
Expanding the reaction toolbox for nanoscale direct-to-biology PROTAC synthesis and biological evaluation†
Rebecca
Stevens
 ab,
Harry J.
Shrives
a,
Jenni
Cryan
c,
Diana
Klimaszewska
c,
Peter
Stacey
c,
Glenn A.
Burley
b,
John D.
Harling
a,
David J.
Battersby
*a and
Afjal H.
Miah
*a
ab,
Harry J.
Shrives
a,
Jenni
Cryan
c,
Diana
Klimaszewska
c,
Peter
Stacey
c,
Glenn A.
Burley
b,
John D.
Harling
a,
David J.
Battersby
*a and
Afjal H.
Miah
*a
aModality Platform Technologies, GSK, Stevenage, SG1 2NY, UK. E-mail: david.j.battersby@gsk.com; afjal.h.miah@gsk.com
bDepartment of Pure and Applied Chemistry, University of Strathclyde, Glasgow, G1 1BX, UK
cDiscovery Biology and Screening, GSK, Stevenage, SG1 2NY, UK
First published on 23rd December 2024
Abstract
High-throughput chemistry (HTC) and direct-to-biology (D2B) platforms allow for plate-based compound synthesis and biological evaluation of crude mixtures in cellular assays. The rise of these workflows has rapidly accelerated drug-discovery programs in the field of targeted protein degradation (TPD) in recent years by removing a key bottleneck of compound purification. However, the number of chemical transformations amenable to this methodology remain minimal, leading to limitations in the exploration of chemical space using existing library-based approaches. In this work, we expanded the toolbox by synthesising a library of degraders in D2B format. First, reaction conditions are established for performing key medicinal chemistry transformations, including reductive amination, SNAr, palladium-mediated cross-coupling and alkylation, in D2B format. Second, the utility of these alternative reactions is demonstrated by rapidly identifying developable PROTACs for a range of protein targets.
Introduction
Targeted protein degraders such as proteolysis targeting chimeras (PROTACs) and molecular glues offer significant promise as a novel drug modality. As a reflection of this, currently there are more than 20 PROTAC candidates that have entered clinical development.1 These targeted degraders exhibit a therapeutic effect by forming a ternary complex with a protein-of-interest (POI) and an E3 ligase, leading to ubiquitin transfer onto a POI and subsequent degradation by the proteasome.2 With a catalytic mechanism that hinges on the induced proximity between POI and E3 ligase rather than occupancy-based inhibition, degraders offer several advantages over small-molecule inhibitors. These include enhanced selectivity, through the strategic selection of E3 ligases with localised tissue expression that may allow for use of PROTACs in precision medicine,3–7 the ability to target proteins with weak or no functional ligands,4,8,9 and the potential for lower doses or dosing frequency.10 However, PROTACs and molecular glues have complex discovery efforts with nuanced structure–activity relationship (SAR) profiles that are unpredictable, thus an empirical approach is often required to identify new degraders.High-throughput chemistry (HTC) and direct-to-biology (D2B) approaches have become increasingly popular in the last five years and have already shown huge potential in accelerating the discovery of PROTACs and molecular glues.11 Applications have included identifying an appropriate exit vector for a POI or E3 ligase ligand,12 choosing the optimal linker length and ligand structures,13 and optimising initial hits by using more rigid linkers to improve physicochemical properties to target oral bioavailability.14 Defining desirable physicochemical characteristics for PROTAC synthesis and downstream development has further been aided by the expansion of assays that are applicable to crude reaction mixtures, from degradation readout, to E3 ligase binding assays14 coupled with measurements of chromatographic log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) D (chromlogD) and exposed/experimental polar surface area (ePSA).15 Whilst D2B has already substantially improved the throughput of degrader synthesis, transformations beyond amide couplings are yet to be well-represented.
D (chromlogD) and exposed/experimental polar surface area (ePSA).15 Whilst D2B has already substantially improved the throughput of degrader synthesis, transformations beyond amide couplings are yet to be well-represented.
Screening of crude reaction mixtures in cells following bioorthogonal transformations such as copper-catalysed azide-alkyne cycloaddition (CuAAC) click chemistry16 and sulfur(VI) fluoride exchange (SuFEX)17,18 have been widely reported for small molecules. In 2018, Merck expanded on these reaction types by using transition metal catalysis for kinase inhibitor synthesis, followed by evaluation by affinity selection mass spectrometry (ASMS),19 but to our knowledge this chemistry is yet to be used in a cellular context. A wide range of amide coupling conditions that are applicable to cellular assays have been used for either small molecule or PROTAC synthesis in D2B format, including the use of coupling reagents such as HATU, or alternative precursors such as N-hydroxysuccinimide (OSu) esters.14,15,20,21 Other transformations include hydrazone and phthalimidine formation,13,22,23 and multi-component reactions,24 yet they remain limited in scope and breadth of applicability, either requiring highly specific starting materials or yielding a restricted chemical space in their products.
A 2016 analysis from Brown and Boström at AstraZeneca found that the five most commonly used reactions in medicinal chemistry were: 1) amide coupling; 2) SNAr; 3) Boc protection and deprotection; 4) ester hydrolysis; and 5) Suzuki–Miyaura coupling.25N-Alkylation, heterocycle formation and reductive amination were also identified within the top-ten, and are highly valuable transformations for PROTAC synthesis. However, these reactions with the greatest utility have not been incorporated in the D2B platform methodology.
The chemical composition of the linker has been shown to be important for PROTAC potency by Ciulli et al. who observed stabilising interactions between the PEG linker and VHL E3 ligase in the ternary complex crystal structure of BRD4 degrader MZ1.26 Beyond the role of the linker in ternary complex stability, linker choice is a crucial component in PROTAC optimisation and is key to identifying developable compounds that can be quickly progressed. For example, a recent paper by Wang et al. showed that in the discovery of an oral CBP/p300 PROTAC switching from a linker containing a piperidine amide to a cyclohexyl ring increased both degradation activity and oral bioavailability.27 Furthermore, introduction of a piperidine or piperazine linker through reductive amination or alkylation chemistry could provide a basic centre that acts as a solubilising group. To investigate this, Goracci et al. performed an analysis of pKa values across a range of PROTAC molecules and found that basicity and protonation state could be tuned significantly by making small changes to neighbouring atoms.28 Whilst amide bond formation is a highly valuable transformation to medicinal chemists, amide linkages can suffer from hydrolytic instability,29 especially at the linker attachment point in PROTAC molecules,30 and thus the ability to synthesise a range of different linker types in an initial optimisation campaign is highly valuable. These literature examples indicate the importance of alternative transformations for PROTAC assembly and the opportunity to improve physicochemical properties for oral exposure. Development of these new transformations in a D2B format will enable the simultaneous identification of more developable PROTACs whilst validating degradation of new POI targets.
