
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Operando infrared and inelastic neutron scattering studies of zeolite catalysed low-density polyethylene degradation†
Emma
Campbell
ab,
Katie S. C.
Morton
bc,
Alexander J.
O’Malley
 bc,
Sandie E.
Dann
d,
Pierre
Moreau
de,
Ian P.
Silverwood
bf,
Simon A.
Kondrat
d,
Andrew M.
Beale
bg and
Matthew E.
Potter
*bc
bc,
Sandie E.
Dann
d,
Pierre
Moreau
de,
Ian P.
Silverwood
bf,
Simon A.
Kondrat
d,
Andrew M.
Beale
bg and
Matthew E.
Potter
*bc
aSchool of Chemistry, Cardiff University, Cardiff CF10 3AT, UK
bUK Catalysis Hub, Research Complex at Harwell (RCaH), Harwell, Didcot, Oxfordshire OX11 0FA, UK. E-mail: mep61@bath.ac.uk
cInstitute of Sustainability and Climate Change, Department of Chemistry, University of Bath, Bath BA2 7AY, UK
dDepartment of Chemistry, Loughborough University, Loughborough LE11 3TU, UK
ePlastic Energy ltd., Charnwood Building, Loughborough LE11 3GB, UK
fISIS Pulsed Neutron and Muon Facility, Science and Technology Facilities Council, Rutherford Appleton Laboratory, Didcot OX11 0QX, UK
gDepartment of Chemistry, University College London, 20 Gordon Street, London WC1H 0AJ, UK
First published on 1st April 2025
Abstract
The degradation of polyethylene (LDPE) by H-ZSM-5 is investigated by vibrational spectroscopy, using diffuse reflectance infrared Fourier transform spectroscopy (DRIFTS) and inelastic neutron scattering (INS). DRIFTS results indicate initial interaction between LDPE and external silanol groups, and hydrogen bonding with some Brønsted acid sites. At low temperatures both techniques suggest minimal interactions between the LDPE and the internal acid site of the zeolite, suggesting initial interactions are primarily with the zeolite surface. At higher reaction temperatures, oligomerisation is observed on Brønsted acid sites.
Plastics are essential in modern life, with over 460 million tons annually produced.1 Their stability, and slow degradation, has created a global build-up of plastic waste, demanding immediate solutions.2 Recycling is typically preferred for disposal, as this minimises environmental contamination. Mechanical recycling blends used polymers into a new material, though eventually yields a lower quality plastic.3 Chemical recycling, through thermal pyrolysis is a promising alternative method.4 Here used polymer is heated under inert gas, to yield light olefins, paraffins, aromatic molecules and longer chain oils (C6+), with some solid residue remaining. This simple method uses high temperatures (∼400 °C), but adding a catalyst can lower the degradation temperature.5
Several studies have shown that solid acid catalysts, such as zeolites are effective for catalytic pyrolysis of low-density polyethylene (LDPE), due to the high density of strong acid sites.6 The most commonly investigated zeolite topologies for this reaction are zeolite Hβ (BEA),7,8 and HZSM-5 (MFI).5,9 HZSM-5 has a range of acid sites that can form on the external surface and internal pore walls, though it is not clear what specific roles each site plays.10 Some suggest that LDPE interacts directly with strong acid sites to form a carbonium ion, with others proposing that the initial interactions occur with silanol groups on the external surface. A common hypothesis is that chains crack on the surface, randomly along the chain (random scission) or at the end (end-chain scission), and cracked species enter the pore, to undergo a further series of reactions towards the final products.11
Few in situ or operando studies have been performed on solid acid catalysed LDPE cracking. Fourier transform infra-red (FT-IR) spectroscopy is often used to probe the acidity and accessibility of zeolites, whilst other reactions, such as fluid catalytic cracking,12 and methanol-to-hydrocarbons,13 have used it to differentiate behaviour on the external surface, pore mouths, and internal pore walls, something that would be hugely beneficial in catalytic LDPE pyrolysis. Primarily FT-IR is used for its fast data collection times (<1 s per scan), and versatile sample environments, with ease of access. Despite there are some limitations. Firstly, selection rules limit the number of visible signals. Also strong signals due to the zeolite's vibrations dominate the spectra below 1200 cm−1, shrouding most other signals.14 Inelastic neutron scattering (INS), a vibrational technique like FT-IR and Raman spectroscopy, is an excellent complement to FT-IR. Hydrogen has one of the largest incoherent neutron scattering cross sections, meaning INS spectra are dominated by hydrocarbons, and not framework modes, allowing measurement below 1500 cm−1.14,15 Cen et al. used INS to identify the reaction pathway of polyethylene with a modified ZSM-5 catalyst over a reaction period of 2 hours.16
In this work we present combine findings from operando diffuse reflectance infrared Fourier transform spectroscopy (DRIFTS) and INS on LDPE catalytic pyrolysis with a commercial HZSM-5 catalyst, focusing on the very initial stages of reaction. We will use these findings to provide further insight into the catalytic mechanism, towards designing a new generation of zeolite catalysts for plastic recycling. Particular attention was given to the lower temperature intermediates, after initial cracking had begun, to determine which products have formed, by combining through on-line mass spectrometry (MS).
