
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Selective and sustainable quinoline hydrogenation with a robust hierarchical catalyst framework†
Azina
Rahmani
 ab,
Diego R.
Javier-Jiménez
ab,
Deborah
Israel
c,
Brian
Butkus
d,
Lei
Zhai
ae,
Parag
Banerjee
bde,
William E.
Kaden
c,
Akihiro
Kushima
*bd and
Titel
Jurca
*abe
ab,
Diego R.
Javier-Jiménez
ab,
Deborah
Israel
c,
Brian
Butkus
d,
Lei
Zhai
ae,
Parag
Banerjee
bde,
William E.
Kaden
c,
Akihiro
Kushima
*bd and
Titel
Jurca
*abe
aDepartment of Chemistry, University of Central Florida, Orlando, Florida 32816, USA. E-mail: Titel.Jurca@ucf.edu
bRenewable Energy and Chemical Transformations Cluster, University of Central Florida, Orlando, Florida 32816, USA
cDepartment of Physics, University of Central Florida, Orlando, Florida 32816, USA
dDepartment of Materials Science and Engineering, University of Central Florida, Orlando, Florida 32816, USA
eNanoScience Technology Center, University of Central Florida, Orlando, Florida 32826, USA
First published on 3rd July 2025
Abstract
A hierarchical heterogeneous palladium on nickel foam-based catalyst system (Al2O3–Pd–D/Ni) was demonstrated for the selective hydrogenation of quinoline and quinoline derivatives under low H2 pressures, with green solvents (ethanol, ethanol water mixture). The catalyst framework features very low palladium loadings and is highly reusable under facile handling, requiring no filtration or other separation aids, and notably demonstrates no loss in reactivity or alteration of selectivity over multiple recycling trials. Theoretical calculations and X-ray photoelectron spectroscopy studies point to a fully-reduced Pd surface as the necessary active site for catalysis, arising from the in situ reduction of the PdOx surface sites of the air-stable hierarchical material system.
Introduction
Heterogeneous catalytic processes utilizing palladium play a crucial role in a myriad of chemical transformations. While Pd-based catalysts are unequivocally important, there is a need for the sustainable use of palladium due to its increasingly high costs, low natural abundance, high demand across other industries, and concentrated global production.1–4 Thus, it is imperative to develop robust catalytic systems that can facilitate chemical transformations with low Pd loadings, are amenable to facile reusability, and ideally, operate under sustainable conditions.5–8We have recently described a hierarchical catalyst framework based on nickel foams as contiguous monolith supports.9 The catalyst relies on bottom-up grown ultralow-loading Pd/PdOx nanoparticles (0.017% w/w; 15.86 ± 6.85 nm) on a carbonized polydopamine interface, with a subsequent ∼2 nm atomic layer deposition (ALD) overcoat of Al2O3; Al2O3–Pd–D/Ni (Fig. 1A). It was demonstrated that the ultrathin Al2O3 overcoat is critical for stabilizing the catalyst framework, and for preferentially blocking low-coordinate sites on the Pd particles which can lead to enhanced selectivity. Al2O3–Pd–D/Ni proved to be an exceptionally robust catalyst for the selective hydrogenation of styrene derivatives, nitroaromatics, and anthracene under mild conditions using ethanol as a sustainable solvent (Fig. 1B). Furthermore, owing to the contiguous porous nature of the Ni foam support, Al2O3–Pd–D/Ni was directly applicable to flow processes.9
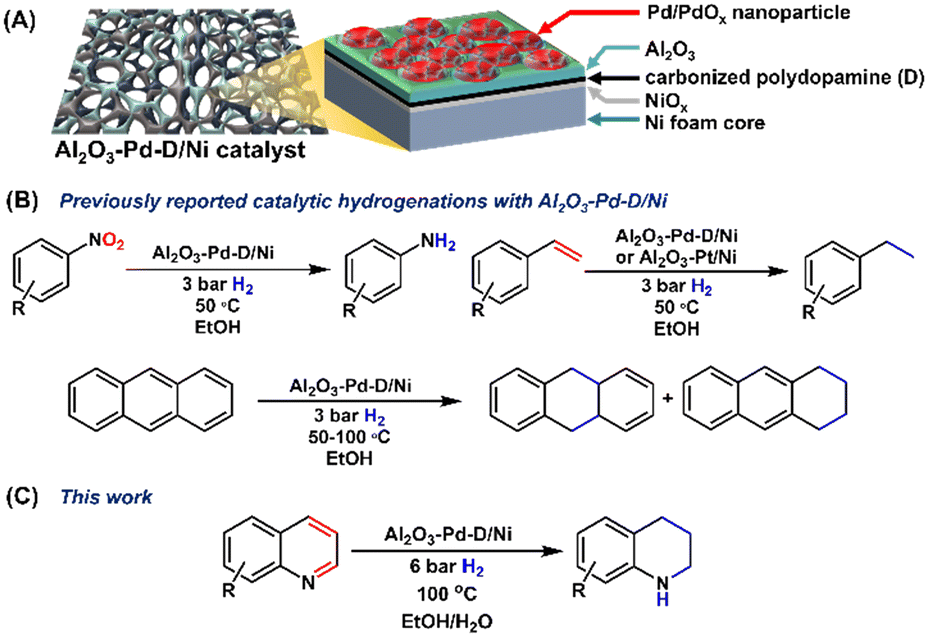 | ||
| Fig. 1 General schematic for the hierarchical Ni-foam supported Pd catalyst framework (A) previously reported for the catalytic hydrogenation of nitroaromatics, styrenes, and anthracene (B) and the hydrogenation of quinolines (C) reported herein. | ||