While the ability to synthesise and evaluate amide-linked PROTACs using previously reported D2B approaches has offered significant acceleration to the discovery of new degraders,12,14,15 the PROTACs in clinical evaluation typically do not contain amide bonds in the linker. These advanced molecules are typically linked by moieties such as aromatic or saturated heterocycles, but crude reaction screening in cell-based assays following reductive amination, SNAr, alkylation or cross-coupling chemistry has not been reported to our knowledge. As such, we envisioned that the development of new transformations for D2B, especially those that are crucial to every medicinal chemist's toolbox, would be a valuable addition to the scientific literature and applicable to a wide range of medicinal molecules beyond PROTACs (Fig. 1). Additionally, these transformations will increase the changes of identifying developable PROTACs using high-throughput approaches, ultimately reducing the timelines to reach orally bioavailable candidate molecules.
 | ||
| Fig. 1 A) Previous PROTAC direct-to-biology (D2B) work in the literature; B) this work describes the expansion of PROTAC D2B to additional chemical transformations with multiple protein-of-interest (POI) targets; C) approach to identifying new reaction conditions for D2B. | ||
In this manuscript, we describe an automated HTC platform using 1536-well plates to access novel PROTAC designs where the reaction to join POI binder and E3 ligase binder components are formed by reductive amination, alkylation, SNAr and a multistep synthetic route involving palladium-mediated cross-coupling chemistry.
Results and discussion
Key considerations in reaction development for D2B
Several criteria were established prior to setting up our D2B platform. Firstly, the reaction conditions must be compatible with different scaffolds and tolerant of existing functionality. Secondly, the reactions used to form PROTACs must be efficient with minimal by-product formation, providing sufficient purity to determine representative biological activity. This is especially crucial given that the cell permeability of the starting materials may not be equivalent to that of the products, and thus chemical analysis of the reaction mixture may not be representative of the concentration of each component in the cellular environment. Furthermore, the reagents or any by-products must be non-toxic to cells, determined using a cell viability assay such as CellTiter-Glo® (CTG). Lastly, the reaction precursors will ideally have common functionality that is commercially available and may also enable the use of one library for multiple sets of compounds through different chemical transformations.Alongside the chemical factors, engineering controls are also important, for example the chemical compatibility of the plate to the reaction conditions and choice of solvent. It should also be noted that a set of reaction conditions for a given transformation may not translate well from batch scale to plate scale, especially in 1536-well format, thus a validation set should be used initially to validate the chemistry in plates followed by LCMS analysis of reaction mixtures. Analysis of the cell viability with reagents alone will also be valuable to assess the suitability of reagent choice. Following the D2B chemistry and direct biological evaluation in a degradation assay, hits across a range of potencies should be resynthesised as purified samples and assessed for correlation with the crude samples.
We first looked to validate three new transformations for D2B format using the BRD4 ligand I-BET469 as a proof-of-concept to synthesise and test BRD4-targeting PROTACs.
Exploration of reaction conditions for PROTAC synthesis
Typically, reductive aminations are carried out using borohydride or borane reducing reagents.31 A range of commonly used reductive amination conditions were considered: sodium cyanoborohydride and related derivatives were avoided due to the potential to release hydrogen cyanide into the glovebox environment; the effect of titanium tetrachloride-based conditions on the subsequent assays were unclear; and hydrogenations were impractical in 1536-well plate format.
Starting this optimisation, a small set of eight amine-functionalised BRD4 ligands were chosen for reaction with three aldehydes based on ligands for E3 ligases, VHL and cereblon (Scheme 1A). The commonly used reagent sodium triacetoxyborohydride (STAB) was trialled first due to its wide commercial availability and release of acetic acid as a by-product which could be quenched by buffer solution in the assay. However, poor conversion was observed by LCMS with significant formation of side products and low conversion to the desired PROTAC. Addition of acetic acid to the reaction mixture did not aid conversion.
 | ||
| Scheme 1 A) Reductive amination library carried out in D2B; B) exemplar resynthesised hits from library. | ||
Picoline borane was trialled next due to its bench stability, ease of weighing, and higher level of solubility in DMSO. Full conversion to the desired PROTAC by LCMS was observed for 13 of the 24 examples (54%) and whilst this was initially considered a modest success rate, the PROTAC purity for the complete reactions was between 50 and 80% by LCMS. It is worth noting that picoline borane is also visible by LCMS and thus these purity values represent full conversion from the respective starting materials with formation of no significant side products. PROTAC purity was measured by percentage area by UV trace utilising LCMS without correction, and reactions with full conversion were deemed successful. Any successful reactions were then tested assuming 100% conversion to desired PROTAC, with no additional dilutions steps to adjust according to observed purity before biological assay.
