. Vinyals, A. W. Senior, K. Kavukcuoglu, P. Kohli and D. Hassabis, Highly accurate protein structure prediction with AlphaFold, Nature, 2021, 596, 583–589 CrossRef CAS PubMed.
- F. Gentile, J. C. Yaacoub, J. Gleave, M. Fernandez, A.-T. Ton, F. Ban, A. Stern and A. Cherkasov, Artificial intelligence–enabled virtual screening of ultra-large chemical libraries with deep docking, Nat. Protoc., 2022, 17, 672–697 CrossRef CAS PubMed.
- N. J. Szymanski, B. Rendy, Y. Fei, R. E. Kumar, T. He, D. Milsted, M. J. McDermott, M. Gallant, E. D. Cubuk, A. Merchant, H. Kim, A. Jain, C. J. Bartel, K. Persson, Y. Zeng and G. Ceder, An autonomous laboratory for the accelerated synthesis of novel materials, Nature, 2023, 624, 86–91 CrossRef CAS PubMed.
- U. Raucci, A. Valentini, E. Pieri, H. Weir, S. Seritan and T. J. Martínez, Voice-controlled quantum chemistry, Nat. Comput. Sci., 2021, 1, 42–45 CrossRef PubMed.
- D. A. Boiko, R. Macknight, B. Kline and G. Gomes, Autonomous chemical research with large language models, Nature, 2023, 624, 570–578 CrossRef CAS PubMed.
- J. Wang, R. M. Wolf, J. W. Caldwell, P. A. Kollman and D. A. Case, Development and testing of a general amber force field, J. Comput. Chem., 2004, 25, 1157–1174 CrossRef CAS PubMed.
- D. A. Pearlman, D. A. Case, J. W. Caldwell, W. S. Ross, T. E. Cheatham, S. DeBolt, D. Ferguson, G. Seibel and P. Kollman, AMBER, a package of computer programs for applying molecular mechanics, normal mode analysis, molecular dynamics and free energy calculations to simulate the structural and energetic properties of molecules, Comput. Phys. Commun., 1995, 91, 1–41 CrossRef CAS.
- S. Seritan, C. Bannwarth, B. S. Fales, E. G. Hohenstein, C. M. Isborn, S. I. L. Kokkila-Schumacher, X. Li, F. Liu, N. Luehr, J. W. Snyder, C. Song, A. V. Titov, I. S. Ufimtsev, L.-P. Wang and T. J. Martínez, TeraChem: A graphical processing unit – accelerated electronic structure package for large-scale ab initio molecular dynamics, Wiley Interdiscip. Rev.:Comput. Mol. Sci., 2021, 11, e1494 CAS.
- R. J. Aguado, A. Mazega, N. Fiol, Q. Tarrés, P. Mutjé and M. Delgado-Aguilar, Durable Nanocellulose-Stabilized Emulsions of Dithizone/Chloroform in Water for Hg2+ Detection: A Novel Approach for a Classical Problem, ACS Appl. Mater. Interfaces, 2023, 15, 12580–12589 CrossRef CAS PubMed.
- K. Gaalswyk and C. N. Rowley, An explicit-solvent conformation search method using open software, PeerJ, 2016, 4, e2088 CrossRef PubMed.
- A. N. Drozdov, A. Grossfield and R. V. Pappu, Role of Solvent in Determining Conformational Preferences of Alanine Dipeptide in Water, J. Am. Chem. Soc., 2004, 126, 2574–2581 CrossRef CAS PubMed.
- J. M. P. Martirez and E. A. Carter, Solvent Dynamics Are Critical to Understanding Carbon Dioxide Dissolution and Hydration in Water, J. Am. Chem. Soc., 2023, 145, 12561–12575 CrossRef CAS PubMed.
