
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHeterogeneous organocatalysis: the proline case
Gustavo Senra G. de Carvalho *,
Douglas C. Alcântara Pinto,
Robson Corrêa da Silva and
Fernando de Carvalho da Silva*
*,
Douglas C. Alcântara Pinto,
Robson Corrêa da Silva and
Fernando de Carvalho da Silva*
Universidade Federal Fluminense, Instituto de Química, Departamento de Química Orgânica, Campus do Valonguinho, Niterói, RJ, Brazil. E-mail: senradcarvalho@gmail.com; fcsilva@id.uff.br
First published on 2nd July 2025
Abstract
Asymmetric catalysis has allowed organic chemists to synthesize chiral molecules, such as amino acids, nucleic acids, sugars, and drugs. To achieve this, it is common to use a chiral catalyst to selectively accelerate the reaction. In the early days, it was believed that there were two main types of effective asymmetric catalysts: enzyme complexes and transition metal complexes. However, with the emergence of organocatalysis and the consequent introduction of a new category of highly effective asymmetric catalysts based solely on organic compounds, there was a revolution in the field of asymmetric catalysis. An excellent example of this innovation is L-proline, a fascinating molecule that demonstrates the transformative impact of organocatalysis. Organocatalysis presents itself as a simpler synthetic tool than other methodologies, however, despite being widely used in the research environment, it has not yet reached large-scale production. This gap occurs mainly due to the methodology working in a homogeneous phase together with the rest of the reagents involved. Although this does not present a problem on a laboratory scale, the recovery and reuse of the catalyst can be an obstacle in industrial processes. In recent years, with a view to some industrial uses, there has been an effort to make the immobilization (also called heterogenization) of organocatalysts possible. Such modified systems have broad catalytic applications in several organic transformations. Taking this into consideration, this review has the general objective of investigating the application of different supports in the immobilization of the classical organocatalyst L-proline, synthesized and characterized by three different methodologies, namely: impregnation, intercalation and grafting.
Introduction
Organocatalysis
Chemists synthesize chiral molecules that behave like objects and mirror images, such as amino acids, nucleic acids, sugars, and drugs. The stereocontrolled construction of chiral carbocycles and heterocycles is a topic of great importance in modern organic synthesis, driven by the predominance of mono- and polycyclic chiral systems in natural products and chiral pharmaceuticals. To achieve this, it is common to use asymmetric catalysis, in which a chiral catalyst selectively accelerates the reaction that leads to a mirror image isomer, also called an enantiomer. For decades, the generally accepted view was that there were two classes of efficient asymmetric catalysts: enzymatic and transition metal complexes. However, this view was challenged by the introduction of organocatalysis, and the use of purely organic catalysts emerged as a third class of powerful asymmetric catalysts (Fig. 1).1–3 | ||
| Fig. 1 The three pillars of asymmetric catalysis: biocatalysis, metal catalysis and organocatalysis. | ||
The word organocatalysis was introduced to the community in 2000 by MacMillan et al., one of the pioneers of the field.4,5 The use of small chiral organic molecules as enantioselective catalysts, with the associated advantages of their easy availability and of carrying out asymmetric transformations in a metal-free environment and under mild and simple reaction conditions, has recently experienced impressive growth. Therefore, asymmetric organocatalysis is today considered the “third pillar” of asymmetric catalysis, together with biocatalysis and metal catalysis, and has been increasingly used in the key stages of the total synthesis of complex natural products.1–3 Recently, this new pillar of asymmetric catalysis was recognized by the academic community through the award of the 2021 Nobel Prize in Chemistry, given to McMillan and List, for their contributions “to the development of asymmetric organocatalysis”.6
Organocatalysts bind to reactant molecules to form short-lived intermediates, which are more reactive than the substrate molecules on their own. Being chiral, the catalyst transfers its laterality to the substrate, controlling which side of the intermediate can react more and better.1–3
In 1912, it was reported that the cinchona alkaloid promoted the addition of hydrogen cyanide to benzaldehyde with low enantioselectivity,7 and in 1960, Pracejus found a cinchona alkaloid catalyzed by asymmetric addition of methanol to ketenes.8 Later, in 1971, the groups of Hajos and Wiechert independently reported the asymmetric aldol reaction catalyzed by L-proline, without a completely satisfactory mechanistic study and without recognizing the important novelty of this finding. However, it was ignored as such an important and independent field of research until the year 2000.9–11 Nevertheless, it was only in 2000 that Benjamin List reported an asymmetric aldol reaction catalyzed by L-proline and officially initiated the “golden age” of organocatalysis.9,12–15
In fact, a very interesting molecule that well exemplifies the innovation brought by organocatalysis is exactly the amino acid proline, which has become a crucial component in examples of all catalytic strategies.16 Proline can be a ligand in asymmetric transition metals, a chiral modifier in heterogeneous catalysts, and most importantly, proline can be an effective organocatalyst of various asymmetric transformations, such as the aldol, Mannich, and Michael reactions (Fig. 2A). In addition to these reasons, and others more specific to each reaction, proline has become an important molecule in asymmetric catalysis. No less important is the fact that proline is an abundant, cheap chiral molecule available in both enantiomeric forms.9,12–15
 | ||
| Fig. 2 (A) Modes of action in proline catalysis. (B) Proline can activate carbonyls to make them more reactive and control reaction geometry – for example, in aldol reactions. | ||
List et al.,9 described proline as a microaldolase, a mimic enzyme that combines a nucleophilic center (the amino group) and an acid–base cocatalyst (the carboxylic acid group). In the aldol reaction, proline binds to the ketone, forming an enamine intermediate that is more reactive than the ketone itself, as its highest occupied molecular orbital (HOMO) is more energetic. The chirality of proline introduces asymmetry into the reaction, allowing the aldehydes to approach each other only at a specific position (Fig. 2B).
L-Proline has been demonstrated to be an effective catalyst in a wide range of chemical reactions. Its versatility is highlighted by its successful application in various processes, including Robinson annulations, aldol reactions, Mannich reactions, and Michael reactions. Additionally, L-proline plays a significant role in direct electrophilic α-aminations, Diels–Alder reactions, and Baylis–Hillman reactions. The utility of L-proline extends to more specialized transformations such as aza-Morita–Baylis–Hillman reactions, α-selenenylation, oxidation, and chlorination.9,17–19
Organocatalysis encompasses a wide variety of chemical reactions, including three-component reactions and catalytic cascades, which makes it a highly versatile tool for total synthesis projects. This broad applicability allows organocatalysis to be utilized in the creation of complex molecular structures across different types of chemical synthesis, providing valuable solutions for constructing intricate compounds.
The field of organocatalysis quickly drew the attention of pharmaceutical companies due to its potential to revolutionize the synthesis of complex bioactive molecules. Pharmaceutical researchers are particularly interested in leveraging organocatalytic methods to explore novel approaches for building molecules with intricate architectures, which are essential for drug discovery and development. The efficiency and selectivity offered by organocatalysis make it an attractive option for generating new therapeutic agents and enhancing the synthesis of existing ones.
The rapid adoption of organocatalysis by the pharmaceutical industry highlights its significant impact on modern drug development. By incorporating organocatalytic techniques, researchers can achieve more efficient and innovative synthesis processes, ultimately leading to the discovery of new drugs with potentially improved efficacy and safety profiles. The ongoing interest and investment in this field underscore its importance in advancing pharmaceutical research and its potential to transform the way complex bioactive molecules are synthesized.20–23
The observation that only one enantiomer of a molecule is present in nature often correlates with the fact that different enantiomers can exhibit distinct biological activities and interact differently in chiral environments. This phenomenon highlights the significance of asymmetric synthesis, which leverages robust generic activation modes to create highly effective and practical synthetic methodologies. Through the systematic screening of substrates, catalysts, and reaction conditions, asymmetric synthesis enables chemists to develop novel drug prototypes efficiently. This approach not only enhances the synthesis of complex molecules but also advances the field of medicinal chemistry by facilitating the creation of new pharmaceuticals with potentially improved efficacy and selectivity. Thus, asymmetric synthesis plays a crucial role in drug discovery and development, showcasing its profound impact on the pharmaceutical industry.20,22–24
Asymmetric organocatalysis is an elegant and simpler tool than other methodologies, so much so that its discovery allowed chemists to think of new ways to bring molecules together. Although organocatalysts are widely used in the laboratory setting in research and development, they have not yet reached large-scale industrial production – which does not mean that they do not have such capabilities.25–28 Asymmetric organocatalysis is on the brink of becoming a broadly utilized technology with significant potential across various industries. Although the widespread adoption of this technology may take some time, it is highly plausible that, in the foreseeable future, organocatalysis will play a crucial role in large-scale industrial applications. Specifically, it is expected to become increasingly important in the production processes of fine chemicals, pharmaceuticals, and other sectors within the chemical industry.29
The gradual integration of asymmetric organocatalysis into large-scale processes is anticipated due to its numerous advantages. These include its ability to facilitate highly selective and efficient transformations under mild conditions, which can lead to cost savings and reduced environmental impact compared to traditional catalytic methods. In fine chemical synthesis, the precision and effectiveness of organocatalysis can significantly enhance the quality and efficiency of production. Similarly, the pharmaceutical industry stands to benefit from the development of organocatalytic methods that offer more sustainable and economical solutions for complex molecular transformations.29
Moreover, the chemical industry will likely experience improvements in process efficiency and sustainability as organocatalysis becomes more prevalent. The transition to using asymmetric organocatalysis on a large scale involves overcoming several challenges, including scaling up laboratory techniques and optimizing processes for industrial applications. However, ongoing research and advancements in this field suggest that these challenges will be addressed over time.20,23
In conclusion, while the transition to widespread use of asymmetric organocatalysis may require patience, the technology's potential to revolutionize large-scale industrial processes in fine chemicals, pharmaceuticals, and chemical industries is substantial and promising.20,23
Recent advancements in asymmetric synthesis have significantly contributed to breakthroughs in medicinal chemistry, enabling the creation of sterically diverse molecules that were previously challenging or even impossible to synthesize. These advancements have broadened the scope of chemical synthesis, allowing chemists to explore new realms of molecular design and development. One area where these improvements are particularly impactful is in the pharmaceutical industry, where the ability to synthesize complex molecules with precise stereochemistry can greatly influence the efficacy and safety of new drugs.29,30
Organocatalysts are in a homogeneous phase along with the rest of the reactants involved in the reactions and their chemical nature is often comparable to the nature of the products. Therefore, adequate separation and purification of the desired compounds can be complicated. However, catalyst recovery and reuse are not necessarily a major problem, at least on a laboratory scale, and catalytically active species are often used in only one run. On a larger scale, all of these factors can be an obstacle in setting up a commercial industrial process based on these systems.25–28
Heterogeneous catalysis
Data from the literature31 informs that catalysis contributes 35% of world gross domestic product (GDP) and is associated with supporting approximately 30% of gross domestic product in European economies, in addition to being involved at some point in the processing of more than 80% of all manufactured products. Catalysts have profoundly changed our understanding and use of chemistry, which is reflected in the fact that seven Nobel Prizes in chemistry have been awarded to discoveries in the field.31,32Given these considerations, recent years have witnessed a concerted effort to advance the industrial application of organocatalysts by focusing on their immobilization, a process also known as heterogenization or anchoring, onto solid support. This development is driven by the need to make catalytic processes more efficient and sustainable for industrial use. Immobilization enhances the practicality of organocatalysts by allowing them to be reused multiple times, thereby reducing costs and minimizing waste. In line with one of the twelve Principles of Green Chemistry, which underscores the importance of catalysis in promoting environmentally friendly chemical processes, the design and implementation of heterogeneous catalysts must meet several crucial criteria. These catalysts should be recyclable, ensuring that they can be recovered and reused without significant loss of performance, which aligns with the principle of minimizing waste. They must also be selective, allowing for precise control over chemical reactions to maximize yields and minimize by-products. Additionally, the catalysts need to be robust, capable of withstanding the rigorous conditions often encountered in industrial settings without degrading or losing efficacy. Cost-effectiveness is another key requirement, making the catalysts economically viable for widespread industrial use. Finally, to achieve a significant advantage over their homogeneous counterparts, these heterogeneous catalysts should exhibit superior activity, demonstrating enhanced performance and efficiency in catalytic processes.28,33 By meeting these standards, the immobilized organocatalysts not only align with the principles of green chemistry but also contribute to the advancement of more sustainable and economically feasible industrial practices.25–28,34 In addition to the advantage of being easily separated from the reaction medium through simple techniques such as simple filtration, decantation and centrifugation, thus allowing multiple reuse and recycling of immobilized compounds when working with expensive materials. Furthermore, studies have shown that heterogenization can increase the stability of incorporated compounds and, in some cases, increase the reactivity and selectivity of catalytic reactions.35
Furthermore, the advantages of heterogeneous catalysis are well-established. Heterogeneous catalysts offer several benefits including ease of separation from the reaction mixture, which simplifies the purification process. They are also amenable to recycling and regeneration, making them economically and environmentally favorable. Heterogeneous catalysts often operate at lower reaction rates, which can be advantageous in controlling reaction conditions. Additionally, they are subject to diffusion control, which can influence reaction kinetics and selectivity. The long catalytic life of heterogeneous catalysts further contributes to their efficiency and sustainability.36
Heterogenization
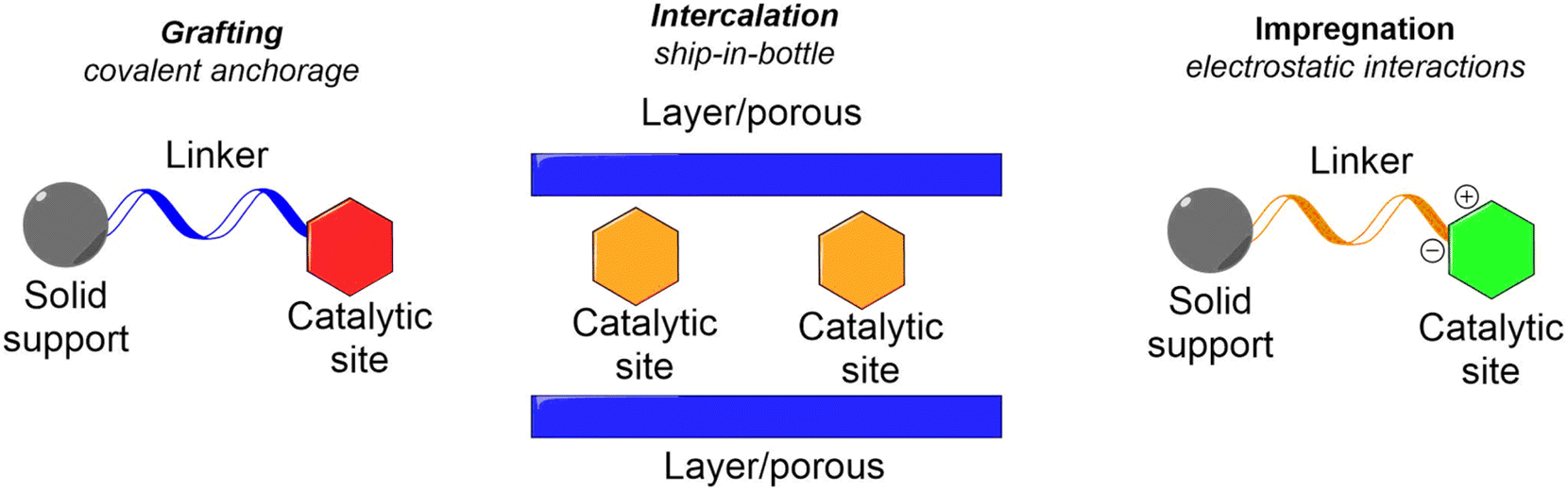
Building on a comprehensive understanding of heterogeneous catalysis, materials chemistry, and organocatalysis, researchers are now developing and testing a wide range of modified catalytic systems for various organic transformations. These systems leverage advanced methods to enhance catalytic efficiency and effectiveness across numerous chemical processes. The immobilization of catalysts, a crucial aspect of these systems, typically depends on intermolecular interactions between the catalytic species and the supporting material.27,37–41These can be broadly categorized into three main types of interactions:
Covalent bonds: this type of interaction involves forming strong, stable covalent bonds between the catalytic species and the supporting material. This method ensures a robust attachment of the catalyst, which can significantly enhance its stability and catalytic performance.
Non-covalent interactions: this category includes several types of interactions that do not involve covalent bonding. Key examples are: electrostatic interactions: these occur due to the attraction or repulsion between charged groups on the catalytic species and the supporting material. London dispersion forces: also known as van der Waals forces, these are weak attractions caused by transient dipoles between molecules. Hydrogen bonding: this involves attractions between a covalent hydrogen atom bonded to an electronegative atom and another electronegative atom.
Intercalation/encapsulation: this involves trapping the catalytic species within a host material, creating a confined environment that can influence catalytic activity and selectivity.
Several well-established methods for the immobilization or heterogenization of homogeneous catalysts are widely used. These methods include:
Impregnation: this method involves the introduction of the catalytic species onto a solid support through electrostatic interactions. In impregnation, the catalyst is dispersed onto or into the support material, which is typically a porous or solid surface. Electrostatic interactions help to anchor the catalytic species, making them accessible for catalytic reactions.
Encapsulation/occlusion/intercalation: in this method, the catalyst is trapped within porous or layered supports. This technique is often referred to as “ship-in-bottle” catalysis. It utilizes the porous structure of the support material to enclose the catalyst, effectively isolating it within a confined space. This approach not only stabilizes the catalyst but also can enhance its selectivity and reactivity.