These compounds were tested in a HiBiT cellular degradation assay using an N-terminal HiBiT tagged-BRD4 cell line derived by CRISPR/Cas9 genome editing of parental HEK293 cells. Seven hits were resynthesised on batch scale and showed excellent correlation with the D2B values for pDC50 and Dmax across a 2–3 log-unit range in compound potency. A drop in Dmax was observed for only one D2B sample (compound 2), which is consistent with literature reports (Fig. 2A).14
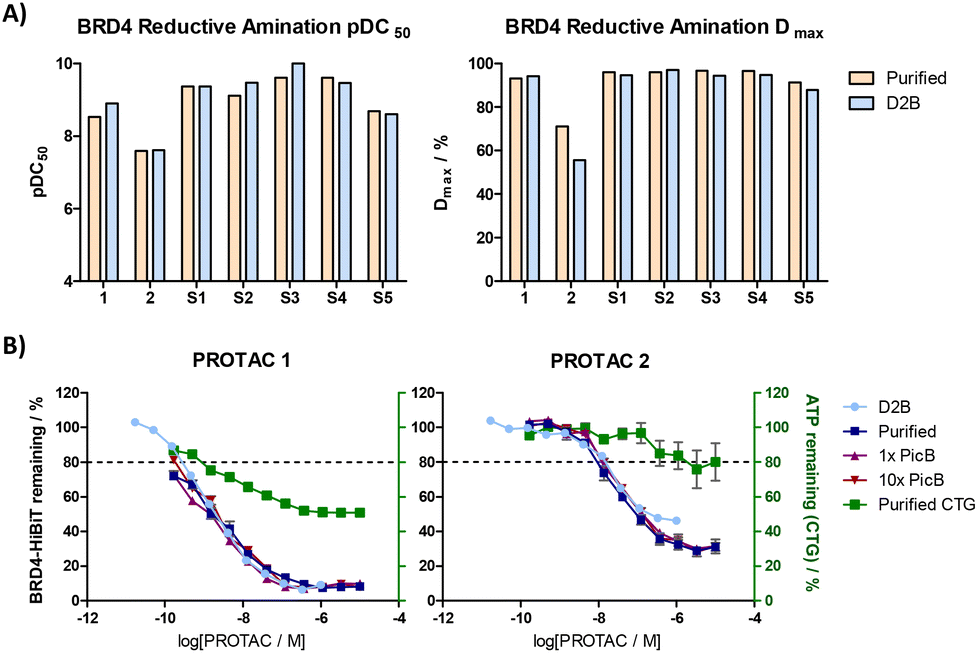 | ||
| Fig. 2 A) Correlation between crude and purified samples; B) curves from HiBiT and CTG assays for D2B samples (N = 1), purified samples (N = 2), and spiking experiments of purified PROTAC with picoline borane (PicB) (N = 2). Error bars represent SEM. | ||
To thoroughly investigate the impact of these reaction conditions on the HiBiT degradation assay, spiking experiments were carried out where reagents were added to the purified PROTACs to mimic the components of the D2B samples (Fig. 2B, more curves shown in ESI†). Spiking the purified PROTACs with picoline borane at a D2B-relevant concentration did not alter the curve shapes or pDC50 and Dmax parameters in the HiBiT assay. Impressively, a 10-fold excess of picoline borane also had no effect on the assay readout, giving confidence that these conditions were robust for crude reaction screening.
Synthesis of this compound set by reductive amination D2B allowed the identification of trends in degradation SAR. Several novel BRD4 PROTACs with picomolar potency were identified, possessing more constrained linkers than most literature examples (Scheme 1B). PROTACs recruiting cereblon such as compound 1 and S1–4 were typically more potent than VHL examples. VHL-recruiting PROTAC 2 was several log units less potent than the cereblon examples shown but a subtle change in the linker to give compound S5 recovered the potency significantly.
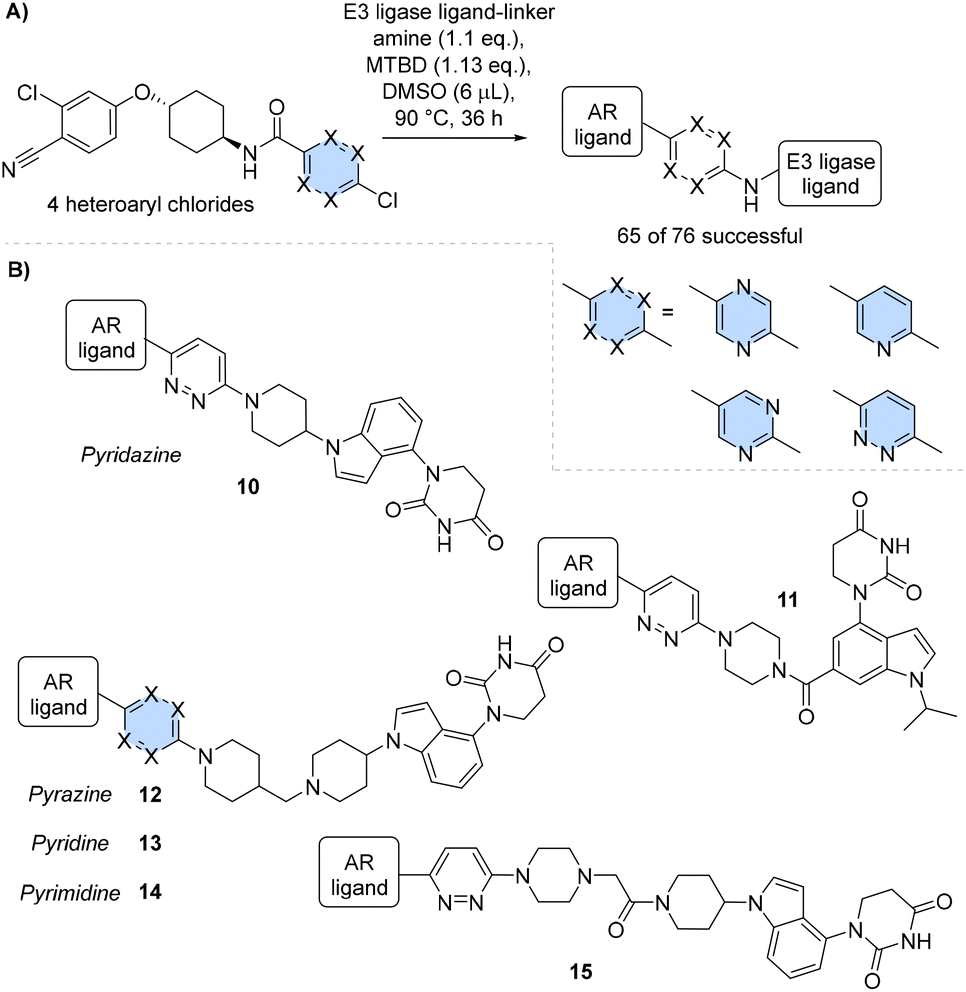
To identify suitable conditions, it was crucial to find a suitable choice of base. Typically SNAr reactions are carried out with organic bases such as triethylamine or Hunig's base32,33 but previous studies had identified that these bases were not miscible with DMSO, and this was a key consideration for the optimisation. Organic superbases such as phosphazene (P2-Et) and 7-methyl-1,5,7-triazabicyclo[4.4.0]dec-5-ene (MTBD) have found uses in ultra-high throughput chemistry due to their non-nucleophilic nature and solubility in most organic solvents, which was ideal for our purposes.19,34 Since reaction dosing for D2B is performed using a mosquito® liquid handler, a range of these superbases were trialled for the SNAr reaction and guanidine base MTBD was identified as a suitable choice due to its high reaction conversion rate.