The expansion of sustainable and selective heterogeneous hydrogenations to increasingly complex fine chemicals is a topic of intense current interest. N-heterocyclic motifs are ubiquitous across fine chemicals, dyes, and pharmaceuticals.9–14 Therein, the selective hydrogenation of quinolines which can afford 1,2,3,4-tetrahydroquinoline (Py-THQ), 5,6,7,8-tetrahydroquinoline (bz-THQ), or decahydroquinoline (DHQ) has drawn significant attention as both a model catalyst test substrate, and an avenue towards diversified quinoline derivatives.9–20 These substrates are typically difficult to hydrogenate due to the high resonance stability of the aromatic rings, as well as the potential for catalyst deactivation upon formation of the respective cyclic amines.11,14,16 Nonetheless, there have been significant contributions towards the utilization of Pd-based quinoline hydrogenation reactions.11–13,17,20–22 However, these transformations typically require high pressure,20,22 high temperature,17 or high catalyst loading.12,17 Herein, we report the selective hydrogenation of quinoline derivatives utilizing Al2O3–Pd–D/Ni, with low Pd loading, low hydrogen pressures, and highly sustainable solvents (EtOH and H2O).
Results and discussion
Catalytic hydrogenation of quinoline was initially tested using a modified protocol previously established for anthracene;9 0.5 mmol quinoline, 5 mL EtOH, ∼100 mg piece of Al2O3–Pd–D/Ni, 6 bar of H2, at 100 °C for 18 h (Fig. 2A). Under these conditions, quinoline was quantitatively converted to Py-THQ. To better understand this system, a conversion and selectivity time profile was constructed by performing individual experiments at t = 2, 4, 6, 8, 12, 16 and 24 h (Fig. 2B). Each experiment utilized a different piece of Al2O3–Pd–D/Ni catalyst of similar weight (97.75 ± 2.05 mg). It is noteworthy that even with the sample-to-sample inhomogeneity inherent to a monolith-based catalyst, the behavior is remarkably consistent. Under the conditions tested, complete hydrogenation is achieved at 6 hours and the high selectivity towards Py-THQ does not change even at longer reaction times of up to 24 h. Using a gravimetric approach (by mass of Pd), at 2 h we observe 32% conversion, which translates to a competitive turnover frequency (TOF) of ca. 500 h−1.23 | ||
| Fig. 2 (A) Reaction scheme for the catalytic hydrogenation of quinoline with Al2O3–Pd–D/Ni. (B) Conversion/selectivity vs. time utilizing 97.75 ± 2.05 mg of Al2O3–Pd–D/Ni (each point = separate reaction); green dotted line denotes reaction completion at the 6 h timepoint. (C) Recycling trials, 6 h reaction times, utilizing 92.0 mg of Al2O3–Pd–D/Ni. | ||
To establish facile reusability, five recycling trials were conducted according to the conditions noted in Fig. 2A, utilizing a 92 mg piece of Al2O3–Pd–D/Ni. Across five trials, no change in the quantitative conversion or selectivity towards Py-THQ was observed (Fig. 2C). Unlike conventional nanocatalysts, the isolation of Al2O3–Pd–D/Ni requires no filtration aids or centrifugation. The solid piece of Ni foam-based monolith is removed by tweezers, rinsed under flowing deionized water, then EtOH, and allowed to dry overnight under ambient. It can be concluded that Al2O3–Pd–D/Ni is remarkably stable under continued use, under refluxing EtOH in a reducing environment, and is physically robust under repeated handling.