Grafting or tethering: this method involves attaching the catalytic active site directly to the supporting material through covalent bonds. Grafting or tethering creates a direct connection between the catalyst and the support, providing a stable and durable attachment. This method allows for precise control over the placement and orientation of the catalytic species, which can improve the overall efficiency of the catalytic process (Fig. 3).
 | ||
| Fig. 3 Catalyst immobilization methods on solid supports. | ||
The process of heterogenization, which involves immobilizing catalytic species onto solid supports, is likely to play a leading role in this transformative shift. In the context of asymmetric synthesis, heterogenization can enhance catalytic efficiency and facilitate the development of novel compounds by providing more stable and reusable catalytic systems. This is especially crucial in the pharmaceutical industry, where the structure–activity relationship (SAR) of compounds is a critical factor in drug discovery and development.
Asymmetry in molecular structure is a key element in determining how a compound interacts with biological targets, and precise control over stereochemistry is essential for optimizing these interactions. Therefore, advances in asymmetric synthesis and the integration of heterogenization techniques are expected to drive further innovation in the field. These developments could lead to the synthesis of new drugs with improved specificity and reduced side effects, ultimately benefiting pharmaceutical research and development.28,33
In summary, the synergy between advancements in asymmetric synthesis and heterogenization is paving the way for significant progress in medicinal chemistry. The ability to synthesize diverse and stereochemically complex molecules is enhancing the drug discovery process, with potential to revolutionize the pharmaceutical industry by refining the structure–activity relationships of therapeutic compounds.22,24,28,33
As previously discussed, the heterogenization of organocatalysts presents both economic and environmental challenges. To address these issues, a variety of strategies have been explored and developed. This subject has generated extensive research, with numerous publications addressing different aspects of the field. Some studies offer a general overview of the heterogenization process,27,37 while others focus on specific types of supports, such as inorganic materials,38 polymers,39 or carbon-based materials.40 Additionally, there is substantial research dedicated to non-covalent methods of immobilization,41 as well as specialized studies on the immobilization of enantioselective catalysts.42–44 These varied approaches reflect the complexity and significance of improving organocatalyst heterogenization for enhanced efficiency and sustainability in catalytic processes.
This review presents strategies described in the literature on L-proline immobilization methods. Work will be reviewed where the three main immobilization methods were used, namely, grafting, impregnation and intercalation. However, it is important to keep in mind that sometimes the separation between methods is not very clear. This is, for example, the case of the transition from a purely ionic (electrostatic) metal–support bond to a covalent (coordinating) bond. Furthermore, more than one effect often explains the immobilization of a homogeneous complex, for example: electrostatic interactions between the complex and the support on a trapped catalyst.
Many of the synthesized enantioselective organocatalysts have also been immobilized on different supports.38,45 Among them, proline occupies a relevant position.44,46 From the first asymmetric reaction using L-proline, to List's initial work 30 years later14 and in current work, proline and its derivatives have been widely applied, especially in asymmetric organocatalysts.29 Although the heterogenization approach requires the use of synthetic derivatives that are more expensive than proline, this commercially available, low-cost amino acid is often used with high catalyst loading. However, attempts to improve or modify their catalytic behavior, taking advantage of specific properties of the support, would justify immobilization in many cases.
In the sections that follow, we will explore a series of representative examples where L-proline is immobilized on various types of support using different immobilization techniques. This comprehensive examination will highlight how each approach impacts the behavior of L-proline as a catalyst. Our primary focus will be on assessing both the catalytic efficiency and the stability of the immobilized L-proline. These factors are crucial because the main goal of the immobilization process is to enable the effective recovery and recycling of the catalytic species, thereby improving its sustainability and practical application. Additionally, we will delve into the unique characteristics and specific challenges that arise from the heterogenization process. By comparing different supports and immobilization methods, we aim to shed light on how these variables influence the performance, longevity, and overall effectiveness of L-proline as a catalyst in various reactions. This analysis will provide valuable insights into optimizing immobilization strategies to enhance the practical utility of L-proline in catalytic applications.
Proline cases
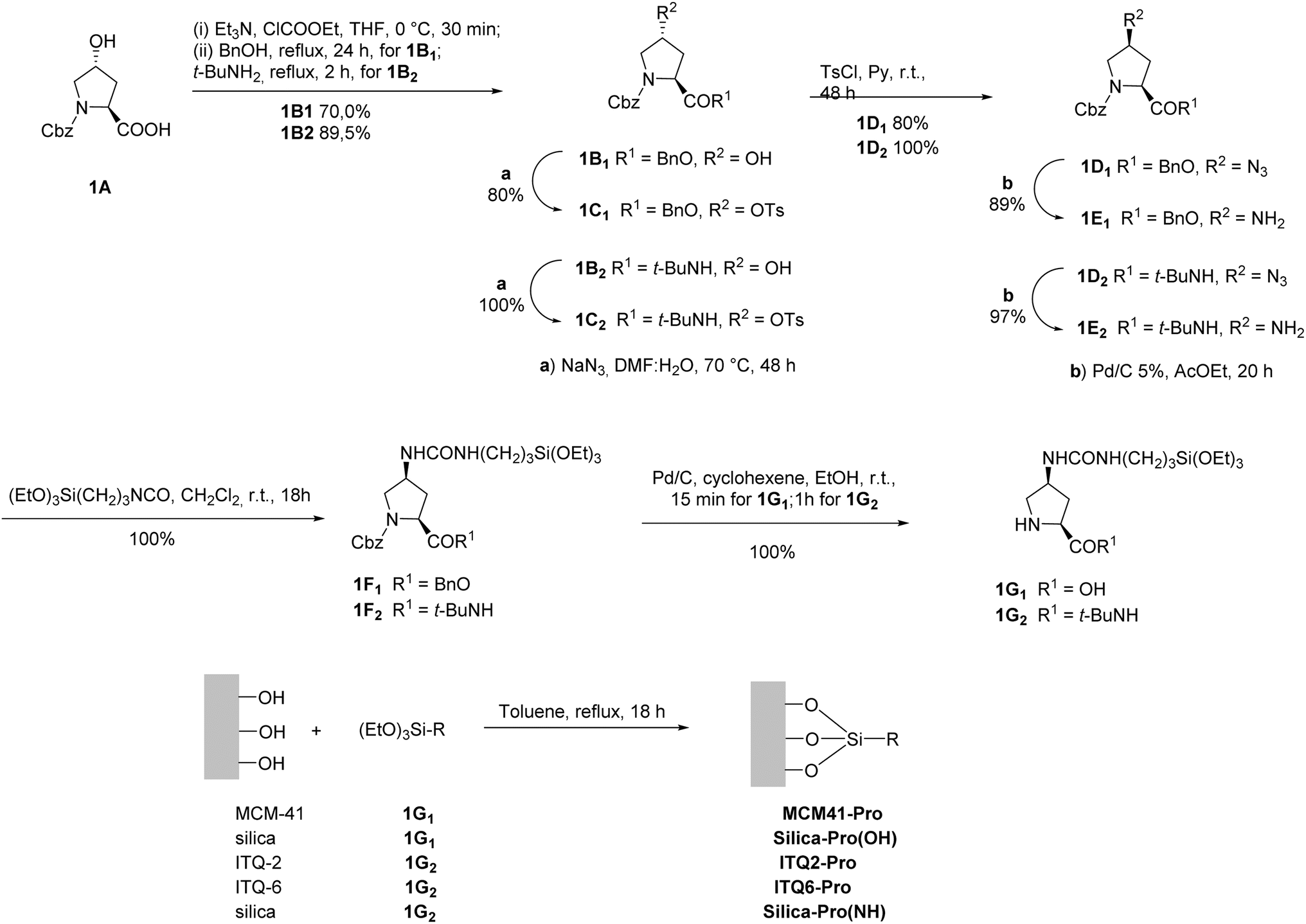 | ||
| Scheme 1 Methodology applied in the synthesis of MCM41-C catalyst. | ||
In this work, the results of a comparative study of aldol reactions of different aldehydes (2A1–5) with hydroxyacetone (2B) using free proline or covalently anchored mesoporous MCM-41 (Scheme 2) were described.
 | ||
| Scheme 2 Aldol condensation between hydroxyacetone (2B) and different aldehydes (2A1–5) catalyzed by L-proline or mesoporous material. | ||
The results demonstrated that MCM-41 functionalized with L-proline (MCM41-Pro) produced the best outcomes. Notably, this heterogeneous catalyst could be reused for a second and third run with only a minor decrease in yield, likely due to some loss of the catalytic material, but without significant loss in stereoselectivity. Changes in stereoselectivity might be attributed to hydrogen bonding interactions between the silanol groups on the support and the oxygen or nitrogen atoms of the R substituent in the aldehyde, which could influence the orientation of the reactant toward the formation of the syn-diol product. This interaction is likely enhanced in non-polar toluene, explaining the higher formation of syn-diol observed when using MCM41-Pro as the catalyst in toluene compared to DMSO. Additionally, the silanol groups on the support might interact synergistically with the aldehyde's carbonyl group, causing polarization and potentially increasing the reaction rate. This effect likely contributes to the higher yields of condensation products obtained with MCM41-Pro compared to the model. However, the harsh conditions required for condensation reactions (90 °C and extended reaction times) may lead to the decomposition of aldehydes or products, which likely explains the lower yields observed in some cases with MCM41-Pro compared to pure proline. To address this issue, the authors tested the reaction under milder conditions using microwave assistance, which significantly reduced reaction times and improved yields, allowing to produce aldols at room temperature within minutes. Overall, the use of microwave heating enhanced the performance of MCM41-Pro, demonstrating an effective heterogeneous methodology for asymmetric aldol condensations with a recoverable and recyclable catalyst.
Despite the already mentioned advantages of organocatalysts, in certain reactions, free proline, depending on the reaction medium and in certain solvents, may have low solubility, which would make it impossible for certain reactions to occur.33 In work carried out by Doyagüez et al.,48 in 2007, asymmetric aldol reactions mediated by a heterogeneous proline catalyst were studied in amorphous silica and structured mesoporous material (MCM41-Pro) (Fig. 4), the choice of the MCM-41 support this was due to its high surface area allowing the attachment of a greater number of moles of catalysts to the surface, with the aim of reducing the non-linear effect commonly seen in other heterogeneous catalysis reactions.48
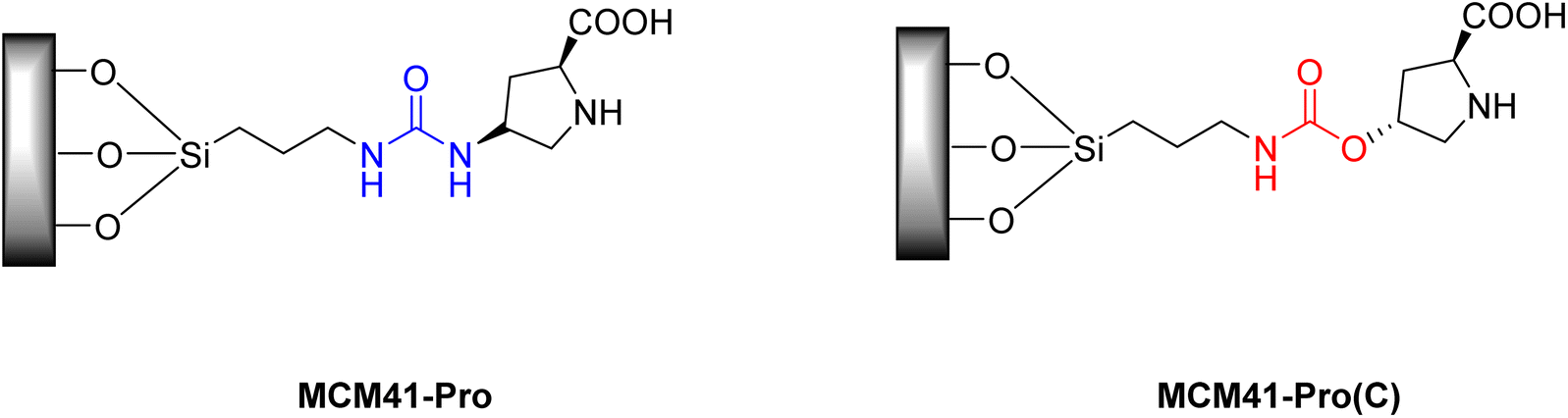 | ||
| Fig. 4 Heterogeneous proline catalysts MCM41-Pro and MCM41-Pro(C) supported on mesoporous material. | ||
The authors evaluated the influence of the solvent on reactions under conditions employing proline organocatalyst. Furthermore, they evaluated the effect of changing the ligand used to attach the proline to the support and its impact on the efficiency of the reactions, using the carbamate ligand (MCM41-Pro(C) catalyst) (Fig. 4).
The nature of the solvent has a significant impact on asymmetric aldol reactions catalyzed by proline, generally, these reactions occur more efficiently in hydrophilic polar solvents, such as formamide and DMF. Therefore, the results of this study demonstrated that the reaction involving 4-nitrobenzaldehyde (3A) and 2,2-dimethyl-1,3-dioxan-5-one (3B) in the presence of MCM41-Pro, high conversion rates to formamide and DMF were obtained 96% (67% of (ee)) and 80% (82% of (ee)) of 3C, respectively (Scheme 3). On the other hand, it was found that for hydrophobic solvents, such as toluene, the addition of a certain amount of water can have a relevant effect on the yield and stereoselectivity of the reaction.
 | ||
| Scheme 3 Reaction of 4-nitrobenzaldehyde (3A) with 2,2-dimethyl-1,3-dioxan-5-one (3B) used as standard model in the presence of MCM41-Pro. | ||
It was found that the efficiency of the reactions using MCM41-Pro was reduced since the hydrophobicity of the solvent was increased, with the solvent toluene a yield of 14% of 3C was obtained after 96 h, in t-butyl ether only traces of product were found, indicating that in hydrophobic solvents there is in fact a considerable loss of efficiency. However, the addition of water to the reaction promotes the formation of aldol products, there is a progressive increase in stereoselectivity promoted by the addition of up to 5 equivalents, which allowed a diastereoselective ratio of 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 and 78% (ee) of 3C. On the other hand, when 10 and 20 equivalents were added, the yield loss was very large, with only traces of the product remaining. This is because this excess water leads to the formation of a two-phase system, being responsible for the reduction in reactivity. According to the authors, the positive effect on reactivity when the addition of 5 equivalents is carried out may be associated with the water molecules that are organized around the solid solution at the interface, very close to the catalytic site, which favors the catalytic cycle of the reaction.
:1 and 78% (ee) of 3C. On the other hand, when 10 and 20 equivalents were added, the yield loss was very large, with only traces of the product remaining. This is because this excess water leads to the formation of a two-phase system, being responsible for the reduction in reactivity. According to the authors, the positive effect on reactivity when the addition of 5 equivalents is carried out may be associated with the water molecules that are organized around the solid solution at the interface, very close to the catalytic site, which favors the catalytic cycle of the reaction.
This work, the ligand group was changed and its effect on the reactions was evaluated. The urea group present in MCM41-Pro can interact with the carbonyl group of the reagents in a solvent-dependent manner and, thus, influences the rate and yield of reactions. Given this fact, a new catalyst was synthesized, in which the urea was replaced by the carbamate ligand (MCM41-Pro(C)) (Scheme 4). With this new catalyst, the solvents formamide, DMF and toluene was tested with the addition of 5 equivalents of water. It was observed that with this catalyst the reaction in toluene occurred more efficiently, generating products with a conversion comparable to that seen in formamide. However, they emphasize that, in the three solvents tested, diastereoselectivity and the enantiomeric excess value (ee) were lower for this catalyst when compared to MCM41-Pro, this may be related to the change in configuration at the C-4 carbon of the proline ring.