Six E3 ligase ligands containing a heteroaromatic ring, either pyrazine, pyrimidine or pyridazine, were synthesised and reacted with a set of eight BRD4 ligands possessing a short linker plus amine, at 60 °C with MTBD in 5 μL of DMSO (Scheme 2A). Quantitative conversion was observed by LCMS with all 48 examples, giving a library success rate of 100%, and indicating that MTBD was a suitable choice of base for plate-based SNAr chemistry. Three PROTAC examples were resynthesised and purified samples were tested in the HiBiT assay for comparison with the crude samples (Fig. 3A).
 | ||
| Scheme 2 A) Nucleophilic aromatic substitution (SNAr) library carried out in D2B; B) exemplar resynthesised hits from library. | ||
 | ||
| Fig. 3 A) Correlation between crude and purified samples; B) curves from HiBiT and CTG assays for D2B samples (N = 1), purified samples (N = 2), and spiking experiments of purified PROTAC with MTBD (N = 2). Error bars represent SEM. | ||
For two of the three samples, close correlation was observed, but compound 3 demonstrated a loss in potency when resynthesised. The appearance of occasional discrepancies between crude and purified samples highlights the importance of hit resynthesis and the authors recommend synthesising purified samples for 5–10% of every library across a range of potencies. Spiking experiments of purified PROTAC with MTBD were performed to mimic the D2B conditions and pleasingly a 10-fold excess of MTBD resulted in no change in the degradation profile by HiBiT assay for any of the compounds, giving confidence that these reagents are suitable for direct use in cellular assays (Fig. 3B, more curves shown in ESI†).
Consistent with our aim to identify more developable PROTACs using the D2B approach, our ideal multistep workflow would replace double amide-linked linkers that are typical in the literature, with single amide linkages. In order to achieve this, we envisioned that a Pd-mediated C(sp2)–C(sp3) cross-coupling performed in small vials would provide initial diversification of the POI binder, followed by Boc deprotection and amide coupling with a large library in plate format. This approach would be well suited to the use of cartridge clean-up after the first step due to the potential cytotoxicity challenges associated with use of metal catalysts within cellular assays.35 However, the throughput would not be limited by the use of cartridges as the final diversification step would be carried out in 1536-well plates using an automated workflow to rapidly assess SAR.
We chose to perform the first cross-coupling step in small microwave vials, as the use of inorganic base and a high catalyst loading was required for the challenging transformation, and this was deemed unsuitable for 1536-well plate format. However, we envision that simpler cross-coupling transformations, such as between two sp2 partners, would be amenable to the use of organic bases and a lower catalyst loading, and thus could be performed in plates.
To explore the applicability of this sp2–sp3 cross-coupling transformation, we used our previously reported system where our prior work utilised purified intermediates from the cross-coupling in a deprotection-amide coupling sequence,12 and investigated the tolerance of the three-step process to cellular assay without chromatographic purification (Scheme 3). As E3 ligase ligand libraries containing elaborated precursors for cross-coupling chemistry were not commercially available, we envisioned that a multistep approach would be highly valuable for the development of cross-coupling chemistry in a D2B format.
 | ||
| Scheme 3 A) Multistep process carried out in D2B; B) exemplar resynthesised hits from library; C) correlation between biological evaluation data obtained for different purification methods, where ‘purified’ data represents entirely purified samples, and other methods indicated correspond to purification method after the sp2–sp3 cross-coupling reaction; D) curves from HiBiT and CTG assays for D2B samples corresponding to each purification method (N = 1) and purified samples (N = 2). Error bars represent SEM. | ||
Bromobenzimidazole BRD4 binder 6 was subjected to Pd-mediated sp2–sp3 cross-coupling conditions to attach a N-Boc protected azetidine with methylene spacer as the first part of the linker. Following the reaction, the intermediate was split into four batches for varied purification processes: HPLC, celite cartridge, solid-phase extraction (SPE) cartridge, and aqueous work up. Several by-products were observed in the cross-coupling reaction, including homo-coupled and des-bromo BRD4 binder, as well as carbazole released from the Pd G3 precatalyst.
Despite the formation of these by-products, 7 was the major product by LCMS following the four purification procedures: HPLC, celite cartridge (59% purity), SPE cartridge (70% purity) and aqueous work up (72% purity). However, LCMS is only capable of capturing UV-absorbent species and thus a full picture of the reaction mixtures is not accurately depicted due to the presence of residual palladium. Accurate quantification of palladium levels on this scale represents a significant challenge, which requires further development of appropriate analytical techniques. Each batch was subsequently deprotected with TFA, passed through a strong cation exchange (SCX) cartridge to give the free amine, and coupled to an E3 ligase ligand-linker library of carboxylic acids using previously described conditions.12 Following all three steps the LCMS purities of the desired PROTAC products were found to be approximately 20–40% across the library. Assessment of the full library by HiBiT assay indicated that of 49 samples, 94% of the pDC50 values lie within one log unit of the HPLC batch and 84% within half a log unit, which was deemed to be within assay error (see ESI† for further analysis of the set). These results are consistent with findings from AstraZeneca where mixtures of starting material and PROTAC product were tested in different ratios to mimic partial reaction conversion.15 Whilst pDC50 values were largely unaffected by partial conversion, Dmax values for the non-HPLC batches were found to be consistently lower when comparing across the full set of 49 samples (see ESI†), indicating that the effect on Dmax is greater than on the pDC50, presumably due to competition of the PROTAC with other reaction components.
When comparing four examples, 8–9 and S6–7 with fully purified samples, the correlation was good to excellent (Scheme 3C). For PROTAC S6, a reduced pDC50 was observed when purifying with celite after the Pd-mediated cross-coupling and reduced Dmax was observed for the SPE cartridge and aqueous work-up examples. However, it is worth noting that despite these discrepancies, the trend in potency remained the same regardless of the purification method chosen, indicating that this is a valuable workflow for hit identification or initial hit optimisation.
These findings suggest that whilst low purity samples can reliably provide accurate pDC50 values, Dmax is likely to be reduced, and thus should not be used for ranking hit compounds following multistep workflows. Accurate Dmax values should be obtained by resynthesising compounds of interest and obtaining degradation data for the purified samples.
Given that clinical candidates ARV-110 and ARV-766 inspired us to find appropriate SNAr conditions for D2B, we synthesised androgen receptor (AR) binder analogues to directly apply this new transformation on clinically relevant PROTAC molecules.
For BRD4 examples, a small library of E3 ligase binders containing heteroaryl chlorides in the linker were synthesised. In this example the position of the functional groups were reversed so that a library of 19 E3 ligase ligands with amine-bearing linkers could be used. An advantage to this approach is the flexibility to use either library depending on precursor availability to minimise the requirement for bespoke synthesis.