- H. Daver, A. G. Algarra, J. J. Rebek, J. N. Harvey and F. Himo, Mixed Explicit–Implicit Solvation Approach for Modeling of Alkane Complexation in Water-Soluble Self-Assembled Capsules, J. Am. Chem. Soc., 2018, 140, 12527–12537 CrossRef CAS PubMed.
- D. van der Spoel, J. Zhang and H. Zhang, Quantitative predictions from molecular simulations using explicit or implicit interactions, Wiley Interdiscip. Rev.:Comput. Mol. Sci., 2022, 12, e1560 Search PubMed.
- J. Zhang, H. Zhang, T. Wu, Q. Wang and D. van der Spoel, Comparison of Implicit and Explicit Solvent Models for the Calculation of Solvation Free Energy in Organic Solvents, J. Chem. Theory Comput., 2017, 13, 1034–1043 CrossRef CAS PubMed.
- A. Eilmes, Solvatochromic probe in molecular solvents: implicit versus explicit solvent model, Theor. Chem. Acc., 2014, 133, 1538 Search PubMed.
- D. R. Turner and J. Kubelka, Infrared and Vibrational CD Spectra of Partially Solvated α-Helices: DFT-Based Simulations with Explicit Solvent, J. Phys. Chem. B, 2007, 111, 1834–1845 Search PubMed.
- C. Merten, Modelling solute–solvent interactions in VCD spectra analysis with the micro-solvation approach, Phys. Chem. Chem. Phys., 2023, 25, 29404–29414 Search PubMed.
- S. Mondal and C. Narayana, Role of Explicit Solvation in the Simulation of Resonance Raman Spectra within Short-Time Dynamics Approximation, J. Phys. Chem. B, 2019, 123, 8800–8813 Search PubMed.
- M. Dračínský and P. Bouř, Computational Analysis of Solvent Effects in NMR Spectroscopy, J. Chem. Theory Comput., 2010, 6, 288–299 Search PubMed.
- C. M. Sterling and R. Bjornsso, Multisteptep Explicit Solvation Protocol for Calculation of Redox Potentials, J. Chem. Theory Comput., 2019, 15, 52–67 Search PubMed.
- A. Pedone, Role of Solvent on Charge Transfer in 7-Aminocoumarin Dyes: New Hints from TD-CAM-B3LYP and State Specific PCM Calculations, J. Chem. Theory Comput., 2013, 9, 4087–4096 CrossRef CAS PubMed.
- B. Auer, A. V. Soudackov and S. Hammes-Schiffer, Nonadiabatic Dynamics of Photoinduced Proton-Coupled Electron Transfer: Comparison of Explicit and Implicit Solvent Simulations, J. Phys. Chem. B, 2012, 116, 7695–7708 Search PubMed.
- I. Tavernelli, Nonadiabatic Molecular Dynamics Simulations: Synergies between Theory and Experiments, Acc. Chem. Res., 2015, 48, 792–800 CrossRef CAS PubMed.
- J. M. Boereboom, P. Fleurat-Lessard and R. E. Bulo, Explicit Solvation Matters: Performance of QM/MM Solvation Models in Nucleophilic Addition, J. Chem. Theory Comput., 2018, 14, 1841–1852 CrossRef CAS PubMed.
- J. J. Varghese and S. H. Mushrif, Origins of complex solvent effects on chemical reactivity and computational tools to investigate them: a review, React. Chem. Eng., 2019, 4, 165–206 Search PubMed.
- J. Chen, Y. Shao and J. Ho, Are Explicit Solvent Models More Accurate than Implicit Solvent Models? A Case Study on the Menschutkin Reaction, J. Phys. Chem. A, 2019, 123, 5580–5589 CrossRef CAS PubMed.
- D. Jurafsky, Speech and Language Processing, 2000 Search PubMed.
- A. Vaswani, N. Shazeer, N. Parmar, J. Uszkoreit, L. Jones, A. N. Gomez, L. Kaiser and I. Polosukhin, Attention is all you need, arXiv, 2017, preprint, arXiv:1706.03762, DOI:10.48550/arXiv.1706.03762.
