
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Recent developments in alkene oxo-functionalization reactions governed by photoredox methods
Pau Sarró
,
Yingmin Ji†
,
Albert Gallego-Gamo†
 ,
Carolina Gimbert-Suriñach
,
Adelina Vallribera
* and
Albert Granados
*
,
Carolina Gimbert-Suriñach
,
Adelina Vallribera
* and
Albert Granados
*
Department of Chemistry and Centro de Innovación en Química Avanzada (ORFEO-CINQA), Universitat Autònoma de Barcelona, Cerdanyola del Vallès, 08193 Barcelona, Spain. E-mail: adelina.vallribera@uab.es; albert.granados@uab.es
First published on 13th March 2025
Abstract
Alkene oxo-functionalization reactions have emerged as a powerful tool in modern synthetic organic chemistry, enabling the sequential formation of carbonyl (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) and various C–R bonds (R = alkyl, aryl, oxygen-, sulfur-, nitrogen- or phosphorus-compounds). Recent advances in photoredox catalysis have propelled the development of these transformations by enabling the use of mild, selective, and sustainable reaction conditions. This review provides an analysis of recent methods for alkene oxo-functionalization under photoredox conditions, with a focus on the synthesis of multifunctionalized products through single and tandem processes. Mechanistic insights into radical generation, photoexcitation pathways and oxygen sources are discussed to highlight the unique role of photoredox catalysis in these transformations. Additionally, we examine the substrate scope, functional group tolerance, and potential applications of these reactions in complex molecule synthesis and drug development. Finally, current challenges and future perspectives in the field are discussed, emphasizing how innovative photoredox approaches continue to expand new opportunities for the selective construction of CO and C–R bonds in organic synthesis.
O) and various C–R bonds (R = alkyl, aryl, oxygen-, sulfur-, nitrogen- or phosphorus-compounds). Recent advances in photoredox catalysis have propelled the development of these transformations by enabling the use of mild, selective, and sustainable reaction conditions. This review provides an analysis of recent methods for alkene oxo-functionalization under photoredox conditions, with a focus on the synthesis of multifunctionalized products through single and tandem processes. Mechanistic insights into radical generation, photoexcitation pathways and oxygen sources are discussed to highlight the unique role of photoredox catalysis in these transformations. Additionally, we examine the substrate scope, functional group tolerance, and potential applications of these reactions in complex molecule synthesis and drug development. Finally, current challenges and future perspectives in the field are discussed, emphasizing how innovative photoredox approaches continue to expand new opportunities for the selective construction of CO and C–R bonds in organic synthesis.
 Pau Sarró | Dr Pau Sarró received his BSc in Chemistry at UAB. In 2017, he defended his Master's thesis under the supervision of Prof. A. Vallribera. Afterwards, he joined Suero's group at ICIQ, where he worked on the development of a Rh-catalyzed carbyne transfer platform for skeletal modifications. He defended his PhD in 2022, and one year later, he joined the CatSyNanoMat group as a postdoctoral fellow. |
 Yingmin Ji | Yingmin Ji received her BSc in Chemistry at UAB in 2023. One year later, she defended her Master's thesis at UAB in the CatSyNanoMat group. Afterwards, she enrolled in the PhD program at UAB under the supervision of Prof. A. Vallribera and Dr A. Granados, where she is studying novel photoinduced transformations. |
 Albert Gallego-Gamo | Albert Gallego-Gamo received his BSc in Chemistry in 2020 at UAB. In 2021, he defended his Master's thesis in the CatSyNanoMat group at UAB. Currently, he is a PhD student under the supervision of Dr C. Gimbert-Suriñach and Prof. A. Vallribera, working on the development of novel organic photoactive materials and photoinduced transformations. In 2024, he spent three months working in the Procter lab as a visiting student. |
 Carolina Gimbert-Suriñach | Dr Carolina Gimbert-Suriñach obtained her PhD at UAB under the supervision of Prof. A. Vallribera. After one year as an assistant professor at the same university, she moved to UNSW to undertake postdoctoral research with Prof. S. B. Colbran. She then worked at ICIQ in Prof. A. Llobet's group as a postdoctoral fellow and research group coordinator. After a short stay at UB as a Serra Húnter professor, she started as a Ramón y Cajal fellow and co-leader of the CatSyNanoMat group at UAB in 2021. She was promoted to Associate Professor of Organic Chemistry in 2023. |
 Adelina Vallribera | Prof. Adelina Vallribera received her PhD in Chemistry in 1993 from UAB under the supervision of Prof. J. Marquet. After a postdoctoral stay at the Laboratoire de Chimie Organique de Synthèse in Belgium in the group of Prof. L. Ghosez, she joined the group of Prof. M. Moreno-Mañas at UAB. She became an assistant professor in 1997 in the Organic Chemistry Unit at the Department of Chemistry at UAB and was promoted to Full Professor in 2017. |
 Albert Granados | Dr Albert Granados received his MSc in Electrochemistry, Science, and Technology in 2014. He then joined the Vallribera group at UAB, where he obtained his PhD with a special award in 2018. Subsequently, he pursued postdoctoral studies with Prof. R. Pleixats and Prof. A. Vallribera at UAB. In 2021, he began another postdoctoral stint in the Molander group at the University of Pennsylvania (UPenn). In early 2023, he started as a Lecturer in Organic Chemistry in the CatSyNanoMat group at UAB and was promoted to Associate Professor in mid-2024. He was appointed as a Member of the Science of Synthesis Early Career Advisory Board for the 2025–2027 period. |
1. Introduction
The carbonyl group is one of the most fundamental and versatile units in organic chemistry because it is present in many functional groups, such as amides, carboxylic acids, carbamates and ketones. Its significance extends from basic concepts taught in undergraduate courses to advanced applications in fields such as materials science, biochemistry, and drug development. Among carbonyl-containing functional groups, ketones play a central role in both industrial and medicinal chemistry. For example, acetone is the simplest ketone and it is obtained along with phenol via the Hock process,1 which is one of the most significant methods in the chemical industry. On the other hand, the ability of ketones to participate in hydrogen bonding and dipole–dipole interactions, along with their polarity,2 makes them essential for modulating the molecular properties of drug candidates.3Typical synthetic routes for preparing ketones include the oxidation of secondary alcohols,4 Friedel–Crafts acylation reactions,5 hydration of alkynes,6 ozonolysis,7 and Wacker-type oxidation of alkenes,8 among others. The continuous development of new methods is crucial for advancing both industrial applications and pharmaceutical research.
The implementation of photoredox catalysis in organic synthesis has undoubtedly contributed to the construction of new chemical bonds, ushering in a new golden era of radical chemistry.9 A key factor driving the rapid growth of this technology is the ability of organic dyes and polypyridyl complexes to facilitate the conversion of visible light into chemical energy under mild reaction conditions, typically using light-emitting diodes (LEDs). Particularly, photocatalytic methods have significantly advanced the field of alkene 1,2-difunctionalization reactions by offering a sustainable and versatile platform for introducing two functional groups across a double bond in a single step.10 These methods, driven by visible light and facilitated by organophotoredox catalysts or transition-metal complexes, have enabled a broad range of transformations, including oxy-amination,11 diamination,12 carbo-amination,13 and dicarbofunctionalization,14 among others. These reactions typically proceed by energy transfer15 or photoredox mechanisms,16 involving radical or radical–polar crossover pathways, all initiated by the excited state of the photocatalyst. This generates reactive intermediates that enable the precise and regioselective incorporation of functional groups under mild reaction conditions. It is worthwhile to recognize innovative and photoactive electron-donor–acceptor (EDA) complexes applied to alkene 1,2-difunctionalization.17
Despite significant advancements in photoredox-mediated alkene 1,2-difunctionalization reactions, recent reviews18 do not cover alkene oxo-functionalization reactions. Remarkably, the preparation of such functionalized ketones governed by photoredox methods has the potential for building high molecular complexity from simple and readily available starting materials, which commonly are the corresponding redox-active species and the olefinic organic skeleton. Importantly, these photoredox approaches are governed by single-electron transfer (SET) processes. Therefore, this review offers a comprehensive guide to designing alkene oxo-functionalization strategies, emphasizing the sequential formation of CO and C–R (R = C, N, P, O or S) bonds through photoredox chemistry (Scheme 1A). Under a suitable set of reaction conditions, a key Csp2-hybridized radical species (Scheme 1A) can be generated and employed to access oxo-functionalized organic molecules. Most studies in this arena are designed around reductive or oxidative quenching photoredox cycles9 to access the Csp2-hybridized radical (Scheme 1B), which is ultimately oxidized to the corresponding carbonyl compound. Typically, carbonyl formation under these photoredox conditions is achieved using non-toxic oxidants such as dimethyl sulfoxide (DMSO) or molecular oxygen (O2) (Scheme 2). Oxidation with DMSO proceeds via a photoredox radical/polar crossover mechanism. Here, the key Csp2-hybridized radical species is first oxidized by the photoredox cycle to the corresponding carbocation, which then reacts with DMSO, followed by a deprotonation process, analogous to the Kornblum oxidation (Scheme 2A). In contrast, oxidations utilizing molecular oxygen are proposed to proceed through distinct mechanistic pathways, as illustrated in Scheme 2B. The C-centered radical can react with O2 (path a) to form the alkyl peroxyl radical intermediate A, which may then be reduced to the anion (path b). Following protonation and dehydration, the desired carbonyl compound is obtained. Alternatively, the anionic species B may also be produced directly from the initial radical through reaction with superoxide (path c). Additionally, the alkyl peroxyl radical A can undergo additional transformations, including trapping with the hydroperoxyl radical (path e) to produce an intermediate that undergoes Russell fragmentation. It can also participate in radical–radical coupling with the initial Csp2 radical to yield the alkoxy radical C (path d) via O–O bond cleavage from peroxide A1, which then undergoes intramolecular 1,2-hydrogen atom transfer (1,2-HAT), ultimately leading to the oxo-functionalized product after SET and deprotonation.
 | ||
| Scheme 1 (A) Reaction design to access oxo-functionalized organic compounds via photoredox catalysis covered in this review. (B) Main reactivity modes for the generation of the key Csp2-hybridized radical intermediate. RP = radical precursor. EA = electron-acceptor. | ||
 | ||
| Scheme 2 Access to oxo-functionalized compounds via (A) a Kornblum-type oxidation process or (B) a molecular oxygen mechanism from the key Csp2-hybridized radical. | ||
This review highlights notable studies that achieved sequential formation of CO and Csp3–R bonds using simple photocatalysts alone. Accordingly, we exclude reactions that proceed via photocatalyst-free systems, photoredox/transition metal dual catalysis, or pathways lacking SET events.
2. Oxo-alkylation reactions
Alkene oxo-alkylation reactions via photoredox catalysis represent a powerful synthetic strategy for C–C bond formation. The section is organized according to the type of carbon-based bond formation: C–CF3, C–CF2R, and C–C(alkyl).2.1 C–CF3 bond formation
In the field of trifluoromethyl chemistry, various radical precursors have emerged over the past decade. In 2014, Akita19 introduced the Togni reagent as an effective precursor for the ˙CF3 species, facilitating the synthesis of α-trifluoromethylated ketones from alkenes and DMSO via an Ir-promoted, oxidative quenching process (Scheme 3, conditions A). This approach generally achieved high yields with styrenes containing electron-donating groups (1–3), while substrates with electron-withdrawing groups showed reduced yields due to the formation of β-trifluoromethylstyrene as a side product. Notably, sensitive boronic esters are well tolerated (5). Endocyclic styrenes provided the desired ketones in good yield (4).