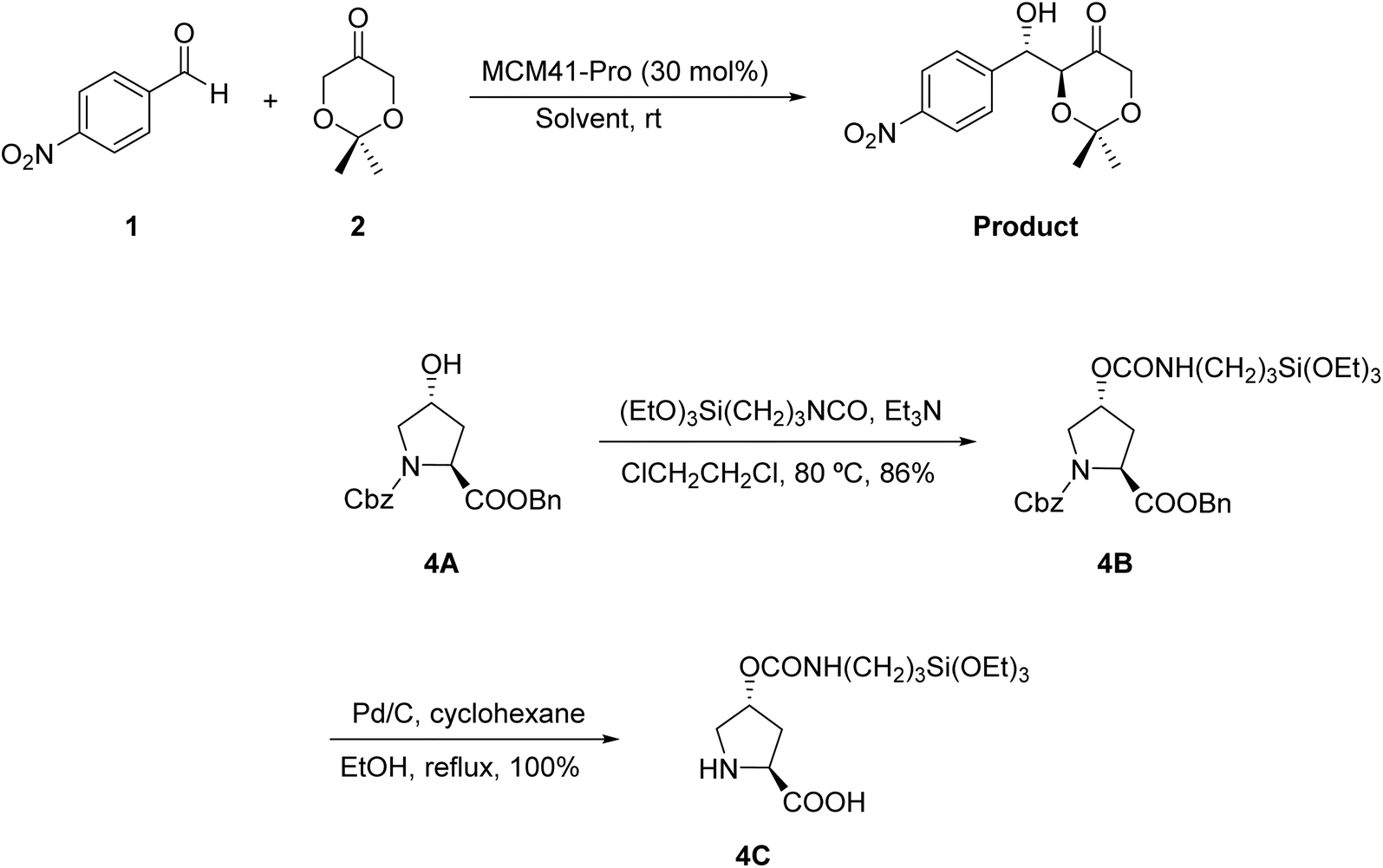 | ||
| Scheme 4 Synthesis of the MCM41-Pro(C) catalyst, exchange of the urea ligand for the new carbamate ligand. | ||
This work demonstrated the efficient use of proline catalysts for asymmetric aldol reactions, with important aspects being the use of catalysts obtained cheaply, without toxicity and available in both enantiomeric forms. Another relevant aspect is that the reactions can take place under mild reaction conditions, without the need for any metal. Another important point highlighted was overcoming the proline solubility problem in hydrophobic solvents, since the change in ligand allowed the reactions could occur efficiently in less polar solvents, with the addition of water contributing to maximum efficiency, this opens a new horizon of possibilities. The authors suggest that this effect of water contribution can be explained by the mechanism they proposed, which involves the generation of an intermediate enamine followed by the formation of a water molecule. Once the aldehyde reacts with the enamine, the formation of a new C–C bond, then the iminium intermediate formed is attacked by a water molecule to regenerate the catalyst. Under hydrophobic conditions, water tends to bind to the silanol groups that make up the support, which results in inhibition of proline regeneration, having a negative impact on the reactions. With the addition of an exact amount of water to the reaction medium, there is an increase in the concentration of water available to act in the catalytic cycle, making it possible to accelerate the reactions. On the other hand, excess water can impair the catalyst/reagent interaction, resulting in a reduction in efficiency.
It is worth noting that both MCM41-Pro and MCM41-Pro(C) heterogeneous catalysts demonstrated considerable efficiency. In the aspect of catalytic efficiency, the catalyst (MCM41-Pro) exhibits greater efficiency in hydrophilic polar solvents. In hydrophobic solvents, such as toluene, this catalyst has significantly increased efficiency with the addition of 5 equivalents of water, while for (MCM41-Pro(C)) it showed good performance in toluene, without the need to add water. Regarding the stereoselectivity of the reactions, the MCM41-Pro catalyst in general resulted in greater diastereoselectivity and enantiomeric excess when compared to MCM41-Pro(C). The effect of the controlled addition of water in toluene favors the increase in stereoselectivity for MCM41-Pro. This demonstrated that the change of ligand in the structure of the MCM41-Pro and MCM41-Pro(C) catalysts has an influence on the interactions with the substrates and the water in the medium, which resulted in the different observations in terms of efficiency and stereoselectivity. MCM41-Pro, with a urea ligand, appears to have greater sensitivity to the nature of the solvent, while MCM41-Pro(C), with a carbamate ligand, shows greater tolerance to more non-polar solvents. The attempt to modify and improve catalytic behavior, associated with the intrinsic properties of the support, proved to be advantageous and would justify its organocatalytic application.
Given the potential of polymeric supports, Cambeiro et al., in 2011,49 carried out a study with the immobilization of hydroxyproline in polystyrene polymeric resin, with the main objective of developing a continuous flow reaction process to obtain greater catalytic efficiency. Polymeric resins with different degrees of crosslinking were used to construct the support system, considering that crosslinking can have a significant effect on mechanical stability and, thus, have an impact on reaction performance. As a ligand group between the resin and the catalytic unit, the triazoyl moiety obtained via a Cu(I)-catalyzed cycloaddition reaction between azides and alkynes was used as an immobilization strategy, which allows for an improvement in conversion efficiency and in the enantioselectivity of the reaction. The scheme below shows the methodology for preparing two different immobilized catalysts. For (5C1), commercial Merrifield resin containing 1% 1,4-divinylbenzene (DVB) was used, and in the case of (5C2), Merrifield resin modified to contain 8% (DVB) prepared by radical copolymerization of styrene, 4-chloromethylstyrene and (DVB) was used (Scheme 5).
 | ||
| Scheme 5 Preparation of the immobilized catalysts 5C1 and 5C2. | ||
The results for catalysts (5C1) and (5C2) based on 4-hydroxyproline immobilized on polystyrene provided expected catalytic activity for the α-aminoxylation reaction of aldehydes. Both resins provided products in good yields and enantiomeric excess in short reaction times (Scheme 6).
 | ||
| Scheme 6 Direct enantioselective α-aminoxylation of propanal (6A) catalyzed by resins 5C1 and 5C2. | ||
The authors conducted continuous-flow reactions based on preliminary results, leveraging flow chemistry to optimize catalyst production. Immobilizing the catalyst in a flow reactor allows it to remain in constant contact with the reaction conditions, which prevents its degradation during operations such as washing, drying, and storage. This eliminates the need for additional processes to separate the catalyst from the mixture, increasing its useful life and productivity.
Although both resins presented a high enantiomeric excess value, resin (5C1) showed a superior catalytic activity compared to resin (5C2) for all substrates tested. Resin (5C1), which contains only 1% DVB as crosslinking agent, achieved a higher conversion rate and produced pure isolated products in yields of 94–96% (ee). This is due to its microporous character and higher degree of swelling in solvents, which facilitate the diffusion of substrates to the catalytic sites. In contrast, resin (5C2), with 8% DVB, is more rigid, resulting in smaller channels and pores that hinder the diffusion of reactants and limit the accessibility to the catalytic sites. This is reflected in lower conversions (95–98% ee), especially affected by the chain length and branching of the aldehyde substrate. Both resins showed moderate stability, with a slow reduction in the conversion rate during continuous use. The loss of activity may be related to oxidation, possibly due to the formation of oxazolidinone between proline and aldehyde, rather than to a destructuring of the polymer network.
In a 2014 study by Guan et al.,50 L-proline was grafted onto mesoporous SBA-15 materials with varying acid–base properties to explore their bifunctional catalytic effects, including L-proline. This work is an example of synergy with active phase and support. The study found that the effectiveness of L-proline as a catalyst varied depending on the specific reaction and the acid–base configuration of the support material. It was observed that the optimal catalytic performance was not solely determined by the levels of acidity or basicity but rather by their synergistic interaction within the material. In their current work, the researchers synthesized both SBA-15 and Al-SBA-15 with different acid–base configurations to further investigate and compare their catalytic behaviors, as shown in Scheme 7.
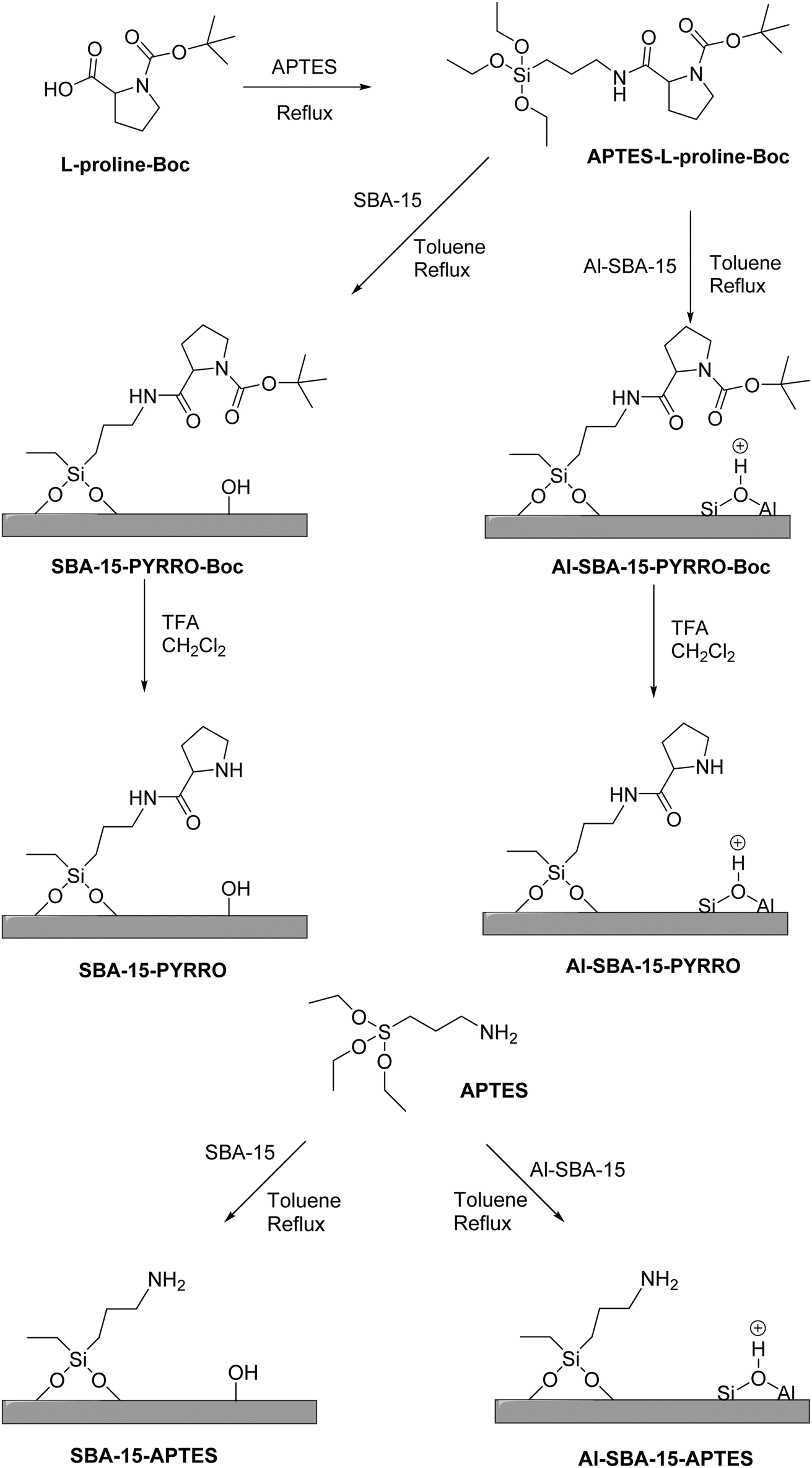 | ||
| Scheme 7 Synthetic route for SBA-15-APTES, Al-SBA-15-APTES, SBA-15-PYRRO, and Al-SBA-15-PYRRO. | ||
The XRD data indicates that the functionalized materials retain a 2D hexagonal mesoporous structure, while TEM images reveal a highly ordered mesostructure. Elemental analysis and IR spectra of pyridine adsorption confirm the incorporation of aluminum species and basic groups into the mesoporous SBA-15.
In the Knoevenagel reaction (Scheme 8), catalysts with weak base properties (SBA-15-APTES and Al-SBA-15-APTES) exhibit better catalytic activity compared to those with strong basic conditions (SBA-15-PYRRO and Al-SBA-15-PYRRO), even when L-proline is grafted. Additionally, catalysts with weak acid (SBA-15-APTES and SBA-15-PYRRO) perform better than those with moderately strong acid and the same base (Al-SBA-15-APTES and Al-SBA-15-PYRRO). Overall, a combination of weak acid and weak base is more favorable for the Knoevenagel reaction.
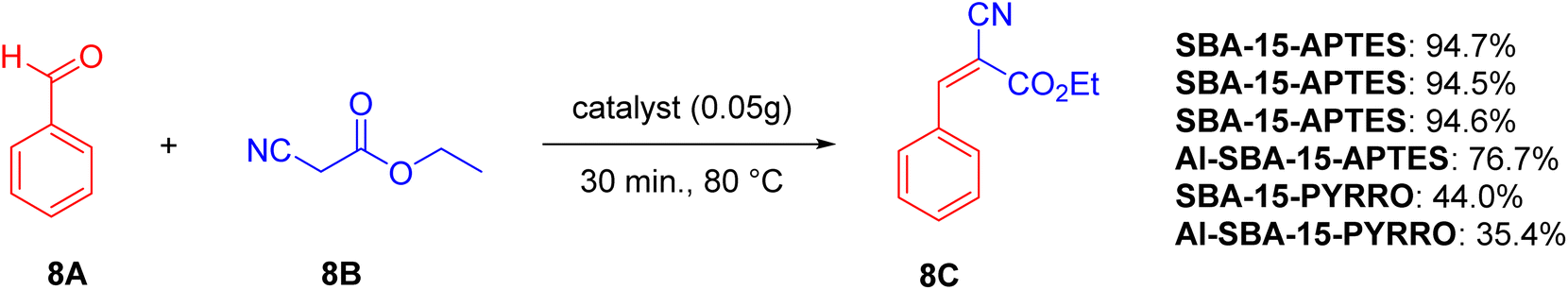 | ||
| Scheme 8 Test reaction for Knoevenagel reaction over different catalysts. | ||
In the one-pot deacetalization-Knoevenagel reaction (Scheme 9), the catalysts are evaluated based on their ability to perform two sequential steps: (1) acid-catalyzed deacetalization of dimethoxymethylbenzene to benzaldehyde, and (2) base-catalyzed base Knoevenagel reaction to produce benzylidene ethyl cyanoacetate. Catalysts with weak acid and weak base (e.g., SBA-15-APTES) are ineffective, as are those with only moderately strong acid (e.g., Al-SBA-15), which lack the necessary base catalysis. Catalysts with moderately strong acid and strong base (e.g., Al-SBA-15-PYRRO) also fail to efficiently catalyze the tandem reaction. The best performance is observed with Al-SBA-15-APTES, which has moderately strong acid and weak base, achieving 75.2% conversion and 72.7% yield of 9C. This catalyst shows minimal deactivation after three uses, with slight decreases in conversion likely due to incomplete removal of absorbed species.
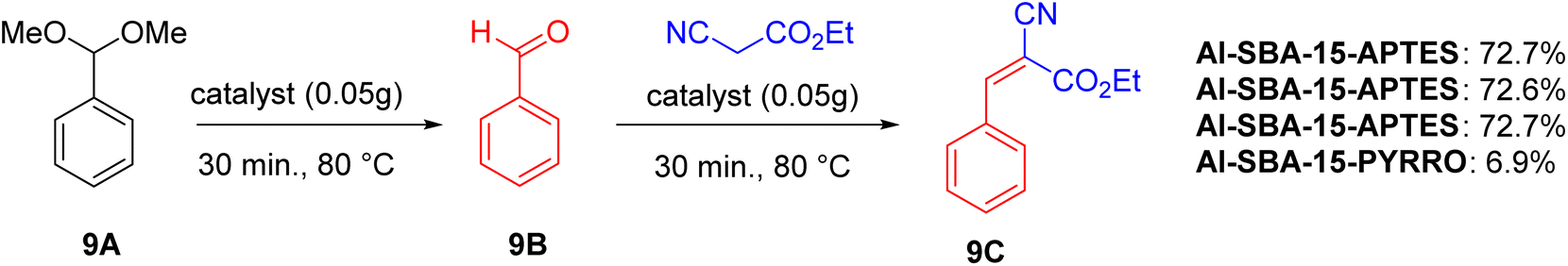 | ||
| Scheme 9 Test reaction for one-pot deacetalization-Knoevenagel. | ||
In the one-pot deacetalization-Henry reaction (Scheme 10), SBA-15, which is a weak acid catalyst, shows no activity. Catalysts that combine weak acid with weak base are also ineffective. Al-SBA-15, featuring a moderately strong acid, results in very low conversion and no final product due to inadequate base presence. In contrast, Al-SBA-15-PYRRO, which has a moderately strong acid and a strong base, achieves a 3.9% yield of 10C, as the strong base helps to accelerate the reaction. Overall, catalysts with moderately strong acid and weak base are more effective for this tandem reaction.
 | ||
| Scheme 10 Test reaction for one-pot deacetalization-Henry reaction. | ||
Strong bases can reduce selectivity for nitroalkenes due to side reactions like dimerization or polymerization, leading to poor yields.
