
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence3D printable polymer foams with tunable expansion and mechanical properties enabled by catalyst-free dynamic covalent chemistry†
Rebecca M.
Johnson‡
 a,
Ariel R.
Tolfree‡
a,
Gustavo
Felicio Perruci
b,
Lyndsay C.
Ayers
a,
Niyati
Arora
a,
Emma E.
Liu
a,
Vijayalakshmi
Ganesh
a,
Hongbing
Lu
b and
Ronald A.
Smaldone
*a
a,
Ariel R.
Tolfree‡
a,
Gustavo
Felicio Perruci
b,
Lyndsay C.
Ayers
a,
Niyati
Arora
a,
Emma E.
Liu
a,
Vijayalakshmi
Ganesh
a,
Hongbing
Lu
b and
Ronald A.
Smaldone
*a
aUniversity of Texas at Dallas, Department of Chemistry and Biochemistry, 800 West Campbell Road, Richardson, Texas 75080, USA. E-mail: ronald.smaldone@utdallas.edu
bUniversity of Texas at Dallas, Department of Mechanical Engineering, 800 West Campbell Road, Richardson, Texas 75080, USA
First published on 28th January 2025
Abstract
Thermoset foams are some of the most common polymer materials in our lives. Despite their prevalence, they are notoriously difficult to form into complex shapes and finding a balance between mechanical strength, pore size and crosslinker density poses a significant challenge in optimizing their performance for specialized applications. 3D printing offers a solution by enabling the production of complex structures that can be foamed on demand using closed cell foaming microspheres, where a post-processing thermal treatment triggers expansion. However, foam expansion is typically constrained by its crosslinking density. This work introduces dynamic phosphodiester bonds into 3D printed polymers embedded with foaming agent microspheres to facilitate dynamic bond exchange during the thermal foaming process. With the inclusion of dynamic bonds, the foaming rate can be increased while also maintaining higher levels of crosslinking. These printed materials exhibit versatility, functioning effectively both before and after foaming, and offer potential for a diverse range of applications. Overall, this dynamic bond approach yields stronger, more expandable foams with improved energy dissipation and allows for the use of the printed foams in multiple lifecycles.
1. Introduction
Polymer foams are integral to many aspects of our lives, with approximately 18 million tons of polyurethane foam produced annually worldwide.1,2 Foams are valued for their lightweight nature, insulating and wide-ranging mechanical properties.3 Despite these benefits, foams face several inherent disadvantages that can limit their performance and recyclability. One issue is their contribution to plastic pollution, as they are often non-biodegradable and can occupy substantial space in landfills.2,4 Mechanical recycling methods such as re-bonding takes waste foam scraps to transform them into materials like carpet padding, but these often suffer from inconsistent mechanical properties.5A major challenge in foam material development lies in the balance between mechanical strength, pore size and crosslinker density.6 Foams are traditionally created by introducing gas into a polymer matrix, resulting in a cellular structure with varying pore sizes.7 In such materials, there is generally a trade-off between density and strength of the material. Larger pores reduce the density, which offers better lightweight and insulative characteristics but at the cost of mechanical properties.8 On the other hand, crosslinking density will enhance the toughness of the foam by creating a more robust network of chemical bonds but results in smaller pores and restricted foam expansion.9 With these trade-offs, it can be difficult to achieve a foam material with both large pores and high crosslinker content.
Dynamic covalent chemistry (DCC) has emerged as a promising solution to address these limitations. By enabling the reversible exchange of covalent bonds, DCC allows for foams to achieve a balance between pore size and mechanical strength. The resulting materials, known as covalent adaptable networks (CANs), benefit from the mechanical strength, thermal stability and chemical resistance of thermosets due to their crosslinked nature while also gaining the recyclability and reprocessability of thermoplastics.10,11 There are many types of dynamic linkages, including esters, imines, urethanes and Diels–Alder cycloadditions.12–17 One example is the phosphodiester bond, a dynamic ester linkage known for its good chemical stability at room temperature. This functional group can undergo exchange without the use of a catalyst, which can be often costly or toxic.18–20 These bonds undergo dynamic exchange at elevated temperatures, with phosphodiester bonds showing healing efficiency at temperatures as low as 50 °C and increased healing efficiency at 125 and 150 °C (reported as percent recovery of mechanical properties), and subsequently have been employed to create reprocessable thermosets.20
The use of DCC in polymer foams offers a unique approach to improving their properties. By incorporating dynamic bonds into the polymer matrix, foams can exhibit controlled pore size, enhanced expandability, and the ability to heal after mechanical damage. These properties are especially valuable when considering the benefit to improving the trade-off between pore size and mechanical properties. Additionally, DCC enables the creation of foams that are easier to recycle, reducing the environmental impact of foam waste and increasing the lifetime of foam-based products.16,21,22
3D printing offers a way to create polymer objects with simple or complex structures.23,24 With 3D printing, foam architectures not possible by traditional manufacturing can be achieved, creating foams with better resolutions and complex shapes.25,26 Extrusion based methods, specifically filament fabrication, involve extruding thermoplastics at elevated temperatures in a layer-by-layer manner. Additives like glass microballoons and cork have been incorporated into filaments to produce 3D printed foams.27,28 Porogens including camphene can be blended into the polymer and then later removed by vacuum to create porous materials.29 Additionally, 3D printing enables the creation of architected polymer foams, where the foam's cell structure can be designed through additive manufacturing to tailor the foam's properties.30,31 Photoprinting methods include stereolithography (SLA) and digital light projection (DLP), where a light source initiates polymerization of a liquid resin that includes monomers, a crosslinker and photoinitiator to create a 3D object layer-by-layer or point-by-point.32–36 However, research into 3D photoprinted foams is still limited.36–39 With photoprinting, blowing agents or foaming microspheres can be added to create a foamed material with a post-printing thermal treatment.