Recently, Xiao20 presented a new trifluoromethylation reagent capable of single-electron reduction to produce ˙CF3. This method tolerates a wide range of functional groups (Scheme 3, conditions B), including nitriles (10), esters (9 and 13), halogens, and silyl groups (12), and has shown promise for late-stage functionalization (15 and 16). Both methods employ Ir(ppy)3 as a photocatalyst to initiate the ˙CF3 radical via SET. Subsequently, this open-shell species undergoes Giese addition with alkenes, forming a key benzylic radical intermediate that is further oxidized to a benzylic carbocation. Finally, in both cases, DMSO served as the oxidant, converting the intermediate to the desired α-trifluoromethylated ketones via Kornblum type oxidation (see Scheme 2A) assisted by the base o-iodobenzoate in Akita's method and a pyridine derivative in Xiao's work.
In 2018, Itoh21 reported a reductive quenching approach to synthesize α-trifluoromethylated ketones using the Langlois reagent in the presence of molecular oxygen (Scheme 4) using a compact fluorescent lamp (CFL). Here, the organic dye 2-benzylamideanthraquinone served as the most convenient photocatalyst, with Ba(OH)2 as base. This reaction proceeds well with both electron-withdrawing and electron-donating group-substituted styrenes (17–20), although strongly electron-withdrawing groups, such as methyl ester or nitrile (20), yield lower amounts. Steric effects were observed among ortho-, meta-, and para-methyl-substituted styrenes (17, 21 and 22). Notably, the reaction tolerates sensitive protecting groups (24–25) including acetals and tert-butyldimethylsilyl ethers (OTBS). The mechanistic investigation of this transformation was supported by radical-trapping experiments and 18O labelling. The authors proposed that after generating the ˙CF3 radical via SET from the excited photocatalyst, the radical undergoes Giese addition with styrenes to yield a benzylic radical intermediate. Finally, this intermediate reacts with molecular oxygen, which is reduced to a peroxyl anion, and the desired product is formed by protonation, followed by decomposition (path a and b, Scheme 2B).
 | ||
| Scheme 4 Photoredox synthesis of α-trifluoromethylketones reported by Itoh.21 | ||
In 2021, Zhu and collaborators22 developed a mild protocol to access CF3-substituted tetrahydrofurans and tetrahydropyrroles from 1,6-dienes and the Togni reagent (Scheme 5). This reaction proceeds under photoredox oxidative quenching conditions using Ir(ppy)3 and DMSO. The scope includes both electron-rich and electron-poor arenes, with yields ranging from 65% to 83% (27–33) and excellent diastereoselectivity. Additionally, the method is compatible with N-protected 1,6-dienes (35) and thiophene-substituted (36) aromatic rings, although it is unsuitable for alkyl 1,6-dienes. The protocol can be scaled up to the gram-scale. Mechanistic studies, including TEMPO radical-trapping and 18O labelling experiments with DMS18O, suggest that the CF3 radical generated via SET undergoes radical addition to the terminal alkene of the 1,6-diene to yield a C-centered radical intermediate, which undergoes an intramolecular cyclization process yielding a benzylic radical species. Subsequently this intermediate is oxidized to the carbonyl as depicted in Schemes 1B and 2A.
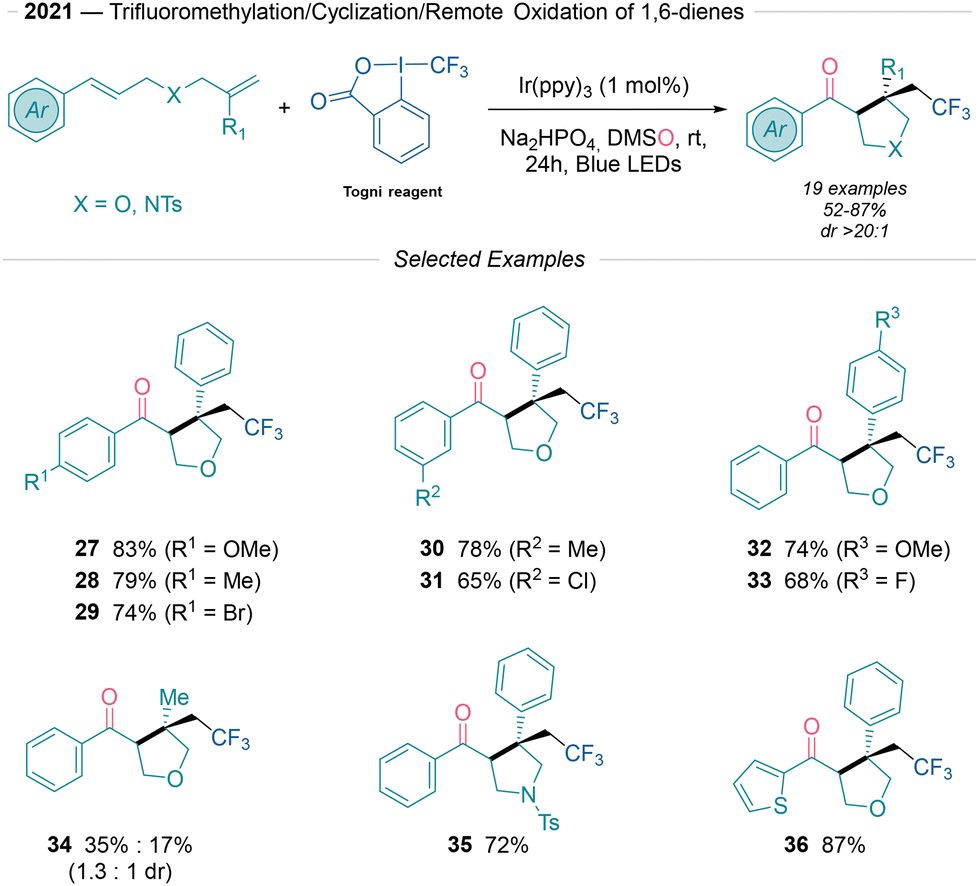 | ||
| Scheme 5 Photoredox trifluoromethylation/cyclization/oxidation of 1,6-dienes.22 | ||
2.2 C–CF2R bond formation
In 2019, Akita and Koike23 reported the synthesis of α-CF2H-substituted ketones through an Ir(ppy)3 photoredox approach using aromatic alkenes and the novel N-tosyl-S-difluoromethyl-S-phenylsulfoximine as the CF2H radical source, with DMSO serving as both a solvent and mild oxidant (Scheme 6). The difluoromethyl group is a fruitful structural motif that acts as a bioisostere for alcohol and thiol functional groups.24 This reaction was carried out on a preparative scale in a photocatalytic flow reaction (MiChS flow reactor) at room temperature and with only 30 minutes of illumination. The substrate scope includes styrenes with para electron-rich substituents with yields ranging between 49 and 69% (for example 67% for 37) and halogens yielding 43–56% yields. Internal alkenes such as trans-stilbene, isosafrole, indene (45) and 2-methylindene required a longer residence time despite giving the CF2H-ketone products in moderate yields (31–64% yields). Electron deficient styrenes and aliphatic alkenes did not react under these conditions. A plausible reaction mechanism is proposed based on radical trapping experiments with TEMPO, intermittent light irradiation experiments and determination of the photochemical quantum yield. The reaction is governed by an oxidative quenching photoredox cycle followed by the oxidation shown in Scheme 2A. Interestingly, when the base is omitted a single-electron reduction of the sulfonium intermediate (Scheme 2A) followed by O–S bond homolysis is also proposed. The resulting alkoxy radical (C in Scheme 2B) undergoes 1,2-HAT and single-electron oxidation to give the CF2H-ketone.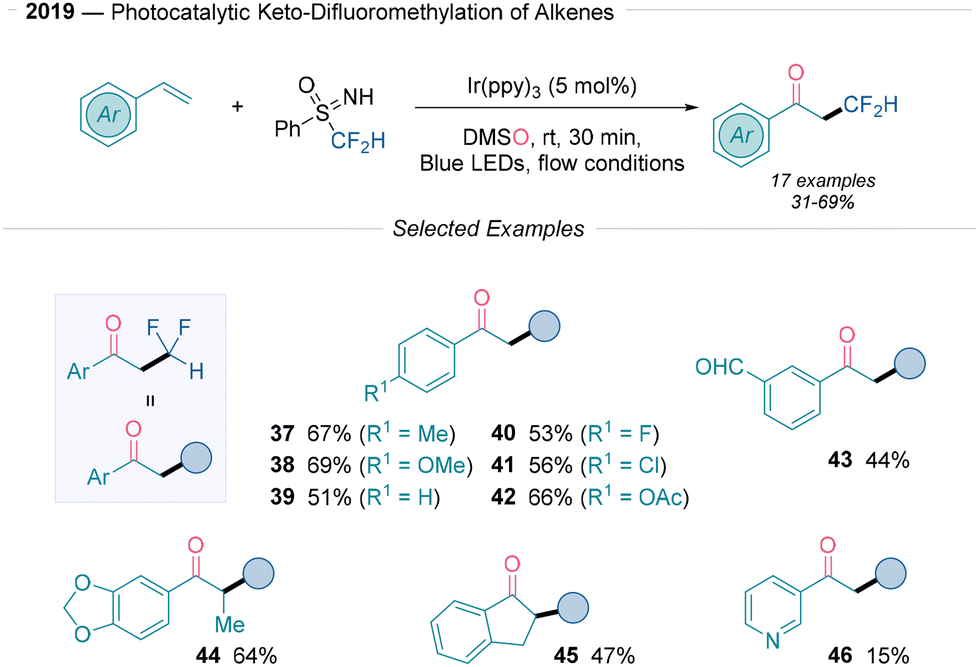 | ||
| Scheme 6 Direct keto-difluoromethylation of alkenes under photoredox conditions reported by Akita and Koike.23 | ||
Subsequently, four more contributions were published in 2019 on visible-light-promoted oxo-difluoroalkylation of alkenes using DMSO. The notable α-difluoroalkylated (α-CF2R) ketones represent valuable building blocks with bioactive properties. The first example details a visible-light-induced oxo-difluoroacetylation to access α,α-difluoroketones from styrenes and bromodifluoroacetates using Ir(ppy)3 under blue LEDs and in the presence of stoichiometric AgTFA (Scheme 7, conditions A).25 This difunctionalization showed moderate to good yields and excellent regioselectivity. Styrenes with para electron-donating groups (47–48) and weakly electron-withdrawing groups (49–50) produced the desired compounds in yields that ranged from 63 to 74%. Internal alkenes also demonstrated good reactivity (53–54), with both meta and ortho substituents being well tolerated. Additionally, a bromodifluoroacetamide example afforded the corresponding ketone in decent yield (55). A scale-up reaction demonstrated the synthetic utility of this approach. The proposed mechanism is supported by radical trapping and labelled DMS18O experiments. It involves the photoexcitation of Ir(ppy)3, which generates a strongly reducing excited state that triggers the generation of the ˙CF2COOEt radical assisted by AgI, likely through AgBr formation. Continuing radical addition to the alkene, the key Csp2-hybridized radical is produced and furnishes the final product via Kornblum oxidation (Scheme 2A). Ye26 described an alternative set of reaction conditions for oxo-difluoroacetylation using 2 equivalents of KH2PO4 as weak base to optimize yields by preventing HF elimination (Scheme 7, conditions B). Styrenes with para electron-donating groups (56 and 57) performed well (66–76% yields), while weakly electron-withdrawing groups (58 and 59) yielded around 46% yield. Moreover, meta (60) and ortho (61) substituents also yielded products within 43–58% yields and moderate yields were obtained for internal alkenes (62 and 63). Bromodifluoroacetamides produced aniline- (64) and indole-derived ketones in moderate yields, although aliphatic alkenes failed under standard conditions. Scale-up of the reaction using 5 mmol of styrene further demonstrated this method's synthetic utility. Mechanistic studies suggest that the reaction proceeds via excitation of Ir(ppy)3, which reduces the BrCF2CO2Et radical precursor and generates a carbon-centered radical. Then, the mechanism follows the pathway depicted in Scheme 2A, were DMSO attacks the generated carbocation, and the final product is yielded in the presence of KH2PO4. Finally, Xi27 extended the scope of the radical precursor to FSO2CF2CO2Me (Scheme 7 conditions C) and obtained the desired compounds (65–68) without additional additives, confirming the reducing ability of the photocatalyst as observed by Akita.23
