
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Calcium catalysed Strecker-type reactions towards α-aminonitriles†
Ashley J.
Basson‡
a,
Michael P.
Cameron‡
b and
Mark G.
McLaughlin
 *ab
*ab
aDepartment of Chemistry, Lancaster University, Bailrigg, UK
bSchool of Chemistry and Chemical Engineering, Queen's University Belfast, Belfast, UK. E-mail: mark.mclaughlin@qub.ac.uk
First published on 14th March 2025
Abstract
We report an operationally simple calcium catalysed Strecker type reaction for the synthesis of α-aminonitriles from readily available N,O-acetals. The reaction is tolerant to a wide range of useful functional groups, including heterocycles, and provides the product in good to excellent yields. Additionally, the reaction does not require the need for anhydrous or air-free conditions, making it a suitable candidate for high throughput experimentation.
α-Amino nitriles are privileged scaffolds in synthetic chemistry,1,2 having found use as a useful building block in the synthesis of a wide range of important scaffolds including α-amino acids and 1,2-diamines. They are also found in bioactive natural products and pharmaceuticals,3,4 with increased usage across medicinal chemistry programmes to treat a range of conditions including diabetes and cancer (Fig. 1).5
 | ||
| Fig. 1 Selected examples of α-aminonitriles. | ||
The traditional approach to synthesise these motifs is via the classical Strecker reaction, in which the treatment of an aldehyde with an amine donor (typically ammonia) in the presence of HCN produces the desired product. Although successful, the use of gaseous HCN and other issues with functional group tolerance and overall applicability has led to a concerted effort from the community to realise an alternative to the Strecker reaction (Scheme 1).1 This has led to several elegant methods employing cyanide surrogates, transition metal6–9 and main group element10,11 catalysed transformations, organocatalysis12,13 and photoredox catalysis.14 Further examples including direct displacement with cyanide surrogates have been shown to improve substrate scope, with indium15 and zinc16,17 both performing well. Our interest in these α-amino nitriles developed from an ongoing medicinal chemistry programme developing type III kinase inhibitors, in which we required ready access to α-functionalised amines. Owing to the group's interest in calcium catalysed direct functionalisation reactions,18–20 we wondered if we could employ a similar tactic to afford this privileged scaffold (Scheme 1(D)).
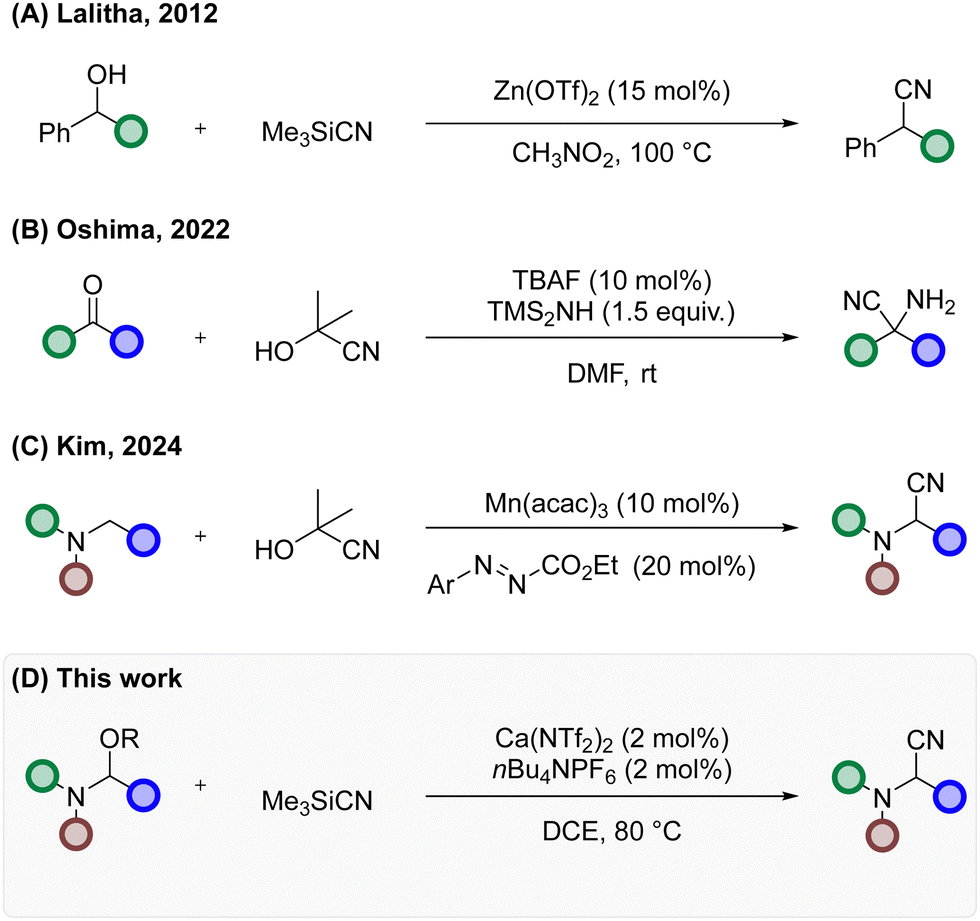 | ||
| Scheme 1 Alternative approaches to a-aminonitrles and derivatives. | ||
As such, we began our study by reacting 1a with TMSCN in the presence of Ca(NTf2)2/nBu4NPF6 in DCM at room temperature for 12 hours, afforded 2a in 22% yield (entry 1). A survey of solvents quickly showed that 1,2-DCE was optimum (entries 2–5). Further optimisation concentrating on catalyst loading, temperature and reagent equivalents (entries 6–10) resulted in 2a being produced in 86% isolated yield (entry 10) (Table 1).
