
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMechanistic understanding of electrochemical hydrogenation of 5-hydroxymethylfurfural (HMF) via scanning electrochemical microscopy (SECM)†
Seokjun Han and
Won Tae Choi*
and
Won Tae Choi*
Department of Chemical Engineering, University of Florida, Gainesville, FL 32611, USA. E-mail: wontae.choi@ufl.edu
First published on 8th April 2025
Abstract
We utilize scanning electrochemical microscopy (SECM) to study the electrocatalytic HMF reduction reaction in aqueous solutions. The surface interrogation mode of SECM is used to quantify the adsorbed intermediates and overall rate constants of elementary chemical reaction steps can be achieved by controlling the time-delay during the measurements. This work provides an in-situ approach to examine the kinetics and mechanisms of competing reactions, namely HMF reduction versus the hydrogen evolution reaction, over applied potentials.
5-Hydroxymethylfurfural (HMF) is a platform chemical derived from biomass, and its selective conversion is essential for sustainable production of commodity chemicals and fuels.1 For example, partial oxidation or reduction of HMF yields valuable chemicals, including 2,5-diformyl furan (DFF), 2,5-furan dicarboxylic acid (FDCA), 2,5-bis(hydroxymethyl)furan (BHMF), and 2,5-dimethylfuran (DMF).2–5 Among these, BHMF is one of the key chemicals used as a precursor for the production of biopolymers (e.g., polyurethane6,7 and polyester8,9) and biofuels (e.g., 2,5-bis(alkoxymethyl)furans10,11). Traditionally, the conversion of HMF to BHMF is achieved by thermal catalytic hydrogenation, which requires elevated temperature (≥100 °C) and high hydrogen pressure (≥10 bar).12 In contrast, electrochemical hydrogenation offers a milder, energy-efficient, and sustainable route as it operates under ambient temperature and pressure without requiring expensive hydrogen gas (H2). Despite these advantages, electrochemical HMF conversion faces significant challenges in achieving high selectivity and yield, hindering its scale-up and practical implementation. A fundamental understanding of the reaction mechanism is critical for designing efficient electrocatalysts and optimizing process conditions to enhance the selectivity and yield of electrochemical HMF hydrogenation.13–15
Two mechanisms have been proposed for the electrochemical reduction of HMF to BHMF in aqueous solutions. In the proton-coupled electron transfer (PCET) pathway, the reaction proceeds via an electron transfer, forming an adsorbed HMF intermediate that is subsequently protonated, followed by the final proton-coupled electron transfer step to produce BHMF. The electrocatalytic hydrogenation (ECH) pathway involves a direct reaction between an adsorbed HMF intermediate and two adsorbed hydrogens (H*), resulting in BHMF generation (Fig. 1).16–21 Previous studies have demonstrated that the HMF reduction proceeds via a PCET mechanism at low potentials where the hydrogen evolution reaction (HER), which involves using adsorbed hydrogens to form H2, is not significant.16,17 Chadderdon et al. conducted isotope labeling experiments and found that proton addition during carbonyl reduction occurs through the PCET pathway.20 Additionally, Guo et al. observed a decrease in charge transfer resistance at low potentials in HMF-containing solutions using electrochemical impedance spectroscopy (EIS), indicating that the facilitated reaction kinetics are attributed to HMF reduction via the PCET pathway.21 However, experimental verification of the ECH mechanism remains a challenge due to the difficulty in distinguishing adsorbed hydrogen consumption by ECH from the HER.15,21,22 Scanning electrochemical microscopy (SECM) is a non-contact electroanalytical scanning probe technique that provides quantitative information on the electrochemical reactions.23 In this study, we utilize SECM to investigate electrochemical HMF hydrogenation on an Ag catalyst in aqueous solution, focusing on the role of adsorbed hydrogen and the competition between ECH and HER. Specifically, we employ substrate generation/tip collection SECM (SG/TC-SECM) to selectively detect reaction products23–26 and surface interrogation SECM (SI-SECM) to in-situ quantify surface-adsorbed reaction intermediates.27–30 Based on the SECM results, we suggest possible electrochemical HMF hydrogenation pathways in aqueous solution depending on the electrode potential.