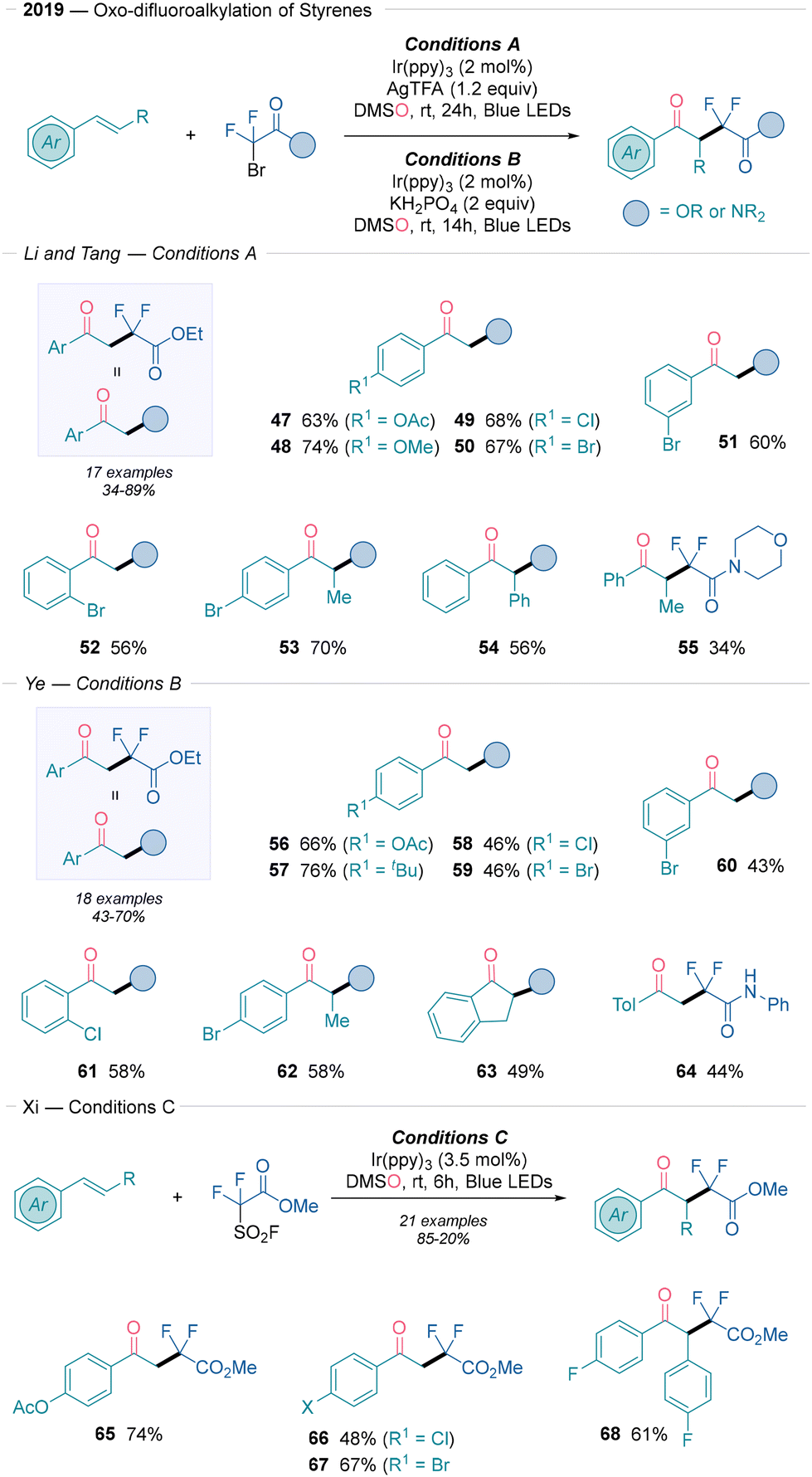 | ||
| Scheme 7 Synthetic methods described by Li, Tang,25 Ye26 and Xi27 to access difluoroalkylated ketones. | ||
Similarly, Li and Ma28 reported the preparation of difunctionalized 1,6-fluoroalkylketones through visible-light-induced photoredox-catalyzed remote oxyfluoroalkylation of alkenes (Scheme 8). This process initiates remote benzyl C–H bond oxidation via a 1,5-HAT. Optimization revealed that organic or inorganic bases were ineffective, whereas the addition of AgOBz significantly improved reactivity. The reaction proceeds via Ag(I)-assisted oxidation of IrIII to IrIV, yielding the ˙CF2COOEt species. Radical addition to the double bond produces an alkyl radical, which undergoes 1,5-HAT to form a stable benzyl radical, ultimately resulting in the oxofunctionalized product. Mechanistic studies involving DMS18O and TEMPO or 1,1-diphenylethene as radical scavengers support this mechanism. Substrates such as 2-ethyl allyl benzene, 2-benzyl allyl benzenes, and 1-aryl-5-hexenes (69–72) gave high yields under these conditions, with the protocol being extendable to bromodifluoroacetamides (73). Perfluorohexyl (74) and perfluorooctyl bromides also proved to be suitable reagents. Notably, the reaction was scalable to 5 mmol of 1-allyl-2-ethylbenzene, demonstrating its practicality.
 | ||
| Scheme 8 Photoredox conditions reported by Li and Ma to synthesize fluorinated ketones via 1,5-HAT.28 | ||
2.3 C–Calkyl bond formation
In 2018, Glorius29 and Ye30 independently reported a similar strategy to access acetophenone derivatives using redox-active esters as radical precursors (Scheme 9). Both methods require the use of the Ir(ppy)3 photocatalyst, although in different catalytic loadings (0.2 mol% and 1 mol%, respectively). In both studies, the substrate scope was remarkably amenable to the incorporation of primary (75 and 82), secondary (76 or 84), and tertiary (79 or 85) radicals without significant changes in yield. Moreover, the functional group tolerance was broad, including ethers, protected amines, and alkenes. Glorius also demonstrated the applicability of the method to naturally occurring and bioactive molecules, such as deoxycholic acid and gemfibrozil. These protocols showed better reactivity with p-substituted styrenes, while steric effects reduced the reactivity by around 15%. The mechanisms of these transformations are based on an oxidative quenching photoredox radical/polar crossover process, where the generated benzylic carbocation is oxidized by DMSO (Scheme 2A).
In the same year, Chen31 reported a keto-alkylation reaction using cycloketone-based oximesters and styrenes under Ir-photoredox conditions (Scheme 10). The resulting products expand the chemical space within ketonitrile chemistry. The reaction mechanism involves an iminyl radical-triggered C–C bond cleavage from an oximester reagent, followed by a radical Giese addition to the alkene and subsequent Kornblum oxidation (Scheme 2A). These conditions showed functional group tolerance for ethers (88) and N-Boc amines (89 and 92).
 | ||
| Scheme 10 Use of oximesters as radical precursors for alkene oxo-alkylation reactions reported by Chen.31 | ||
Additionally, it was applied to a broad range of cyclobutanone-derived O-acyl oximes (93–98). Moreover, the reaction enabled efficient product formation with various styrenyl-based radical acceptors, including biologically relevant derivatives (98).
The synthesis of γ-keto esters from styrenes under photoredox conditions has been independently reported by Liu32 and Maity33 (Scheme 11). Liu first introduced an Ir-based oxidative quenching photoredox method using α-bromoesters as convenient radical precursors and DMSO as the oxidant (Scheme 11, conditions A). Notably, other electron-withdrawing groups, such as NO2 (100), CF3 (101), and CN (102) derivatives, could also be incorporated using the same reaction conditions. Primary, secondary, and tertiary radicals (99, 103 and 104) were successfully introduced into the styrenyl radical acceptor. This protocol was also applicable to various styrenes (107 and 108), including β-methylstyrene (109). Radical trapping experiments and the use of tetramethylene sulfoxide confirmed that the reaction proceeds via a photoredox cycle, where the cationic intermediate is oxidized by DMSO (Scheme 2A). Later, in 2020, Maity described an organophotoredox approach under aerobic conditions using α-bromocarbonyls (Scheme 11, conditions B). The combination of eosin Y as a photocatalyst and DIPEA as a base in DMF was crucial for the reaction's success. The substrate scope included α-bromoesters (110–113), α-bromoketoesters, α-bromomalonates (114), α-bromoketones, and α-bromoamides. Various styrene derivatives, including endocyclic alkenes and β-substituted styrenes, were well tolerated. The reaction mechanism was investigated through radical trapping studies, Stern–Volmer fluorescence quenching, and radical-clock experiments. Photoexcited eosin Y reduces the corresponding α-bromocarbonyl to a carbon-centered radical via SET. Oxidized eosin Y is then regenerated by DIPEA that acts as a sacrificial electron-donor. The generated radical undergoes Giese addition to the double bond, forming a benzylic radical intermediate. This intermediate reacts with O2 to form the peroxyl radical, which follows path d in Scheme 2B. The authors proposed that the generated α-hydroxy-C-centered radical is oxidized to the final product by either DIPEA˙+ (chain termination) or by another substrate molecule (chain propagation). The authors also noted the possibility of a reductive quenching photoredox cycle, since Stern–Volmer experiments indicate that DIPEA can quench the excited state of eosin Y.
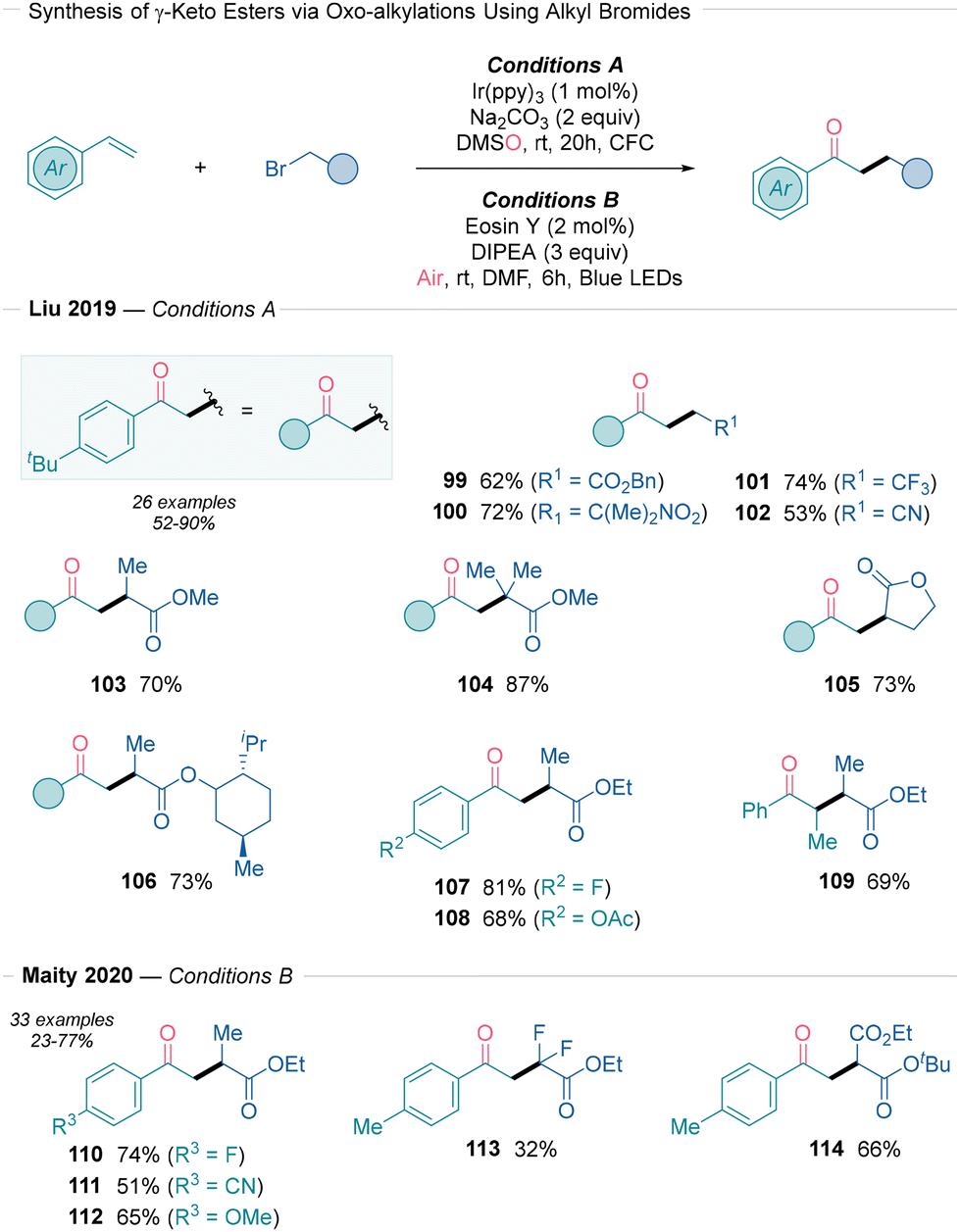 | ||
| Scheme 11 Photoredox approaches32,33 for accessing γ-keto esters using alkyl bromides and alkenes. | ||
The trifluoroethanol moiety is significant in medicinal chemistry.34 Recently, access to 2-hydroxy-1,1,1-trifluoroethylacetophenones has been reported via the oxo-alkylation of alkenes under oxidative quenching photoredox conditions (Scheme 12).35 Interestingly, this chemical space is explored using N-trifluoroethoxyphthalimide as a radical precursor. This redox-active ether can be reduced to the trifluoroethoxyl radical using the cost-effective and readily available 1,3-dicyano-2,4,5,6-tetrakis(diphenylamino)benzene (4DPAIPN) photocatalyst in the presence of DMSO. Notably, the reaction is completed in 90 minutes, with a broad substrate scope, including heteroaromatics (122 and 123) and complex molecules (124), making it ideal for late-stage functionalization.