|
|
|||||
|---|---|---|---|---|---|
| Entry | Cat. loading (mol%) | Solvent | TMSCN (equiv.) | Temp (°C) | Yielda (%) |
| a NMR yield 1,3,5-trimethoxybenzene as an internal standard. b Isolated yield after flash column chromatography. | |||||
| 1 | 2 | DCM | 3 | 25 | 22 |
| 2 | 2 | DCM | 3 | 40 | 29 |
| 3 | 2 | 1,2-DCE | 3 | 40 | 30 |
| 4 | 2 | EtOAc | 3 | 40 | <5 |
| 5 | 2 | HFIP | 3 | 40 | <5 |
| 6 | 2 | 1,2-DCE | 3 | 80 | 66 |
| 7 | 2 | 1,2-DCE | 6 | 80 | 89 |
| 8 | 1 | 1,2-DCE | 6 | 80 | 66 |
| 9 | 0 | 1,2-DCE | 6 | 80 | nr |
| 10 | 2 | 1,2-DCE | 6 | 80 | 86b |

With these conditions now in hand, our attention turned to establishing the limits of the reaction in terms of functional group tolerance (Fig. 2). As shown in Fig. 2, the reaction is tolerant to a range of groups with differential electronics and sterics. Regarding the groups derived from the amide starting material, electron donating groups worked well (2b–c), however a moderate reduction in yield was observed when electron withdrawing groups were present on the aromatic ring (2d–g). However, this drop is reversed if an electron withdrawing group was present in the ortho-position of the aryl ring (2h–i). Heteroatoms were also tolerated well, with furan (2j), thiophene (2k) and pyrazine (2l) motifs providing the desired product in good yields.
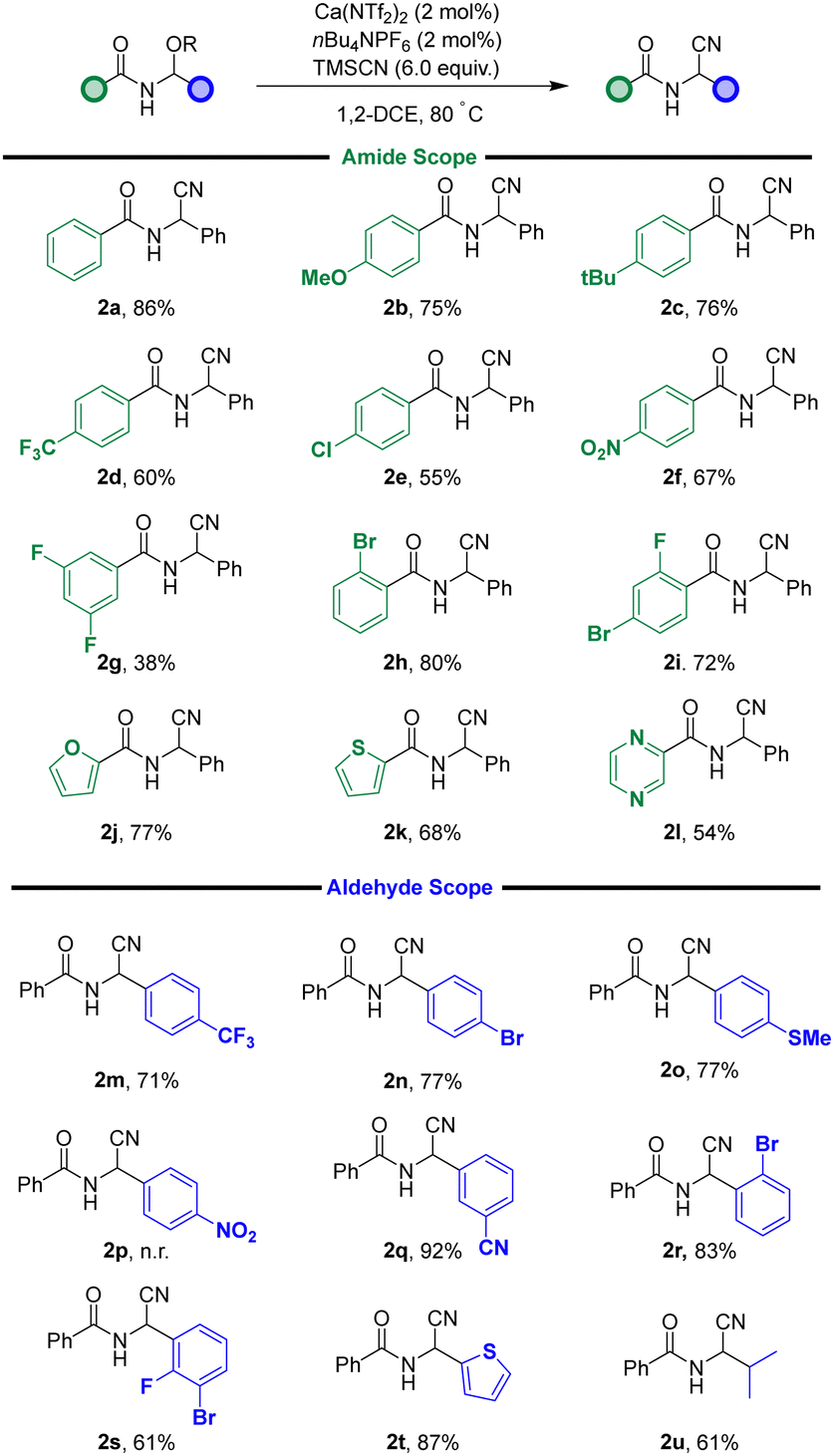 | ||
| Fig. 2 Substrate scope. | ||
Moving onto substrates derived from altering the aldehyde portion of the starting material, the reaction was, in general, much less variable. Each substrate performed admirably, with electron withdrawing (2m–n) and donating groups reacting similarly (2o). Interestingly, one repeated failure was the para-NO2 group (2p), which resulted in a complex mixture of products after each attempt. Substitution around the aromatic ring did not alter the efficiency, with ortho-(2r), meta-(2q) and doubly substituted working well (2s). Thiophene was also tolerated (2t), as was iPr (2u), which provided the leucine-type derivative in good yields.