In this work, we demonstrate the potential of dynamic covalent chemistry to broaden the range of accessible properties of 3D photoprinted foams. By employing a phosphodiester crosslinker that facilitates dynamic bond exchange at temperatures used for expansion via foaming agent microspheres, we achieve higher rates of expansion and increased crosslinking densities without the need for a catalyst. This provides 3D printed foams with larger pore sizes and higher mechanical properties in comparison to traditional crosslinked non-dynamic foams. We analyze the expansion behavior for dynamic versus non-dynamic foams and investigate the role the dynamic chemistry plays in mechanical properties. Our approach includes multiple lifecycle stages, with 3D printed materials usable both before and after foaming.
2. Results and discussion
2.1 Resin formulations and printing
The first consideration in designing the resin was to ensure a homogenous dispersion of the microspheres. To achieve this, the monomer composition was selected to match the density of the foaming microspheres (∼ 1 g cm−3), which helps maintain uniform dispersion and consistent foaming after printing. If the density is mismatched, the microspheres will settle during printing leading to uneven foaming of the printed parts. The chosen monomer ratio, previously reported by Seo and coworkers,32 was a 4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2:4 ratio of butyl acrylate (BA, density 0.89 g cm−3): N,N-diethylacrylamide (NDA, density 0.92 g cm−3): N-(2-hydroxyethyl) acrylamide (HEAm, density 1.11 g cm−3) (Fig. 1) with tetraethylene glycol diacrylate (TEGDA) as the non-dynamic crosslinker.32 TEGDA was chosen as it has a similar molecular weight as the dynamic crosslinker, Bis[2-(methacryloyloxy)ethyl] phosphate (DPE). The dynamic crosslinker features a phosphodiester group,19 which can undergo dynamic bond exchange through an associative mechanism. However, unlike conventional ester exchange reactions, the phosphodiester does not require a catalyst for activation which is beneficial as many catalysts can be harmful to human health or the environment.20 The resins were prepared with varying crosslinker concentrations, ranging from 0 to 1.50 mol% of the non-dynamic TEGDA or dynamic DPE crosslinker.
:2:4 ratio of butyl acrylate (BA, density 0.89 g cm−3): N,N-diethylacrylamide (NDA, density 0.92 g cm−3): N-(2-hydroxyethyl) acrylamide (HEAm, density 1.11 g cm−3) (Fig. 1) with tetraethylene glycol diacrylate (TEGDA) as the non-dynamic crosslinker.32 TEGDA was chosen as it has a similar molecular weight as the dynamic crosslinker, Bis[2-(methacryloyloxy)ethyl] phosphate (DPE). The dynamic crosslinker features a phosphodiester group,19 which can undergo dynamic bond exchange through an associative mechanism. However, unlike conventional ester exchange reactions, the phosphodiester does not require a catalyst for activation which is beneficial as many catalysts can be harmful to human health or the environment.20 The resins were prepared with varying crosslinker concentrations, ranging from 0 to 1.50 mol% of the non-dynamic TEGDA or dynamic DPE crosslinker.
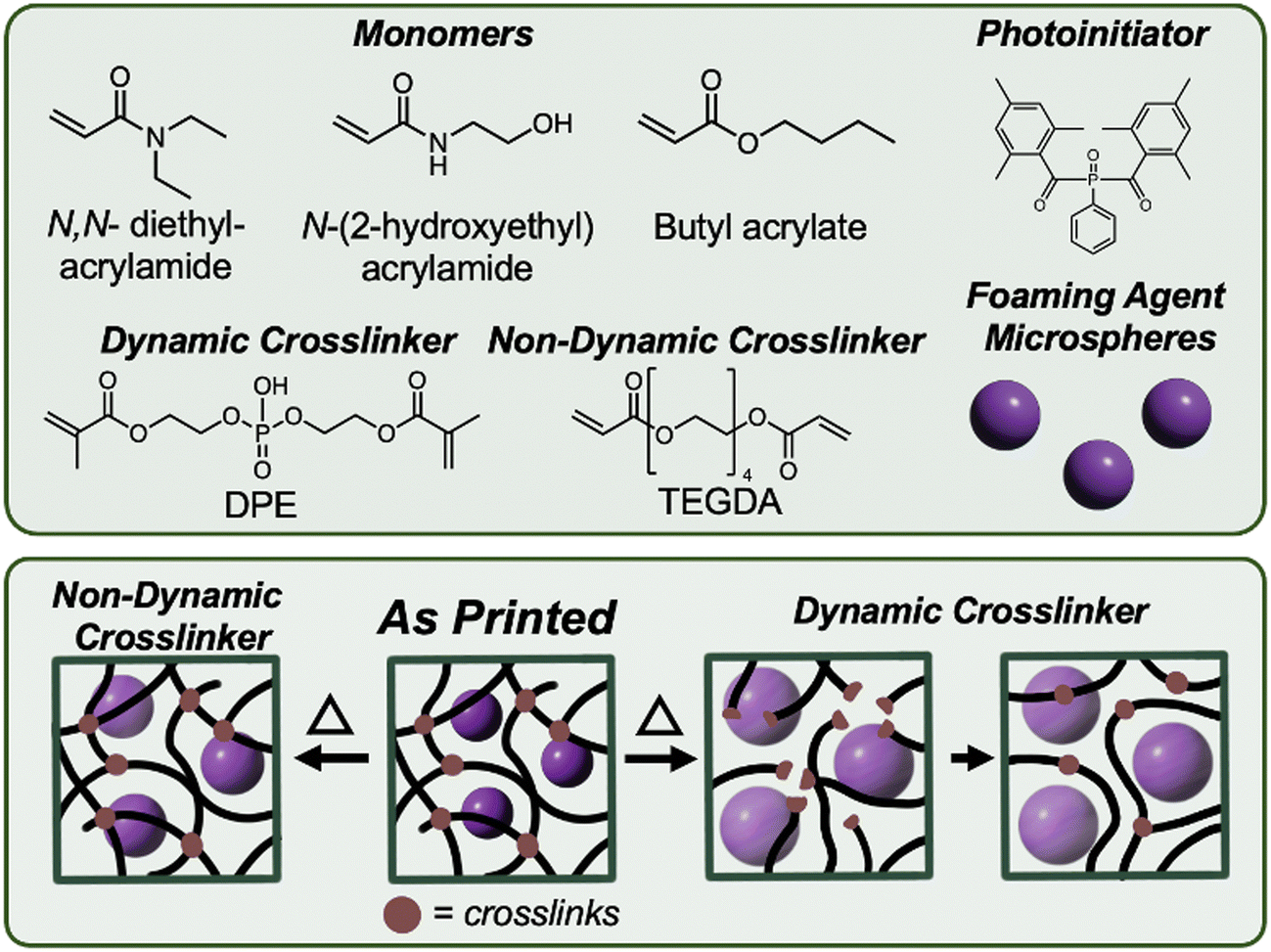 | ||
| Fig. 1 (Top) The resin components for the printed foams including the monomers, and the dynamic (DPE) or non-dynamic (TEGDA) crosslinker. (Bottom) As printed, the foaming agent microspheres remain trapped within the crosslinked network. Upon heating, the microspheres expand to form a closed cell foam material. With a non-dynamic crosslinker, the expansion of the spheres is limited by the space available within the polymer chains. In contrast, with a dynamic network, the bonds can rearrange during foaming, allowing for greater expansion while maintaining crosslinking density. | ||