- T. B. Brown, Language models are few-shot learners, arXiv, 2020, preprint, arXiv:2005.14165.
- Z. Ji, N. Lee, R. Frieske, T. Yu, D. Su, Y. Xu, E. Ishii, Y. J. Bang, A. Madotto and P. Fung, Survey of hallucination in natural language generation, ACM Comput. Surv., 2023, 55, 1–38 CrossRef.
- D. Y. Hancock, J. Fischer, J. M. Lowe, W. Snapp-Childs, M. Pierce, S. Marru, J. E. Coulter, M. Vaughn, B. Beck, N. Merchant, E. Skidmore and G. Jacobs, Practice and Experience in Advanced Research Computing, Boston, MA, USA, 2021 Search PubMed.
- R. M. Hanson, J. Prilusky, Z. Renjian, T. Nakane and J. L. Sussman, JSmol and the next-generation web-based representation of 3D molecular structure as applied to proteopedia, Isr. J. Chem., 2013, 53, 207–216 CrossRef CAS.
- A. Jakalian, D. B. Jack and C. I. Bayly, Fast, efficient generation of high-quality atomic charge s. AM1-BCC model: I I. Parameterization and validation, J. Comput. Chem., 2002, 23, 1623–1641 Search PubMed.
- C. I. Bayly, P. Cieplak, W. Cornell and P. A. Kollman, A well-behaved electrostatic potential based method using charge restraints for deriving atomic charges: the RESP model, J. Phys. Chem., 1993, 97, 10269–10280 CrossRef CAS.
- A. Marini, A. Muñoz-Losa, A. Biancardi and B. Mennucci, What is solvatochromism?, J. Phys. Chem. B, 2010, 114, 17128–17135 CrossRef CAS PubMed.
- M. J. Minch and S. S. Shah, Merocyanin dye preparation for the introductory organic laboratory, J. Chem. Educ., 1977, 54, 709 CrossRef CAS.
- B. R. Osterby and R. D. McKelvey, Convergent Synthesis of Betaine-30, a Solvatochromic Dye: An Advanced Undergraduate Project and Demonstration, J. Chem. Educ., 1996, 73, 260 Search PubMed.
- D. González-Arjona, G. López-Pérez, M. Domínguez and A. González, Solvatochromism: a comprehensive project for the final year undergraduate chemistry laboratory, J. Lab. Chem. Educ., 2016, 2016, 45–52 Search PubMed.
- C. Amovilli and B. Mennucci, Self-Consistent-Field Calculation of Pauli Repulsion and Dispersion Contributions to the Solvation Free Energy in the Polarizable Continuum Model, J. Phys. Chem. B, 1997, 101, 1051–1057 Search PubMed.
- S. Hirata and M. Head-Gordon, Time-dependent density functional theory within the Tamm–Dancoff approximation, Chem. Phys. Lett., 1999, 314, 291–299 CrossRef CAS.
- R. Cammi and B. Mennucci, Linear response theory for the polarizable continuum model, J. Chem. Phys., 1999, 110, 9877–9886 CrossRef CAS.
- R. Cammi, S. Corni, B. Mennucci and J. Tomasi, Electronic excitation energies of molecules in solution: State specific and linear response methods for nonequilibrium continuum solvation models, J. Chem. Phys., 2005, 122, 104513 CrossRef CAS PubMed.
- C. Adamo and V. Barone, Toward reliable density functional methods without adjustable parameters: The PBE0 model, J. Chem. Phys., 1999, 110, 6158–6170 CrossRef CAS.
- J. P. Perdew, M. Ernzerhof and K. Burke, Rationale for mixing exact exchange with density functional approximations, J. Chem. Phys., 1996, 105, 9982–9985 CrossRef CAS.