 | ||
| Fig. 1 Scheme of mechanisms of the electrochemical aldehyde hydrogenation of HMF to BHMF on the Ag catalyst; proton-coupled electron transfer (PCET) and electrocatalytic hydrogenation (ECH) pathways. | ||
Silver-based catalysts have demonstrated effective conversion of HMF to BHMF with a high selectivity (>90%) at negative potentials (≤−1.3 V vs. Ag/AgCl) in a borate buffer solution (pH 9.2).16,17,31,32 Fig. 2a shows the linear sweep voltammograms (LSVs) of the Ag ultramicroelectrode (UME) in borate buffer solutions, with and without HMF. The LSV in the absence of HMF shows negligible current until an applied potential of −1.3 V. In the presence of HMF, however, a distinct cathodic current was observed in the potential range from −1.0 to −1.3 V, confirming predominant HMF reduction.
 | ||
| Fig. 2 Production rate analysis of BHMF and H2 on the Ag UME (radius 12.5 μm, RG = 2 for BHMF, and RG = 4 for H2) by SG/TC-SECM. (a) Linear sweep voltammograms (LSVs) of the Ag UME in borate buffer (pH 9.2) with and without HMF (scan rate, 10 mV s−1). (b) Scheme of the SG/TC-SECM for BHMF and H2 titrations. (c) LSVs of HMF and BHMF oxidation on the carbon UME (radius 15 μm), (scan rate, 10 mV s−1). (d) LSVs of the carbon tip (radius 15 μm, RG = 1.5) for titration of BHMF in borate buffer (pH 9.2) with 2.5 mM HMF, with a tip-substrate gap of ca. 5 μm. (e) Substrate potential dependent tip current corresponding to the BHMF production rate. Chronoamperomograms (CAs) of the platinum tip (radius 12.5 μm, RG = 2) for titration of H2 in borate buffer (pH 9.2) (f) without HMF and (g) with 2.5 mM HMF; with a tip-substrate gap of ca. 6 μm. (h) Charge density of H2 collection on a platinum tip (radius 12.5 μm, RG = 2) in borate buffer (pH 9.2) with and without 2.5 mM HMF. | ||
Beyond −1.3 V, a sharp increase in cathodic current is shown, which overlaps with the increase in the activity of the HER on the Ag surface. To delve into the potential-dependent competition between HMF reduction and HER on the Ag surface, we employed SG/TC-SECM (Fig. 2b). The Ag UME was used as a substrate electrode for all SECM experiments. A C UME was chosen as a tip electrode for selective detection of BHMF (Fig. 2b). We confirmed by LSV that only BHMF can be oxidized on the C UME within the applied potential range from +1.0 to +1.4 V (Fig. 2c). When the Ag electrode was held at discrete constant potentials from –1.0 to –1.7 V, the C tip electrode potential was scanned to detect BHMF (Fig. 2d). The measured tip current at ETip = 1.4 V was taken to compare the BHMF production rate at each applied substrate potential, ESub (Fig. 2e). A Pt UME was chosen as the tip electrode for the detection of by-product H2 (Fig. 2b). The Ag electrode was pulsed to constant discrete potentials ranging from –1.0 to –1.7 V for 15 s, then the produced H2 was detected by the tip electrode through chronoamperometry at ETip = 0.1 V.23 The amount of charge passed during the chronoamperometry was used to compare the H2 production rates at the Ag surface depending on the substrate potentials. Significant increases in H2 production rates were observed at negative potentials at −1.3 V and beyond in both solutions with and without HMF. However, the presence of HMF led to slightly lower H2 production rates compared to its absence (Fig. 2h), indicating that the hydrogen evolution reaction and HMF reduction reaction are competing with each other.16,17 If the HER and HMF reduction were purely competing reactions, an increase in HER should have led to a drastic suppression of BHMF production. Despite the rapid increase in H2 production beyond −1.3 V, the BHMF production rate reached its maximum at −1.3 V and exhibited only a slight decrease between −1.3 V and −1.5 V (Fig. 2e). Thus, based on the observed trend we speculate that the adsorbed hydrogen is participating in BHMF formation via the ECH mechanism at potentials beyond −1.3 V.21,22
To find proof of the ECH mechanism at negative potentials (≤−1.3 V), we in-situ examined the Ag surface by SI-SECM (Section S2.2 of the ESI†). Fig. 3a demonstrates a scheme of SI-SECM on the Ag electrode. When the substrate is pulsed to a negative potential for 7 s, surface intermediates (e.g., adsorbed hydrogen and/or adsorbed hydroxyalkyl species) are generated. Subsequently, the substrate electrode is held at open circuit, while a positive potential is applied at the tip electrode to oxidize the molecular probe, FcMeOH, dissolved in the solution (eqn (1)). Then, the oxidized molecular probes, FcMeOH+, diffuse and react with the adsorbed intermediates (A*) at a substrate electrode, regenerating the reduced form of the molecular probes (eqn (2)). The regeneration of the molecular probe can amplify the tip current during SI-SECM.