 | ||
| Scheme 12 Visible light-mediated synthesis of 2-hydroxy-1,1,1-trifluoroethylacetophenones.35 | ||
Importantly, the reaction can be scaled up to 2.5 mmol without loss of efficiency, and access to CF2H analogs is also feasible under the same conditions. The authors noted that the solvent not only facilitates the formation of carbonyl through Kornblum oxidation but also is crucial for generating the key C-centered radical via intramolecular 1,2-HAT from the trifluoroethoxyl radical. This radical then undergoes Giese addition to the olefin, followed by a SET event, leading to Kornblum oxidation (Scheme 2A). This plausible mechanism has been proposed after extensive mechanistic investigation, including Stern–Volmer luminescence quenching studies, radical trapping experiments and cyclic voltammetry investigations.
The preparation of β-trifluoromethyl β-amino ketones is feasible under organophotoredox conditions, starting from styrenes and a fluorinated redox-active hydroxylamine (Scheme 13).36 This reaction is catalyzed by 4DPAIPN as the photocatalyst, which upon photoexcitation, can reduce the radical precursor to the corresponding p-CF3-benzoate anion and the nitrogen-centered radical species. Subsequently, a carbon-centered radical species is formed via intramolecular 1,2-HAT, which then undergoes a Giese addition to the alkene. The resulting Csp2-hybridized species is oxidized to the corresponding carbonyl, as depicted in Scheme 2A. The mechanism is supported by Stern–Volmer luminescence quenching studies, cyclic voltammetry and isotope labelling experiments. This three-component reaction proceeds efficiently (125–133), with yields ranging from 41% to 94%, demonstrating its utility in synthesizing these valuable compounds. Notably, the reaction's scalability retains its efficiency, and the method is not restricted to the preparation of CF3 derivatives, but other fluorinated groups such as pentafluoroethyl (CF2CF3, 134), difluoromethyl (CF2H, 135), and monofluoromethyl (CH2F, 136) derivatives are also accessible.
 | ||
| Scheme 13 Photoredox synthesis of β-trifluoromethyl β-amino ketones.36 | ||
The Katayev lab37 recently developed a carbo-heterofunctionalization method via Ir-photoredox radical/polar crossover, enabling the synthesis of 2-nitroketones and other nitro derivatives (Scheme 14). In this protocol, geminal bromonitroalkanes serve as precursors to generate key carbon-centered radical intermediates, which undergo Giese addition with olefins. The presence of DMSO facilitates the formation of the final ketone via Kornblum-type oxidation (Scheme 2A). The reaction demonstrates broad substrate compatibility, performing well with various styrene derivatives, including ortho- (138), meta- (139), and para-substituted (140) examples; however, it does not extend to heterocycles. Furthermore, both secondary and tertiary β-nitro ketones (140) can be accessed by modifying the geminal reagent under identical reaction conditions. The proposed radical/polar crossover mechanism is supported by extensive mechanistic investigations, including radical-clock and control experiments and radical trapping studies.
 | ||
| Scheme 14 Photoredox keto-alkylation developed by Katayev.37 | ||
3. C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/h3_char_e001.gif) O and C–Csp2 bond formation
O and C–Csp2 bond formation
Cai38 presented a mild protocol for the preparation of 1,2-diarylethan-1-ones through oxidative arylation of styrenes using aryldiazonium salts under photoreductive conditions (Scheme 15). This transformation mandates the use of [Ru(bpy)3]Cl2·6H2O as a photocatalyst under an oxygen atmosphere, resulting in good yields. The difunctionalization reaction is effective, although limited by the electronic nature of the aryldiazonium salt used. Electron-poor aryldiazonium salts efficiently produced the desired 1,2-diarylethan-1-ones in yields ranging from 51% to 88%, tolerating nitro (141), nitrile (142), and ester (144) groups, as well as halides (145). In contrast, electron-donating substituents did not work under the established reaction conditions (146). Notably, a heteroaryl system also produced the corresponding ketone successfully (151). Steric effects had little impact on yields (141, 148), and a range of additional organic backbones, including variously substituted styrenes, were evaluated, with tolerance for both electron-donating and electron-withdrawing groups on the phenyl ring. The mechanistic operation of this transformation was studied through radical trapping experiments using TEMPO, 2,3-dimethyl-2-butene, and 1,4-diazabicyclo[2.2.2]octane (DABCO). The authors propose that after the generation of the aryl radical via SET, the radical undergoes Giese addition to the styrene to yield a benzylic radical intermediate. This intermediate reacts with molecular oxygen that follows path d in Scheme 2B. The produced α-hydroxy-carbon-centered radical yields the final carbonyl compound after single electron oxidation from the RuIII species and the subsequent deprotonation event.
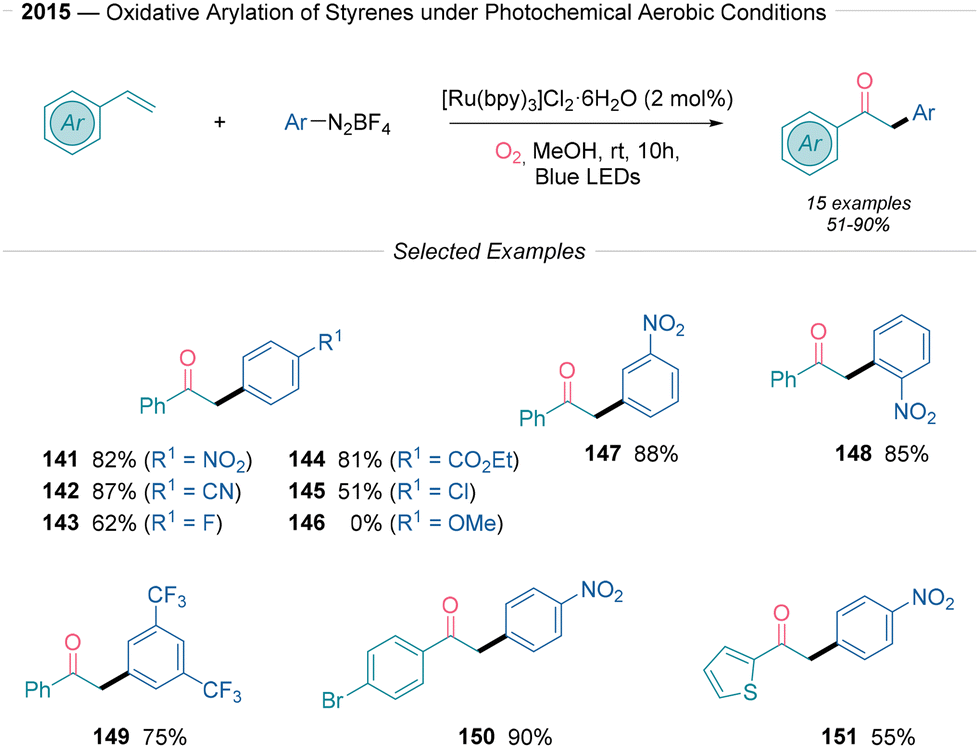 | ||
| Scheme 15 Keto-arylation of alkenes using aryldiazonium salts under photoredox conditions described by Cai.38 | ||
In 2017, Zhu39 introduced a metal-free method for synthesizing ketones via oxidative radical addition of arylhydrazines as radical precursors to styrenes (Scheme 16). This aerobic process employs blue LEDs to activate methylene blue as a photocatalyst, achieving complete transformation in 8 hours. Using DABCO as a non-nucleophilic base was crucial to optimize yields. This protocol remained highly efficient when scaled up to 10 mmol, and a broad substrate scope was explored, yielding excellent results across aryl hydrazines with diverse substituents, including halides (153 and 154), ethers (155), and strongly electron-withdrawing groups (156). ortho-Chlorine substituents (158) led to slightly lower yields due to steric hindrance. The reaction tolerated various substituents on the radical acceptor, including halides or nitriles (159 and 160) and selectively produced mono-ketones from divinylbenzene substrates in high yields (163 and 164). Additionally, the reaction was not restricted to terminal alkenes, but also β-methyl styrene proved to be efficient toward the synthesis of 165 in 76% yield. Mechanistic studies, including Stern–Volmer quenching, suggest that methylene blue's excited state is quenched by the redox-active aryl hydrazine, forming an aryl radical via oxidation and deprotonation. This sp2-hybridized radical undergoes Giese addition with the olefin to produce a benzyl radical intermediate. The final ketone formation may proceed via two possible pathways; in the first, the benzyl radical is intercepted by a hydroperoxyl radical, forming a hydroperoxyl intermediate that decomposes to yield the ketone; in the second, the benzyl radical follows path a and e shown in Scheme 2B.
 | ||
| Scheme 16 Aryl hydrazines as aryl radical precursors for the keto-arylation of alkenes by Zhu.39 | ||
4. CO and C–N bond formation reactions
In 2017, Yu40 presented the first example of oxo-functionalization involving the construction of a C(sp3)–N bond (Scheme 17). In their study, a hydroxylamine derivative was used as a N radical source, initiated via a SET process by an Ir photocatalyst, followed by homolytic cleavage of the O–N bond. After a Giese addition and a Kornblum-type oxidation, this method enabled the synthesis of various α-amino ketones in moderate to excellent yields. Notably, both para- and ortho-substituted arenes demonstrated high efficiency, although yields were slightly lower for meta-substituted arenes. Additionally, the scope was extended to complex scaffolds, including thiophenes (169), naphthalenes, indenes (172), and estrone (173–174) derivatives. Importantly, this work also explored alternative hydroxylamine precursors bearing Ph or Me groups (170–171), showing how these modifications impacted final yields. Experimental findings confirmed the involvement of radical species, with DMSO acting as the oxidant. The authors proposed the general mechanism described in Scheme 2A.
 | ||
| Scheme 17 Oxo-amination photoredox method described by Yu and collaborators.40 | ||
Shao41 recently developed an aerobic method for synthesizing aminoketones using photoredox catalysis, leveraging molecular oxygen as a green oxidant in conjunction with a Ru catalyst to achieve moderate to good yields (Scheme 18). The method allows the incorporation of medicinally relevant nitrogen containing heteroaromatics under mild conditions. Different heteroaromatics (175, 180–183) were well tolerated, although indoles, imidazoles or pyrroles are shown to be unproductive under these conditions. Mechanistic investigations demonstrated a β-scission step via a labelling experiment with α-deuterated methylstyrene, which yielded the non-deuterated product. Additionally, EPR experiments detected radical adducts of singlet O2 or O2˙− confirming the presence of key radical intermediates. Based on these findings, the authors propose two potential pathways for the generation of the nitrogen radical, converging into a unified mechanism. On the one hand, energy transfer (EnT) from the excited photocatalyst to O2 generates a singlet oxygen state, which then engages in a SET with the heteroaromatic reagent and proton loss. Alternatively, a direct SET between the reagent and the excited photocatalyst followed by proton release is also proposed. Subsequently, this N-centered radical species undergoes Giese addition to the α-methylstyrene derivative. The generated key-benzylic radical reacts with in situ formed O2˙− and H+ to form an alkyl hydroperoxide, which subsequently undergoes homolytic cleavage of the O–O bond to release ˙OH. Finally, β-scission induced by the resulting alkoxy radical produces the final ketones after losing a ˙Me radical.