An AR binder based on literature PROTAC ARV-110 was synthesised with a series of attached heteroaryl chlorides. Increasing the reaction temperature to 90 °C allowed the inclusion of a pyridine scaffold in this set alongside pyrimidine, pyridazine, and pyrazine (Scheme 4A). High library success rates of 74% to 95% were observed for the scaffolds with minimal variation between choice of heteroaryl chloride: 14 of 19 for pyridazine, 18 of 19 for pyrazine, 15 of 19 for pyridine, and 18 of 19 for pyrimidine.
 | ||
| Scheme 4 A) Nucleophilic aromatic substitution (SNAr) library carried out in D2B; B) exemplar resynthesised hits from library. | ||
A set of 6 PROTACs were resynthesised and excellent correlation between crude and purified samples was observed across a range of potencies (Fig. 4A). Spiking experiments of excess reagent with purified PROTAC were also consistent with observations from the BRD4 assays, and 10x excess MTBD gave no interference in the AR HiBiT assay readout (Fig. 4B). Matched pairs 12, 13 and 14 demonstrated comparable activity regardless of the ring present, whilst the loss of a second saturated ring in the case of 10 resulted in a significant loss of potency. PROTAC 11 bearing a comparable linker length to 10 was several log units more potent, potentially due to use of a different exit vector from the cereblon binder.
 | ||
| Fig. 4 A) Correlation between crude and purified samples; B) curves from HiBiT assay for D2B samples, purified samples, and spiking experiments of purified PROTAC with MTBD – data is N = 2 and error bars represent SEM. | ||
The receptor-interacting protein kinase 2 (RIPK2) was used as a case study for developing D2B chemistry as RIPK2 ligand 16 had previously been incorporated into IAP and VHL-based PROTACs.38,39 However, previous optimisation efforts suffered from lengthy synthetic routes where each unique analogue required up to seven synthetic steps to prepare from intermediate 16. This challenge is underpinned by the presence of two reactive functional groups on the POI ligand, a phenol and a diarylamine. It was envisioned that a selective alkylation of the binder would be highly advantageous for exploring degradation SAR.
Furthermore, the potential to differentiate between two N- and O-linked PROTACs simultaneously adds an additional exploration element to a D2B experiment without further synthetic derivatisation of the binder. This approach would also enable the exploration of amide-free linear linkers with a range of different lengths, without the requirement for any pre-functionalisation of RIPK2 ligand 16.
Utilising the knowledge gained from SNAr development, organic superbases were trialled for the reaction between RIPK2 ligand 16 and a series of VHL ligands with linear PEG- or C-linkers bearing alkyl chlorides at 90 °C (Scheme 5A). Interestingly, the choice of base was found to be crucial to the alkylation selectivity and close-running isobaric products were detected in LCMS of the reaction mixtures (Scheme 5B). In microtitre plates, two chemoselective libraries were prepared using MTBD and P2-Et, where one major product was formed in each well, with minor formation of the alternative regioisomer.
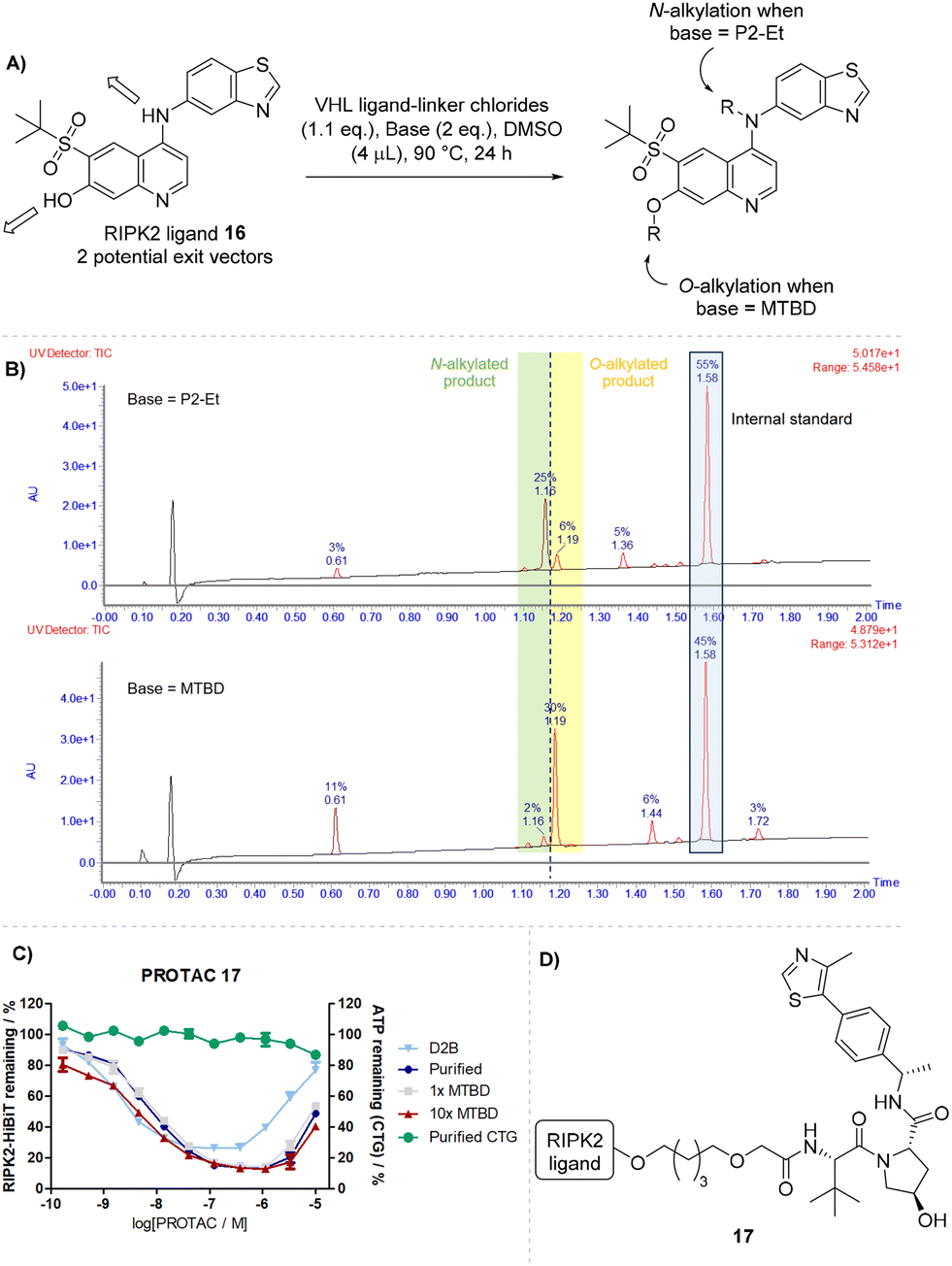 | ||
| Scheme 5 A) Alkylation library carried out in D2B; B) selective alkylation observed by LCMS when using P2-Et and MTBD as bases in 1536-well plate format; C) curves from HiBiT and CTG assays for D2B samples (N = 3), purified samples (N = 2), and spiking experiments of purified PROTAC with MTBD (N = 2); error bars represent SEM; D) structure of RIPK2 PROTAC 17. | ||
The degradation activity of these two isomeric libraries was assessed by RIPK2 HiBiT assay and analysis of the D2B data revealed that PROTACs synthesised with MTBD i.e. alkylation at the phenol O- were typically more potent, indicating that this exit vector was well tolerated. Assessment of the library also indicated that linker lengths of 8 to 12 atoms were well tolerated in the compounds tested and results from the D2B experiment highlighted that these should be the focus of any future optimisation efforts for RIPK2 PROTACs.