| FcMeOH → FcMeOH+ + e− | (1) |
| FcMeOH+ + A* → FcMeOH + A+ + * | (2) |
 | ||
| Fig. 3 Kinetic analysis of surface-adsorbed intermediates on Ag UME (radius 12.5 μm, RG =2) by SI-SECM. (a) Scheme of SI-SECM with time delay; A represents intermediates and R/O is the molecular probe. CAs of the carbon tip (radius 15 μm, RG = 1.5) at Esub = −1.6 V (vs. Ag/AgCl) in borate buffer (pH 9.2) (b) without HMF and (c) with 5 mM HMF. (d) Two possible pathways occurring at the Ag electrode at open circuit: the final step of ECH of HMF hydrogenation and the Tafel step of the HER. (e) Simulated contribution of kECH and kTafel at kECH/kTafel = 4. (f) Simulated profile of koverall vs. CHMF*/H* for different kECH/kTafel ratios (4, 1, 0.25). (g) Simulated reaction rates of ECH and Tafel steps depending on the ratio of adsorbed intermediates, assuming starting concentration [H*] = 0.01 and [HMF*] = 0.99 with kECH/kTafel = 4. | ||
The tip current was measured by chronoamperometry (CA), where the passed charge is related to the amount of intermediates adsorbed on the Ag surface. The surface concentrations of adsorbed intermediates were estimated by assuming that one electron transfer is required for intermediate titration (Table 1). Then, we introduced a time delay between the substrate-pulse and tip-titration steps to monitor changes in the intermediate concentration with time. As the time delay interval increased, the recorded tip current profiles decreased (Fig. 3b and c). During the time delays, open circuit was held at the substrate electrode to prevent electron transfer to the intermediates. The smaller tip current profiles primarily reflect the dissipation of surface intermediates by chemical reactions, with the rate of decay corresponding to the reaction kinetics (Fig. 3d). The decay follows a combination of two second-order chemical reactions, where the intermediates react through both the Tafel step of the HER and ECH of HMF hydrogenation (eqn (3)).
| r = kTafel[H*]2 + kECH[H*][HMF*] | (3) |
| [A*] = [H*] + [HMF*] | (4) |
| CHMF*/H* = [HMF*]/[H*] | (5) |
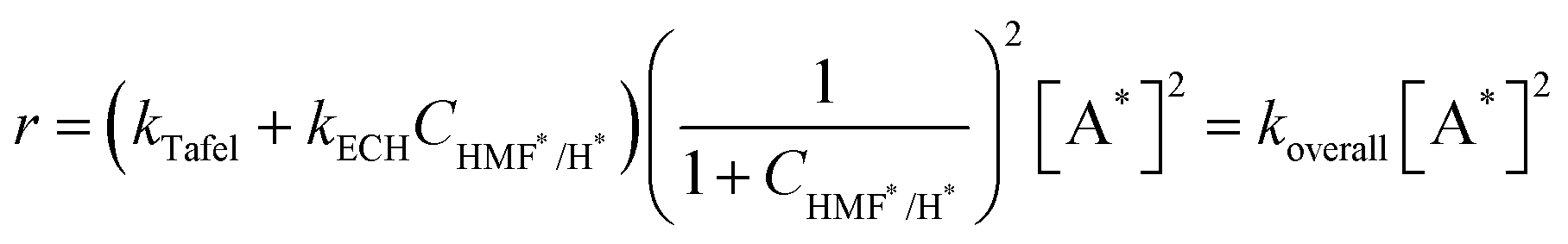 | (6) |
| Substrate potentials (V vs. Ag/AgCl) | Surface concentration [mol m−2] | koverall [mol−1 m2 s−1] | ||||
|---|---|---|---|---|---|---|
| 5 mM | 2 mM | 0 mM | 5 mM | 2 mM | 0 mM | |
| −1.3 | 3.29 × 10−4 | 3.45 × 10−4 | — | 424 | 352 | — |
| −1.4 | 3.78 × 10−4 | 4.10 × 10−4 | 3.47 × 10−4 | 401 | 318 | 297 |
| −1.5 | 4.33 × 10−4 | 4.47 × 10−4 | 4.14 × 10−4 | 374 | 308 | 297 |
| −1.6 | 4.56 × 10−4 | 4.88 × 10−4 | 4.36 × 10−4 | 356 | 302 | 301 |
The temporal decays of intermediate concentrations were fitted to the second order reaction, and Table 1 summarizes the calculated koverall values. In the presence of HMF, the calculated rate constant exceeds the Tafel rate constant (kTafel = 300 mol−1 m2 s−1), indicating the involvement of an additional pathway for consuming adsorbed intermediates, the ECH mechanism. A further increase in the koverall value was observed in 5 mM HMF solution compared to 2 mM HMF solution, supporting that an increased amount of adsorption of HMF intermediate on the Ag surface increases the koverall value. Different CHMF*/H* ratios can be yielded upon pulsing with varied potentials, leading to varied koverall. As more negative potentials were applied, decreases in koverall were observed in the presence of HMF. This suggests that the H* surface population becomes dominant over HMF*, decreasing the contribution of ECH while increasing the HER. Using the equations describing each contribution (Eqn (7) and (8)), we simulated each contribution's dependency on the ratio of adsorbed intermediates, CHMF*/H*, confirming the opposing trends (Fig. 3e).