In the nitroaldol reaction (Scheme 11), base-free catalysts like SBA-15 and Al-SBA-15 are ineffective. Acid–basic bifunctional catalysts are required for catalysis. Among these, the catalytic activity follows the order: SBA-15-APTES > Al-SBA-15-APTES > SBA-15-PYRRO > Al-SBA-15-PYRRO. Strong bases are less suitable for this reaction, while catalysts with weak acid and weak base (e.g., SBA-15-APTES) can achieve nearly 100% yield of 11B.
 | ||
| Scheme 11 Test reaction from nitroaldol reaction. | ||
In the aldol reaction of acetone with 4-nitrobenzaldehyde (Scheme 12), catalysts lacking base sites, such as SBA-15 and Al-SBA-15, or those with protected base sites, like SBA-15-PYRRO-Boc and Al-SBA-15-PYRRO-Boc, prove ineffective. The catalytic activity of bifunctionalized materials ranks as follows: Al-SBA-15-APTES (moderately strong acid and weak base) performs best, followed by SBA-15-PYRRO (weak acid and strong base), Al-SBA-15-PYRRO (moderately strong acid and strong base), and SBA-15-APTES (weak acid and weak base). The distribution of products is also affected by the catalyst's acidity and basicity, with weaker acidity leading to a higher proportion of alcoholic products compared to dehydrated ones.
 | ||
| Scheme 12 Test reaction from nitroaldol reaction. | ||
In their paper, Khalafi-Nezhad and co-authors51 introduce a straightforward and effective approach for creating a new silica-supported L-proline-based organocatalyst system (SSLP) through a grafting technique. The strategy applied for the preparation of the SSLP catalyst is shown in Scheme 13.
 | ||
| Scheme 13 Synthetic route for the preparation of SSLP catalyst. | ||
Given that the substrate SO can interact with numerous reagents to produce various functional groups and catalytic sites on the silica surface, it seems that SO is an excellent candidate for developing new silica-based materials.
In the structure of the SSLP catalyst there are portions of L-proline that are covalently linked to silica. These groups provide active catalytic sites on the silica surface.
Following the preparation and characterization of the SSLP catalyst, its catalytic performance was assessed in a multicomponent reaction involving isatin and dicarbonyl compounds to produce a range of spirooxindole derivatives under environmentally friendly conditions (Scheme 14). To optimize the synthesis of these compounds, the reaction of isatin, barbituric acid, and malononitrile was chosen as a model. This reaction was investigated using various catalysts and solvents at different temperatures to determine the best conditions (Scheme 14A).
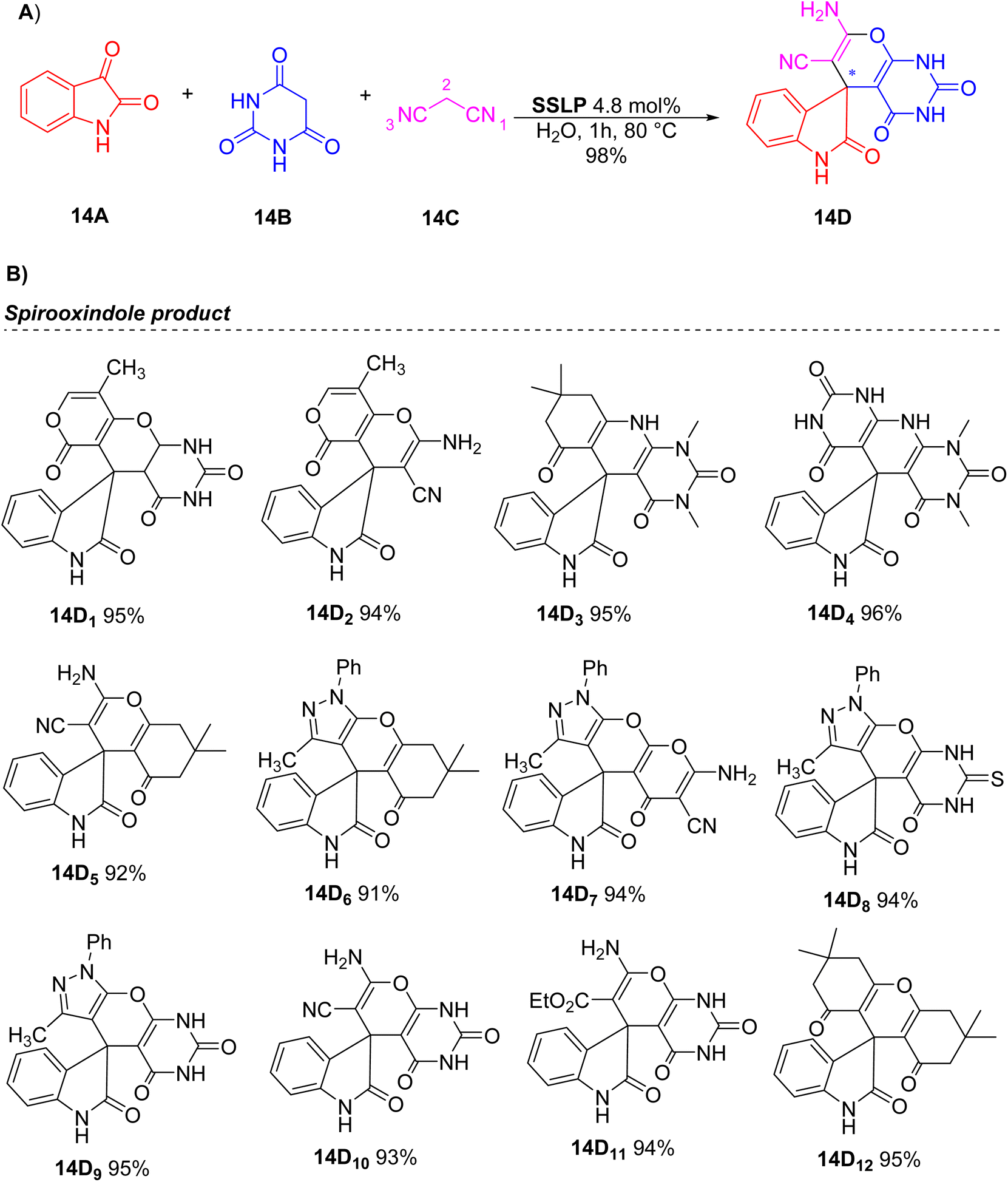 | ||
| Scheme 14 (A) Test reaction between isatin, barbituric acid, malononitrile to form spirooxindole product. (B) Spirooxindole derivatives analogues. | ||
Initially, the model reaction was tested in water without a catalyst, yielding 51% of the desired product 14D after 5 hours. Using L-proline as a catalyst, the yield increased to 80%. Reducing the amount of L-proline decreased the yield, confirming its role as a catalyst. The reaction with SiO2 showed no significant reactivity difference, but the yield improved to 79% of 14D with a SiO2/L-proline mixture. Using the SSLP catalyst resulted in a 95% yield, which increased to 98% of 14D at 80 °C, although it dropped to 71% at room temperature. The best results were obtained in water, likely due to the organic–inorganic nature of SSLP. Optimizing the catalyst quantity, 4.8 mol% was found to be optimal. The SSLP catalyst, being heterogeneous, can be easily recovered by filtration. After optimization, the catalyst's versatility was evaluated with diverse reagents (Scheme 14B). Notably, all starting materials afforded the target spirooxindole derivatives (14D1–12) in excellent yields.
The authors proposed a reaction mechanism involving the SSLP catalyst based on prior research (Scheme 15). The grafted L-proline on silica activates isatin by forming an iminium ion and serves as a bifunctional organocatalyst, enhancing substrate activation through hydrogen bonding. Additionally, the SSLP catalyst acts as a Brønsted acid/base, promoting the enolization of the dicarbonyl compound, which then reacts with the activated isatin. Despite the importance of grafted L-proline in the reaction, it does not affect the creation of a chiral center, resulting in a lack of stereoselectivity in the synthesis of the target compounds.
 | ||
| Scheme 15 Proposed mechanism for the synthesis of spirooxindole derivatives using SSLP catalyst. | ||
To evaluate the practical application of the heterogeneous organocatalyst, its reusability was tested. Following the model reaction, the catalyst was filtered, washed with acetone, dried, and then reused. It was found that the catalyst could be recycled over 8 times without additional treatment. To ensure the SSLP catalyst's activity remained consistent over multiple uses, nitrogen content was measured after 8 reaction cycles. Elemental analysis revealed only a 0.5% loss of nitrogen, indicating stable catalytic performance and durability.
Overall, the novel organocatalyst exhibited high efficiency with various isatins and dicarbonyl compounds, producing spiroindolones with yields from good to excellent (up to 90%). The SSLP catalyst's performance was evaluated in a three-component system for synthesizing spirooxindoles through the condensation of isatins and dicarbonyl compounds in water. Reactions were mild conditions, yielding products in good to excellent amounts. Furthermore, the SSLP catalyst could be recovered and reused up to eight times without additional treatment.
In their 2014 study, Zhi and colleagues52 introduced an innovative approach using immobilized organocatalysts and flow chemistry. They explored the benefits of catalyst heterogenization and leveraging flow chemistry. By immobilizing L-proline on silica gel, they developed a heterogeneous system that allows aldol reaction in a silica-filled chromatographic column using eluent flow.
The authors chose silica to immobilize L-proline due to its thermal and kinetic stability as well as chemical inertness. The silica gel-supported proline catalyst was prepared, via a covalent bond, using reaction conditions and procedures reported in Scheme 16. The synthesis involved: activating silica gel with sodium hydroxide to introduce hydroxyl groups, modifying it with aminopropyl triethoxysilane to incorporate amino groups (Si-L), activating the surface with ethyl chloroformate, reacting with N-Boc-L-proline to form Si-L-N-Boc-L-proline, and deprotecting with trifluoroacetic acid to yield the final silica gel-supported catalyst Si-LP.
 | ||
| Scheme 16 Illustration of proline functionalized silica gel. (1) Overnight (2) ethyl chloroformate, triethylamine, CH2Cl2, 0 °C to rt, overnight (3) trifluoroacetic acid, CH2Cl2, rt. | ||
Under optimized conditions for synthesizing α–β-unsaturated cyclohexenone from benzaldehyde and acetone (Scheme 17), Si-LP exhibits high catalytic efficiency across various aldehyde substrates. Aromatic and heterocyclic aldehydes yielded between 74.1% and 98% conversion. In contrast, batch reactions under identical conditions yielded lower product yields compared to continuous flow methods.
 | ||
| Scheme 17 Illustration of the aldol reaction triggered by Si-LP (the catalyst was in a silica-packed chromatographic column eluted with dehydrated ethanol at rt and at atmospheric pressure at a flow rate of around 0.1 mL min−1). | ||
To gain insights into the catalytic mechanism (Scheme 18), a study was conducted using p-cyanobenzaldehyde and acetone with Si-LP as the catalyst over time. Initially, the concentration of intermediate compound “18C” increased during the first 18 hours, subsequently decreasing as it converted into cyclohexenone “18D”. The reaction reached completion within 60 hours. Compound “18C”, an unsaturated α,β-ketone, was identified as an intermediate in the reaction between the aldehyde and acetone, capable of further reacting with acetone via Michael addition and Robinson cyclization to form cyclohexenone “18D”.
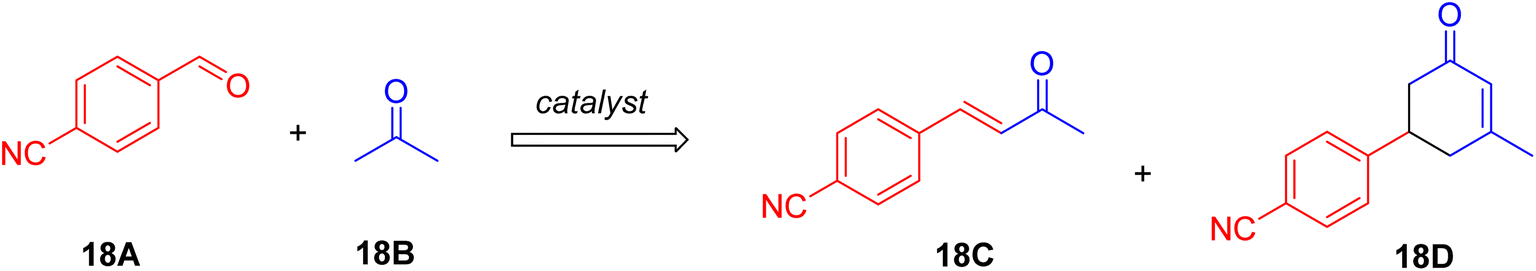 | ||
| Scheme 18 Illustration of reaction to obtain mechanism considerations. | ||
This method supports catalyst recycling, simplifies product isolation, and maintains mild reaction conditions, representing a significant advancement in sustainable organic synthesis.
In an interesting work, Dong and collaborators53 present a mechanistically distinct approach to heterogenizing linear chiral catalysts in MOF cavities, rather than synthetic modification of MOFs. This study developed highly porous chiral polymer/MOF composites via in situ polymerization of pre-impregnated chiral monomers within MOFs, creating hybrids with both local homogeneity and global heterogeneity. The flexible chiral catalytic sites in the pores allowed for cooperative interactions with the catalyst near metal nodes, demonstrating a synergistic effect between the chiral sites and metal Lewis acid nodes in enhancing catalytic performance.
The synthesis of chiral polymer/MOF composite MIL-101-PP1 includes: (1) impregnating a toluene solution of S1 vinyl monomer with L-proline, methyl acrylate, and AIBN into porous structure of Cr-MIL-101, and (2) in situ radical copolymerization within the MOF to form MIL-101-PP1-Boc, followed by N-Boc group removal to yield MIL-101-PP1, as showed in Scheme 19.
 | ||
| Scheme 19 Schematic diagram for the heterogenization of homogeneous linear chiral catalysts within the cavities of MOFs via in situ polymerization to afford chiral polymer/MOF composites (R) (taken from Dong, 2018).53 | ||
The method involves embedding linear chiral polymer chains into mesopores of Cr-MIL-101, which aids in dissolving and separating reaction mixtures efficiently, ensuring effective catalyst exposure and sustainability. These composites stand out from homochiral MOFs and traditional cross-linked polyethylene resins due to their unique structure and catalytic capabilities. Additionally, the combination of local homogeneous domains within globally heterogeneous hosts enhances both homogeneous and heterogeneous characteristics, offering advantages in catalysis.
In this work, MIL-101-PP1 was tested as a heterogeneous catalyst in the asymmetric direct Aldol reaction (Scheme 20), yielding the desired product 20C with 91% efficiency, featuring an impressive antidiastereoselectivity of 12:1 and enantioselectivity of 92% ee. Compared to homogeneous L-proline and CPP1 resin, MIL-101-PP1 demonstrated superior catalytic performance, leveraging the synergistic effects of high porosity of Cr-MIL-101 and Lewis acidity. Filtration experiments validated the crucial role of MIL-101-PP1 in maintaining reaction conversion. The catalyst retained its structure and activity over multiple cycles, showcasing sustainability and recyclability. Subsequent studies with diverse aldehydes confirmed excellent yields and selectivities, underscoring the catalyst's robustness and specificity within MOF pores.
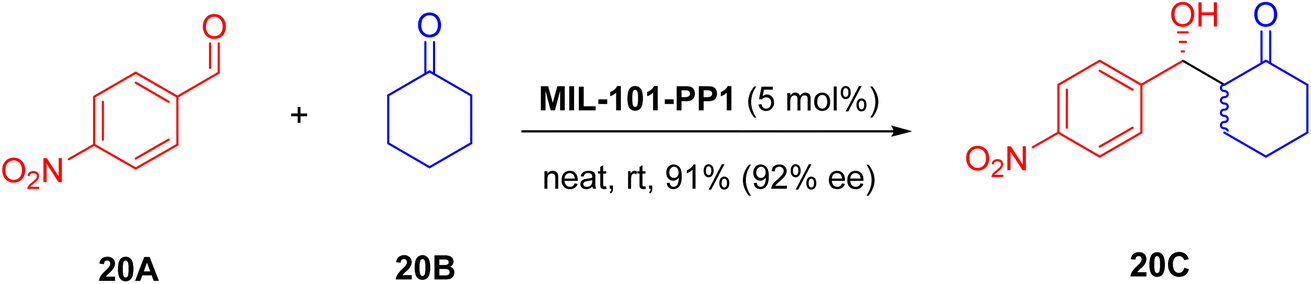 | ||
| Scheme 20 Test asymmetric direct aldol reaction. | ||
The study highlighted the superior performance of chiral polymer/MOF composites in asymmetric Aldol reactions, exhibiting excellent diastereo- and enantioselectivities alongside recyclability. By integrating Lewis acids with L-proline using Cr-MIL-101 as a cocatalyst, the authors achieved enhanced reaction outcomes. This approach capitalized on extensive surface area of Cr-MIL-101 and distributed Lewis acid sites, synergizing with L-proline to yield higher yields, improved enantiomeric excess, and better diastereomeric ratios compared to L-proline alone, which exhibited lower yield and selectivity.