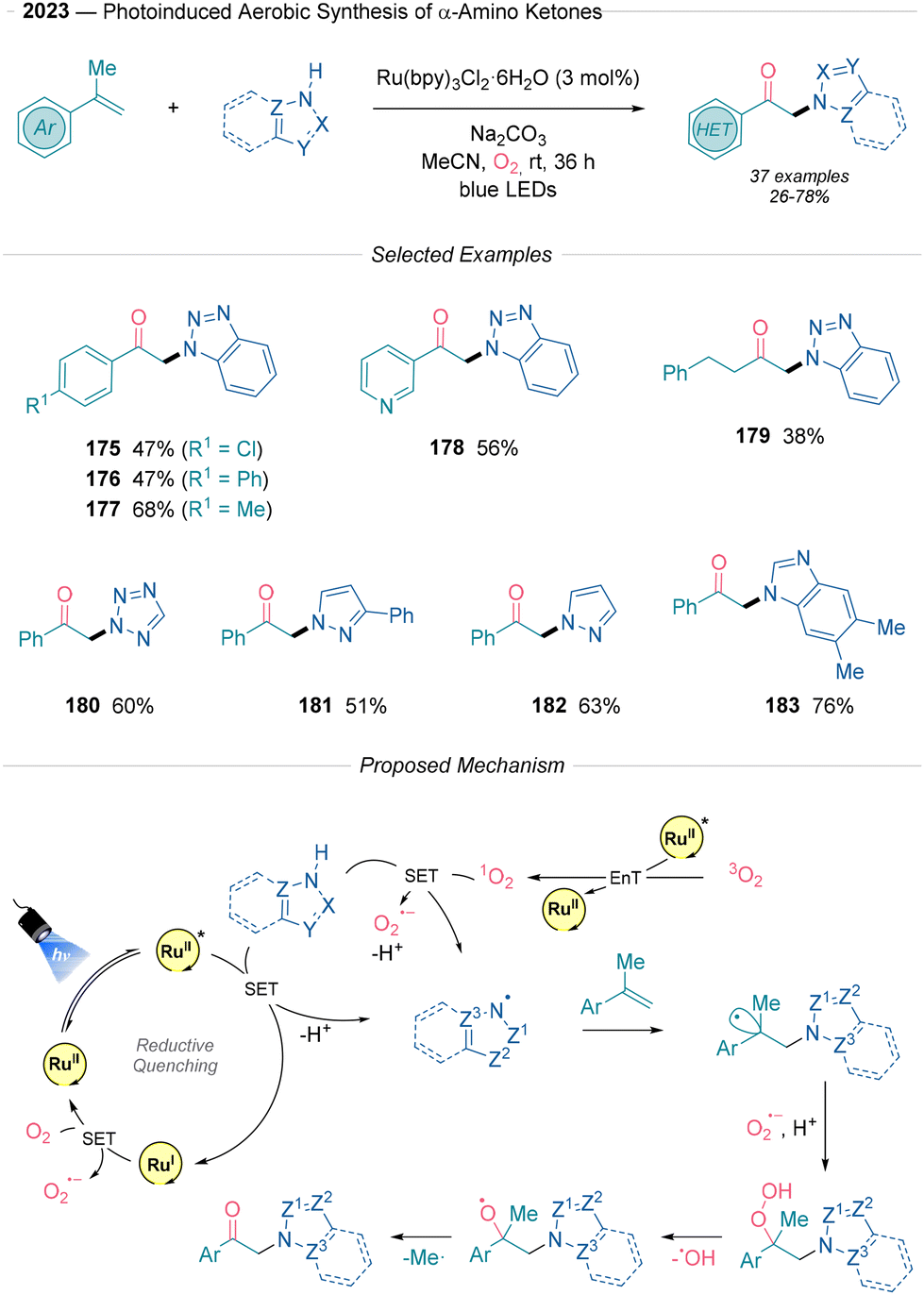 | ||
| Scheme 18 Photochemical synthesis of α-aminoketones described by Shao.41 | ||
5. CO and C–P bond formation reactions
β-Ketophosphine oxides are valuable intermediates in organic synthesis due to their versatility in constructing complex molecular structures. All the presented methods below utilize disubstituted phosphine oxides as radical progenitors. Importantly, these organophosphorous compounds exist in equilibrium with the minor tautomer disubstituted phosphinous acid, which is able to undergo SET under a suitable set of reaction conditions. Then this P-centered radical undergoes addition to the olefin (Scheme 19).
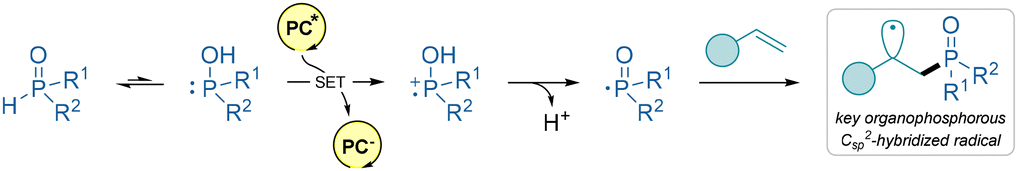 | ||
| Scheme 19 Main mechanistic pathway for the generation of the organophosphorous Csp2-hybridized radical in this section. | ||
Zhu42 introduced the first metal-free oxo-phosphorylation of alkenes, enabling aerobic oxidative incorporation of a phosphoxide unit into a variety of activated alkenes (Scheme 20). The reaction utilizes rhodamine B as the photocatalyst, diaryl phosphine oxides, DMSO as the solvent, and white light as the energy source. Notably, the authors do not confirm that O2 is solely responsible for the oxidation step since no control experiments were conducted under an inert atmosphere, thus leaving open a possible Kornblum-type oxidation process (Scheme 2A). Nonetheless, the method demonstrates impressive scope and high yields, successfully transforming various 2-, 3-, and 4-substituted styrenes (184–187), as well as internal alkenes (188–189) and thiophenes (190), which highlights its compatibility with heteroaromatic systems. Diarylphosphine oxides showed the best reactivity, although a single example using ethyl phenylphosphinate (192) was investigated and dialkyl phosphites were ineffective (193). Mechanistic investigations included only a radical scavenger experiment. The authors suggest that the minor organophosphorous tautomer undergoes SET with the excited state of the photocatalyst, generating a phosphorus-centered radical cation. After deprotonation, the P-centered radical adds to the styrene skeleton and subsequently reacts with preformed O2˙− following the general mechanism described in Scheme 2A path c.
 | ||
| Scheme 20 Photoredox alkene oxo-phosphorylation conditions presented by Zhu.42 | ||
In 2018, Zou and collaborators43 described a pioneering method for the oxidative phosphorylation of alkenyl carboxylic acids via an organophotoredox relay approach. This reaction uses cinnamic acid derivatives with diarylphosphine oxides, rose bengal as an organophotocatalyst, DMSO as the solvent, under an oxygen atmosphere and at 40 °C (Scheme 21). This transformation is effective using different styrene derivatives, including both electron-rich (194) and electron-deficient (195) systems. Some heteroaromatics also showed compatibility (197), although furans suffered from decreased yields or were entirely unreactive. The method allows the use of some diverse phosphorus oxides, including a dithiophene derivative (199), although alkyl and alkoxy substituents on phosphorus were incompatible (200). Radical scavengers suppressed the desired transformation and the use of an acrylamide derivative instead of the aromatic olefin led to the formation of a phosphorus oxide oxindole product. These experiments confirmed the involvement of the phosphoryl radical species. Furthermore, EPR spectroscopy allowed the detection of both O2˙− and ˙POPh2 species. However, given the presence of DMSO it is important to consider a Kornblum-type oxidation mechanism since there are not enough control experiments. Based on the authors’ observations, an oxidative quenching photoredox cycle operates this transformation. The PC* species is quenched by O2 to form O2˙− and the oxidized state of the photocatalyst. Subsequently, the diarylphosphinous acid tautomer undergoes SET yielding the radical phosphoxide after deprotonation and adds to the cinnamic acid derivative. The generated intermediate follows the general mechanism described in Scheme 2B, path c, where an additional decarboxylation event is promoted by SET.
 | ||
| Scheme 21 Rose bengal promoted synthesis of β-oxophosphine oxides described by Zou.43 | ||
Kim presented two oxo-phosphorylation methods utilizing different substituted vinyls. First in 2020,44 the installation of diphenylphosphine oxides onto vinyl azides under eosin Y catalysis was enabled using open-air conditions (Scheme 22). Although requiring pre-functionalization of the olefin, its implementation into non-styrene derivatives (207) is remarkable, broadening the substrate scope. These conditions tolerated various styrene derivatives, including halogen- and alkyl-substituted 2-, 3-, and 4-styrenes (201–204), and demonstrated scalability, retaining efficiency at the gram-scale. Mechanistically, the authors proposed a general oxidative quenching photoredox pathway based on a TEMPO experiment and related literature. The excited eosin Y yields the P-centered radical after deprotonation, which adds to the alkene generating nitrogen gas and an iminyl radical. Lastly, this intermediate is reduced to the corresponding imine anion, and upon protonation, it is hydrolyzed to the targeted carbonyl.
 | ||
| Scheme 22 Visible-light mediated synthesis of β-oxophosphine oxides reported by Kim.44 | ||
Recently, the same group45 expanded this reactivity by introducing phosphine oxides into bromovinyls under similar organophotocatalyzed reaction conditions (Scheme 23). This method successfully incorporated phosphine oxides into electron-rich styrenes, although it struggled with electron-poor systems (209 and 211). Notably, the transformation retained efficiency at the gram-scale and it could be performed on allylbenzene (213), albeit in lower yields. The proposed mechanism parallels the initial method, starting with the phosphine oxide's equilibrium and undergoing SET with eosin Y, generating the phosphorus-centered radical. Following Giese addition and oxygen incorporation, a SET regenerates the catalyst, and the formed anionic intermediate is protonated. A hydroxyl transfer to a phosphine oxide radical cation yields a bromoalcohol intermediate, which, after the loss of HBr, forms the desired acetophenone derivative.