Comparison of the degradation profiles of the D2B and purified samples for O-alkylated PROTAC 17 showed comparable degradation activity and curve shape, which was not altered when stress tested with 1× and 10× spikes of excess MTBD base (Scheme 5C). This proof of concept demonstrated the feasibility of a selective POI binder alkylation in D2B format. The addition of these new conditions to the reaction toolbox offers significant opportunity for the acceleration of PROTAC projects via an alkylation approach.
Conclusions
Reductive amination, SNAr, Pd-mediated cross-coupling and alkylation are four of the top chemical transformations for any medicinal chemist. In this work, these key reactions have been developed for nanoscale synthesis in 1536-well plates, adding them to the toolbox of chemistry for PROTAC D2B experiments. Whilst the power of this approach lies in the ability to synthesise hundreds of compounds simultaneously, here we exemplify proof-of-concept for a range of new transformations in 1536-well plates. For each transformation described, excellent correlation between pure and crude samples has been exemplified and the effect of the reagents is shown to be minimal on degradation readout, showing that the chemistry is robust and can reliably identify new hits. Furthermore, the PROTACs synthesised in these transformations are not only identified as potent hits, but offer developable structures for the lead optimisation stages of a medicinal chemistry campaign. These examples highlight the key value in using D2B as a tool in the drug discovery process; compounds which are identified as potent degraders when tested as crude reaction mixtures can be resynthesised and then be progressed to follow up studies, whilst limiting the resource that would be required to carry out bespoke synthesis for all examples.Several key learnings were identified during this work. Although the increased throughput for hit-finding using these D2B approaches is advantageous, it is not advisable to use them as a replacement for all bespoke synthesis. Dmax values were reduced when increasing the number of synthetic transformations, resulting in lower purity samples, likely due to suppression of degradation level by competition with reactants or side-products. As such, it is important to remain cautious in selecting top compounds of interest for resynthesis based on Dmax values rather than pDC50. In contrast, reduced purity had a negligible effect on pDC50, presumably due to its logarithmic scale, and thus in a set of PROTACs with LCMS purities of 20–40%, 84% of compounds gave pDC50 values that were within assay error.
PROTACs targeting a range of POIs have been identified using single-step reductive amination, SNAr and alkylation chemistry. A case study for the multistep synthesis of BRD4 PROTACs including a Pd-mediated cross-coupling with no chromatographic purification has also been demonstrated, and further expanding the scope of conditions and scaffolds for metal-mediated cross-couplings without purification before cellular assay remains an area of interest, and we anticipate that additional multistep D2B workflows will be developed in future.
Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
RS, HJS, JC, DJB, AHM and DK conducted experiments and contributed to presentation of the data; RS prepared and edited draft manuscripts; PS and GAB provided supervision and manuscript review; JDH, DJB and AHM were responsible for project conceptualisation, manuscript review and editing, and supervision.Conflicts of interest
The authors declare the following competing financial interest(s): several of the authors are employees and/or shareholders at GSK.Acknowledgements
The authors thank the GSK/University of Strathclyde Collaborative Ph.D. program for funding and scientific resources. RS also thanks the Royal Commission for the Exhibition of 1851 for funding via an Industrial Fellowship and the Society of Chemical Industry for funding via an SCI Scholarship. Lastly, the authors would like to thank Robert Watson, Heather Barnett, Julie Fournier and James Thompson for their contributions to compound synthesis.Notes and references
- M. N. O'Brien Laramy, S. Luthra, M. F. Brown and D. W. Bartlett, Delivering on the promise of protein degraders, Nat. Rev. Drug Discovery, 2023, 22(5), 410–427 CrossRef PubMed.
- K. M. Sakamoto, K. B. Kim, A. Kumagai, F. Mercurio, C. M. Crews and R. J. Deshaies, Protacs: chimeric molecules that target proteins to the Skp1-Cullin-F box complex for ubiquitination and degradation, Proc. Natl. Acad. Sci. U. S. A., 2001, 98(15), 8554–8559 CrossRef CAS PubMed.
- B. E. Smith, S. L. Wang, S. Jaime-Figueroa, A. Harbin, J. Wang, B. D. Hamman and C. M. Crews, Differential PROTAC substrate specificity dictated by orientation of recruited E3 ligase, Nat. Commun., 2019, 10(1), 131 CrossRef.
- D. P. Bondeson, B. E. Smith, G. M. Burslem, A. D. Buhimschi, J. Hines, S. Jaime-Figueroa, J. Wang, B. D. Hamman, A. Ishchenko and C. M. Crews, Lessons in PROTAC Design from Selective Degradation with a Promiscuous Warhead, Cell Chem. Biol., 2018, 25(1), 78–87 CrossRef CAS.
- B. Jiang, E. S. Wang, K. A. Donovan, Y. Liang, E. S. Fischer, T. Zhang and N. S. Gray, Development of Dual and Selective Degraders of Cyclin-Dependent Kinases 4 and 6, Angew. Chem., Int. Ed., 2019, 58(19), 6321–6326 CrossRef CAS.
- D. Lv, P. Pal, X. Liu, Y. Jia, D. Thummuri, P. Zhang, W. Hu, J. Pei, Q. Zhang, S. Zhou, S. Khan, X. Zhang, N. Hua, Q. Yang, S. Arango, W. Zhang, D. Nayak, S. K. Olsen, S. T. Weintraub, R. Hromas, M. Konopleva, Y. Yuan, G. Zheng and D. Zhou, Development of a BCL-xL and BCL-2 dual degrader with improved anti-leukemic activity, Nat. Commun., 2021, 12(1), 6896 CrossRef CAS PubMed.