We next wanted to explore if protecting groups other than benzoyl were applicable in the reaction (Fig. 3). To this end, we synthesised a range of Fmoc protected substrates and subjected them to the reaction conditions. Pleasingly, all but the aliphatic substrate worked well (4a–4d), thus providing access to a range of Fmoc-protected amino acids if required.
 | ||
| Fig. 3 Protecting group substrate scope. | ||
We also attempted the same reaction with the Boc protected amine, however the major product under the optimised conditions was the deprotected amine, which was isolated in 29% yield. We reasoned that the deprotection was a result of the well-established thermal degradation of Boc carbamates in the presence of protic solvents.21,22 We therefore attempted to retard the deprotection by simply lowering the temperature to 60 °C, which proved successful, affording the desired product (4f) in 57% yield.
Finally, we wanted to show the utility of the products towards further derivatisation. A such, we subjected 4b to catalytic hydrolysis employing the highly active, yet easily accessible, Ghaffar-Parkins catalyst,23,24 which afforded Fmoc-protected primary amide 5 in quantitative yield (Scheme 2).
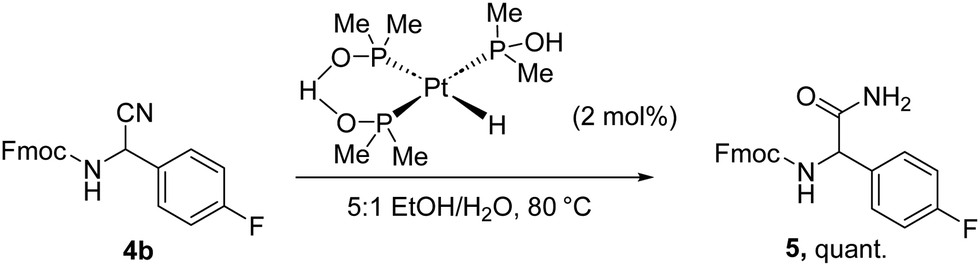 | ||
| Scheme 2 Ghaffar-Parkins hydrolysis. | ||
Moving on to other derivatisations, we took Bz protected amino nitrile 2a and produced the corresponding ester 6 and primary amine 8 in good yields. Furthermore, imidazole 7 can be produced using PPh3/CCl4 in MeCN in synthetically useful yields (Scheme 3).25
 | ||
| Scheme 3 Further derivatisations. | ||
In conclusion, we have developed a high yielding synthesis of α-aminonitriles employing a calcium catalysed Strecker type reaction. The transformation is tolerant to a wide range of functional groups including heterocycles as well as having the ability to use useful amine protecting groups.