- W. J. Hehre, R. Ditchfield and J. A. Pople, Self-Consistent Molecular Orbital Methods. XII. Further Extensions of Gaussian-Type Basis Sets for Use in Molecular Orbital Studies of Organic Molecules, J. Chem. Phys., 1972, 56, 2257–2261 CrossRef CAS.
- P. C. Hariharan and J. A. Pople, The influence of polarization functions on molecular orbital hydrogenation energies, Theor. Chim. Acta, 1973, 28, 213–222 Search PubMed.
- D. Hegarty and M. A. Robb, Application of unitary group-methods to configuration-interaction calculations, Mol. Phys., 1979, 38, 1795–1812 Search PubMed.
- E. Hruska, A. Gale and F. Liu, Bridging the Experiment-Calculation Divide: Machine Learning Corrections to Redox Potential Calculations in Implicit and Explicit Solvent Models, J. Chem. Theory Comput., 2022, 18, 1096–1108 CrossRef CAS PubMed.
- A. Moreno-Ceballos, M. E. Castro, N. A. Caballero, L. Mammino and F. J. Melendez, Implicit and Explicit Solvent Effects on the Global Reactivity and the Density Topological Parameters of the Preferred Conformers of Caespitate, Computation, 2024, 12, 5 CrossRef CAS.
- R. J. Aguado, A. Mazega, Q. Tarrés and M. Delgado-Aguilar, The role of electrostatic interactions of anionic and cationic cellulose derivatives for industrial applications: A critical review, Ind. Crops Prod., 2023, 201, 116898 CrossRef CAS.
- I. Gustin, C. W. Kim, D. W. McCamant and I. Franco, Mapping electronic decoherence pathways in molecules, Proc. Natl. Acad. Sci. U. S. A., 2023, 120, e2309987120 CrossRef CAS PubMed.
- M. Jorge, Theoretically grounded approaches to account for polarization effects in fixed-charge force fields, J. Chem. Phys., 2024, 161, 180901 CrossRef CAS PubMed.
- I. Leontyev and A. Stuchebrukhov, Accounting for electronic polarization in non-polarizable force fields, Phys. Chem. Chem. Phys., 2011, 13, 2613–2626 RSC.
- D. Seiferth, S. J. Tucker and P. C. Biggin, Limitations of non-polarizable force fields in describing anion binding poses in non-polar synthetic hosts, Phys. Chem. Chem. Phys., 2023, 25, 17596–17608 RSC.
- M. D. Wilkinson, M. Dumontier, I. J. Aalbersberg, G. Appleton, M. Axton, A. Baak, N. Blomberg, J.-W. Boiten, L. B. da Silva Santos and P. E. Bourne, The FAIR Guiding Principles for scientific data management and stewardship, Sci. Data, 2016, 3, 1–9 Search PubMed.
- Intuition Machines, Inc., hCaptcha Search PubMed.
- W. Reese, Nginx: the high-performance web server and reverse proxy, Linux J., 2008, 2008, 2 Search PubMed.
- G. M. Barca, C. Bertoni, L. Carrington, D. Datta, N. De Silva, J. E. Deustua, D. G. Fedorov, J. R. Gour, A. O. Gunina and E. Guidez, Recent developments in the general atomic and molecular electronic structure system, J. Chem. Phys., 2020, 152, 154102 CrossRef CAS PubMed.
- J. Hursey, E. Mallove, J. M. Squyres and A. Lumsdaine, An Extensible Framework for Distributed Testing of MPI Implementations, Proceedings of the 14th European Conference on Recent Advances in Parallel Virtual Machine and Message Passing Interface, Springer, Berlin Heidelberg, 2007, pp. 64–72 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Details of the conversation between the chatbot and the user, all pages used in the conversation, installation guide of AutoSolvateWeb, scaling analysis, list of all output files, list of solvents and their force field, lists of solutes used in the scaling analysis, description of the embedded job queue, and the example conversation for the innovative course design. See DOI: https://doi.org/10.1039/d4sc08677e |
| This journal is © The Royal Society of Chemistry 2025 |