 | (7) |
 | (8) |
Assuming that the CHMF*/H* ratio decreases with the application of more negative potentials, the observed decline in koverall at more negative potentials suggests that kECH is greater than that of kTafel (Fig. 3f). Given that kECH > kTafel, Fig. 3g displays simulated elementary second order reaction rates as a function of 1/CHMF*/H*. A maximum ECH rate (rECH) was observed, which aligns with the trends of BHMF production, showing the highest reaction rates of BHMF production at −1.3 V followed by a gradual decrease (Fig. 2e). Notably, the H2 production rate is moderate at −1.3 V (Fig. 2h). This highlights the significant role of hydrogen coverage in facilitating HMF conversion and the HMF reduction pathway depends on the applied potential:
1. Up to −1.2 V: HMF reduction is likely dominated by the PCET mechanism due to a limited amount of adsorbed hydrogen.
2. −1.3 V to −1.5 V: ECH becomes viable as moderate adsorbed hydrogen facilitates the formation of BHMF, with maximum selectivity at −1.3 V.
3. Beyond −1.6 V: adsorbed hydrogen is increasingly consumed by the HER, reducing its availability for ECH and leading to a decrease in BHMF production.
In summary, we investigated HMF electroreduction by using SECM. We found that an optimal level of adsorbed hydrogen is crucial for selective and efficient electrochemical HMF hydrogenation: neither too high, which favors the HER, nor too low, which limits the ECH reaction rate. This finding provides valuable insights on catalyst design and optimization of the electrochemical process towards biomass conversion.
This research was financially supported by Prof. Choi's startup funds provided by the Department of Chemical Engineering and Herbert Wertheim College of Engineering at the University of Florida.
Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts of interest to declare.Notes and references
- A. Mittal, H. M. Pilath and D. K. Johnson, Energy Fuels, 2020, 34, 3284–3293 CrossRef CAS.
- D. Carvajal, V. Bolos-Sánchez, J. Solera-Rojas, C. Mejuto, F. Fabregat-Santiago and E. Mas-Marzá, Electrochim. Acta, 2024, 475, 143676 CrossRef CAS.
- D. K. Lee, S. R. Kubota, A. N. Janes, M. T. Bender, J. Woo, J. Schmidt and K. S. Choi, ChemSusChem, 2021, 14, 4563–4572 CrossRef CAS PubMed.
- B. Zhang, Z. Li, Y. Zhou, Z. Yang, Z. Xue and T. Mu, Small, 2024, 20, 2306663 CrossRef CAS PubMed.
- Y. Wu, Y. Jiang, W. Chen, X. Yue, C. L. Dong, M. Qiu, T. T. T. Nga, M. Yang, Z. Xia and C. Xie, Adv. Mater., 2024, 36, 2307799 Search PubMed.
- Z. Mou, S. K. Feng and E. Y. Chen, Polym. Chem., 2016, 7, 1593–1602 RSC.
- L. Zhang, F. C. Michel Jr and A. C. Co, J. Polym. Sci., Part A: Polym. Chem., 2019, 57, 1495–1499 CrossRef CAS.
- C. Post, P. van den Tempel, P. Herrera Sánchez, D. Maniar, R. K. Bose, V. S. Voet, R. Folkersma, F. Picchioni and K. Loos, ACS Sustainable Chem. Eng., 2025, 13(9), 3543–3553 CrossRef CAS PubMed.