We extended the methodology (with 18 h reaction times) to a variety of functionalized derivatives (Fig. 3). The presence of –CH3 or –OH at the 8 position (b, c) yielded near quantitative conversion with similar selectivity for hydrogen addition at the 1,2,3,4 positions. Switching to –OCH3 at the 8 position (d) lowered the conversion to 71%, while maintaining high selectivity. Introduction of –Cl at the 6 position (e) yielded a lower overall conversion, but maintained high selectivity with no evidence for significant hydrodehalogenation. The presence of –CH3 groups at the 2 and 4 positions (f) did not impede complete conversion, however selectivity for the 1,2,3,4 was observed to be ca. 26%, with addition at the 5,6,7,8 being preferred at 74%, likely due to steric factors and the interaction with the catalyst surface; this result is further evaluated by density functional theory (DFT) studies (vide infra). In the case of 9-CH3 acridine (g), near quantitative conversion was achieved with 97% selectivity for hydrogenation at the 1,2,3,4 and 5,6,7,8 positions. Attempted hydrogenation of quinoline with an aldehyde at the 2 position (h) yielded complete conversion with preferred selectivity towards the hydrogenation of the aldehyde, and only 7% for both the aldehyde and the 1,2,3,4 of the quinoline. Hydrogenation of quinoxaline functionalized with a terminal alkyne at the 7 position (i) resulted in a 65% conversion, favoring the semi-hydrogenation product by 94%; no hydrogens were added to the quinoxaline core. Addition of hydrogen to 4-aminoisoquinoline (j) only furnished 25% conversion, with approximately equal preference for the 1,2,3,4 and 5,6,7,8 positions. Hydrogenation of 7,8-benzoquinoline (k) yielded complete conversion, with 80% selectivity for hydrogenation at 1,2,3,4. Reduction of temperature to 50 °C lowered the conversion to 43%, with near quantitative selectivity at the 1,2,3,4 position.
 | ||
| Fig. 3 Scope of catalytic hydrogenation of quinoline derivatives utilizing 95.8 ± 7.4 mg of Al2O3–Pd–D/Ni for a–l. Reaction time = 18 hours. | ||
To further improve the sustainability of this process, we explored the introduction of H2O as a reaction co-solvent (Fig. 3. l). First, conducting the reaction under similar conditions noted above with a 4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 EtOH:H2O mixture resulted in complete consumption of the quinoline with near quantitative yield of Py-THQ (l). Switching to 100% H2O as solvent similarly furnished quantitative conversion, but hampered facile isolation and characterization, with the presence of secondary intractable products (Fig. S19†). However, the result validates the potential to run such reactions with “wet” EtOH, e.g. bioethanol, which precludes further processing to generate dry solvent, or other efforts to exclude H2O from the reaction process. Overall, this is promising for enhanced process sustainability.24
:1 EtOH:H2O mixture resulted in complete consumption of the quinoline with near quantitative yield of Py-THQ (l). Switching to 100% H2O as solvent similarly furnished quantitative conversion, but hampered facile isolation and characterization, with the presence of secondary intractable products (Fig. S19†). However, the result validates the potential to run such reactions with “wet” EtOH, e.g. bioethanol, which precludes further processing to generate dry solvent, or other efforts to exclude H2O from the reaction process. Overall, this is promising for enhanced process sustainability.24
In our prior work it was surmised that the PdOx was reduced in situ to provide a reactive Pd0 surface.9 Herein, to examine the fate of the surface under reaction-like conditions, in situ X-ray photoelectron spectroscopy (XPS) was conducted at 100 °C in the presence of H2. Fig. 4 provides in situ Pd 3d XPS spectra collected from the Al2O3–Pd–D/Ni interface as a function of sample environment and temperature. The binding energy (BE) scale used to present the Pd 3d XPS data has been adjusted by +0.7 eV to place the primary adventitious C 1s peak at a BE of 284.8 eV.25 The pristine catalyst exhibits a bimodal peak structure indicative of both PdO (red fits with Pd 3d5/2 BE = 336.8 eV) and metallic Pd0 (green fits with Pd 3d5/2 BE = 335.0 eV).25 The relative abundance of these species is unaffected by introduction of 1 × 10−3 mBar Ar/H2 3% at room temperature, but the Pd 3d:Ni 2p XPS peak area ratio attenuates by ∼20% indicating subtle attrition of Pd surface site density. Heating to 100 °C within the Ar/H2 3% environment leads to near complete reduction of PdO to metallic Pd0 and further loss of Pd surface site concentration via some combination of sintering, adlayer formation and/or partial support encapsulation resulting from strong metal–support interactions. Independent of mechanism, the diminished surface site concentration of Pd is present in its fully reduced metallic state when exposed to conditions approximating those described above for the catalytic hydrogenation of quinoline(s).