 | ||
| Scheme 23 Synthesis of β-oxophosphine oxides from α-bromostyrenes described by Kim.45 | ||
In a nice contribution by Huang and Darcel,46 the installation of phosphine oxides onto vinyl arenes through a photoinduced iron-catalyzed method is detailed (Scheme 24). This work highlighted the potential of Earth-abundant iron complexes as cost-effective alternatives to traditional Ir- or Ru-based photocatalysts. The reaction involved combining diaryl phosphine oxides with styrenes as radical acceptors in the presence of Fe(OTf)2 in 1,4-dioxane without additives, leveraging ambient aerobic conditions. The scope was broad among alkyl- and halide-substituted arenes (216–222), although reactivity was limited when strongly electron-donating or withdrawing groups were present. Additionally, the reaction was effective for β-methylstyrenes (223). Mechanistic studies using radical scavengers confirmed the presence of phosphine oxide and benzylic radicals, and control experiments highlighted the necessity of aerobic conditions for product formation. The authors proposed a mechanism involving an initial photoinduced SET between the diaryl phosphine oxide and FeII, producing an FeIII species and a radical cation. This cation releases a proton, generating a phosphine oxide radical that undergoes Giese addition with styrene to form a benzylic radical. This radical then couples with an O2˙− species generated from FeIII, oxygen, and light, leading to the formation of the final ketones via path c shown in Scheme 2B.
 | ||
| Scheme 24 Light-induced and iron-catalyzed oxo-phosphorylation of alkenes described by Huang and Darcel.46 | ||
6. CO and C–O/S bond formation
In this section the sequential formation of CO and C–O/S bonds is discussed. All the upcoming methods have in common the use of O2 as an oxidant that yields the carbonyl moiety. Wu and Feng47 addressed the unique oxo-alkoxylation method of (gem-difluoro)alkenes (Scheme 25), overcoming significant challenges related to β-fluoride elimination and high redox potentials. These difluorovinyl species, especially in the electron-deficient context, often fail to form stable benzylic radicals, instead leading to anionic species that can trigger side reactions. The optimized method requires an Ir-based photocatalyst, TEMPO as an additive and o-dichlorobenzene (o-DCB) as solvent. The reaction demonstrated high yields for a variety of mono-, di-, and tri-substituted styrenes (224–227), as well as compatibility with heteroaromatic (228 and 229) compounds like benzothiophenes and indoles. However, electron-poor alkenes remain challenging organic backbones. Secondary alcohols showed good reactivity (230–231), while tertiary alcohols (232) did not react due to steric hindrance. Stern–Volmer luminescence quenching experiments indicated that the difluoroalkene quenched the Ir photocatalyst more effectively than the alcohol, while isotope labelling confirmed the role of O2 as the oxygen source. The proposed mechanism involves an initial reductive quenching photoredox cycle where the IrIII* species oxidizes the organofluorinated styrenes to the radical cation. Subsequently, this species reacts with the desired alcohols followed by proton release yielding a benzylic radical. This intermediate reacts with the generated O2˙− species generated by IrII to yield the final ketone through the general mechanism depicted in Scheme 2B path c.
 | ||
| Scheme 25 Method disclosed by Wu and Feng for photoredox oxo-alkoxylation of alkenes.47 | ||
In 2016, Yang and Wang48 pioneered the synthesis of β-ketosulfones from alkenes and sulfinic acids under photoredox conditions, utilizing eosin Y as an organophotocatalyst and di-tert-butyl peroxide (TBHP) as a sacrificial oxidant (Scheme 26, conditions A). This protocol showed broad substrate compatibility, yielding β-ketosulfones from a variety of substituted alkenes. Both electron-donating and electron-withdrawing groups (233–236) on the aryl ring delivered moderate to good yields, including nitro- and cyano-substituents. Naphthyl-substituted alkenes were also well tolerated (237). A minimal effect from steric hindrance was observed (233, 238), although aliphatic alkenes such as but-1-ene (239) proved unreactive. Mechanistic insights from Stern–Volmer luminescence quenching experiments and photochemical quantum yield measurements suggested a photoredox cycle. Isotope-labelling with H218O revealed that carbonyl formation derives from both H2O and TBHP, with products containing 52% O18-labeled carbonyls and 48% unlabeled. The proposed mechanism involves SET between TBHP and excited eosin Y* to generate hydroxyl anions and tert-butoxy radicals, with the latter facilitating HAT with sulfinic acid to produce the S-centered radical. This radical adds to the alkene, forming a key C-centered radical intermediate that is then oxidized by eosin Y˙+ to form a carbocation (Scheme 1B, left cycle), followed by carbonyl formation via water or hydroxyl addition and subsequent oxidation.
 | ||
| Scheme 26 Synthetic methods enabling β-ketosulfones via photoredox reported by Yang, Wang48 and Wang.49 | ||
In 2020, Wang49 extended this methodology using a naphthalimide-based photocatalyst (ND-O-EAc in Scheme 26, conditions B) with sulfinic acids as sulfone sources, expanding the β-keto sulfone synthesis to a broader range of alkenes, including styrenes (242–247), stilbenes (248), and heteroaromatics, achieving yields up to 95% and scalability to gram-scale reactions. However, challenges arose with aliphatic alkenes and vinyl-pyridine, which yielded hydroxylated or non-oxidized products. Mechanistic studies indicated a reductive quenching photoredox cycle, with the excited photocatalyst accepting an electron from the sulfinic acid derivative to generate the sulfonyl radical, similar to Yang and Wang's mechanism discussed above. Notably, a Russell fragmentation pathway (Scheme 2B) was also proposed for this system, adding depth to the mechanistic understanding.
In 2018, Yue and Nan50 described a photocatalytic formation of CO and C–S bonds, introducing a method for β-thiocyanato ketone synthesis through styrene difunctionalization (Scheme 27). This reaction utilizes eosin Y as the photocatalyst with oxygen serving as the oxidant and NH4SCN as the thiocyanide source. The substrate scope of this transformation is amenable to different styrenes containing alkyls (254), ethers, halides (255) or acetates (256). No steric effects were observed when assessing the methyl group at different positions of the aromatic ring (257 and 258). Extension to β-methylstyrene (261) is also feasible although stilbenes and aliphatic alkenes were incompatible (262 and 263). The mechanistic investigation included radical trapping experiments and Stern–Volmer quenching studies. The excited eosin Y* species undergoes SET with the thiocyanate anion to yield the corresponding open-shell species, which adds to the alkene via Giese addition. Then, the authors suggested that the generated C-centered radical species reacts with either O2 or O2˙− via paths a and b or c shown in Scheme 2B, with NH4+ serving as the proton source.
 | ||
| Scheme 27 Synthetic method presented by Yue for accessing β-thiocyanato ketones.50 | ||
One year later, Zhu's team51 synthesized β-keto sulfones via alkene difunctionalization using sulfonyl hydrazides as precursors and methylene blue as an organic photocatalyst (Scheme 28). The olefin scope is applicable to different alkyls, halides and ethers attached to the phenyl ring. Of note, divinylbenzene substrates could efficiently react at only one side. On the other hand, the arylsulfonylhydrazide substrate scope is limited to only five examples, yet a heteroaromatic thiophene derivative can be accessed in excellent yield (266). Mechanistically, the reaction involves three initial single electron oxidation processes of the sulfonyl hydrazine derivative through the photoredox cycle, which ultimately releases N2 and forms a sulfonyl radical that adds to the olefin. Finally, the authors suggest that the produced benzyl radical intermediate proceeds through the general mechanism shown in Scheme 2B path a and e, or directly reacts with HOO˙ species to form the final ketone after dehydration.
 | ||
| Scheme 28 Photoredox β-ketosulfone synthesis described by Zhu.51 | ||
In 2021, Singh52 developed a method for synthesizing β-keto dithiocarbamates using styrenes, carbon disulfide and secondary amines (Scheme 29). Rhodamine B catalyzes this transformation via reductive quenching, with the reaction showing remarkable broad substrate compatibility and achieving moderate to good yields. Different electron-donating (268, 270) and electron-withdrawing (269) groups tethered to the aromatic olefin showed good reactivity, including bulky mesitylene (271). The amenability of this transformation to (hetero)cyclic, symmetric non-cyclic and asymmetric non-cyclic secondary amines is remarkable (272–275). The use of TBHP as a sacrificial electron-acceptor significantly improved the yield. The mechanism initiates with amine nucleophilic attack on carbon disulfide to form dithiocarbamic acid, which, after SET with excited rhodamine B, yields a sulfur-centered radical that adds to styrene, producing a benzyl radical. The authors suggest that this radical either reacts with tBuOO˙ followed by Kornblum–DeLaMare rearrangement or follows the general radical mechanism shown in paths a and e in Scheme 2B.
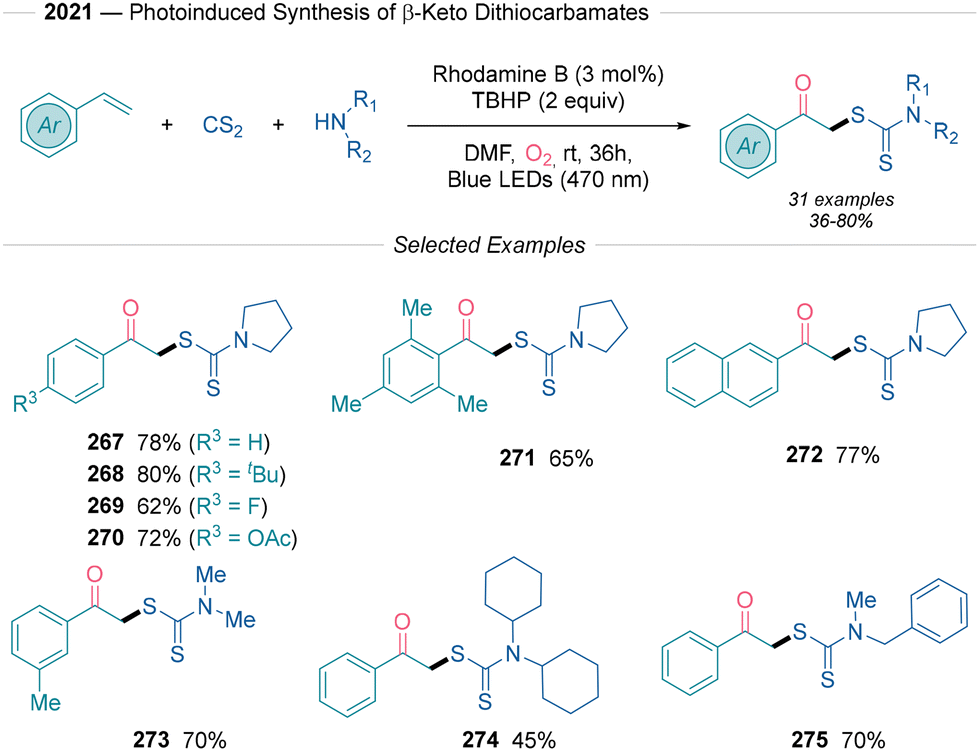 | ||
| Scheme 29 Visible-light mediated synthesis of β-keto dithiocarbamates described by Singh.52 | ||
Finally, in 2022, Maiti53 advanced this field using aromatic thiols as sulfur sources in alkene difunctionalization (Scheme 30). Among the different methods presented, access to β-keto sulfides (276–280) is possible using eosin Y under an oxygen atmosphere after 24–30 hours of illumination at 50 °C. Despite showing a limited scope of 8 examples, the protocol demonstrated promising yields (75–89%). The authors propose that the synthesis of these organosulfur compounds proceeds via the Russell fragmentation pathway (see path a and e in Scheme 2B).