Hajipour and co-authors present interesting work where L-proline was covalently anchored in carbon nanotubes.54 Multi-walled carbon nanotubes (MWCNTs) are one of the nanomaterials of greatest interest due to their unique thermal, mechanical and chemical characteristics. Furthermore, MWCNTs have a high surface area and are insoluble in almost solvents, which makes them a good candidate for support for heterogenization of organocatalysts. The Pro/MWCNTs catalyst was prepared using a multistep synthesis process demonstrated in Scheme 21.
 | ||
| Scheme 21 Preparation of the Pro/MWCNTs nanocatalyst. | ||
In the work, the authors carried out comparative studies and showed that Pro/MWCNTs can be more effective under certain optimized conditions, being able to catalyze the synthesis of different 2-amino-3-cyano-4H-pyran derivatives 22C efficiently (Scheme 22).
 | ||
| Scheme 22 Schematic reaction tests for synthesis of 2-amino-3-cyano-4H-pyrans analogues 22C. | ||
These derivatives are obtained in high yields and in relatively short reaction times. The catalyst proved to be versatile, working well with aldehydes and acetophenones that have different chemical groups, such as electron donors or withdrawers. It even tolerated specific functionalities such as chlorine and cyano in aldehydes, as well as heterocyclic variants. Compared to pure L-proline, Pro/MWCNTs proved to be faster in completing the reaction and more selective in forming the desired product, without generating significant byproducts. These results highlight the potential of Pro/MWCNTs as a promising choice for industrial and research applications in organic synthesis.
To verify the recyclability of Pro/MWCNTs as a heterogeneous catalyst, it was recovered by centrifugation and washed with diethyl ether to remove impurities before reuse. The catalyst can be recycled up to 10 times without losing significant activity. The catalyst was tested with different recovery methods and reused. The scale-up synthesis at 10.0 mmol was successful using the Pro/MWCNTs catalyst while maintaining stoichiometry, producing 22C at 89% efficiency, crucial for industrial applications.
The catalyst Pro/MWCNTs, prepared and evaluated in the reaction involving malononitrile and α-substituted chalcones devoid of substituents, aimed to synthesize multisubstituted 2-amino-3-cyano-4H-pyrans 22C. A novel approach was devised to stabilize proline chemically within multi-walled carbon nanotubes via direct grafting amination. Pro/MWCNTs demonstrated effective catalytic performance as a heterogeneous organocatalyst under gentle and eco-friendly conditions in this synthesis.
In an article published in 2018, Maleki et al. showing the first report of the catalytic application of magnetic nanoparticles functionalized with L-proline in organic reactions as a magnetic nanocatalyst.26 This pioneering study addresses the design, synthesis, functionalization and characterization of this new magnetic nanocomposite, in addition to its use as a heterogeneous catalyst in organic reactions. In this work, the method for the immobilization of L-proline on magnetic nanoparticles was devised since magnetic nanoparticles (MNPs) have recently been considered as a new type of catalytic support for organocatalysts due to their price, high dispersion, good stability, easy synthesis and method of functionalization, high surface area and easy separation using external magnetic fields. To this end, the developed nanomagnetic organocatalyst, called LPSF (Fe3O4/SiO2/propyltriethoxysilane/L-proline), was synthesized and its preparation involved a series of sequential steps, as indicated in the Scheme 23.
 | ||
| Scheme 23 Preparation of the magnetic nanocatalyst. | ||
The synthesized magnetic nanocatalyst was evaluated in the synthesis of 2,4,6-triarylpyridine derivatives 24D through a three-component reaction in a single vessel (Scheme 24). After optimizing the reaction conditions, using acetophenone, 4-chloro-benzaldehyde 24B10 and ammonium acetate 24C at a temperature of 60 °C and without solvent, it was observed that few amounts of the LPSF catalyst were sufficient to achieve high product yields. The effectiveness of ethanol as a solvent was compared, showing that solvent-free conditions provided better results. Compared to previous works, the present study stood out for its energy savings, high product yield and reusability of the nanocatalyst. Furthermore, the nanocatalyst has been shown to be effective in synthesizing a variety of 2,4,6-triarylpyridine derivatives 24D1–11 using different aromatic aldehydes 24A1–11 and acetophenones 24B1–11 with various electronic substitutions.
 | ||
| Scheme 24 Synthesis of 2,4,6-triarylpyridines analogues in the presence of LPSF nanocatalyst. | ||
The LPSF magnetic nanocatalyst was evaluated for recyclability by separating it from the reaction mixture using an external magnet. After washing with ethanol and water, drying and subsequent reuse, the catalyst maintained its effectiveness over seven cycles without a significant decrease in product yield. Characterization of the recycled nanocatalyst using FT-IR spectroscopy and FE-SEM imaging indicated that its structure and morphology were well retained.
 | ||
| Scheme 25 Methodology for obtaining supported proline material. | ||
The effectiveness of proline as a catalyst in aldol reactions depends on the support used. Proline performs better with ionic liquids than with silica gel alone. When proline is supported on silica gel modified with the ionic liquid 1-butyl-3-methylimidazolium tetrafluoroborate ([BMIMBF4]), it achieves a 51% yield and 64% enantiomeric excess (ee) in the reaction between acetone and benzaldehyde. This performance is similar to homogeneous conditions (60% ee) and slightly lower than using pure ionic liquid (71–76% ee). The [BMIMBF4] modified silica gel also offers better catalyst regeneration and reproducibility over three cycles compared to proline on PEG or in pure ionic liquid, with similar improvements seen with other aldehydes (Fig. 5).
 | ||
| Fig. 5 Supported asymmetric ionic liquid catalysis. | ||
This study found that proline supported on silica gel functionalized with the ionic liquid 1-butyl-3-methylimidazolium tetrafluoroborate ([BMIMBF4]) achieved better performance compared to proline on silica gel with adsorbed ionic liquid or without any ionic liquid. The [BMIMBF4] support led to higher enantiomeric excess and better catalyst regeneration, maintaining reproducibility over three cycles. The choice of support affects the catalyst's activity, selectivity, and recyclability, with the BF4 anion proving more effective than PF6. This emphasizes that optimizing support characteristics is essential for creating efficient and sustainable catalytic systems.
Based on these premises, Gruttadauria et al.,56 carried out a subsequent study with the aim of further evaluating the effect of the nature of the ionic portion of the modified silica gel on aldol reactions. Imidazole-modified silica gel was prepared with anion exchange, the silica gel (26C) having a different charge distribution and the silica gel (26F) having different steric properties and charge distribution. Finally, silica gel (26G) was prepared, with the absence of an ionic portion in the structure (Scheme 26).
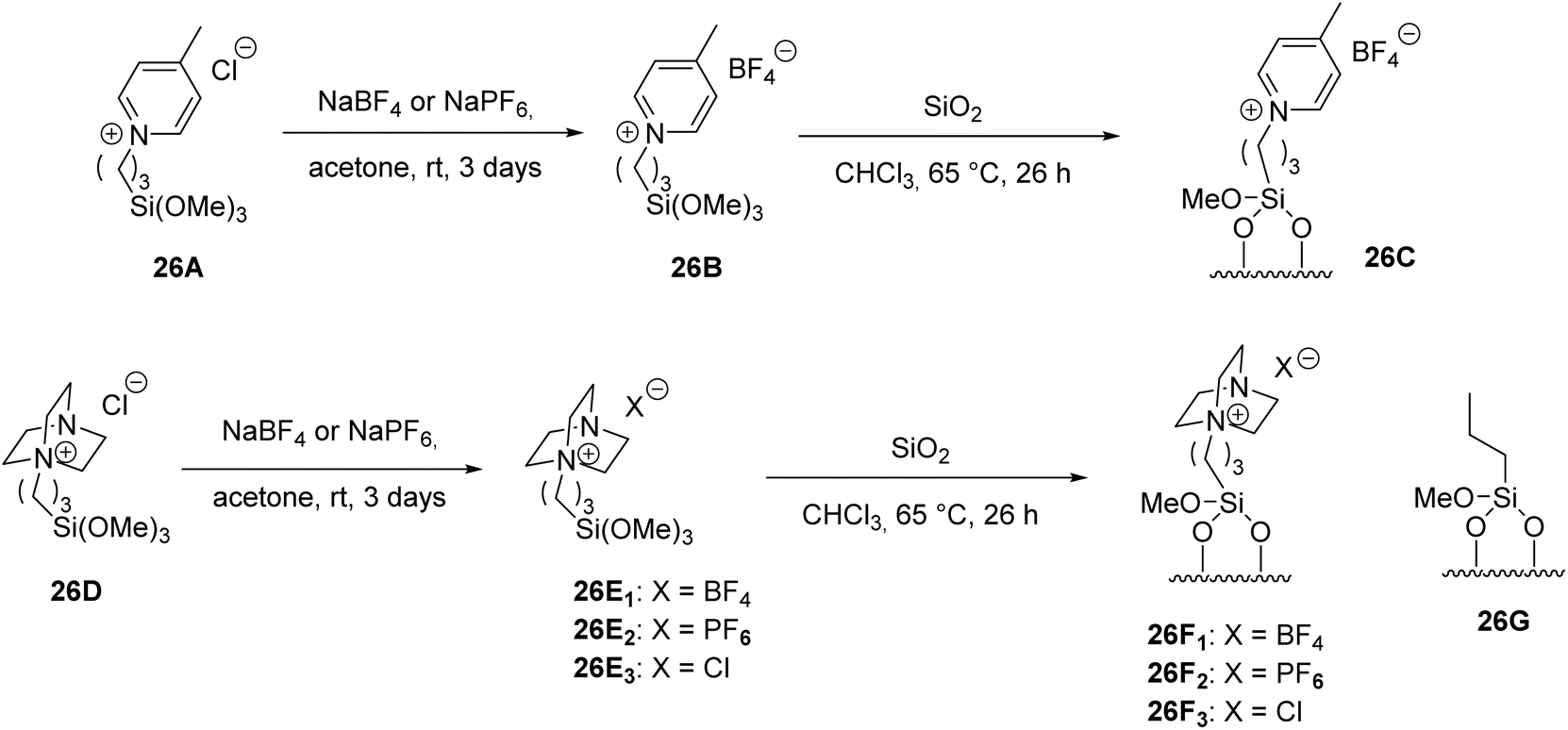 | ||
| Scheme 26 Reagents and conditions for synthesis of supported ionic liquids. | ||
The next step was to prepare various L-proline supported materials for testing. Two series were created: one with L-proline on modified silica gels and propyl (26E1/pro, 26E2/pro, 26E3/pro, 26C/pro, 26F1/pro, 26G/pro), and another with L-proline on silica gels modified with ionic liquids (25C1/BMIMBF4/pro, 26C/4mbpBF4/pro) or with adsorbed ionic liquids (SiO2/BMIMBF4/pro, SiO2/4MBPBF4/pro). These materials were made by mixing silica gel with L-proline solutions and then drying (Fig. 6).
 | ||
| Fig. 6 Catalytic systems studied. | ||
To evaluate the catalyst systems, a series of aldehydes were tested, benzaldehyde was used as an evaluation parameter for aldol reactions under the influence of these systems. The reaction occurred using supported L-proline (30 mol%) with acetone and benzaldehyde, room temperature for 24 h. According to the observed data, the 25C1/pro system presented a very relevant result, although a moderate yield was obtained, but the (ee) was 64%, like that found for homogeneous reaction conditions. Similar results were observed when the reactions were carried out in pure ionic liquid, the same was observed with L-proline dissolved in PEG and in a PEG-proline/DMF system with yields ranging from 58–76%. Therefore, to verify the influence of the anion, systems 25C2/pro and 25C2/pro were tested, both showed lower values of (ee) 42%, in addition to greater formation of elimination products. Therefore, it became evident that the BF4 anion has the best effect. Then, the 1-propyl-2,3-dimethylimidazolium cation was exchanged for 1-propyl-4-methylpyridinium, thus, for the 26C/pro system there was an increase in the enantiomeric excess value (70% ee). Finally, with the change in the nature of the ionic portion, in the cases of 26F1/pro and 26G/pro, the yields were small with very low enantioselectivity. Given this evidence, in which the type of cation has a considerable effect and, as these results clearly present the relevance of the nature of the supported ionic liquid phase, it is likely that the π–π interactions between the heterocyclic ring and the carbonyl play an important role.
To evaluate the catalyst systems, benzaldehyde was used in aldol reactions with supported L-proline and acetone. The 25C1/pro system showed a moderate yield and a high enantiomeric excess (64% ee), like homogeneous conditions. The BF4 anion proved most effective and changing the cation to 1-propyl-4-methylpyridinium increased the ee to 70% for the 26C/pro system. However, systems 26F1/pro and 26G/pro had poor yields and enantioselectivity, emphasizing the importance of ionic liquid structure and π–π interactions (Fig. 7).
 | ||
| Fig. 7 Reaction carried out using the systems (26C1/bmimBF4/pro) or (26C/4MBPBF4/pro). | ||
In terms of system regeneration, the 25C1/BMIMBF4/pro modified silica gel system showed high yields (84–94%) and good enantiomeric excess (ee) of 66–70% for the first four cycles, but the ee dropped in subsequent cycles. After regeneration, the system achieved yields of 50–86% and improved ee values, demonstrating easy recovery and highlighting the importance of the ionic surface for high enantiomeric excess.
In a study from 2013 by Tan and collaborators,57 they showed in their paper that L-proline was non-covalently loaded onto graphene oxide (GO) sheets by simply mixing them in an aqueous solution (Scheme 27). GO, with its layered structure and functional groups such as hydroxyl, epoxy, and carboxyl groups, effectively adsorbs L-proline. The method preserves the integrity of L-proline's structure, which is crucial for effective asymmetric induction, as it maintains both the carboxylic acid and pyrrolidine groups. The interaction between L-proline and GO sheets primarily involves hydrogen bonding between the hydroxyl and epoxy groups on GO and the carboxyl and secondary amine groups of L-proline. Additionally, ionic interactions at the carboxylic group sites on the edges of the GO layers may also play a role in the binding process.
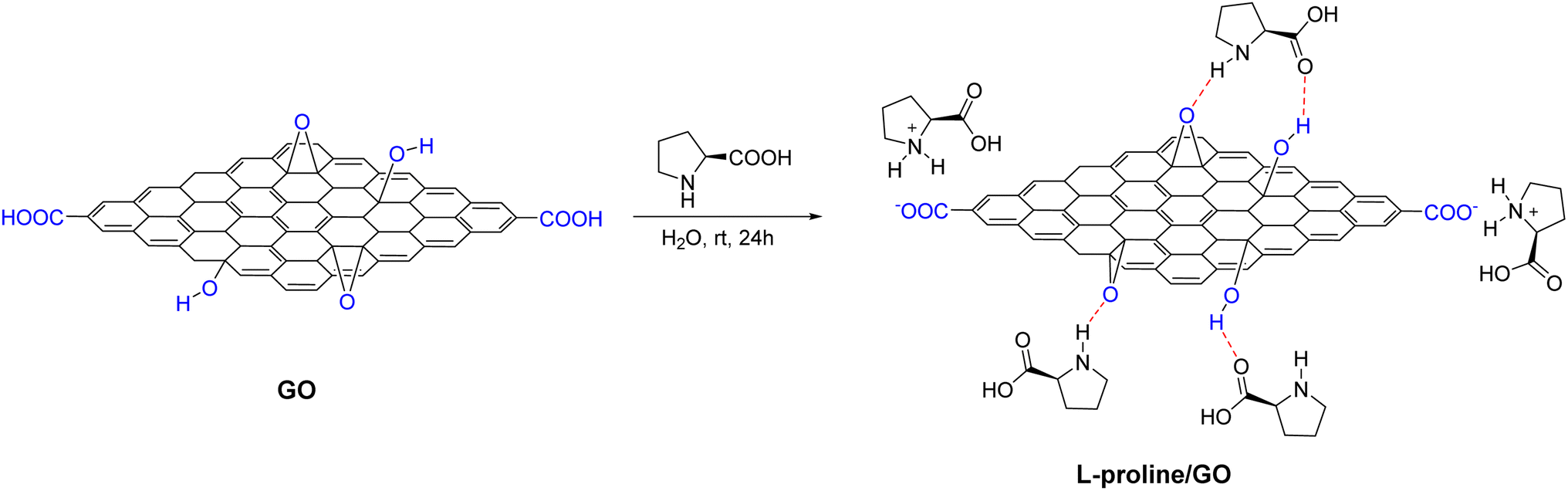 | ||
| Scheme 27 Illustration of the preparation of the L-proline/GO hybrid. The red arrows demonstrate hydrogen bonds. | ||
The study compared the loading of L-proline onto graphene oxide (GO) with other carbon supports like activated carbon and graphite. It was found that L-proline loading on GO (4.06 mmol g−1) was significantly higher than on activated carbon (1.49 mmol g−1) and graphite (0.59 mmol g−1). This higher loading is attributed to the abundant oxygen functional groups on the GO surface and its efficient use of surface area.