 | ||
| Fig. 4 (Left) In situ Pd 3d XPS collected from the as prepared Al2O3–Pd–D/Ni interface at 25 °C within UHV (lower), at 25 °C within 1 × 10−3 mBar Ar/H2 3% (middle), and at 100 °C within 1 × 10−3 mBar Ar/H2 3% (upper). (Right) Variation in the Pd 3d:Ni 2p (black) and Pd0:Pd 3d (green) XPS peak area ratios as a function of sample environment and temperature. | ||
To better understand the prevailing surface interactions that can be in effect at lower temperatures where PdOx species dominate, and elevated temperatures where the entire surface is Pd0 (vide supra), DFT simulations were conducted (Fig. 5). The calculations focused on quinoline “Q” where hydrogenation proceeded as expected at the 1,2,3,4 position, and 2,4-dimethylquinoline “Q-2,4”, where hydrogen addition at the 5,6,7,8 was significantly preferred. DFT simulations were conducted to calculate the adsorption energies and analyze the atomic structures of the molecules on the surface of both oxidized and reduced Pd surfaces.
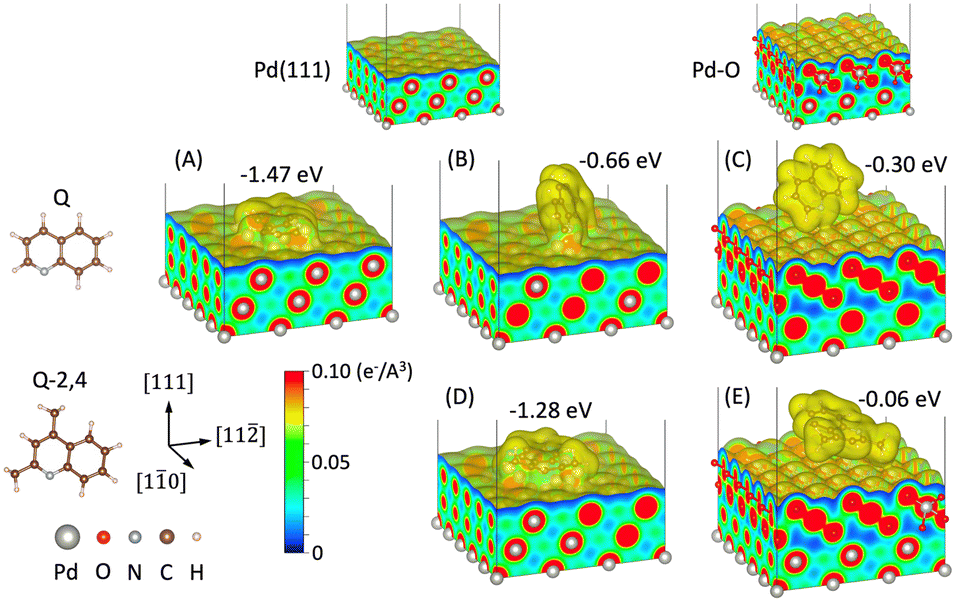 | ||
| Fig. 5 Adsorption energy and structure of quinoline (Q) on (A and B) Pd(111) and (C) PdO surface and 2,4-dimethylquinoline (Q-2,4) on (D) Pd(111) and (E) PdO surface. Charge density isosurface of 0.005 e− Å−3 is shown. | ||
First, a lattice constant of Pd was obtained using an FCC unit cell containing four Pd atoms. Post structural optimization, using this lattice constant, a slab model with a Pd(111) surface was constructed, containing 90 Pd atoms and consisting of 3 layers of (111) planes with orthogonal lattice vectors corresponding to [1![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) 0], [11
0], [11![[2 with combining macron]](https://www.rsc.org/images/entities/char_0032_0304.gif) ], and [111], with 15 Å of vacuum layer added in [111]. For PdO, two layers of oxygen atoms were added on and below the top Pd surface at cubic PdO sites containing 90 Pd and 60 O atoms. After a Q or Q-2,4 molecule was placed on the surface, molecular dynamics (MD) simulations were conducted (300 K, 1 ps) followed by structural optimization to calculate the molecular adsorption energies using ET − (Es + Em); ET, Es, and Em are calculated potential energies of the molecule-adsorbed surface, surface slab model, and free-standing molecule, respectively. During MD and structural optimization process, the Pd atoms at the bottom layer and the size/shape of the simulation box were fixed. The simulations were performed using Vienna ab initio simulation package (VASP).26,27 A plane-wave energy cut-off of 520 eV was employed, and a generalized gradient approximation parameterized by Perdew, Burke and Ernzerho28 was used for the exchange-correlation functional. The ionic core was represented with a projector augmented wave potential.29,30 11 × 11 × 11 and 2 × 2 × 1 Monkhorst–Pack31k-point mesh were used for the bulk and the slab models, respectively.