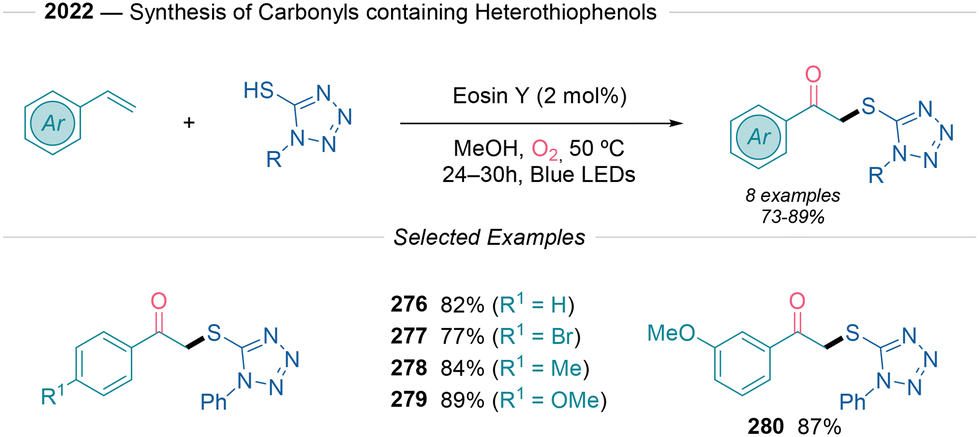 | ||
| Scheme 30 Eosin Y facilitated photoredox difunctionalization process reported by Maiti.53 | ||
7. Conclusions
In conclusion, recent developments in alkene oxo-functionalization through photoredox catalysis have significantly expanded the toolkit of synthetic organic chemists, providing versatile and sustainable pathways for creating complex molecular architectures. Photoredox methods have demonstrated exceptional ability to promote selective formation of CO and C–R bonds under mild conditions, with broad substrate scope and tolerance to diverse functional groups using mainly DMSO or O2 as mild and non-toxic oxidants. Understanding the mechanisms behind radical generation and photoexcitation pathways highlights the ability to precisely control reaction dynamics, enabling the synthesis of complex multifunctional molecules and opening new avenues in medicinal chemistry. Despite considerable progress, some challenges remain. Reaction optimization for more challenging substrates beyond styrenes, stereoselective methods, and scalability for industrial applications are areas where additional advancements could enhance the impact of photoredox-driven alkene oxo-functionalization. As researchers continue to innovate within this field, it is anticipated that new catalytic systems, novel oxygen sources, and refined reaction conditions will drive further development, expanding the scope and applicability of these reactions in organic synthesis. Overall, photoredox catalysis stands poised to play an increasingly central role in the future of sustainable and efficient synthetic strategies for alkene difunctionalization.
Author contributions
YJ and AGG contributed equally. All authors have given approval to the final version of the manuscript.Data availability
No primary research results, software or code have been included and no new data were generated or analyzed as part of this review.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Support for this work under grants PID2021-124916NB-I00, PID2021-128496OB-I00 and RED2022-134287-T funded by MCIN/AEI/10.13039/501100011033 (Spain) and 2021SGR00064 from AGAUR Generalitat de Catalunya is gratefully acknowledged.References
- H. Hock and S. Lang, Autoxydation von Kohlenwasserstoffen, IX. Mitteil.: Über Peroxyde von Benzol-Derivaten, Ber. Dtsch. Chem. Ges. B, 1944, 77, 257 CrossRef.
- (a) T. Shikata and N. Yoshida, Dielectric Behavior of Some Small Ketones as Ideal Polar Molecules, J. Phys. Chem. A, 2012, 116, 4735 CrossRef CAS PubMed.
- (a) R. L. Hoffman, R. S. Kania, M. A. Brothers, J. F. Davies, R. A. Ferre, K. S. Gajiwala, M. He, R. J. Hogan, K. Kozminski, L. Y. Li, J. W. Lockner, J. Lou, M. T. Marra, L. J. Mitchell Jr., B. W. Murray, J. A. Nieman, S. Noell, S. P. Planken, T. Rowe, K. Ryan, G. J. Smith III, J. E. Solowiej, C. M. Steppan and B. Taggart, Discovery of Ketone-Based Covalent Inhibitors of Coronavirus 3CL Proteases for the Potential Therapeutic Treatment of COVID-19, J. Med. Chem., 2020, 63, 12725 CAS; (b) A. Citarella and N. Micale, Peptidyl Fluoromethyl Ketones and Their Applications in Medicinal Chemistry, Molecules, 2020, 25, 4031 CAS.
- D. Crich and S. Neelamkavil, The fluorous Swern and Corey–Kim reactions: scope and mechanism, Tetrahedron, 2002, 58, 3865 CrossRef CAS.
- R. H. Vekariya and J. Aubé, Hexafluoro-2-propanol-Promoted Intermolecular Friedel–Crafts Acylation Reaction, Org. Lett., 2016, 18, 3534 CAS.
- T. Tachinami, T. Nishimura, R. Ushimaru, R. Noyori and H. Naka, Hydration of Terminal Alkynes Catalyzed by Water-Soluble Cobalt Porphyrin Complexes, J. Am. Chem. Soc., 2013, 135, 50 CAS.
- X. Li, H. Hua, Y. Liu and L. Yu, Iron-Promoted Catalytic Activity of Selenium Endowing the Aerobic Oxidative Cracking Reaction of Alkenes, Org. Lett., 2023, 25, 6720 CAS.
- M. Miyazaki and Y. Ura, Palladium/Iron-Catalyzed Wacker-Type Oxidation of Aliphatic Terminal and Internal Alkenes Using O2, ACS Omega, 2023, 8, 41983 CAS.
- Selected examples: (a) J. K. Matsui, S. B. Lang, D. R. Heitz and G. A. Molander, Photoredox-Mediated Routes to Radicals: The Value of Catalytic Radical Generation in Synthetic Methods Development, ACS Catal., 2017, 7, 2563 CAS; (b) M. H. Shaw, J. Twitton and D. W. C. MacMillan, Photoredox Catalysis in Organic Chemistry, J. Org. Chem., 2016, 81, 6898 CrossRef CAS; (c) N. A. Romero and D. A. Nicewicz, Organic Photoredox Catalysis, Chem. Rev., 2016, 116, 10075 CrossRef CAS.
- Selected examples: (a) D.-K. Wang, L. Li, Q. Xu, J. Zhang, H. Zheng and W.-T. Wei, 1,3-Difunctionalization of alkenes: State-of-the-art and future challenges, Org. Chem. Front., 2021, 8, 7037 RSC; (b) Y. Ji, A. Jafaar, C. Gimbert-Suriñach, M. Ribargorda, A. Vallribera and A. Granados, Photocatalyst-free light-mediated three-component alkoxy-, hydroxy-, and azidotrifluoromethylation of alkenes, Org. Chem. Front., 2024, 11, 6660 RSC; (c) R. K. Dhungana, A. Granados, M. Sharique, J. Majhi and G. A. Molander, A three-component difunctionalization of N-alkenyl amides via organophotoredox radical-polar crossover, Chem. Commun., 2022, 58, 9556 RSC; (d) Z.-L. Zhou, Y. Zhang, P.-Z. Cui and J.-H. Li, Photo-/Electrocatalytic Difunctionalization of Alkenes Enabled by C–H Radical Functionalization, Chem. – Eur. J., 2024, 30, e202402458 CrossRef CAS PubMed.
- Selected examples: (a) N. L. Reed, M. I. Herman, V. P. Miltchev and T. P. Yoon, Photocatalytic Oxyamination of Alkenes: Copper(II) Salts as Terminal Oxidants in Photoredox Catalysis, Org. Lett., 2018, 20, 7345 CAS; (b) X.-Q. Mou, M. Wang, L.-C. Ren, Y.-R. Jian, X.-Y. Fu, H.-Y. Zheng, X. Zha, B.-D. Cui, Y. Zhang and Y.-Z. Chen, Photoredox-catalyzed intramolecular oxy- and aminoacylation of alkenes with acyl oxime esters: facile synthesis of acylated saturated heterocycles, Org. Chem. Front., 2024, 11, 74 CAS.
- Selected examples: (a) J. Liu, L. Guo, Z. Chen, Y. Guo, W. Zhang, X. Peng, Z. Wang and Y.-F. Zeng, Photoredox-catalyzed unsymmetrical diamination of alkenes for access to vicinal diamines, Chem. Commun., 2024, 60, 3413 CAS; (b) S. Govaerts, L. Angelini, C. Hampton, L. Malet-Sanz, A. Ruffoni and D. Leonori, Photoinduced Olefin Diamination with Alkylamines, Angew. Chem., Int. Ed., 2020, 59, 15021 CAS.
- Selected examples: (a) W. Tang, D.-Y. Yan, K.-C. Liang, M. Su and F. Liu, Radical-mediated alkene carboamination/dearomatization of arylsulfonyl-o-allylanilines via photoredox catalysis, Org. Chem. Front., 2022, 9, 6535 CAS; (b) H. Jiang, G. Seidler and A. Studer, Carboamination of Unactivated Alkenes through Three-Component Radical Conjugate Addition, Angew. Chem., Int. Ed., 2019, 58, 16528 CAS.
- Selected examples: (a) M. W. Campbell, J. S. Compton, C. B. Kelly and G. A. Molander, Three-Component Olefin Dicarbofunctionalization Enabled by Nickel/Photoredox Dual Catalysis, J. Am. Chem. Soc., 2019, 141, 20069 CAS; (b) B. Zhang, Y. Yi, Z.-Q. Wu, C. Chen and C. Xi, Photoredox-catalyzed dicarbofunctionalization of styrenes with amines and CO2: a convenient access to γ-amino acids, Green Chem., 2020, 22, 5961 CAS.
- Selected examples: (a) R. Laskar, S. Dutta, J. C. Spies, P. Mukherjee, Á. Rentería-Gómez, R. E. Thielemann, C. G. Daniliuc, O. Gutierrez and F. Glorius, γ-Amino Alcohols via Energy Transfer Enabled Brook Rearrangement, J. Am. Chem. Soc., 2024, 146, 10899 CAS; (b) F. Strieth-Kalthoff and F. Glorius, Triplet Energy Transfer Photocatalysis: Unlocking the Next Level, Chem, 2020, 6, 1888 CAS.
- Selected examples: (a) T. Zhang, J. Rabeah and S. Das, Red-light-mediated copper-catalyzed photoredox catalysis promotes regioselectivity switch in the difunctionalization of alkenes, Nat. Commun., 2024, 15, 5208 CAS; (b) R. J. Wiles and G. A. Molander, Photoredox-Mediated Net-Neutral Radical/Polar Crossover Reactions, Isr. J. Chem., 2020, 60, 281 CAS.
- Selected examples: (a) E. de Pedro Beato, D. Spinnato, W. Zhou and P. Melchiorre, A General Organocatalytic System for Electron Donor–Acceptor Complex Photoactivation and Its Use in Radical Processes, J. Am. Chem. Soc., 2021, 143(31), 12304 CAS; (b) H.-Y. Wu, X. Tang, R. Guo, H. Ting Ang, J. Nie, J. Wu, J.-A. Ma and F.-G. Zhang, Catalytic photoactivation of a triarylamine electron donor-acceptor complex for difunctionalization of alkenes, Cell Rep. Phys. Sci., 2024, 5, 102135 CAS.