The L-proline/GO hybrid catalyst, made by non-covalently loading L-proline onto graphene oxide (GO), demonstrates excellent performance for direct asymmetric aldol reactions (Scheme 28). The multilayer structure of GO provides ample interlayer space, facilitating the diffusion of reactants to the chiral L-proline and enhancing catalytic efficiency. This hybrid catalyst benefits from GO's high L-proline loading capacity and the accessibility of active sites on its surface, making it highly effective for the direct asymmetric aldol reaction.
 | ||
| Scheme 28 Direct asymmetric aldol reaction between acetone and 2-nitrobenzaldehyde over different L-proline catalysts. | ||
The graphene oxide (GO) material alone is inactive for the direct asymmetric aldol reaction, yielding no products. However, when L-proline is used as a catalyst, it achieves a 95% yield and 79% enantiomeric excess (ee) at 30 mol% loading. Despite typically reduced activity when homogeneous catalysts are immobilized, L-proline's catalytic activity remains unaffected when immobilized on GO via hydrogen bonding and ionic interactions. The L-proline/GO hybrid shows comparable performance to pure L-proline, with a 96% yield and 79% ee. The unique multilayer structure of GO provides a stable anchoring site and accessibility for reactants, allowing the hybrid to perform as effectively as a homogeneous catalyst. Reducing the amount of the L-proline/GO hybrid catalyst to 20 mol% or even 10 mol% still results in high yields and maintains the ee. In contrast, hybrids with lower L-proline loading, such as those with activated carbon or graphite, perform less effectively. The L-proline/GO hybrid can be easily recovered and recycled up to seven times with minimal loss in reactivity, demonstrating its stability and efficiency.
In 2017, Nazari and Keshavarz used Amberlite as a support for the immobilization of L-proline, the objective of the work was to obtain an organocatalyst capable of overcoming one of the major challenges with immobilization, which is maintaining the activity and stereoselectivity of the immobilized catalyst, in addition to the separation and reuse of the catalyst after several reaction cycles.58 Pyrans was the class of molecules chosen for this study. Several methodologies applying different catalysts have already been reported in the literature, however, these methods face disadvantages such as low yields, long time of reaction, high temperatures, extreme reaction conditions and use of expensive reagents. In the search for a high-efficiency, easily applicable methodology using cheap catalysts, reusable and commercially accessible, the authors sintered an organocatalyst by immobilizing the ion pair of the L-prolinate anion in the cationic polymer resin Amberlite. The heterogeneous catalyst was structured through the construction of an L-prolinate catalyst supported by a heterogeneous macroporous polymer of Amberlite IRA900OH. The synthesis of this catalyst occurs easily and directly, basically, the Amberlite was treated with a solution of L-proline in a mixture of ethanol and water. L-Proline is supported on the anion exchange resin through immobilization of ion pairs. This process is understood via the interaction of the L-prolinate anion on the surface of the Amberlite anion exchange resin (AmbIRA900OH). This structuring occurs through the ionic interaction between the carboxylate group of L-prolinate and the quaternary ammonium cation of the cationic support Amb. (Scheme 29).
 | ||
| Scheme 29 Preparation of the catalyst via immobilization of the L-prolinate anion cationic polymer resin. | ||
FT-IR analysis confirmed the successful immobilization of L-proline in the [Amb]L-Prolinate hybrid by revealing new characteristic bands. This hybrid is essential for synthesizing 4H-pyrano[2,3-c]pyrazole derivatives, acting as a catalyst in the multicomponent reactions of Knoevenagel condensation, Michael addition, and intramolecular cyclization.
To evaluate the potential of the [Amb]L-Prolinate catalyst, benzaldehyde (30A), 3-methyl-1-phenyl-4,5-dihydro-1H-pyrazol-5-one (30B) and malononitrile (30C) were used (Scheme 30). This reaction was studied under different conditions, and it was evident from the results that the best yield for the formation of 6-amino-3-methyl-1,4-dihydropyrano[2,3-c]pyrazole-5-carbonitrile (30D) with 98%, was established with the use of 10 mol% [Amb]L-Prolinate under reflux in ethanol, increasing the catalyst load did not increase the yields, this condition being ideal. Therefore, the study was conducted for a series of aromatic aldehydes with different substituents, using the pre-established ideal condition. All analogues were isolated by filtration and recrystallized from ethanol with yields ranging from (85–98%) (Scheme 30).
 | ||
| Scheme 30 Optimal condition for reaction with a series of aromatic aldehydes containing different substituent groups using 10 mol% [Amb]L-Prolinate in ethanol under reflux. | ||
The reaction mechanism (Scheme 31) involving the [Amb]L-Prolinate catalyst, it abstracts a proton from the methylene group of malononitrile and gives rise to the formation of a carbanium which in turn attacks the carbon of the aromatic aldehyde, with the subsequent loss of a molecule of water. The catalyst reacts with 3-methyl-1-phenyl-4,5-dihydro-1H-pyrazol-5-one, forming the enamine intermediate, this intermediate undergoes a Michael addition to the Knoevenagel condensation product, of the first stage. Finally, after addition, intramolecular cyclization and deprotonation occur with the concomitant formation of the 4H-pyrano[2,3-c]pyrazole of interest.
 | ||
| Scheme 31 Mechanistic proposal for the formation of 4H-pyrano[2,3-c]pyrazole using the [Amb]L-Prolinate catalyst. A = benzaldehyde; B = malononitrile and C = 3-methyl-1-phenyl-4,5-dihydro-1H-pyrazol-5-one. | ||
The [Amb]L-Prolinate catalyst for synthesizing 4H-pyrano[2,3-c]pyrazole derivatives have shown superior performance compared to other methods, with shorter reaction times and better or comparable yields. Its heterogeneous nature allows for easy recovery and reuse up to eight cycles with minimal activity loss and high stability. This catalyst also improves sustainability by reducing waste and ensuring high efficiency and stereoselectivity without significant leaching. The use of inexpensive and commercially available L-proline further enhances its cost-effectiveness. Overall, this methodology offers significant advantages in reaction time and yield over previous methods.
Modifications to the chosen support are not required for the heterogenization process. Vaid and colleagues59 describe a straightforward method for impregnating L-proline onto silica. In their study, they present a process for creating L-proline supported on silica (SiO2-L-proline) where L-proline is first dissolved in methanol, then activated silica gel is added to the solution. After stirring the mixture for a few minutes, it is left to evaporate for 25 hours to yield a dry powder. The detailed preparation method for the silica-supported L-proline is illustrated in Scheme 32.
 | ||
| Scheme 32 Preparation of silica-supported L-proline (SiO2-L-proline). | ||
The characterization revealed that SiO2-L-proline had a fine, transparent powder appearance with a porous structure, and it was observed that L-proline particles are adsorbed on the surface of the silica, as illustrated in Scheme 32.
This article presents a simple method for synthesizing different malononitrile-substituted benzylidenes using amino acid-functionalized silica composites as heterogeneous catalysts (Scheme 33). The effectiveness of SiO2-L-proline as a catalyst was demonstrated by the absence of reaction without it after 8 h at 80 °C in acetonitrile. Additionally, a microwave-assisted reaction with SiO2-L-proline also did not achieve complete conversion.
 | ||
| Scheme 33 Benzylidene test reactions. | ||
To confirm the heterogeneity of the catalyst, its recyclability was evaluated with 4-nitrobenzaldehyde (33A3) and malononitrile (33B), showing only a small decrease in activity over five cycles. Furthermore, a hot filtration test confirmed that there was no leakage of L-proline from the SiO2-L-proline catalyst, as the reaction stopped as soon as the catalyst was removed.
This method developed by the authors presented several advantages, including good, isolated yields of the products and easy evaluation. The catalyst also showed good activity and could be recovered and recycled up to the fourth run with little drop-in activity after each run.
In work carried out by Li et al.,60 a new heterogeneous proline catalytic system was developed based on strong non-covalent interactions between polymeric ionic liquids (PIL) and L-proline. The synthesis of pyridine PILs was carried out using reversible addition–fragmentation chain transfer (RAFT) polymerization, allowing effective complexation with L-proline monomers and the formation of catalytic polymer networks by additional radical polymerization. The network structures varied significantly due to the different proportions of PIL, chiral monomer and PIL anions used in the reactions. These variations directly influenced the catalytic properties of PILCs, demonstrating a promising method for developing tailored functional materials (Scheme 34).
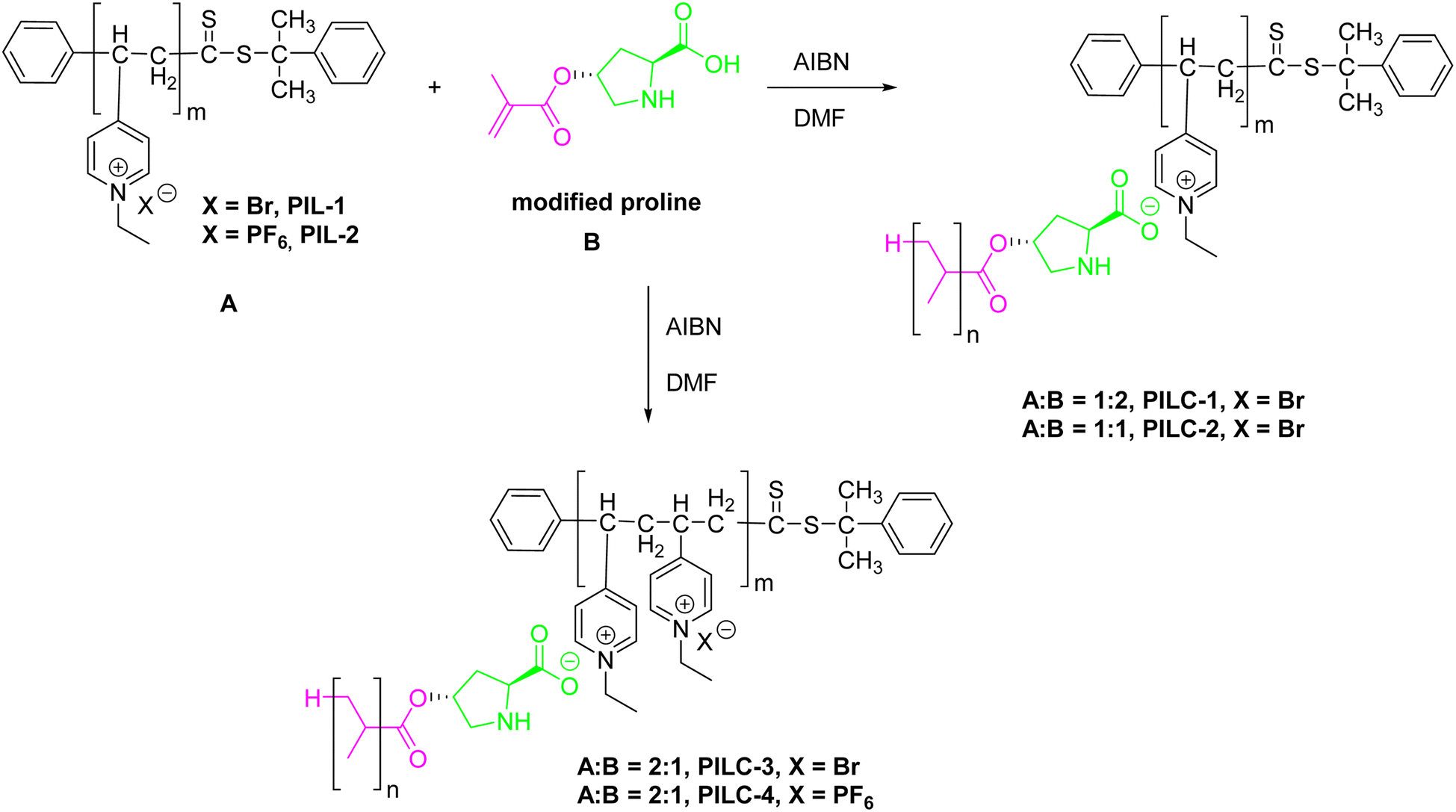 | ||
| Scheme 34 Synthesis of heterogeneous catalyst-supported L-proline based on supramolecular interactions and free radical polymerization. | ||
In aldol reaction experiments using 4-nitrobenzaldehyde and acetone, catalyzed by PILCs in DMF at 25 °C for 24 hours, it was observed that PILC-1, PILC-2 and PILC-3 achieved conversion rates greater than 80%, while L-proline and proline pyridinium salt only reached 55% and 35%, respectively (Scheme 35). PILC-3 maintained the optical activity of L-proline at 77% ee within the polymeric network, demonstrating the effectiveness of the incorporation of the chiral catalyst. The interaction between PIL and L-proline in free solution did not negatively affect the catalytic performance, suggesting that the complex network structure of PILC-3 and the synergistic effects with PIL contribute to homogeneous dispersion and improved mass transfer in catalysis.
 | ||
| Scheme 35 Aldol reaction between acetone and 4-nitrobenzaldehyde catalyzed by different catalyst systems for 24 h in DMF (catalysts 10 mol%). | ||
In addition to aldol reactions, the system was successfully applied to complex three-component reactions involving 4-chlorobenzaldehyde, 2-hydroxy-1,4-naphthoquinone and 3-amino-5-4-methylpyrazole, crucial in the synthesis of heterocyclic compounds important for medicine, pesticides and other materials (Scheme 36). In a solvent system containing acetonitrile, ethanol, and THF, PILC-1 demonstrated superior performance, achieving optimal conversion of 79% and enantioselectivity of 91% in EtOH, the chosen solvent for other steps. Evaluation of several PILCs in three-component reactions highlighted that while L-proline and proline pyridinium salt served as references with moderate conversions and enantioselectivities, the addition of PIL to L-proline did not provide significant improvements in catalytic performance. PILC-1 showed enhanced activity (75% conversion, 91% ee) due to its layered structure, while PILC-2 exhibited greater selectivity (72% conversion, 97% ee). PILC-3, however, showed the best overall performance with high conversion (92%) and enantioselectivity (98%), highlighting the synergistic effects between PIL and the polymer network. PILC-4, although it presented lower activity, exhibited excellent enantioselectivity (60% conversion, 99% ee), exemplifying different catalytic behaviors within the PILC series (Scheme 36).
 | ||
| Scheme 36 Effect of solvents on reactions at 80 °C for 21 h (catalysts 20 mol%). | ||
The hydrophobic structure of PIL influenced the efficiency of the reaction, making it difficult for the substrate to enter in certain contexts. However, PILCs offer tunable catalytic properties through modification of the PIL anion. PILC-3 demonstrated superior catalytic activity in single-pot multicomponent reactions, facilitated by a synergistic mechanism, where the bromide anion of IL activated O–H bonds and the cation activated C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bonds, promoting efficient substrate in Michael addition reactions. This synergistic effect likely contributed to the greater efficacy of PILC-3 in catalyzing MCRs compared to other PILs.
O bonds, promoting efficient substrate in Michael addition reactions. This synergistic effect likely contributed to the greater efficacy of PILC-3 in catalyzing MCRs compared to other PILs.
PILC-3 was successful in catalyzing three-component reactions using several substituted benzaldehydes, such as 4-nitrobenzaldehyde, 2-hydroxy-1,4-naphthoquinone, and 3-amino-5-4-methylpyrazole, with products achieving high conversion (97%) and enantioselectivity (87%) in ethanol, compared to MeCN.
Demonstrating stability over five cycles in a three-component reaction, PILC-3 maintained its catalytic activity and enantioselectivity without deterioration. Elemental analysis confirmed that the L-proline structure remained intact during consecutive cycles, reinforcing the catalyst's durability and efficiency in practical applications.
The study introduced PILCs with layered structures and synergistic PIL effects, enhancing catalytic activity and enantioselectivity for heterocyclic compound synthesis in a single vessel. PILCs demonstrated reusable capability for five cycles without loss of catalytic efficiency, attributed to L-proline's efficient incorporation via supramolecular interactions. This underscores PILCs' role in high-performance catalyst synthesis and their potential in advancing organic catalysis through innovative polymer network structures and PIL synergies.
In the article by Keshavarz et al.,61 the author presents an interesting graphene oxide/Fe3O4/L-proline (GO/Fe3O4/L-proline) superparamagnetic nanohybrid obtained from the non-covalent immobilization of L-proline on graphene oxide/Fe3O4 nanocomposite that was used as a new magnetically separable catalyst. The work's strategy is to combine the advantages of having support such as graphene and magnetic materials in a single composite, all associated with the properties of organocatalysts.
The method for preparing GO/Fe3O4/L-proline is illustrated in Scheme 37. Hydrogen bond interaction between hydroxyl, epoxy and carboxyl groups on GO sheet in GO/Fe3O4 nanohybrid with carboxyl and secondary amine groups of L-proline are the driving force for the binding of L-proline to the GO/Fe3O4 nanohybrid, which was also presented in Scheme 37.
 | ||
| Scheme 37 Preparation pathway of graphene oxide GO/Fe3O4/L-proline nano hybrid. | ||
After obtaining the catalyst, its catalytic properties were examined in a pseudo-three-component synthesis reaction carried out in a single container for the production of 4,4′-(arylmethylene)bis(1H-pyrazol-5-ol) (38C) (Scheme 38). To determine the ideal conditions, the reaction between benzaldehyde and two equivalents of 3-methyl-1-phenyl-2-pyrazolin-5-one was chosen as a model to investigate the impact of the GO/Fe3O4/L-proline catalyst in various configurations. Optimization analysis indicated that the best performance was obtained when conducting the reaction in the presence of 0.05 g of GO/Fe3O4/L-proline under reflux in ethanol. Increasing the amount of catalyst (0.1 g) did not result in an improvement in yield, while reducing its amount led to a decrease in efficiency.
 | ||
| Scheme 38 Test reaction for investigation of catalytic activity of GO/Fe3O4/L-pro for the synthesis of bis-pyrazole under various conditions. | ||
Under the optimized occurrence conditions, the effectiveness of this strategy in obtaining a diversity of 4,4′-(arylmethylene)bis(1H-pyrazol-5-ol) derivatives was investigated. All reactions resulted in high-yield products and could incorporate a wide variety of aromatic aldehydes with electron-donating and electron-withdrawing groups.