Incorporating foaming microspheres into the resin formulation enables the printing of well-defined shapes and parts that can later be transformed into a closed cell foam by using thermally expandable microspheres mixed in with the print resin. Developed by Dow Chemical in the early 1970s, these microspheres consist of a thermoplastic shell that encases liquid hydrocarbon.40 The microspheres remain stable within the resins and form a closed cell foam after the printing and foaming process. When placed in an oven at 165 °C for 15 minutes, the thermoplastic shell softens, and the liquid hydrocarbon vaporizes causing the microspheres to expand. After foaming, the shell re-hardens in the expanded state and the hydrocarbon remains encapsulated. The resin composition consisted of microspheres (15 wt%) and photoinitiator (1 wt%). The viscosity of the resins were approximately 50–60 cps, based on the known values for the monomer components. The addition of the spheres did not affect the resin viscosity, printing time of the layers or the resolution of the parts. We were able to print a variety of different samples including 10 × 10 mm cylinders, ASTM D638 type V dog-bone samples and complex shapes (Fig. 2). Time-lapse videos of the foaming process are included in the ESI† for both printed cylinders and a complex balloon dog shape.
 | ||
| Fig. 2 (A) Scanning electron microscopy (SEM) images of loose microspheres before and after thermal foaming, illustrating free expansion. Printed cylinders before and after foaming for (B) dynamic and (C) non-dynamic crosslinkers. (D) Complex 3D prints before and after foaming, demonstrating compatibility with 3D printing and isotropic foam expansion. (E) Comparison of dynamic and non-dynamic foams at the same crosslinker concentration, showing that dynamic foams exhibit greater expansion with the same crosslinker content. | ||
2.2 Rate of foaming and dynamic crosslinker exchange
After the printing and post-curing process, the samples were foamed by heating at 165 °C for 15 minutes (Fig. 2). During foaming, the shape expands isotropically to form a larger, foamed version of the printed object. The increase in volume expansion is reported in Table 1. While longer heating times slightly increased expansion, signs of over-foaming began to show such as discoloration and layer separation, particularly in the polymers with the non-dynamic TEGDA crosslinker (Fig. S1†). As the crosslinking density increased for both the dynamic DPE and non-dynamic TEGDA systems, the expansion decreased. Notably, samples printed using the TEGDA crosslinker resulted in less expansion in comparison to those printed with same loading of dynamic DPE crosslinker. In formulations with no crosslinker, over-foaming occurred leading to surface bubbling (Fig. S1†).| Formulation | Foaming volume expansion (%) | Crosslinker (mol%) | Average pore area (μm2) |
|---|---|---|---|
| A-TEGDA-0.1 | 186 ± 12 | 0.10 | 87 ± 32 |
| A-DPE-0.5 | 179 ± 7 | 0.50 | 80 ± 27 |
| B-TEGDA-0.5 | 146 ± 8 | 0.50 | 41 ± 4 |
| B-DPE-1.0 | 142 ± 6 | 1.00 | 36 ± 6 |
| C-TEGDA-1.0 | 95 ± 3 | 1.00 | 32 ± 8 |
| C-DPE-1.5 | 101 ± 5 | 1.50 | 30 ± 1 |
To compare materials with similar foam expansion volumes, we selected three non-dynamic TEGDA and three dynamic DPE crosslinker formulations. The notations A, B, and C groups with similar expansion values regardless of crosslinker density or composition. Specifically, we compared the 0.10 mol% non-dynamic TEGDA to 0.50 mol% phosphodiester (186 vs. 179% volume expansion, respectively, A), 0.50 mol% non-dynamic TEGDA to 1.00 mol% dynamic phosphodiester (146 vs. 142%, B), and 1.00 mol% non-dynamic TEGDA to 1.50 mol% dynamic phosphodiester (95 vs. 101%, C), with these values seen in Table 1. This approach allowed us to focus on the properties of the materials with similar foam pore sizes, with average pore areas listed in Table 1, demonstrating that dynamic foams exhibit greater expansion than non-dynamic at higher concentrations of crosslinker.