The authors wish to thank QUB/EPSRC for a studentship (MPC) and Lancaster University for a studentship (AJB). We want to thank QUB for infrastructural and financial assistance. We also thank Mr Conor McGrath for HRMS.
Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Notes and references
- V. V. Kouznetsov and C. E. P. Galvis, Tetrahedron, 2018, 74, 773–810 CAS.
- D. Enders and J. P. Shilvock, Chem. Soc. Rev., 2000, 29, 359–373 RSC.
- E. J. Martinez, T. Owa, S. L. Schreiber and E. J. Corey, Proc. Natl. Acad. Sci. U. S. A., 1999, 96, 3496–3501 CrossRef CAS PubMed.
- L. Juillerat-Jeanneret, J. Med. Chem., 2014, 57, 2197–2212 CAS.
- F. F. Fleming, L. Yao, P. C. Ravikumar, L. Funk and B. C. Shook, J. Med. Chem., 2010, 53, 7902–7917 CAS.
- L. Zhao, B. Liu, Q. Tan, C.-H. Ding and B. Xu, Org. Lett., 2019, 21, 9223–9227 CAS.
- Y. Takahashi, R. Yoshii, T. Sato and N. Chida, Org. Lett., 2018, 20, 5705–5708 CAS.
- S. B. Kim, G. Park, E. S. Park, S. Maiti and J. Kim, J. Org. Chem., 2024, 89, 14543–14548 CAS.
- J. Jarusiewicz, Y. Choe, K. S. Yoo, C. P. Park and K. W. Jung, J. Org. Chem., 2009, 74, 2873–2876 CAS.
- P. Fontaine, A. Chiaroni, G. Masson and J. Zhu, Org. Lett., 2008, 10, 1509–1512 CAS.
- Y. Kondo, Y. Hirazawa, T. Kadota, K. Yamada, K. Morisaki, H. Morimoto and T. Ohshima, Org. Lett., 2022, 24, 6594–6598 CrossRef CAS PubMed.
- H. Wang, K. Wang, Y. Ren, N. Li, B. Tang and G. Zhao, Adv. Synth. Catal., 2017, 359, 1819–1824 CrossRef CAS.
- Y. Wen, B. Gao, Y. Fu, S. Dong, X. Liu and X. Feng, Chem. – Eur. J., 2008, 14, 6789–6795 CrossRef CAS PubMed.
- M. Rueping, S. Zhu and R. M. Koenigs, Chem. Commun., 2011, 47, 12709–12711 CAS.
- G. Chen, Z. Wang, J. Wu and K. Ding, Org. Lett., 2008, 10, 4573–4576 CAS.
- P. Theerthagiri and A. Lalitha, Tetrahedron Lett., 2012, 53, 5535–5538 CAS.
- Y. Nagase, T. Sugiyama, S. Nomiyama, K. Yonekura and T. Tsuchimoto, Adv. Synth. Catal., 2014, 356, 347–352 CAS.
- A. J. Basson, N. R. Halcovitch and M. G. McLaughlin, Chem. – Eur. J., 2022, 28, e202201107 CAS.
- A. J. Basson and M. G. McLaughlin, Cell Rep. Phys. Sci., 2023, 4, 101234 CAS.
- A. J. Basson and M. G. McLaughlin, ChemSusChem, 2021, 14, 1696–1699 CAS.
- J. Wang, Y.-L. Liang and J. Qu, Chem. Commun., 2009, 5144–5146, 10.1039/B910239F.
- W. Medina-Ramos, M. A. Mojica, E. D. Cope, R. J. Hart, P. Pollet, C. A. Eckert and C. L. Liotta, Green Chem., 2014, 16, 2147–2155 CAS.
- T. Ghaffar and A. W. Parkins, Tetrahedron Lett., 1995, 36, 8657–8660 CAS.
- D. W. Turner, A. T. Hands and N. T. Garg, Org. Synth., 2024, 101, 327–341 CAS.
- Y.-L. Zhong, J. Lee, R. A. Reamer and D. Askin, Org. Lett., 2004, 6, 929–931 CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5cc00539f |
| ‡ These authors contributed equally to this work and inclusion is alphabetical. |
| This journal is © The Royal Society of Chemistry 2025 |