The investigation of the reusability of the catalyst was carried out in eight consecutive cycles in a model reaction. The results revealed that the catalyst maintained its effectiveness throughout the cycles, with only a 5% reduction in yield from the first to the eighth run. Furthermore, the superparamagnetic properties of the catalyst facilitated its recovery using an external magnet, without compromising its functionality over multiple cycles.
The GO/Fe3O4/L-proline catalyst combines three widely used catalysts: GO, Fe3O4 nanoparticles and pure L-proline. Thus, the authors compared their catalytic efficiency with each isolated component in the synthesis of 4,4′-(arylmethylene)bis(1H-pyrazol-5-ol) derivatives, using the reaction between benzaldehyde and 3-methyl-1-phenyl-2-pyrazolin-5-one as a model. Compared with pure L-proline, it was observed that the form immobilized in the nanocomposite exhibits greater catalytic activity, with better performance in terms of reaction time and product yield. Furthermore, the efficiency of this method in the synthesis of 38C was demonstrated, comparing it with other procedures, where the proposed method by the authors presents shorter reaction times and higher product yields.
In general, it can be said that it increases catalytic robustness and thermal stability through the hydrogen bond with GO/Fe3O4. The L-proline immobilized in the nanocomposite shows superior catalytic activity than pure L-proline and, also, that the event is easily recoverable with a magnetic and reusable for at least six cycles in model interactions.
In 2020, Prabhakara et al.,62 developed a new method for Mannich reactions, which are important in the formation of carbon–carbon bonds in organic synthesis to produce β-amino carbonyl derivatives. They described the synthesis of a L-proline organocatalyst immobilized in ionic liquid, a novel approach that allows easy recycling and recovery of the catalyst. This method is advantageous because the reactions are carried out without solvents, making the process environmentally friendly and non-toxic.
In the present study, the authors describe a methodological development with better performance for the case of these multi-component reactions using the L-proline. The initial strategy was to immobilize proline in an appropriate ionic liquid. For this, methylimidazole reacted with 2-chloroethanol without the use of solvent, giving rise to the compound [HEMIM][Cl−]. This [HEMIM][Cl−] compound, under the action of NaBF4 in acetonitrile, provided the desired hydroxyl-functionalized ionic liquid [HEMIM][BF4−] (Scheme 39).
 | ||
| Scheme 39 Synthesis of alcohol-functionalized ionic liquid [HEMIM][BF4−]. | ||
The next step was the synthesis of the desired organocatalyst IL-P. To obtain it, the previously generated ionic liquid, [HEMIM][BF4−], was coupled to protected L-proline, boc-L-proline, in the presence of DCC/DMAP coupling reagent in ACN, providing the proline organocatalyst immobilized in ionic liquid IL-P-Boc. Finally, IL-P-Boc undergoes deprotection reaction by 30% TFA/ACN to give rise to L-proline immobilized in ionic liquid IL-P (Scheme 40).
 | ||
| Scheme 40 Synthesis of the organocatalyst IL-P immobilized in L-proline ionic liquid. | ||
After the synthesis of the organocatalyst IL-P, its activity in Mannich reactions was evaluated (Scheme 41). Using 10 mol% of IL-P with acetophenone (41A), benzaldehyde (41B) and aniline (41C) in various solvents, it was observed that polar aprotic solvents such as THF, CH2Cl2, DMF and DMSO produced Mannich product 41D with yields of 10–20% in 24 h. However, the solvent CH3CN resulted in a significantly higher yield of 65%. Tests with polar protic solvents such as EtOH, IPA and H2O showed even better yields, between 60–70%.
 | ||
| Scheme 41 Optimization of the Mannich reaction. | ||
To optimize the methodology, the reactions were carried out without solvent, at room temperature and for short times. After 2 h, the yield of 41D increased significantly to 96%. However, the temperature of 80 °C reduced the yield to 40%. The loading of the organocatalyst IL-P was varied from 0 to 20 mol%. Without IL-P, there was no formation of 41D, but with 5 mol%, 85% yield was obtained. With 7 mol% and a time of 2 h, the yield of 41D was 96%, and higher loadings (10, 15 and 20 mol%) also resulted in 96% yield. Therefore, the optimal condition was established with 7 mol% of IL-P, since further increases in the amount of catalyst did not improve the yield.
To investigate the influence of the catalyst, 7 mol% IL-P, 7 mol% L-proline and 7 mol% 4-hydroxy-L-proline were tested. While L-proline and 4-hydroxy-L-proline gave yields of 20% and 25% in 48 h, respectively, IL-P achieved a yield of 96% in only 2 h, showing superior catalytic performance. The authors performed reactions with acetophenones, aromatic aldehydes and substituted arylamines.
Both electron-donating and electron-withdrawing substituents did not negatively affect the reactions, which proceeded efficiently in 2–3 h, resulting in excellent yields. The catalytic potential of IL-P is attributed to its proline-immobilized ionic liquid fraction (Scheme 42). The mechanistic proposal suggests that the aromatic aldehyde, activated by IL-P, reacts with the arylamine to form an imine intermediate (III). Simultaneously, acetophenone reacts with IL-P to form enamines (I) and (II). The imine (III) then reacts with the enamines to form transition states (IV) and (V), where attacks by the Re and Si faces of the enamines lead to the formation of the carbon–carbon bond. The IL-P catalyst can be recycled and reused up to five times without loss of activity.
 | ||
| Scheme 42 Proposed mechanism for the transition state for Mannich reaction using organocatalyst IL-P. | ||
The traditional method of organocatalyst attachment uses supports such as polymers, oxides, or magnetic particles, which limit the loading of catalysts due to the reduced number of available functional groups, resulting in lower activity and selectivity. In contrast, Xu et al.,63 developed an innovative method by attaching L-proline to multiwalled carbon nanotubes (MWCNTs), which offer exceptional loading capacity. Although MWCNTs are stable and conduct electricity and heat well, their cylindrical structure limits the availability of functional groups for attachment, which can reduce catalytic efficiency. However, Xu and colleagues showed that L-proline can be attached to MWCNTs at a content of 67% while maintaining good catalytic efficiency and reusability in various chemical reactions.
The incorporation of L-proline into MWCNTs carbon nanotubes was confirmed by thermogravimetric (TGA), elemental and catalytic performance analyses. TGA showed little weight loss for MWCNTs up to 800 °C, indicating structural stability. For pure L-proline, the weight loss occurs at 229 °C and approaches zero at 347 °C. The L-proline/MWCNTs catalyst showed a slight loss of 11% at about 200 °C, with a significant weight loss similar to that of pure L-proline, suggesting an L-proline loading of 67% by weight. Elemental analysis confirmed a percentage of 8.17% nitrogen, equivalent to 67.3% L-proline in the catalyst (Fig. 8).
 | ||
| Fig. 8 The schematic model of L-proline/MWCNTs. | ||
The high adsorption rate of L-proline on MWCNTs is surprising, given the high solubility of L-proline in water and the difficulty in removing excess L-proline during washing. The initial loss of 11% observed in the TGA of the loaded MWCNTs can be attributed to the presence of trapped water, indicating that L-proline is adsorbed internally in the MWCNT cavities. Elemental analysis revealed 27.4% oxygen in the catalyst, suggesting that the difference in oxygen content corresponds to about 12% water, consistent with the initial loss observed. The exceptional loading of 67 wt% of L-proline suggests that L-proline is not only on the surface but also in the internal cavities of the MWCNTs. Experiments with NaOH, which promotes the deprotonation of the carboxyl group of L-proline, significantly reduced the L-proline content, indicating that this group plays a crucial role in the adsorption.
The high loading capacity of L-proline on MWCNTs results from efficient adsorption on the surface and internal cavities of the nanotubes due to their large surface area. The hydrophobic interaction and adsorption by the carboxylic group of L-proline are crucial, as demonstrated by the cyclohexane emulsion tests, which significantly remove L-proline from MWCNTs.
The catalytic activity profile and recycling of the L-proline/MWCNTs catalyst were evaluated in the aldol reaction with acetone and 4-nitrobenzaldehyde (43B), using 29 mol% of catalyst for 8 h (Scheme 43). The product 43C yield was 91%, similar to the 89% obtained with free L-proline, and the enantiomeric excesses were comparable. After seven cycles, the L-proline/MWCNTs maintained high efficiency and excellent stability, showing good recyclability.
 | ||
| Scheme 43 Aldol reaction of acetone and 4-nitrobenzaldehyde catalyzed by L-proline/MWCNTs. | ||
The L-proline/MWCNTs catalyst demonstrated faster reaction evolution, reaching equilibrium with a yield of approximately 91%, compared to free L-proline. This is due to the high adsorbed catalyst load, maintaining most of the active molecules and with similar enantioselectivity.
The article by Mutiah et al.,64 provides an example of immobilization of L-proline in MOFs, in this case a zirconium type. This strategy represents an innovative method for creating efficient heterogeneous catalysts for the aldol reaction. The zirconium MOFs used in this process are porous and stable structures that incorporate mixed ligands, forming a solid network around the zirconium centers. This porous structure provides an ideal environment for proline, allowing it to act effectively as a catalyst, but on a solid support.
The immobilization process was performed by impregnating the MOFs with L-proline via solvothermal method. Proline was dissolved in a suitable solvent and mixed with the MOFs, and then adsorbed or chemically bound to the surface of the MOFs, as described in the following scheme (Scheme 44).
 | ||
| Scheme 44 Proline immobilization by directly solvothermal method. | ||
After immobilization, the resulting materials were characterized using techniques such as spectroscopy (FT-IR, NMR) and surface area analysis (BET) to confirm the presence and distribution of L-proline, as well as the integrity of the MOF structure.
The heterogeneous catalyst, containing immobilized proline, was tested in the aldol reaction, more specifically in the asymmetric aldol reaction between acetone and two different aldehydes: 2-chlorobenzaldehyde (2-CBA) and 4-nitrobenzaldehyde (4-NBA) (Scheme 45).
 | ||
| Scheme 45 Aldol reaction test. | ||
The results showed that UiO-66-S30Pr achieved good conversions and enantiospecific excesses (ee) for the aldol products, ranging from 60–80% in ee and 59–73% in conversion. The ee obtained with UiO-66-S30Pr was comparable to that achieved with the homogeneous L-proline catalyst (64–83%), although the conversion was slightly lower with the MOF for both aldehydes. The UiO-66-S30Pr catalyst showed excellent reusability, although it showed a significant decrease in activity with 4-NBA, possibly due to the mild toxicity of nitrogen to proline. The conversion and ee decreased slightly after three cycles, confirming the stability and efficacy of the L-proline-functionalized MOF for recyclable use. Overall, the results showed high catalytic activity and demonstrated that immobilized proline is effective in forming the desired products. The porous structure of MOFs facilitates reagent accessibility and catalyst regeneration, providing a practical and sustainable approach for chemical synthesis.
The study by Xu et al.,65 addressed the synthesis and application of chiral porous organic cages (POCs) in asymmetric organocatalysis, developing two types with chiral profilin units. These cages, formed by self-assembly of organic molecules, have cavities accessible to small molecules, providing highly active and efficient catalytic sites. Although promising for asymmetric catalysis, the construction of chiral POCs is challenging due to changes in crystallinity and self-assembly processes.
The authors developed functionalized chiral POCs, CPOC-401-Pro and CPOC-302-Pro, using proline due to its catalytic capacity in asymmetric organic reactions. The synthesis involved the attachment of N-Boc-L-proline to diamine ligands by amide coupling, followed by removal of the protecting group and formation of enantiopure ligands. CPOC-401-Pro, with a tetrameric [4 + 8] cage, was produced by the reaction of a ligand with C4RACHO, presenting a larger diameter and lower steric hindrance. CPOC-302-Pro, an octahedral [6 + 12] cage, was formed by the reaction of ligand B3 with C4RACHO (Fig. 9). The structures of the POCs were confirmed by NMR and circular dichroism spectroscopy.
 | ||
| Fig. 9 (a) The synthesis and structure of chiral [4 + 8] tetrameric cage CPOC-401-Pro. (b) The synthesis and structure of chiral [6 + 12] hexameric cage CPOC-302-Pro. Hydrogen atoms have been omitted for clarity (taken from Xu, 2022).65 | ||
The adsorption isotherms for CPOC-401-Pro and CPOC-302-Pro exhibit high porosity with BET surface areas of 648 and 861 m2 g−1, respectively, improving the catalytic activity. However, CPOC-302-Pro has lower N2 adsorption capacity and BET value due to the blockage by the proline-occupied channels. The permanent porosity of chiral POCs promotes a more efficient interaction between the catalytic centers and the reactants, accelerating the reactions.
In comparison of the catalytic activity of POCs, the reaction with 4-nitrobenzaldehyde and cyclohexanone was used with 10 mol% chiral proline in CPOC-401-Pro and CPOC-302-Pro (Scheme 46). CPOC-302-Pro achieved 99% yield, 92% ee and 93:7 dr, while CPOC-401-Pro showed lower ee and dr values despite a faster reaction. CPOC-302-Pro showed high enantioselectivity with a lower reaction rate. Compared to the model organocatalyst BPP, which had lower enantioselectivity and diastereoselectivity after 4 days, and to L-proline, which was inert and took 10 days with low selectivity.
 | ||
| Scheme 46 Asymmetric aldol reactions catalyzed by chiral POCs. | ||
The three-dimensional arrangement of CPOC-401-Pro and CPOC-302-Pro affects their catalytic activity due to the spatial distribution of the catalytic sites. CPOC-401-Pro has a tetrameric prism structure with chiral proline distributed in its backbone, while CPOC-302-Pro, with a hexameric octahedral structure, has proline anchored internally. These differences in spatial arrangement influence the interaction of the catalytic sites with the reagents, resulting in better performance of CPOC-302-Pro, which showed greater catalytic activity and selectivity in the asymmetric aldol reaction. Thus, POCs are promising as organocatalysts with high regeneration and well-defined catalytic cavities.
 | ||
| Scheme 47 The schematic representation of aldol reaction between benzaldehyde and acetone. | ||
Furthermore, the reconstruction method based on the “memory effect” of Mg–Al LDHs proved to be an effective means of achieving the intercalation of L-proline anions. Furthermore, the proline content could be adapted to a certain range. The structural model for L-Pro LDHs (Mg/Al molar ratio = 3:1, intercalated yield = 59%). The model shows the L-proline anions in a roughly horizontal orientation relative to the hydroxide layers, with the carboxylate groups binding directly to the hydroxide layers containing four water molecules and one carbonate. Water molecules alternately interspersed with L-proline anions promote hydrogen bonds with hydroxide layers, which can have a significant effect on the catalytic activity of L-Pro LDHs (Fig. 10).
 | ||
| Fig. 10 The schematic model of L-Pro LDHs (molar ratio of Mg/Al = 3:1). | ||
This new solid catalyst has several advantages, including high activity under mild liquid phase conditions, easy separability by simple filtration, and lack of side products, and is therefore environmentally friendly. The application of LDHs L-Pro in the aldol reaction reveals that immobilization in LDHs has no adverse effect on the catalytic activity of L-proline. Furthermore, immobilization by LDHs can effectively prevent the racemization of L-proline under heat treatment and UV irradiation. Thus, the catalytic activity of L-Pro LDHs decreases markedly due to the lower mobility of proline anions. The stabilization of enantiomeric selectivity in heat treatment provides advantages to L-Pro LDHs in storage, delivery and application processes.
In their paper, Arya and co-workers67 explore a new concept of immobilization, in which an organocatalyst, such as L-proline, is immobilized in pure silica mesoporous materials. In this case, proline is confined in the channels of mesoporous and microporous materials. The central concept is to encapsulate organocatalysts, such as proline, in a solid matrix formed by silicate hydrolysis. The matrix has pores and channels tuned to retain the catalyst and allow efficient transport of reactants and products. This approach, which prevents leaching while allowing efficient flow, distinguishes this system from conventional supported or immobilized catalysts.
FAU zeolite is an excellent base for organocatalysts due to its uniform distribution of large mesopores (3–10 nm) and high surface area. Its crystalline structure provides high hydrothermal stability, superior to other mesoporous materials without crystalline structure. In addition, water is increasingly valued as a solvent in organic synthesis for its economic, environmental and polar advantages, encouraging the development of reactions in water instead of conventional organic solvents.
The L-proline-confined zeolite catalyst was synthesized by adding 10 mol% L-proline to an aluminosilicate gel, followed by crystallization at 100 °C for 24 h. UV-Raman characterization showed a change in the vibration of the carboxylate group of L-proline, indicating strong interaction with the zeolite, while X-ray diffraction revealed that the FAU zeolite structure remained unchanged, and X-ray fluorescence confirmed the expected Si–Al ratio. High-resolution TEM showed that L-proline forms uniform spots of 1.3 ± 0.2 nm in the zeolite supercages, indicating that it is physically confined and does not leach.
Focusing on green chemistry and aqueous reactions, the authors investigated the reaction of ketones/lactams, amines and formaldehydes using FAU zeolite confined with L-proline in water at room temperature (Scheme 48).
 | ||
| Scheme 48 Preparation of spiroheterocycles through the Mannich reaction. | ||
The Mannich reaction with indole, 4-methylaniline and formaldehyde demonstrated that the new catalytic system produced the desired product in much shorter times and with higher yields than FAU zeolite without catalyst or solvent. By adjusting the reaction ratio, better results were obtained with a 2:4 ratio of arylamines and formaldehyde, confirming the effectiveness of the catalyst system with L-proline and FAU zeolite (L-proline/FAU).