- D. A. Nalawansha and C. M. Crews, PROTACs: An Emerging Therapeutic Modality in Precision Medicine, Cell Chem. Biol., 2020, 27(8), 998–1014 CrossRef CAS.
- X. Han, L. Zhao, W. Xiang, C. Qin, B. Miao, T. Xu, M. Wang, C.-Y. Yang, K. Chinnaswamy, J. Stuckey and S. Wang, Discovery of Highly Potent and Efficient PROTAC Degraders of Androgen Receptor (AR) by Employing Weak Binding Affinity VHL E3 Ligase Ligands, J. Med. Chem., 2019, 62(24), 11218–11231 CrossRef CAS.
- J. Liu, H. Chen, H. U. Kaniskan, L. Xie, X. Chen, J. Jin and W. Wei, TF-PROTACs Enable Targeted Degradation of Transcription Factors, J. Am. Chem. Soc., 2021, 143(23), 8902–8910 CrossRef CAS PubMed.
- A. Mares, A. H. Miah, I. E. D. Smith, M. Rackham, A. R. Thawani, J. Cryan, P. A. Haile, B. J. Votta, A. M. Beal, C. Capriotti, M. A. Reilly, D. T. Fisher, N. Zinn, M. Bantscheff, T. T. MacDonald, A. Vossenkamper, P. Dace, I. Churcher, A. B. Benowitz, G. Watt, J. Denyer, P. Scott-Stevens and J. D. Harling, Extended pharmacodynamic responses observed upon PROTAC-mediated degradation of RIPK2, Commun. Biol., 2020, 3(1), 140–153 CrossRef CAS.
- R. Stevens, J. D. F. Thompson, J. C. L. Fournier, G. A. Burley, D. J. Battersby and A. H. Miah, Innovative, combinatorial and high-throughput approaches to degrader synthesis, Chem. Soc. Rev., 2024, 53, 4838–4861 RSC.
- R. Stevens, E. Bendito-Moll, D. J. Battersby, A. H. Miah, N. Wellaway, R. P. Law, P. Stacey, D. Klimaszewska, J. M. Macina, G. A. Burley and J. D. Harling, Integrated Direct-to-Biology Platform for the Nanoscale Synthesis and Biological Evaluation of PROTACs, J. Med. Chem., 2023, 66(22), 15437–15452 CrossRef CAS.
- L. Guo, Y. Zhou, X. Nie, Z. Zhang, Z. Zhang, C. Li, T. Wang and W. Tang, A platform for the rapid synthesis of proteolysis targeting chimeras (Rapid-TAC) under miniaturized conditions, Eur. J. Med. Chem., 2022, 236, 114317 CrossRef CAS.
- C. E. Hendrick, J. R. Jorgensen, C. Chaudhry, I. I. Strambeanu, J. F. Brazeau, J. Schiffer, Z. Shi, J. D. Venable and S. E. Wolkenberg, Direct-to-Biology Accelerates PROTAC Synthesis and the Evaluation of Linker Effects on Permeability and Degradation, ACS Med. Chem. Lett., 2022, 13(7), 1182–1190 CrossRef CAS PubMed.
- M. P. Plesniak, E. K. Taylor, F. Eisele, C. M. B. K. Kourra, I. N. Michaelides, A. Oram, J. Wernevik, Z. S. Valencia, H. Rowbottom, N. Mann, L. Fredlund, V. Pivnytska, A. Novén, M. Pirmoradian, T. Lundbäck, R. I. Storer, M. Pettersson, G. M. De Donatis and M. Rehnström, Rapid PROTAC Discovery Platform: Nanomole-Scale Array Synthesis and Direct Screening of Reaction Mixtures, ACS Med. Chem. Lett., 2023, 14(22), 1882–1890 CrossRef CAS PubMed.
- A. Brik, J. Muldoon, Y. C. Lin, J. H. Elder, D. S. Goodsell, A. J. Olson, V. V. Fokin, K. B. Sharpless and C. H. Wong, Rapid diversity-oriented synthesis in microtiter plates for in situ screening of HIV protease inhibitors, ChemBioChem, 2003, 4(11), 1246–1248 CrossRef CAS PubMed.
- L. Garnar-Wortzel, T. R. Bishop, S. Kitamura, N. Milosevich, J. N. Asiaban, X. Zhang, Q. Zheng, E. Chen, A. R. Ramos, C. J. Ackerman, E. N. Hampton, A. K. Chatterjee, T. S. Young, M. V. Hull, K. B. Sharpless, B. F. Cravatt, D. W. Wolan and M. A. Erb, Chemical Inhibition of ENL/AF9 YEATS Domains in Acute Leukemia, ACS Cent. Sci., 2021, 7(5), 815–830 CrossRef CAS PubMed.
- S. Kitamura, Q. Zheng, J. L. Woehl, A. Solania, E. Chen, N. Dillon, M. V. Hull, M. Kotaniguchi, J. R. Cappiello, S. Kitamura, V. Nizet, K. B. Sharpless and D. W. Wolan, Sulfur(VI) Fluoride Exchange (SuFEx)-Enabled High-Throughput Medicinal Chemistry, J. Am. Chem. Soc., 2020, 142(25), 10899–10904 CrossRef CAS.
- N. J. Gesmundo, B. Sauvagnat, P. J. Curran, M. P. Richards, C. L. Andrews, P. J. Dandliker and T. Cernak, Nanoscale synthesis and affinity ranking, Nature, 2018, 557(7704), 228–232 CrossRef CAS PubMed.
- R. P. Thomas, R. E. Heap, F. Zappacosta, E. K. Grant, P. Pogany, S. Besley, D. J. Fallon, M. M. Hann, D. House, N. C. O. Tomkinson and J. T. Bush, A direct-to-biology high-throughput chemistry approach to reactive fragment screening, Chem. Sci., 2021, 12(36), 12098–12106 RSC.
- R. P. Thomas, E. K. Grant, E. R. Dickinson, F. Zappacosta, L. J. Edwards, M. M. Hann, D. House, N. C. O. Tomkinson and J. T. Bush, Reactive fragments targeting carboxylate residues employing direct to biology, high-throughput chemistry, RSC Med. Chem., 2023, 14(4), 671–679 RSC.
- B. L. Roberts, Z. X. Ma, A. Gao, E. D. Leisten, D. Yin, W. Xu and W. Tang, Two-Stage Strategy for Development of Proteolysis Targeting Chimeras and its Application for Estrogen Receptor Degraders, ACS Chem. Biol., 2020, 15(6), 1487–1496 Search PubMed.