During the foaming process, phosphodiester bonds undergo dynamic exchange, which contributes to increased foam expansion. This exchange can occur in several ways, with the first being a direct exchange between two phosphodiester linkages (type 1) (Fig. 3A).19 The second involves a rearrangement in which two phosphodiesters form triester and monoester products (type 2). Finally, dynamic exchange can occur between a phosphodiester and an ester group (type 3). These multiple pathways for dynamic bond exchange allow for easy rearrangement of the polymer network during the foaming. Additionally, the phosphodiesters can undergo a condensation reaction, with either free hydroxyls in the polymer network or other phosphodiesters.41 These condensation reactions can increase the crosslinking density of the phosphodiester networks, and repair missing crosslinks that could occur during the foaming process.
 | ||
| Fig. 3 (A) There are several possible ways the phosphodiester bond can undergo dynamic bond exchange. Two types include direct phosphate ester exchanges and a third exchange between a phosphate ester and an ester group. (B) Condensation reactions that are possible with phosphodiester bonds. (C) Left-fourier transform infrared spectroscopy (FTIR) from 3000 cm−1 to 3700 cm−1 showing the characteristic hydroxyl peaks Middle-FTIR from 1670–1780 cm−1 showing the characteristic carbonyl peak. Right-FTIR from 800–1300 cm−1 showing the characteristic phosphate ester peaks. (D) Differential scanning calorimetry (DSC) of as printed and annealed samples showing the glass transition temperatures. | ||
To better understand the dynamic phosphodiester bond exchange occurring in the polymer network, small tile-shaped samples were printed without foaming agent and heated overnight at 165 °C and analyzed using Fourier transform infrared spectroscopy (FTIR). Changes in the characteristic phosphodiester peaks confirmed dynamic bond exchange occurred as a mixed transesterification and condensation mechanism (Fig. 3B).19,41 Depending on the concentration of the phosphodiester crosslinker, different responses were observed in the peak intensities for both the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretch at 1730 cm−1 and P–OH stretch at 850 and 1050 cm−1. At the lower concentration 0.50 mol% (DPE-0.5), there was a more significant decrease in peak intensity, whereas the higher concentration of 1.50 mol% (DPE-1.5) showed minimal changes. We hypothesize that the concentration of the phosphodiester crosslinker affects the type of dynamic bond exchange, influencing whether the same bond type reforms or new bonds are created. With higher crosslinker density, the phosphate diesters are more likely to participate in a direct phosphate diester exchange, shown as type 1 in Fig. 3. This is because there are more phosphodiester crosslinks available for a direct exchange indicated by a lack of change in peak intensity. With less crosslinker in the networks, the phosphate esters are more likely to exchange with esters in the polymer backbone sidechains (e.g., butyl acrylate), or undergo condensation with free hydroxyls, as these are more abundant than the lower concentration of phosphodiester bonds.41 This is observed by a decrease in the peak intensity of the hydroxyl, carbonyl and phosphonic acid signals after heating. The complete FTIR spectra before and after the thermal treatment, along with spectra of the foamed prints are included in the ESI (Fig. S3†).
O stretch at 1730 cm−1 and P–OH stretch at 850 and 1050 cm−1. At the lower concentration 0.50 mol% (DPE-0.5), there was a more significant decrease in peak intensity, whereas the higher concentration of 1.50 mol% (DPE-1.5) showed minimal changes. We hypothesize that the concentration of the phosphodiester crosslinker affects the type of dynamic bond exchange, influencing whether the same bond type reforms or new bonds are created. With higher crosslinker density, the phosphate diesters are more likely to participate in a direct phosphate diester exchange, shown as type 1 in Fig. 3. This is because there are more phosphodiester crosslinks available for a direct exchange indicated by a lack of change in peak intensity. With less crosslinker in the networks, the phosphate esters are more likely to exchange with esters in the polymer backbone sidechains (e.g., butyl acrylate), or undergo condensation with free hydroxyls, as these are more abundant than the lower concentration of phosphodiester bonds.41 This is observed by a decrease in the peak intensity of the hydroxyl, carbonyl and phosphonic acid signals after heating. The complete FTIR spectra before and after the thermal treatment, along with spectra of the foamed prints are included in the ESI (Fig. S3†).
To further understand the effects of heating upon the dynamic polymer network, differential scanning calorimetry (DSC) was conducted on both printed samples without foaming agent and samples without foaming agent, that were heat treated at 150 °C overnight to anneal them. The non-dynamic TEGDA networks all show a consistent 5 °C increase in the glass transition temperature (Tg), which can be attributed to the removal of water from the polymer network. The dynamic DPE networks, however, show an increase of Tg with the increase of crosslinker concentration. A-DPE-0.5 increases 6.5 degrees, B-DPE-1.0 increases 9.7 °C and C-DPE-1.5 increases 12.1 °C. The increase of Tg with increasing crosslinker content indicates several behaviors of the dynamic ability. This includes the annealing step providing the dynamic bonds the ability to rearrange to decrease stress and improve network homogeneity, and the potential for the network to undergo condensation to increase crosslinking density.