After optimizing the conditions, we extended the reaction of aqueous formaldehyde with arylamines and several cyclic lactams/ketones to construct a library and verify the synthesis of novel spiroheterocyclic derivatives. Arylamines with electron-withdrawing groups showed low activity, resulting in average yields.
This new procedure represents the first efficient and eco-friendly method for the exclusive synthesis of spironolone [indoline-3,5′-pyrimidin]-2-one and spironolone [indene-2,5′-pyrimidin]-1(3H)-one derivatives, achieving higher yields and shorter reaction times. Economically, the stability and extended activity of the catalyst are crucial. We tested the recovery and reusability of L-proline/FAU catalyst in a one-pot reaction involving indole-2-one, 4-methyl aniline, and aqueous formaldehyde. The results demonstrated that the catalyst retained its performance after five cycles, with no loss of activity, and was effectively washed with water and dried at 300 °C between uses.
An example of intercalation in LDH's is the study carried out by Ju et al.,68 which investigated the use of LDHs for the immobilization of L-proline. In this work, MxAl-LDHs catalysts with OH− anions were prepared and used in the aldol condensation reaction of formaldehyde with propionaldehyde to produce methacrolein. L-Proline was successfully intercalated into the LDH layers through co-precipitation and rehydration, taking advantage of its amphotericity.
Six different catalysts were synthesized by varying the metals and their ratios: re-Ca2Al-LDH, re-Ca3Al-LDH, re-Ca4Al-LDH, re-Mg2Al-LDH, Mg3Al-LDH and Mg4Al-LDH. The characterizations showed that the metal atoms in the LDHs are covalently bonded and connected to the L-proline layers by electrostatic forces and hydrogen bonds. The carboxyl group of L-proline is inclinedly arranged between the metal layers. The spacing between the intercalated materials varies with the anion used: NO3− in the coprecipitation process and OH− in the rehydration process, with NO3− being larger than OH (Fig. 11).
 | ||
| Fig. 11 Structural model of L-proline-intercalated LDHs. | ||
Solid-based catalysts (re-MxAl-LDHs) with OH− anions demonstrated high catalytic activity for the aldol condensation of formaldehyde (49A) and propionaldehyde (49B) (Scheme 49).
 | ||
| Scheme 49 Test reaction using re-MxAl-LDHs catalysts. | ||
Although calcium-based catalysts improved the conversion and selectivity with higher Ca/Al ratios, magnesium-based catalysts, especially re-Mg3Al-LDHs, showed the best performance with 82.59% propionaldehyde conversion and 36.01% methacrolein (49C) selectivity. Using re-MgxAl-LDHs, several products were observed, including methacrolein, 2-methyl-2-pentenal, cis-2-butene-1,4-diol and 1,1,1-tris(hydroxymethyl)ethane, all catalyzed by bases. Despite modifications in metal ions and their ratios, the selectivity for methacrolein was unsatisfactory. This led to the incorporation of L-proline to improve cross-condensation selectivity.
Intercalation of L-proline into LDHs improved the selectivity for methacrolein compared to re-MxAl-LDHs due to the facilitation of the Mannich reaction mechanism (Fig. 12).
 | ||
| Fig. 12 Proposed catalytic cycle for the aldol condensation reaction with the Mannich pathway. | ||
However, the LDHs catalysts intercalated with L-proline by rehydration showed lower catalytic performance than those obtained by coprecipitation, due to less efficient intercalation and lower density of active sites.
The results show that increasing the substrate amount decreases the propionaldehyde conversion but increases the selectivity for methacrolein. The optimum conditions were 91.39% propionaldehyde conversion and 51.48% methacrolein selectivity at 60 °C for 30 min. Despite the maintenance of methacrolein selectivity after four cycles, the catalytic activity decreased due to L-proline leaching and catalyst loss. Despite the challenges, the use of intercalated L-proline in LDHs offers advantages such as simpler synthesis, lower L-proline consumption, and reduced separation costs, integrating homogeneous and heterogeneous catalysis.
Summary
The available data show that the immobilization of proline and its derivatives has generated considerable interest, revealing how these simple molecules have stimulated innovation in the synthesis of new organocatalysts. Proline and its derivatives, such as prolinamide and pyrrolidine, have been the focus of current studies. However, there is a growing demand for more complex catalysts that are stereoselective and highly recyclable. This trend underscores the need for the development of more sophisticated organocatalysts that overcome the limitations of simple catalysts.The proposed review aims to highlight the relevance of organic molecules in heterogeneous organocatalysis and emphasize the potential of these catalysts for heterogenization. Although the review discusses fundamental concepts of organocatalysts, heterogenization strategies, preparation methods, and their various applications, it does not attempt to cover all aspects of this emerging field. A notable aspect is the choice of immobilization method, which may include techniques such as grafting, impregnation, or inclusion. The choice of immobilization method is crucial for the final performance of the catalyst, directly affecting the efficiency and stability of the catalytic system. In addition, as in other heterogeneous catalytic systems, the role of the materials or supports used is essential for the overall performance of the catalysts. The synergistic effect between the catalyst and the support can significantly influence the effectiveness of the catalytic process. The interaction between the catalyst and the support material can affect the distribution of active sites and their accessibility, directly impacting the efficiency and selectivity of the catalyzed reactions. Therefore, it is crucial to continue exploring new approaches and methods in heterogeneous organocatalysis to overcome challenges and expand the possibilities of this area. The continuous development and investigation of new techniques promise to bring significant advances, expanding the scope and application of organocatalysts in the near future.
Data availability
No primary research results, software or code have been included, and no new data were generated or analyzed as part of this review.Author contributions
Gustavo Senra G. de Carvalho and Douglas Alcântara C. Pinto searched references, wrote the manuscript, drawing figures/schemes and sorting tables. Robson C. da Silva searched for references and drawing figures. Fernando de Carvalho da Silva revised the manuscript. All of the authors have read and approved the final manuscript.Conflicts of interest
The authors declare no conflicts of interest.Acknowledgements
This study was supported by FAPERJ (E-26/211.343/2021 and E-26/204.023/2024) and CNPq (307736/2023-7).References
- T. Kitanosono, K. Masuda, P. Xu and S. Kobayashi, Chem. Rev., 2018, 118, 679–746, DOI:10.1021/acs.chemrev.7b00417.
- A. Moyano and R. Rios, Chem. Rev., 2011, 111, 4703–4832, DOI:10.1021/cr100348t.
- M. M. Mukherjee, S. K. Maity and R. Ghosh, RSC Adv., 2020, 10, 32450–32475, 10.1039/d0ra05355d.
- K. A. Ahrendt, C. J. Borths and D. W. C. MacMillan, J. Am. Chem. Soc., 2000, 122, 4243–4244, DOI:10.1021/ja000092s.
- W. S. Jen, J. J. M. Wiener and D. W. C. MacMillan, J. Am. Chem. Soc., 2000, 122, 9874–9875, DOI:10.1021/ja005517p.
- B. List and D. W. C. Mac Millan, MLA Style: Press Release, NobelPrize.Org, Nobel Prize Outreach, 2025, Thu. 20 Mar 2025, https://www.nobelprize.org/prizes/chemistry/2021/press-release/.
- Y. Shi, Acc. Chem. Res., 2004, 37, 488–496, DOI:10.1021/ar030063x.
- H. Pracejus, Justus Liebigs Ann. Chem., 1960, 634, 23–29, DOI:10.1002/jlac.19606340104.
- B. List, Tetrahedron, 2002, 58, 5573–5590, DOI:10.1021/ja994280y.
- V. d. G. Oliveira, M. F. D. C. Cardoso and L. d. S. M. Forezi, Catalysts, 2018,(8), 1–29, DOI:10.3390/catal8120605.
- B. List, J. Am. Chem. Soc., 2000, 122, 9336–9337, DOI:10.1021/ja001923x.
- L. Hoang, S. Bahmanyar, K. N. Houk and B. List, J. Am. Chem. Soc., 2003, 125, 16–17, DOI:10.1021/ja028634o.
- B. List, P. Pojarliev and H. J. Martin, Org. Lett., 2001, 3, 2423–2425, DOI:10.1021/ol015799d.
- B. List, L. Hoang and H. J. Martin, PNAS, 2004, 101, 5839–5842, DOI:10.1073/pnas.030797910.
- K. N. Rankin, J. W. Gauld and R. J. Boyd, J. Phys. Chem. A, 2002, 106, 5155–5159, DOI:10.1021/jp020079p.
- B. List, Chem. Rev., 2007, 107, 5414–5415, DOI:10.1021/cr078412e.
- T. Ahmad and N. Ullah, R. Soc. Chem., 2021, 8, 1329–1344, 10.1039/d0qo01312a.
- C. F. Nising and S. Bräse, Chem. Soc. Rev., 2008, 37, 1218–1228, 10.1039/b718357g.
- D. X. Zhu, J. G. Liu and M. H. Xu, J. Am. Chem. Soc., 2021, 143, 8583–8589, DOI:10.1021/jacs.1c03498.
- M. H. Aukland and B. List, Pure Appl. Chem., 2021, 93, 1371–1381, DOI:10.1515/pac-2021-0501.
- A. Carlone, L. Bernardi and De Gruyter, Phys. Sci. Rev., 2019, 4, 20180097, DOI:10.1515/psr-2018-0097.
- B. Han, X. H. He, Y. Q. Liu, G. He, C. Peng and J. L. Li, Chem. Soc. Rev., 2021, 50, 1522–1586, 10.1039/d0cs00196a.
- S. H. Xiang and B. Tan, Nat. Commun., 2020, 1–5, DOI:10.1038/s41467-020-17580-z.
- M. P. van der Helm, B. Klemm and R. Eelkema, Nat. Rev. Chem., 2019, 3, 491–508, DOI:10.1038/s41570-019-0116-0.
- R. T. Baker, S. Kobayashi and W. Leitner, Adv. Synth. Catal., 2006, 348, 1337–1340, DOI:10.1002/adsc.200600344.
- A. Maleki and R. Firouzi-Haji, Sci. Rep., 2018, 8, 1–8, DOI:10.1038/s41598-018-35676-x.
- I. R. Shaikh, J. Catal., 2014, 2014, 1–35, DOI:10.1155/2014/402860.
- C. Yu and J. He, Chem. Commun., 2012, 48, 4933–4940, 10.1039/c2cc31585h.
- E. Reyes, L. Prieto and A. Milelli, Molecules, 2023, 28, 2–24, DOI:10.3390/molecules28010271.
- D. W. C. MacMillan, Nature, 2008, 455, 305–308, DOI:10.1038/nature07367.
- C. Richard Catlow, M. Davidson, C. Hardacre and G. J. Hutchings, Philos. Trans. R. Soc., 2016, 1–2, DOI:10.1098/rsta.2015.0089.
- C. Vogt and B. M. Weckhuysen, Nat. Rev., 2022, 6, 89–111, DOI:10.1038/s41570-021-00340-y.
- B. S. Vachan, M. Karuppasamy, P. Vinoth, S. Vivek Kumar, S. Perumal, V. Sridharan and J. C. Menéndez, Adv. Synth. Catal., 2020, 1–23, DOI:10.1002/adsc.201900558.
- M. Rumyantsev and S. Rumyantsev, Polymer, 2018, 136, 101–108, DOI:10.1016/j.polymer.2017.12.058.
- T. Fulgheri, F. Della Penna, A. Baschieri and A. Carlone, Curr. Opin. Green Sustainable Chem., 2020, 25, 1–9, DOI:10.1016/j.cogsc.2020.100387.
- R. Schlögl, Angew. Chem., Int. Ed., 2015, 54, 2–58, DOI:10.1002/anie.201410738.
- M. Gruttadauria, F. Giacalone and R. Noto, Chem. Soc. Rev., 2008, 37, 1666–1688, 10.1039/b800704g.
- C. E. Song and S. G. Lee, Chem. Rev., 2002, 102, 3495–3524, DOI:10.1021/cr0103625.
- R. Shajahan, R. Sarang, R. Ramakrishnan and A. Saithalavi, Curr. Organocatal., 2023, 10, 81–112, DOI:10.2174/2213337210666230224115814.
- E. Wojaczyńska and M. Zielińska-Błajet, Arkivoc, 2023, 5, 54–68, DOI:10.24820/ARK.5550190.P011.894.
- E. Lopez-Fontal, A. Grochmal, T. Foran, L. Milanesi and S. Tomas, Chem. Sci., 2018, 9, 1760–1768, 10.1039/C7SC04553K.
- M. Bartók, Catal. Rev.: Sci. Eng., 2015, 57, 192–255, DOI:10.1080/01614940.2015.1039432.
- G. Di Carmine, C. D'Agostino, O. Bortolini, L. Poletti, C. De Risi, D. Ragno and A. Massi, Molecules, 2024, 29, 2–28, DOI:10.3390/molecules29102166.
- M. Maciá, I. Muñoz, R. Porcar, F. G. Cirujano, B. Altava, S. V. Luis and E. García-Verdugo, Adv. Synth. Catal., 2024, 366, 892–899, DOI:10.1002/adsc.202301221.
- F. Zaera, Coord. Chem. Rev., 2021, 448, 2–15, DOI:10.1016/j.ccr.2021.214179.
- M. Sanchez-Fuente, J. L. Alonso-Gómez, L. M. Salonen, R. Mas-Ballesté and A. Moya, Catalysts, 2023, 13, 2–24, DOI:10.3390/catal13071042.
- F. Calderón, R. Fernández, F. Sánchez and A. Fernández-Mayoralas, Adv. Synth. Catal., 2005, 347, 1395–1403, DOI:10.1002/adsc.200505058.
- E. G. Doyagüez, F. Calderón, F. Sánchez and A. Fernández-Mayoralas, J. Org. Chem., 2007, 72, 9353–9356, DOI:10.1021/jo070992s.
- X. C. Cambeiro, R. Martín-Rapún, P. O. Miranda, S. Sayalero, E. Alza, P. Llanes and M. A. Pericàs, Beilstein J. Org. Chem., 2011, 7, 1486–1493, DOI:10.3762/bjoc.7.172.
- J. Guan, B. Liu, X. Yang, J. Hu, C. Wang and Q. Kan, ACS Sustain. Chem. Eng., 2014, 2, 925–933, DOI:10.1021/sc4005247.
- A. Khalafi-Nezhad, E. S. Shahidzadeh, S. Sarikhani and F. Panahi, J. Mol. Catal. A: Chem., 2013, 379, 1–8, DOI:10.1016/j.molcata.2013.07.009.
- C. Zhi, J. Wang, B. Luo, X. Li, X. Cao, Y. Pan and H. Gu, RSC Adv., 2014, 4, 15036–15039, 10.1039/c4ra01231c.
- X. W. Dong, Y. Yang, J. X. Che, J. Zuo, X. H. Li, L. Gao, Y. Z. Hu and X. Y. Liu, Green Chem., 2018, 20, 4085–4093, 10.1039/c8gc01323c.
- A. R. Hajipour and Z. Khorsandi, ChemistrySelect, 2017, 2, 8976–8982, DOI:10.1002/slct.201700847.
- M. Gruttadauria, S. Riela, P. Lo Meo, F. D’Anna and R. Noto, Tetrahedron Lett., 2004, 45, 6113–6116, DOI:10.1016/j.tetlet.2004.06.066.
- M. Gruttadauria, S. Riela, C. Aprile, P. Lo Meo, F. D’Anna and R. Noto, Adv. Synth. Catal., 2006, 348, 82–92, DOI:10.1002/adsc.200505227.
- R. Tan, C. Li, J. Luo, Y. Kong, W. Zheng and D. Yin, J. Catal., 2013, 298, 138–147, DOI:10.1016/j.jcat.2012.11.024.
- S. Nazari and M. Keshavarz, Russ. J. Gen. Chem., 2017, 87, 539–545, DOI:10.1016/j.fuel.2015.08.055.
- R. Vaid and M. Gupta, Monatsh. Chem., 2015, 146, 645–652, DOI:10.1007/s00706-014-1331-5.
- X. Li, S. Wang, K. Wang, X. Jia and Z. Hu, RSC Adv., 2018, 8, 42292–42299, 10.1039/c8ra08712a.
- M. Keshavarz, A. Z. Ahmady, L. Vaccaro and M. Kardani, Molecules, 2018, 23, 2–16, DOI:10.3390/molecules23020330.
- M. D. Prabhakara and B. Maiti, Res. Chem. Intermed., 2020, 46, 2381–2401, DOI:10.1007/s11164-020-04096-w.
- J. Xu, J. Liang, S. Huang, G. Yang, K. Tian, R. Chen, H. Chen and Y. Zhang, Catalysts, 2020, 10, 1–10, DOI:10.3390/catal10111246.
- M. Mutiah, S. Rochat, I. Amura, A. D. Burrows and E. A. C. Emanuelsson, Chem. Eng. Process. Process Intensif., 2021, 2–6, DOI:10.1016/j.cep.2021.108315.
- N. Xu, K. Su, E. S. M. El-Sayed, Z. Ju and D. Yuan, Chem. Sci., 2022, 13, 3582–3588, 10.1039/d2sc00395c.
- Z. An, W. Zhang, H. Shi and J. He, J. Catal., 2006, 241, 319–327, DOI:10.1016/j.jcat.2006.04.035.
- K. Arya, U. C. Rajesh and D. S. Rawat, Green Chem., 2012, 14, 3344–3351, 10.1039/c2gc35822k.
- L. Ju, G. Li and H. Luo, Catalysts, 2022, 2–12, DOI:10.3390/catal12010042.
| This journal is © The Royal Society of Chemistry 2025 |