- J. Li, C. Li, Z. Zhang, Z. Zhang, Z. Wu, J. Liao, Z. Wang, M. McReynolds, H. Xie, L. Guo, Q. Fan, J. Peng and W. Tang, A platform for the rapid synthesis of molecular glues (Rapid-Glue) under miniaturized conditions for direct biological screening, Eur. J. Med. Chem., 2023, 258, 115567 Search PubMed.
- Z. Wang, S. Shaabani, X. Gao, Y. L. D. Ng, V. Sapozhnikova, P. Mertins, J. Kronke and A. Domling, Direct-to-biology, automated, nano-scale synthesis, and phenotypic screening-enabled E3 ligase modulator discovery, Nat. Commun., 2023, 14(1), 8437 CrossRef PubMed.
- D. G. Brown and J. Bostrom, Analysis of Past and Present Synthetic Methodologies on Medicinal Chemistry: Where Have All the New Reactions Gone?, J. Med. Chem., 2016, 59(10), 4443–4458 CrossRef.
- M. S. Gadd, A. Testa, X. Lucas, K.-H. Chan, W. Chen, D. J. Lamont, M. Zengerle and A. Ciulli, Structural basis of PROTAC cooperative recognition for selective protein degradation, Nat. Chem. Biol., 2017, 13(5), 514–521 CrossRef PubMed.
- Z. Chen, M. Wang, D. Wu, L. Bai, T. Xu, H. Metwally, Y. Wang, D. McEachern, L. Zhao, R. Li, J. Takyi-Williams, M. Wang, L. Wang, Q. Li, B. Wen, D. Sun and S. Wang, Discovery of CBPD-268 as an Exceptionally Potent and Orally Efficacious CBP/p300 PROTAC Degrader Capable of Achieving Tumor Regression, J. Med. Chem., 2024, 67(7), 5275–5304 CrossRef PubMed.
- J. A.-O. X. Desantis, A. A.-O. Mammoli, M. A.-O. Eleuteri, A. A.-O. Coletti, F. A.-O. Croci, A. Macchiarulo and L. A.-O. Goracci, PROTACs bearing piperazine-containing linkers: what effect on their protonation state?, RSC Adv., 2022, 12, 21968–21977 RSC.
- L. Di, E. H. Kerns, Y. Hong and H. Chen, Development and application of high throughput plasma stability assay for drug discovery, Int. J. Pharm., 2005, 297(1–2), 110–119 CrossRef PubMed.
- L. Goracci, J. Desantis, A. Valeri, B. Castellani, M. Eleuteri and G. Cruciani, Understanding the Metabolism of Proteolysis Targeting Chimeras (PROTACs): The Next Step toward Pharmaceutical Applications, J. Med. Chem., 2020, 63(20), 11615–11638 CrossRef.
- E. W. Baxter and A. B. Reitz, Reductive Aminations of Carbonyl Compounds with Borohydride and Borane Reducing Agents, in Organic Reactions, John Wiley and Sons, Inc., 2002, pp. 1–714 Search PubMed.
- S. Challenger, Y. Dessi, D. E. Fox, L. C. Hesmondhalgh, P. Pascal, A. J. Pettman and J. D. Smith, Development of a Scaleable Process for the Synthesis of the A2a Agonist, UK-371,104, Org. Process Res. Dev., 2008, 12(4), 575–583 CrossRef.
- M. M. Hansen, N. J. Kallman, T. M. Koenig, R. J. Linder, R. N. Richey, J. R. Rizzo, J. A. Ward, H. Yu, T. Y. Zhang and D. Mitchell, Double Heck Route to a Dibenzoxepine and Convergent Suzuki Cross-Coupling Strategy for the Synthesis of an MR Antagonist, Org. Process Res. Dev., 2017, 21(2), 208–217 CrossRef.
- A. Buitrago Santanilla, M. Christensen, L. C. Campeau, I. W. Davies and S. D. Dreher, P2Et Phosphazene: A Mild, Functional Group Tolerant Base for Soluble, Room Temperature Pd-Catalyzed C-N, C-O, and C-C Cross-Coupling Reactions, Org. Lett., 2015, 17(13), 3370–3373 CrossRef PubMed.
- C. Petrarca, E. Clemente, L. Di Giampaolo, R. Mariani-Costantini, K. Leopold, R. Schindl, L. V. Lotti, R. Mangifesta, E. Sabbioni, Q. Niu, G. Bernardini and M. Di Gioacchino, Palladium nanoparticles induce disturbances in cell cycle entry and progression of peripheral blood mononuclear cells: paramount role of ions, J. Immunol. Res., 2014, 295092 Search PubMed , (2314-7156 (Electronic)).
- F. M. Ferguson and N. S. Gray, Kinase inhibitors: the road ahead, Nat. Rev. Drug Discovery, 2018, 17(5), 353–377 CrossRef PubMed.
- F. Yu, M. Cai, L. Shao and J. Zhang, Targeting Protein Kinases Degradation by PROTACs, Front. Chem., 2021, 9, 679120 Search PubMed , (2296-2646 (Print)).
- D. P. Bondeson, A. Mares, I. E. D. Smith, E. Ko, S. Campos, A. H. Miah, K. E. Mulholland, N. Routly, D. L. Buckley, J. L. Gustafson, N. Zinn, P. Grandi, S. Shimamura, G. Bergamini, M. Faelth-Savitski, M. Bantscheff, C. Cox, D. A. Gordon, R. R. Willard, J. J. Flanagan, L. N. Casillas, B. J. Votta, W. den Besten, K. Famm, L. Kruidenier, P. S. Carter, J. D. Harling, I. Churcher and C. M. Crews, Catalytic in vivo protein knockdown by small-molecule PROTACs, Nat. Chem. Biol., 2015, 11(8), 611–617 CrossRef.
- A. H. Miah, I. E. D. Smith, M. Rackham, A. Mares, A. R. Thawani, R. Nagilla, P. A. Haile, B. J. Votta, L. J. Gordon, G. Watt, J. Denyer, D. T. Fisher, P. Dace, P. Giffen, A. Goncalves, I. Churcher, P. Scott-Stevens and J. D. Harling, Optimization of a Series of RIPK2 PROTACs, J. Med. Chem., 2021, 64(17), 12978–13003 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: All experimental procedures, analysis and compound characterisation data. Plots showing HiBiT data for key compounds, including spiking pure PROTACs with excess reagents. See DOI: https://doi.org/10.1039/d4md00760c |
| This journal is © The Royal Society of Chemistry 2025 |