], and [111], with 15 Å of vacuum layer added in [111]. For PdO, two layers of oxygen atoms were added on and below the top Pd surface at cubic PdO sites containing 90 Pd and 60 O atoms. After a Q or Q-2,4 molecule was placed on the surface, molecular dynamics (MD) simulations were conducted (300 K, 1 ps) followed by structural optimization to calculate the molecular adsorption energies using ET − (Es + Em); ET, Es, and Em are calculated potential energies of the molecule-adsorbed surface, surface slab model, and free-standing molecule, respectively. During MD and structural optimization process, the Pd atoms at the bottom layer and the size/shape of the simulation box were fixed. The simulations were performed using Vienna ab initio simulation package (VASP).26,27 A plane-wave energy cut-off of 520 eV was employed, and a generalized gradient approximation parameterized by Perdew, Burke and Ernzerho28 was used for the exchange-correlation functional. The ionic core was represented with a projector augmented wave potential.29,30 11 × 11 × 11 and 2 × 2 × 1 Monkhorst–Pack31k-point mesh were used for the bulk and the slab models, respectively.
Fig. 5 shows adsorption energy and atomic/electronic structure of the Pd(111) and PdO surface with Q and 2,4-Q molecules. Two different configurations were obtained for Q on Pd(111) surface depending on the initial structures. The molecule lying flat on the surface has a higher adsorption energy at −1.47 eV compared with the tilted configuration at −0.66 eV. The adsorption energy of 2,4-Q on Pd(111) was −1.28 eV. On the other hand, the values for Q and 2,4-Q on PdO were −0.30 eV and −0.06 eV, respectively. Both molecules favor the reduced Pd surface. This is consistent with the literature which points to a preferential flat configuration on Pd0 (e.g. Pd(111)) sites.32–34 This can be also seen from the charge density distributions. Electrons are conforming the molecules on the bare Pd(111) surface, while they are bound only at the edge of the molecules as shown in (C) and (E). Differential charge density distributions were compared to analyze the bonding between the molecule and the surface in more detail (Fig. S20†). The result confirms that the molecular adsorption on bare Pd surface was chemisorption, while it was physisorption on PdO, which agrees well with the difference in the adsorption energies. Here, the rearrangement of electrons for chemisorbed molecules were localized at the binding atoms for tilted configurations. Hahn and Baiker report similar chemisorption and flat Q orientation with Pd0 while in their case, tilted Q configuration and physisorption with an Au0 surface.34 This change in the electronic structure contributes to accelerating the catalytic reaction of the molecules on the bare Pd, which is consistent with our experimental measurement. Although the flat configuration is more energetically favorable, other molecules adsorbed on the surface such as H2/C2H5OH or surface roughness may prevent the molecules from lying flat. In addition, a partially oxidized surface is expected to promote tilted adsorption, and the chemical bond between the adsorbed molecule and Pd atoms enhances the catalytic reactions. However, a fully oxidized Pd surface hinders activity as only physisorption is allowed.