2.3 Characterization of foams
The tensile and compressive properties of the printed samples were evaluated in three conditions: no foam, unfoamed, and foamed. This allowed for a comparison of the polymer networks alone (without microspheres), as well as before and after foaming. For compression, the unfoamed non-dynamic TEGDA polymers exhibited similar performance to one another, while the unfoamed dynamic DPE polymers also showed consistent behavior, with the dynamic formulations overall having higher compressive strength than the non-dynamic materials. The no foam and unfoamed materials behaved like a ductile material, with a clear linear elastic initial behavior. From the stress–strain curves, the Young's modulus, plateau stress (for foamed samples), compressive yield stress (for non-foamed samples), energy loss, and energy absorption were calculated. Specifically, the unfoamed TEGDA polymers had compressive strengths ranging from 130–150 MPa, while the dynamic DPE polymers ranged from 180–216 MPa (Fig. 4). Polymers, both with dynamic and non-dynamic crosslinker, without any foaming agent were generally the strongest materials among the three states, with the C-DPE-1.5 and C-TEGDA-1.0 having the highest compressive strengths (Fig. S13 and Table S4†).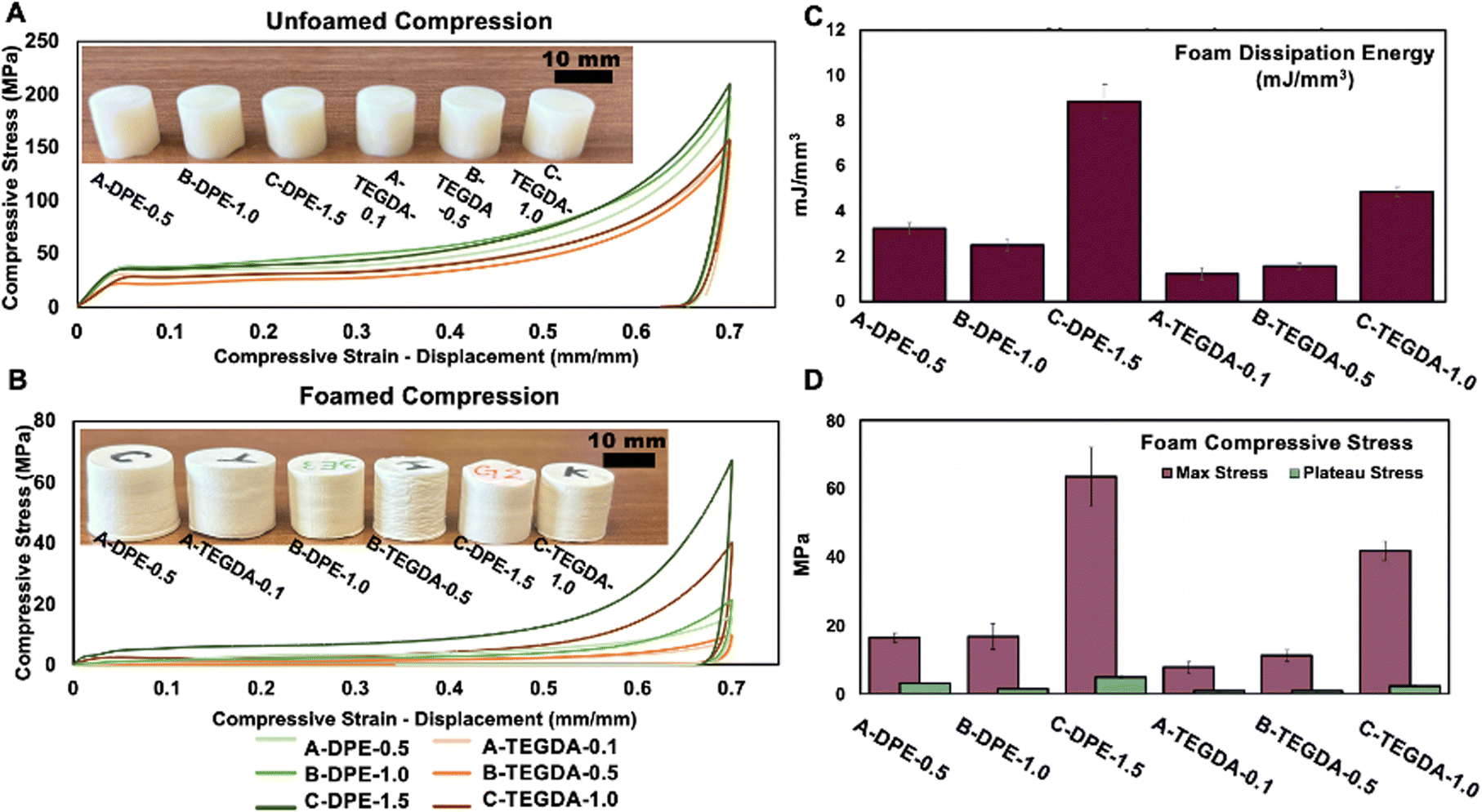 | ||
| Fig. 4 (A) Compression curves of unfoamed samples comparing similar foaming volume expansion rates up to 70% strain. (B) Compression tests for the foamed samples comparing similar volume expansion values up to 70% strain. (C) Energy dissipation values for foamed samples. (D) Plateau and max stress values. | ||
After foaming, the compressive properties of the polymers changed significantly. Unlike the unfoamed samples, which showed similar compressive strengths, the foamed polymers showed a significant increase in compressive strengths with the increase of crosslinking density (Fig. 4). In all cases, the dynamic foams exhibited higher compressive strengths compared to non-dynamic foams with comparable expansion. Not only are the compressive strengths and moduli increased for the dynamic foams, but they also have significantly increased energy dissipating properties in comparison to the non-dynamic foams. Energy dissipation is calculated as the area under the loading and unloading curves All the dynamic foam formulations dissipate nearly twice as much energy as their non-dynamic counterparts.
The compressive stress–strain curves indicate that for foams with comparable expansion, the dynamic phosphodiester polymers exhibit higher compressive strength, attributed to the increased crosslinking density. By employing dynamic bonds, higher rates of foaming are achieved along with increased mechanical strength in comparison to a traditionally crosslinked foam. The presence of phosphodiester bonds in the polymer can also promote hydrogen bonding, which can further strengthen the network. These bonds can form between the P–OH groups of the phosphodiester and CO carbonyl groups from both the monomers and phosphodiester crosslinker.19 Detailed compressive property data can be found in the ESI (Tables S4, S5 and S6†), while the densities of the foamed cylinders, which increase with crosslinking density, are reported in Table S3.†
The tensile properties of the no foam, unfoamed and foamed polymers were analyzed for comparison. The no foam polymers are the most strong and tough (Fig. S16†) in comparison to the unfoamed and foamed polymers in tensile behavior. These properties decrease for the unfoamed polymers, due to the incorporation of the microspheres (Fig. S17†) and decrease more after foaming to yield a lower overall strain at break and ultimate tensile strength (Fig. S18†). Overall, with increasing crosslinker content the tensile strength increases, and the dynamic and non-dynamic counterparts have similar tensile properties.