- Y. Jiang, A. J. Woortman, G. O. Alberda van Ekenstein, D. M. Petrovic and K. Loos, Biomacromolecules, 2014, 15, 2482–2493 CrossRef CAS PubMed.
- S. Fulignati, C. Antonetti, T. Tabanelli, F. Cavani and A. M. Raspolli Galletti, ChemSusChem, 2022, 15, e202200241 CrossRef CAS PubMed.
- L. Hu, Y. Jiang, X. Wang, A. He, J. Xu and Z. Wu, Biomass Convers. Biorefin., 2020, 1–16 Search PubMed.
- X. Tang, J. Wei, N. Ding, Y. Sun, X. Zeng, L. Hu, S. Liu, T. Lei and L. Lin, Renewable Sustainable Energy Rev., 2017, 77, 287–296 CrossRef CAS.
- S. Li, Z. Kan, J. Bai, A. Ma, J. Lu and S. Liu, ChemSusChem, 2024, 17, e202400869 CrossRef CAS PubMed.
- P. Zhu, M. Shi, Z. Shen, X. Liao and Y. Chen, Chem. Sci., 2024, 15, 4723–4756 RSC.
- Y. Gao, L. Ge, H. Xu, K. Davey, Y. Zheng and S.-Z. Qiao, ACS Catal., 2023, 13, 11204–11231 CrossRef CAS.
- J. J. Roylance, T. W. Kim and K.-S. Choi, ACS Catal., 2016, 6, 1840–1847 CrossRef CAS.
- X. H. Chadderdon, D. J. Chadderdon, T. Pfennig, B. H. Shanks and W. Li, Green Chem., 2019, 21, 6210–6219 RSC.
- X. Yuan, K. Lee, J. Schmidt and K.-S. Choi, J. Am. Chem. Soc., 2023, 145, 20473–20484 CrossRef CAS PubMed.
- X. Yuan, K. Lee, M. T. Bender, J. Schmidt and K. S. Choi, ChemSusChem, 2022, 15, e202200952 CrossRef CAS PubMed.
- X. H. Chadderdon, D. J. Chadderdon, J. E. Matthiesen, Y. Qiu, J. M. Carraher, J.-P. Tessonnier and W. Li, J. Am. Chem. Soc., 2017, 139, 14120–14128 CrossRef CAS PubMed.
- X. Guo, H. Fu, J. Yang, L. Luo, H. Zhou, M. Xu, X. Kong, M. Shao, H. Duan and Z. Li, ACS Catal., 2023, 13, 13528–13539 CrossRef CAS.
- K. Ji, M. Xu, S. M. Xu, Y. Wang, R. Ge, X. Hu, X. Sun and H. Duan, Angew. Chem., Int. Ed., 2022, 61, e202209849 CrossRef CAS PubMed.
- D. T. Jantz and K. C. Leonard, Ind. Eng. Chem. Res., 2018, 57, 7431–7440 CrossRef CAS.
- C. M. Sánchez-Sánchez, J. Rodríguez-López and A. J. Bard, Anal. Chem., 2008, 80, 3254–3260 CrossRef PubMed.
- C. Jung, C. M. Sanchez-Sanchez, C.-L. Lin, J. Rodríguez-López and A. J. Bard, Anal. Chem., 2009, 81, 7003–7008 CrossRef CAS PubMed.
- A. Salverda, S. Abner, E. Mena-Morcillo, A. Zimmer, A. Elsayed and A. Chen, J. Phys. Chem. C, 2023, 127, 7151–7161 CrossRef CAS.
- H. S. Park, K. C. Leonard and A. J. Bard, J. Phys. Chem. C, 2013, 117, 12093–12102 CrossRef CAS.
- H. S. Ahn and A. J. Bard, J. Am. Chem. Soc., 2015, 137, 612–615 CrossRef CAS PubMed.
- Z. Jin and A. J. Bard, Angew. Chem., 2021, 133, 807–812 CrossRef.
- S. Han, J. Yoo and W. T. Choi, J. Mater. Chem. A, 2024, 12, 12026–12033 RSC.
- Y. Kwon, E. de Jong, S. Raoufmoghaddam and M. T. Koper, ChemSusChem, 2013, 6, 1659–1667 CrossRef CAS PubMed.
- L. Zhang, F. Zhang, F. C. Michel Jr and A. C. Co, ChemElectroChem, 2019, 6, 4739–4749 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5cc01465d |
| This journal is © The Royal Society of Chemistry 2025 |