Thus, a reduced Pd0 surface of sufficient dimension to facilitate chemisorption and subsequent reactivity with surface-bound H2 is required and falls in line with experimental observations from both catalysis and in situ XPS (vide supra). The presence of steric bulk and other functional groups impede both the initial physisorption on PdO, and lead to lower energy interactions on Pd0, and in the case of 2,4-Q lead to preferential hydrogenation on the phenyl portion of the quinoline framework, which is directly interfacing with the surface.
Conclusions
Hierarchical nickel foam-based Al2O3–Pd–D/Ni was demonstrated as an excellent catalyst framework for the selective hydrogenation of quinoline derivatives utilizing low H2 pressures and green solvents (EtOH and H2O), and leveraging an inherently low Pd loading. The selectivity observed towards 1,2,3,4-tetrahydroquinoline may prove advantageous compared to very common Pd-based hydrogenation catalysts, such as Pd/C which has been shown to fully hydrogenate quinoline to decahydroquinoline even under very mild conditions (room temperature and H2 balloon).35 The catalyst framework is highly reusable under facile handling, requiring no filtration media or other separation aids, and notably demonstrates no loss in reactivity or alteration of selectivity over multiple trials. Thus, while the catalyst system uses Pd, the extremely low loading, and high degree of reusability drastically enhance it's sustainability.Data availability
All relevant information is provided in the ESI.† Further information and/or clarification can be provided by the authors upon request.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Departments of Chemistry and Physics (College of Sciences), the Department of Materials Science and Engineering (College of Engineering and Computer Science) and the Faculty Cluster Initiative at the University of Central Florida. The authors thank the PREM Center for Quantum Materials Innovation and Education Excellence funded by the National Science Foundation (NSF) with Award No. 2424976. In situ XPS analysis was completed within the University of Central Florida's Materials Characterization Facility using equipment procured using NSF Grant No. 2018319.Notes and references
- C. Torborg and M. Beller, Adv. Synth. Catal., 2009, 351, 3027–3043 CrossRef CAS.
- L. B. Vitaliy, S. S. Peter, H. C. James and L. Rafael, Curr. Org. Synth., 2010, 7, 614–627 CrossRef.
- P. Devendar, R. Y. Qu, W. M. Kang, B. He and G. F. Yang, J. Agric. Food Chem., 2018, 66, 8914–8934 CrossRef CAS PubMed.
- S. Xu, E. H. Kim, A. Wei and E. Negishi, Sci. Technol. Adv. Mater., 2014, 15, 044201 CrossRef CAS PubMed.
- S. McCarthy, D. C. Braddock and J. D. E. T. Wilton-Ely, Coord. Chem. Rev., 2021, 442, 213925 CrossRef CAS.
- S. Mitchell, E. Vorobyeva and J. Pérez-Ramírez, Angew. Chem., Int. Ed., 2018, 57, 15316–15329 CrossRef CAS PubMed.
- J. Liu, ACS Catal., 2017, 7, 34–59 CrossRef CAS.
- L. R. Shultz-Johnson, A. Rahmani, J. Frisch, T. E. Hsieh, L. Hu, J. Sosa, M. Davy, S. Xie, M. J. Beazley, Z. Gao, P. Golvari, T. H. Wang, T. G. Ong, N. G. Rudawski, F. Liu, P. Banerjee, X. Feng, M. Bär and T. Jurca, ACS Appl. Mater. Interfaces, 2024, 16, 39387–39398 CrossRef CAS PubMed.
- A. Rahmani, T. M. Currie, L. R. Shultz, J. T. Bryant, M. J. Beazley, F. J. Uribe-Romo, L. Tetard, N. G. Rudawski, S. Xie, F. Liu, T. H. Wang, T. G. Ong, L. Zhai and T. Jurca, Catal. Sci. Technol., 2022, 12, 6992–6997 RSC.
- Y. Hu, M. Liu, S. Bartling, H. Lund, H. Atia, P. J. Dyson, M. Beller and R. V. Jagadeesh, Sci. Adv., 2023, 48, eadj8225 CrossRef PubMed.