Both the TEGDA and DPE polymers exhibit similar thermal stability, with no significant thermal degradation at the temperatures required for foaming and dynamic bond exchange, as confirmed by thermogravimetric analysis (TGA) (Fig. S4†). Mass loss below 200 °C is likely due to the foaming spheres, as further thermal treatment after 165 °C for 15 minutes seems to affect the foams interlayer adhesion. The glass transition temperatures were determined by differential scanning calorimetry of the polymers without any foaming agent. The dynamic DPE polymers had glass transition temperatures from 68–73 °C and the non-dynamic TEGDA had glass transition temperatures from 67–74 °C (Fig. S20†). Stress relaxation tests were performed on both as printed no foam samples and annealed no foam samples from 50 °C to 110 °C (Fig. S28–31†), showing the rate of relaxation. Gel content and swelling experiments were performed on the no foam, unfoamed and foamed polymers to confirm crosslinking and demonstrate completion of the polymerization before and after foaming (Fig. S11 and 12†). For all samples, swelling in both methanol and THF revealed a trend of decreased swelling with increasing crosslinking concentration, indicating the polymer network remains intact for both the TEGDA and DPE containing formulations. This shows that the phosphodiester bonds remain crosslinked after dynamic exchange and foaming. Additionally, gel content exceeded 90% for all formulations, demonstrating good monomer-to-polymer conversion and effective crosslinking.
2.4 Foam recycling
Both 3D photoprinted thermoset polymers and foams are known for their poor recyclability.12,22,42 Current literature shows that dynamic chemistry is a promising method to help overcome the issues in recycling and reprocessing these materials.10,12,13,16,21,22,42–44 Bearing this in mind, we leveraged the dynamic bonds in the foams that not only created a higher expansion rate, but also to extend the lifecycle of thermoset foams. We set out to show that these materials can be used in multiple forms, first unfoamed and then after use they could be foamed for a second life. In this process, we compressed unfoamed printed cylinders to 70% of their height. The compressed cylinder returned to nearly its original shape after resting for 2–3 days and appeared visually like an uncompressed cylinder (Fig. 5). The height and diameter recovery values for the dynamic DPE cylinders are 95–96 and 105–107% respectively. The height and diameter recovery values for the TEGDA cylinders are 93–96 and 104–108% respectively. Nearly all the height is recovered for the cylinders, and they are slightly wider than before compression. | ||
| Fig. 5 (A) Workflow for reverse foam recycling, shown with all C-DPE-1.5 cylinders. (B) Compression curves of foamed samples comparing as foamed to reverse foamed samples to 70% strain. (C) When compressed to 70% of their initial height, mechanical damage occurs to the polymer network. This mechanical damage is unable to be repaired in the TEGDA polymers during the foaming process, but with the dynamic DPE crosslinker, bonds can rearrange to provide consistent mechanical properties in comparison to an as-printed foam. | ||
The cylinders were thermally expanded after the recovery period to produce a pre-compressed (PC) foam (Fig. S19†). The compressive properties were then compared to the as-printed foams. The dynamic DPE PC foams performed more consistently in comparison to the non-dynamic TEGDA PC foams, while all generally showed a decrease in the Young's modulus indicating a softening of the elastic region of the foams (Fig. 5B). The presence of multiple moduli in some, but not all, of the foamed materials is likely due to regions with varying foam densities. However, the reported modulus represents the majority of the material properties. While the additional moduli are provided in the ESI,† they will not be analyzed in detail in this work. The non-dynamic TEGDA foams had varying and inconsistent compressive strengths however, in comparison to their as-foamed counterparts. The high non-dynamic TEGDA had the highest compressive strength, and performed more similarly to an unfoamed sample, likely due to poor foaming. The dynamic DPE foams retained their plateau stress better in comparison to the non-dynamic TEGDA crosslinked foams. The B-DPE-1.0 PC performed better than the as-printed B-DPE-1.0 with an increase in energy dissipation, plateau stress and max stress. PC A-DPE-0.5 did not recover its properties as well as the other dynamic foams, likely due to its lower concentration of reversible crosslinks available for damage repair. The values and percent change for as printed to PC foams are reported in Table S10.† The dynamic phosphodiester can provide damage repair during foaming through bond rearrangement, while the non-dynamic TEGDA foam cannot. Some of the repair can also be attributed to the ability for condensation to occur within the network, replacing broken crosslinks to repair damage. This shows that the dynamic phosphodiester polymers can withstand a significant amount of compressive damage in their as-printed state and then be foamed to create polymers that behave similarly compared to an as-foamed print. This shows the potential to create complex, 3D printed foams that have multiple uses both pre- and post-foaming and can be effectively diverted from landfills as waste through use in multiple manners.
3. Conclusion
In this work, we have shown that dynamic covalent phosphodiester crosslinkers can be used to produce 3D photoprintable polymer foams with a wider range of mechanical properties and life cycle control compared to non-dynamic TEGDA crosslinked foams. The incorporation of a thermally activated dynamic crosslinker enables greater foam expansion, even in materials with increased crosslinking densities. This dynamic crosslinking also results in improved compressive strength and energy dissipation compared to conventional, non-dynamic TEGDA thermoset foams. Furthermore, these materials demonstrate significant potential for extending the lifetime of 3D printed phosphodiester foams, as they can be re-used, both pre- and post-foaming with minimal change in mechanical properties resist damage.4. Experimental
4.1 Materials
All chemicals are used as received unless otherwise noted. N-(2-Hydroxyethyl) acrylamide, butyl acrylate, tetraethylene glycol diacrylate, N,N-diethylacrylamide were purchased from TCI. Phenylbis(2,4,6-trimethylbenzoyl) phosphine oxide and Bis[2-(methacryloyloxy)ethyl] phosphate were purchased from Sigma-Aldrich. Isopropanol was purchased from Fisher Scientific. Unexpanded microspheres (920DU40) were obtained from Nouryon.4.2 Resin formulations
The composition of all resin formulations is shown in Fig. 1. The resins were made with varying amounts of tetraethylene glycol diacrylate or Bis[2-(methacryloyloxy)ethyl] phosphate as the crosslinker from 0.05 mol% to 1.50 mol percent of the total resin composition with amounts listed in Table 1, and the monomers in a 4:2:4 ratio of butyl acrylate: N,N-diethylacrylamide: N-(2-hydroxyethyl) acrylamide. The resins also included 1 wt% of BAPO as the photoinitiator and 15 wt% of the foaming agent microspheres, 920DU40. All materials were combined in an amber vial and sonicated for 30 minutes to allow the photoinitiator to dissolve and a homogenous dispersion of the microspheres.