- (a) S. Gupta, A. Kundu, S. Ghosh, A. Chakraborty and A. Hajra, Visible light-induced organophotoredox-catalyzed difunctionalization of alkenes and alkynes, Green Chem., 2023, 25, 8459 RSC; (b) Y. Liu, H. Liu, X. Liu and Z. Chen, Recent Advances in Photoredox-Catalyzed Difunctionalization of Alkenes, Catalysts, 2023, 13, 1056 CrossRef CAS; (c) S. Mondal, S. Ghosh and A. Hajra, Visible-light-induced redox-neutral difunctionalization of alkenes and alkynes, Chem. Commun., 2024, 60, 9659 RSC; (d) B. C. Lee, C.-F. Liu, L. Q. Hao Lin, K. Zheng Yap, N. Song, C. H. Min Ko, P. H. Chan and M. J. Koh, N-Heterocyclic, carbenes as privileged ligands for nickel-catalysed alkene functionalisation, Chem. Soc. Rev., 2023, 52, 2946 RSC; (e) B. Dong, J. Shen and L.-G. Xie, Recent developments on 1,2-difunctionalization and hydrofunctionalization of unactivated alkenes and alkynes involving C–S bond formation, Org. Chem. Front., 2023, 10, 1322 CAS; (f) A. Vignesh, J. Liu, Z. Wang, Y. Liu and Z. Ke, Nascent developments in main group elementcatalyzed hydrosilylation and dehydrogenative silylation of alkenes and alkynes, Org. Chem. Front., 2024, 11, 576 CAS; (g) J. Liu, J.-P. Wan and Y. Liu, Electrochemical difunctionalization of alkenes and alkynes for the synthesis of organochalcogens involving C–S/Se bond formation, Org. Chem. Front., 2024, 11, 597 CAS; (h) M.-Y. Qian, K.-X. Zhang and L.-J. Xiao, Catalytic transformations of alkenes via nickelacycles, Org. Chem. Front., 2024, 11, 4602 CAS.
- R. Tomita, Y. Yasu, T. Koike and M. Akita, Combining Photoredox-Catalyzed Trifluoromethylation and Oxidation with DMSO: Facile Synthesis of α-Trifluoromethylated Ketones from Aromatic Alkenes, Angew. Chem., Int. Ed., 2014, 53, 7144 CAS.
- Z. Wang, J.-H. Lin and J.-C. Xiao, Photocatalytic Keto- and Amino-Trifluoromethylation of Alkenes, Org. Lett., 2024, 26, 1980 CAS.
- E. Yamaguchi, Y. Kamito, K. Matsuo, J. Ishihara and A. Itoh, Photooxidative Keto-Trifluoromethylation of Styrenes by Means of an Anthraquinone-Based Organocatalyst, Synthesis, 2018, 3161 CAS.
- J. Chun, H. Zhang, F. Meng, K. Guo, S. Cao, Q. Fang, J. Li and Y. Zhu, Visible-Light Photoredox-Catalyzed Tandem Trifluoro-methylation/Cyclization/Remote Oxidation of 1,6-Dienes: Access to CF3-Containing Five-Membered Heterocycles, Adv. Synth. Catal., 2021, 363, 751 CAS.
- Y. Nakayama, G. Ando, M. Abe, T. Koike and M. Akita, Keto-Difluoromethylation of Aromatic Alkenes by Photoredox Catalysis: Step-Economical Synthesis of α-CF2H-Substituted Ketones in Flow, ACS Catal., 2019, 9, 6555 CAS.
- Y. Zafrani, G. Sod-Moriah, D. Yeffet, A. Berliner, D. Amir, D. Marciano, S. Elias, S. Katalan, N. Ashkenazi, M. Madmon, E. Gershonov and S. Saphier, CF2H, a Functional Group-Dependent Hydrogen-Bond Donor: Is It a More or Less Lipophilic Bioisostere of OH, SH, and CH3?, J. Med. Chem., 2019, 62, 5628 CAS.
- L. Li, Y.-N. Ma, M. Tang, J. Guo, Z. Yang, Y. Yan, X. Ma and L. Tang, Photoredox-Catalyzed Oxydifluoroalkylation of Styrenes for Access to Difluorinated Ketones with DMSO as an Oxidant, Adv. Synth. Catal., 2019, 361, 3723 CAS.
- Z.-H. Xia, Z.-H. Gao, L. Dai and S. Ye, Visible-Light-Promoted Oxo-difluoroalkylation of Alkenes with DMSO as the Oxidant, J. Org. Chem., 2019, 84, 7388 CrossRef CAS PubMed.
- X. Luo, Z. Fan, B. Zhang, C. Chen and C. Xi, Visible-light-triggered direct keto-difluoroacetylation of styrenes with (fluorosulfonyl)difluoroacetate and dimethyl sulfoxide leads to a-difluoroacetylated ketones, Chem. Commun., 2019, 55, 10980 RSC.
- L. Li, H. Luo, Z. Zhao, Y. Li, Q. Zhou, J. Xu, J. Li and Y.-N. Ma, Photoredox-Catalyzed Remote Difunctionalizations of Alkenes To Synthesize Fluoroalkyl Ketones with Dimethyl Sulfoxide as the Oxidant, Org. Lett., 2019, 21, 9228 CrossRef CAS PubMed.
- A. Tlahuext-Aca, R. A. Garza-Sanchez, M. Schäfer and F. Glorius, Visible-Light-Mediated Synthesis of Ketones by the Oxidative Alkylation of Styrenes, Org. Lett., 2018, 20, 1546 CrossRef CAS PubMed.
- Z.-H. Xia, C.-L. Zhang, Z.-H. Gao and S. Ye, Switchable Decarboxylative Heck-Type Reaction and Oxo-alkylation of Styrenes with N-Hydroxyphthalimide Esters under Photocatalysis, Org. Lett., 2018, 20, 3496 CrossRef CAS PubMed.
- B.-Q. He, X.-Y. Yu, P.-Z. Wang, J.-R. Chen and W.-J. Xiao, A photoredox catalyzed iminyl radical-triggered C–C bond cleavage/addition/Kornblum oxidation cascade of oxime esters and styrenes: synthesis of ketonitriles, Chem. Commun., 2018, 54, 12262 RSC.
- X.-X. Fang, P.-F. Wang, W. Yi, W. Chen, S.-C. Lou and G.-Q. Liu, Visible-Light-Mediated Oxidative Coupling of Vinylarenes with Bromocarboxylates Leading to γ-Ketoesters, J. Org. Chem., 2019, 84, 15677 CrossRef CAS PubMed.
- S. R. Chowdhury, D. Singh, I. U. Hoque and S. Maity, Organic Dye-Catalyzed Intermolecular Radical Coupling of α-Bromocarbonyls with Olefins: Photocatalytic Synthesis of 1,4-Ketocarbonyls Using Air as an Oxidant, J. Org. Chem., 2020, 85, 13939 CrossRef PubMed.
- A. Gallego-Gamo, R. Pleixats, C. Gimbert-Suriñach, A. Vallribera and A. Granados, Hydroxytrifluoroethylation and Trifluoroacetylation Reactions via SET Processes, Chem. – Eur. J., 2024, 30, e202303854 CrossRef CAS PubMed.
- A. Gallego-Gamo, P. Sarró, Y. Ji, R. Pleixats, E. Molins, C. Gimbert-Suriñach, A. Vallribera and A. Granados, Direct Synthesis of 2-Hydroxytrifluoroethylacetophenones via Organophotoredox-Mediated Net-Neutral Radical/Polar Crossover, J. Org. Chem., 2024, 89, 11682 CrossRef CAS PubMed.
- M. Gil-Ordóñez, A. Gallego-Gamo, P. Sarró, R. Pleixats, C. Gimbert-Suriñach, A. Vallribera and A. Granados, Organophotoredox-Driven Three-Component Synthesis of β-Trifluoromethyl β-Amino Ketones, J. Org. Chem., 2025, 90, 2500 CrossRef PubMed.
- S. Patra, V. Valsamidou, B. N. Nandasana and D. Katayev, Chem. Commun., 2025, 61, 1689 RSC.
- M. Bu, T. F. Niu and C. Cai, Visible-light-mediated oxidative arylation of vinylarenes under aerobic conditions, Catal. Sci. Technol., 2015, 5, 830 RSC.
- J. Wu, Y. Zhang, X. Gong, Y. Meng and C. Zhu, Metal-free synthesis of ketones by visible-light induced aerobic oxidative radical addition of aryl hydrazines to alkenes, Green Chem., 2017, 19, 2941 RSC.
- Q. Qin, Y.-Y. Han, Y.-Y. Jiao, Y. He and S. Yu, Photoredox-Catalyzed Diamidation and Oxidative Amidation of Alkenes: Solvent-Enabled Synthesis of 1,2-Diamides and α-Amino Ketones, Org. Lett., 2017, 19, 2909 CrossRef CAS PubMed.
- J. Wang, B. Shao, H. Ge, Y. Li, H. Qi and Li Xiao, Visible-Light-Induced Regioselective Radical Oxo-Amination of Alkenes with O2 as the Oxygen Source, Org. Lett., 2023, 25, 5333 CrossRef CAS PubMed.
- Y. Shi, R. Chen, K. Guo, F. Meng, S. Cao, C. Gu and Y. Zhu, Visible light-promoted metal-free aerobic oxyphosphorylation of olefins: A facile approach to β-ketophosphine oxides, Tetrahedron Lett., 2018, 59, 2062 CrossRef CAS.
- H.-F. Qian, C.-K. Li, Z.-H. Zhou, Z.-K. Tao, A. Shoberu and J.-P. Zou, Visible Light-Mediated Photocatalytic Metal-Free Cross-Coupling Reaction of Alkenyl Carboxylic Acids with Diarylphosphine Oxides Leading to β-Ketophosphine Oxides, Org. Lett., 2018, 20, 5947 CrossRef CAS PubMed.
- H. I. Jung and D. Y. Kim, Visible light-mediated photocatalytic phosphorylation of vinyl azides: A mild synthesis of β-ketophosphine oxides, Synth. Commun., 2020, 50, 380 CrossRef CAS.
- R. Kim and D. Y. Kim, Visible light photoredox-catalyzed phosphorylation of α-bromostyrenes: a mild synthesis of β-ketophosphine oxides, Phosphorus, Sulfur Silicon Relat. Elem., 2024, 199, 162 CAS.
- Y. Yuan, Q. Huang and C. Darcel, Blue-Light Driven Iron-Catalyzed Oxy-phosphinylation of Activated Alkenes for β-Ketophosphine Oxide Synthesis, Chem. – Eur. J., 2023, 29, e202302358 CrossRef CAS PubMed.
- Y. Liang, N. Zhou, G. Ma, L. Wen, X. Wu and P. Feng, Tunable alkoxy-nucleophilic addition under photochemical condition: Dioxidation of gem-difluoroalkenes with O2, Mol. Catal., 2022, 528, 112373 CAS.
- D. Yang, B. Huang, W. Wei, J. Li, G. Lin, Y. Liu, J. Ding, P. Sun and H. Wang, Visible-light initiated direct oxysulfonylation of alkenes with sulfinic acids leading to β-ketosulfones, Green Chem., 2016, 18, 5630 RSC.
- X. Yang, J. Yang, K. Yan, H. Qin, W. Dong, J. Wen and H. Wang, A Naphthalimide-Based ND-O-EAc Photocatalyst for Sulfonation of Alkenes to Access β-Ketosulfones Under Visible Light, Eur. J. Org. Chem., 2020, 3456 CrossRef CAS.
- G. Nan and H. Yue, Visible-Light-Promoted Difunctionalization of Olefins Leading to α-Thiocyanato Ketones, Synlett, 2018, 1340 CAS.
- J. Wu, Y. Zhang, X. Gong, Y. Meng and C. Zhu, Visible-light promoted aerobic difunctionalization of alkenes with sulfonyl hydrazides for the synthesis of β-keto/hydroxyl sulfones, Org. Biomol. Chem., 2019, 17, 3507 CAS.
- R. K. Vishwakarma, S. Kumar and K. N. Singh, Visible-Light-Induced Photocatalytic Synthesis of β-Keto Dithiocarbamates via Difunctionalization of Styrenes, Org. Lett., 2021, 23, 4147 CrossRef CAS PubMed.
- R. Rahaman, M. T. Hoque and D. K. Maiti, Organophotoredox-Catalyzed Sulfurization of Alkenes and Alkynes: Selective and Controlled Synthesis of Sulfoxides, β-Hydroxysulfoxides, and β-Keto Sulfides, Org. Lett., 2022, 24, 6885 CrossRef CAS PubMed.
Footnote |
| † These authors contributed equally. |
| This journal is © the Partner Organisations 2025 |