- M. M. Dell'Anna, V. F. Capodiferro, M. Mali, D. Manno, P. Cotugno, A. Monopoli and P. Mastrorilli, Appl. Catal., A, 2014, 481, 89–95 CrossRef.
- Y. Zhang, J. Zhu, Y. T. Xia, X. T. Sun and L. Wu, Adv. Synth. Catal., 2016, 358, 3039–3045 CrossRef CAS.
- X. Ding, Y. Chen, J. Nan, H. Dai, Y. Wang, G. Bai and W. Qiu, ACS Sustainable Chem. Eng., 2022, 10, 14011–14023 CrossRef CAS.
- F. Chen, A. E. Surkus, L. He, M. M. Pohl, J. Radnik, C. Topf, K. Junge and M. Beller, J. Am. Chem. Soc., 2015, 137, 11718–11724 CrossRef CAS PubMed.
- L. Bai, X. Wang, Q. Chen, Y. Ye, H. Zheng, J. Guo, Y. Yin and C. Gao, Angew. Chem., Int. Ed., 2016, 55, 15656–15661 CrossRef CAS PubMed.
- H. Cho, F. Török and B. Török, Org. Biomol. Chem., 2013, 11, 1209–1215 RSC.
- A. Mollar-Cuni, S. Martín, G. Guisado-Barrios and J. A. Mata, Carbon, 2023, 206, 314–324 CrossRef CAS.
- M. N. Shaikh, RSC Adv., 2019, 9, 28199–28206 RSC.
- S. Li, Y. Yang, Y. Wang, H. Liu, J. Tai, J. Zhang and B. Han, Catal. Sci. Technol., 2018, 8, 4314–4317 RSC.
- S. Zhang, Z. Xia, T. Ni, H. Zhang, C. Wu and Y. Qu, J. Mater. Chem. A, 2017, 5, 3260–3266 RSC.
- M. Guo, C. Li and Q. Yang, Catal. Sci. Technol., 2017, 7, 2221 RSC.
- Y. Ren, Y. Wang, X. Li, Z. Zhang and Q. Chi, New J. Chem., 2018, 42, 16694 RSC.
- D. Chandra, S. Saini, S. Bhattacharya, A. Bhaumik, K. Kamata and M. Hara, ACS Appl. Mater. Interfaces, 2020, 12, 52668–52677 CrossRef CAS PubMed.
- D. Prat, A. Wells, J. Hayler, H. Sneddon, C. R. McElroy, S. Abou-Shehada and P. J. Dunn, Green Chem., 2016, 18, 288–296 RSC.
- J. F. Moulder, W. F. Stickle, P. E. Sobol and K. D. Bomben, in Handbook of X-ray Photoelectron Spectroscopy, ed. J. Chastain, Perkin-Elmer Corporation Physical Electronics Division, Minnesota, 1992 Search PubMed.
- G. Kresse and J. Hafner, Phys. Rev. B, 1993, 47, 558 CrossRef CAS PubMed.
- G. Kresse and J. Furthmüller, Phys. Rev. B, 1996, 54, 11169 CrossRef CAS PubMed.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865 CrossRef CAS PubMed.
- G. Kresse and D. Joubert, Phys. Rev. B, 1999, 59, 1758 CrossRef CAS.
- P. E. Blöchl, Phys. Rev. B, 1994, 50, 17953 CrossRef PubMed.
- H. J. Monkhorst and J. D. Pack, Phys. Rev. B, 1976, 13, 5188 CrossRef.
- J. M. Bonello, R. Lindsay, A. K. Santra and R. M. Lambert, J. Phys. Chem. B, 2002, 106, 2672 CrossRef CAS.
- G. Santarossa, M. Iannuzzi, A. Vargas and A. Baiker, ChemPhysChem, 2008, 9, 401 CrossRef CAS PubMed.
- K. R. Hahn and A. Baiker, J. Phys. Chem. C, 2022, 126, 20840 CrossRef CAS.
- N. Tanaka and T. Usuki, Eur. J. Org. Chem., 2020, 5514 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, 1H NMR of catalytic experiments, and additional computational details. See DOI: https://doi.org/10.1039/d5cy00675a |
| This journal is © The Royal Society of Chemistry 2025 |