4.3 3D printing
All samples were printed using an Anycubic Photon Mono 4K or an Anycubic Photon Mono 2. The printed specimens included cylindrical discs with a diameter of 10 mm and a height of 10 mm, ASTM D638 type V dog-bones and complex structures such as gyroid lattices and a balloon animal dog. The printers use a 405 nm light source, and the general printing parameters are as follows: 6 base layers, 22 seconds base exposure, 10 seconds regular exposure, 0.25 s light off between layers. Once printed, unreacted excess resin was washed off using isopropanol and they were post-cured under a 405 nm lamp for 24 h.4.4 Foaming of 3D printed materials and microspheres
After post curing, the printed materials were foamed by placing in an oven at 165 °C for 15 minutes. Free microsphere foaming agent was also foamed in a Petri dish at 165 °C for 15 minutes.4.5 Characterization
Differential scanning calorimetry (DSC) was performed using a TA DSC 2500 using 5–10 mg samples loaded into Tzero aluminum pans with Tzero Hermetic lids. The heating and cooling rate was 10 °C min−1 from −40 to 200 °C for three heating/cooling cycles with a one-minute isothermal before each ramp. The data from the second cycle was selected for all experiments. Annealed samples were prepared by an overnight thermal treatment at 150 °C.
4.6 Mechanical testing
Two main techniques were used for the foamed samples to identify the modulus of elasticity and the plateau stress, following previously reported methods.45–48 The calculation of the modulus of elasticity was made by fitting a linear equation up to the peak in the derivative of the stress and strain curve (dσ/dε) which measures the changes in stress in relation to the respective strain and can show when there is an inflection in the curve.44 The main plateau stress was identified by the intersection of the linear fit of the plateau region and the linear fit of the elastic region.40–48 Finally, the energy dissipation was calculated by the hysteresis of the load and unload cycle (the area under the loading phase minus the area under the unloading phase), the energy absorption of the samples were measured by the area under the loading phase up until the maximum energy efficiency stress defined by  .45
.45
4.7 Foam recycling
Unfoamed cylinders used for compression testing following the procedures listed above were foamed after the samples returned to their original height, usually after two to three days. They were foamed using the same foaming process as listed above, the surfaces of the specimens were lightly sanded to remove any imperfections so cylindrical samples would have flat surfaces for secondary compression testing. They were then compressed using the same process. The dimensions of the cylinders as printed, immediately after compression and after returning to their original height were recorded to determine the percent recovery of both height and diameter. This was done in triplicate for each crosslinking density to obtain averages and standard deviation.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge the support of the Welch Foundation (X-AT-0004-20230731), the National Science Foundation (2323729, 2219347) and Department of Energy (DE-NA0003962, DE-EE0011016, DE-EE0010200). The dynamic mechanical analyzer was obtained with support from the Army Research Office (W911NF-24-1-0063). H. Lu also thanks the Louis A. Beecherl Jr. Chair for additional support.References
- P. Kanchanapiya, N. Intaranon and T. Tantisattayakul, J. Environ. Manage., 2021, 280, 111638 CrossRef CAS PubMed.
- W. Yang, Q. Dong, S. Liu, H. Xie, L. Liu and J. Li, Procedia Environ. Sci., 2012, 16, 167 CrossRef CAS.
- Y. Deng, R. Dewil, L. Appels, R. Ansart, J. Baeyens and Q. Kang, J. Environ. Manage., 2021, 278, 111527 CrossRef CAS PubMed.
- D. K. Schneiderman and M. A. Hillmyer, Macromolecules, 2017, 50, 3733 CrossRef CAS.
- K. M. Zia, H. N. Bhatti and I. A. Bhatti, React. Funct. Polym., 2007, 67, 675 CrossRef CAS.
- Z. Lan, R. Daga, R. Whitehouse, S. McCarthy and D. Schmidt, Polymer, 2014, 55, 2635 CrossRef CAS.
- V. Srivastava and R. Srivastava, J. Mater. Sci., 2014, 49, 2681 CrossRef CAS.
- X. C. Xia, X. W. Chen, Z. Zhang, X. Chen, W. M. Zhao, B. Liao and B. Hur, J. Magnesium Alloys, 2013, 1, 330 CrossRef CAS.
- S. Aney and A. Rege, Math. Mech. Solids, 2023, 28, 1624 CrossRef.
- C. J. Kloxin and C. N. Bowman, Chem. Soc. Rev., 2013, 42, 7161 RSC.
- B. Krishnakumar, R. V. S. P. Sanka, W. H. Binder, V. Parthasarthy, S. Rana and N. Karak, J. Chem. Eng., 2020, 385, 123820 CrossRef CAS.
- R. M. Johnson, K. P. Cortés-Guzmán, S. D. Perera, A. R. Parikh, V. Ganesh, W. E. Voit and R. A. Smaldone, J. Polym. Sci., 2024, 62, 2585–2596 CrossRef CAS.
- K. P. Cortés-Guzmán, A. R. Parikh, M. L. Sparacin, A. K. Remy, L. Adegoke, C. Chitrakar, M. Ecker, W. E. Voit and R. A. Smaldone, ACS Sustainable Chem. Eng., 2022, 10, 13091 CrossRef.
- K. P. Cortés-Guzmán, A. R. Parikh, M. L. Sparacin, R. M. Johnson, L. Adegoke, M. Ecker, W. E. Voit and R. A. Smaldone, Polym. Chem., 2023, 14, 2697 RSC.
- A. Durand-Silva, K. P. Cortés-Guzmán, R. M. Johnson, S. D. Perera, S. D. Diwakara and R. A. Smaldone, ACS Macro Lett., 2021, 10, 486 CrossRef CAS PubMed.
- D. T. Sheppard, K. Jin, L. S. Hamachi, W. Dean, D. J. Fortman, D. J. Fortman, C. J. Ellison and W. R. Dichtel, ACS Cent. Sci., 2020, 6, 921 CrossRef CAS PubMed.
- J. Smith-Jones, N. Ballinger, N. Sadaba, X. Lopez de Pariza, Y. Yao, S. L. Craig, H. Sardon and A. Nelson, RSC Appl. Polym., 2024, 2, 434 RSC.
- K. Deng, X. Feng, H. Yang and C. Yan, Eur. Polym. J., 2023, 196, 112286 CrossRef CAS.
- X. Feng and G. Li, J. Chem. Eng., 2021, 417, 129132 CrossRef CAS.
- X. Feng and G. Li, ACS Appl. Mater. Interfaces, 2020, 12, 57486 CrossRef CAS PubMed.
- M. Sun, D. T. Sheppard, J. P. Brutman, A. Alsbaiee and W. R. Dichtel, Macromolecules, 2023, 56, 6978 CrossRef CAS.
- S. Kim, K. Li, A. Alsbaiee, J. P. Brutman and W. R. Dichtel, Adv. Mater., 2023, 35, 2305387 CrossRef CAS PubMed.
- A. Bagheri and J. Jin, ACS Appl. Polym. Mater., 2019, 1, 593 CrossRef CAS.
- R. M. Johnson and R. A. Smaldone, Polym. Interact., 2024 Search PubMed . Article ASAP.
- P. Bedarf, A. Dutto, M. Zanini and B. Dillenburger, Autom. Constr., 2021, 130, 103861 CrossRef.
- T. R. Klein, A. Kirillova, K. Gall and M. L. Becker, RSC Appl. Polym., 2023, 1, 73 RSC.
- B. Dileep and M. Doddamani, Compos., Part C: Open Access, 2021, 6, 100181 CAS.
- N. Gama, A. Ferreira and A. Barros-Timmons, Mater. Des., 2019, 179, 107905 CrossRef CAS.
- J. L. Self, H. Xiao, M. M. Hausladen, R. A. Bramanto, S. S. Usgaonkar and C. J. Ellison, ACS Appl. Mater. Interfaces, 2022, 14, 49244 CrossRef CAS PubMed.
- H. Jiang, H. Ziegler, Z. Zhang, H. Meng, D. Chronopoulos and Y. Chen, Mater. Des., 2020, 194, 108946 CrossRef CAS.
- H. Jiang, A. Coomes, Z. Zhang, H. Ziegler and Y. Chen, Composites, Part B, 2021, 224, 109183 CrossRef CAS.
- S. E. Seo, Y. Kwon, N. D. Dolinski, C. S. Sample, J. L. Self, C. M. Bates, M. T. Valentine and C. J. Hawker, ACS Appl. Polym. Mater., 2021, 3, 4984 CrossRef CAS.
- N. Corrigan, A. L. Mutch, C. Boyer and S. C. Thickett, RSC Appl. Polym., 2024, 2, 914 RSC.
- A. Priester, J. Yeng, Y. Zheng, R. Wang and A. J. Convertine, RSC Appl. Polym., 2024, 2, 612 RSC.
- F. Kröger, R. Eichelmann, G. Sauter, A. Pollien, P. Tegeder, L. H. Gade and E. Blasco, RSC Appl. Polym., 2024, 2, 847 RSC.
- L. Hodásová, I. Isarn, F. Bravo, C. Alemán, N. Borrás, G. Fargas and E. Armelin, RSC Appl. Polym., 2024, 2, 284 RSC.
- Y. Kwon, S. E. Seo, J. Lee, S. Berezvai, J. Read de Alaniz, C. D. Eisenbach, R. M. McMeeking, C. J. Hawker and M. T. Valentine, Compos. Commun., 2023, 37, 101453 CrossRef.
- D. M. Wirth, A. Jaquez, S. Gandarilla, J. D. Hochberg, D. C. Church and J. K. Pokorski, ACS Appl. Mater. Interfaces, 2020, 12, 19033 CrossRef CAS PubMed.
- D. M. Wirth, J. D. Hochberg and J. K. Pokorski, ACS Appl. Mater. Interfaces, 2022, 14, 54245 CrossRef CAS PubMed.
- M. Jonsson, O. Nordin and E. Malmström, J. Appl. Polym. Sci., 2011, 121, 369 CrossRef CAS.
- A. Durand-Silva and R. A. Smaldone, ACS Cent. Sci., 2020, 6, 836 CrossRef CAS PubMed.
- P. Banaszek and M. Podgórski, SSRN, 2024, preprint. DOI:10.2139/ssrn.5002108.
- H. Sardon and A. P. Dove, Science, 2018, 360, 380–381 CrossRef CAS PubMed.
- E. Sanchez-Rexach, T. G. Johnston, C. Jehannno, H. Sardon and A. Nelson, Chem. Mater., 2020, 32, 7105–7119 CrossRef CAS.
- R. Q. Albuquerque, J. Meuchelböck and H. Ruckdäschel, J. Polym. Sci., 2024, 62, 1034–1043 CrossRef CAS.
- D. T. Morton, A. Reyes, A. H. Clausen and O. S. Hopperstad, Mater. Today Commun., 2020, 23, 100917 CrossRef CAS.
- M. F. Ashby, Philos. Trans. R. Soc., A, 2006, 364, 15–30 CrossRef CAS PubMed.
- R. Bouix, P. Viot and J. L. Lataillade, Int. J. Impact Eng., 2009, 36, 329–342 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4lp00374h |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2025 |