The H-ZSM-5 is from Zeolyst International (NH4-ZSM-5, CBV 8014, Si/Al 40), and was calcined for 6 hours at 550 °C (ramp rate 1 °C min−1) under static conditions to yield the proton form H-ZSM-5. Physicochemical characterisation is provided in the ESI† (Fig. S1 and S2). LDPE is from Sigma Aldrich (427![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 772, average Mw ∼ 4000). Thermogravimetric analysis (TGA) was performed on a TGA 5500 from TA instruments, by heating the sample in a flow of nitrogen at 5 °C min−1 from room temperature to 700 °C. During DRIFTS measurements, the zeolite was fully dehydrated in a stream of dry oxygen in argon, in a Praying Mantis reaction chamber from Harrick Scientific, and the reactor transported into an argon filled glove box, where the zeolite was physically mixed at room temperature in a 3:1 mass ratio with LDPE. The mixture was transferred back into the reaction chamber in the glove box to prevent any water adsorption. The reaction chamber was placed back into the Praying Mantis accessory in the sample compartment of a Bruker Vertex 80 to measure DRIFTS spectra. Dry argon was flowed over the zeolite and plastic mixture, and outlet gases were monitored by mass spectrometry (Pfieffer Omnistar). Measurements were collected at temperature, during reaction. Samples were prepared for INS measurements by first drying the zeolite in a flow of dry oxygen and helium, then combining the zeolite with plastic in a 3:1 ratio by mass. The mixture of plastic and zeolite was separated into portions which were heated to 80, 130, 170, or 200 °C under flowing helium. After heating, the samples were cooled and transferred into an aluminium can for measurement on TOSCA at ISIS Neutron and Muon Source, to measure at −253 °C. Further experimental details are given in the ESI.†
772, average Mw ∼ 4000). Thermogravimetric analysis (TGA) was performed on a TGA 5500 from TA instruments, by heating the sample in a flow of nitrogen at 5 °C min−1 from room temperature to 700 °C. During DRIFTS measurements, the zeolite was fully dehydrated in a stream of dry oxygen in argon, in a Praying Mantis reaction chamber from Harrick Scientific, and the reactor transported into an argon filled glove box, where the zeolite was physically mixed at room temperature in a 3:1 mass ratio with LDPE. The mixture was transferred back into the reaction chamber in the glove box to prevent any water adsorption. The reaction chamber was placed back into the Praying Mantis accessory in the sample compartment of a Bruker Vertex 80 to measure DRIFTS spectra. Dry argon was flowed over the zeolite and plastic mixture, and outlet gases were monitored by mass spectrometry (Pfieffer Omnistar). Measurements were collected at temperature, during reaction. Samples were prepared for INS measurements by first drying the zeolite in a flow of dry oxygen and helium, then combining the zeolite with plastic in a 3:1 ratio by mass. The mixture of plastic and zeolite was separated into portions which were heated to 80, 130, 170, or 200 °C under flowing helium. After heating, the samples were cooled and transferred into an aluminium can for measurement on TOSCA at ISIS Neutron and Muon Source, to measure at −253 °C. Further experimental details are given in the ESI.†
TGA (Fig. S3, ESI†) compared the pyrolysis temperature of pure LDPE, with a 3:1 mixture by mass of HZSM-5 (catalyst) and LDPE, under nitrogen. This ratio was used throughout this study to ensure there is LDPE signals are visible, while maximising the zeolite-LDPE interactions. Without a catalyst, the majority of LDPE degradation occurs between 400 and 500 °C (Fig. S3a, ESI†), however, in the 3:1 mixture the degradation temperature was drastically lowered to between 200 and 300 °C. Beyond 300 °C the LDPE in the mixed system has been fully converted (roughly 100% conversion). This is consistent with previous works showing the influence of the zeolite on lowering the degradation temperature.5 The associated MS data (Fig. S3b, ESI†) confirms that almost exclusively the 3:1 ZSM-5 and LDPE mixture are small chain alkenes and alkanes, particularly in the 200 to 300 °C region. There was little evidence of any evolved water (beyond 100 °C), hydrogen, or CO2, confirming the polymer is indeed being cracked, making the ZSM-5 catalyst a suitable model system for further investigation.
The DRIFTS O–H region of the dehydrated zeolite (black, Fig. 1a and Fig. S4, ESI†) shows signals (Table S1, ESI†) associated with Brønsted acid sites (BAS; 3605 cm−1), external (3736 cm−1) and internal silanol groups (3725 cm−1).17–19 A broad feature below 3500 cm−1 indicates that the zeolite contains internal defects, such as silanol nests. The changes in the O–H region, on physically mixing ZSM-5 with the LDPE, show a clear interaction between the components (Table S2, ESI†). There is a notable loss in intensity for the external silanol groups at 3737 cm−1, which is accompanied by a new feature at 3694 cm−1, showing an interaction between the zeolite surface and the LDPE. This peak shift (Δν) of −43 cm−1 is similar to previous observations of external silanols interacting with products of butene oligomerisation (−48 cm−1), and heptane adsorption (−46 cm−1),20 but smaller than measured in other studies of isobutane (−80 cm−1) and n-heptane (−95 cm−1) adsorption.19 We also observe a the BAS signal (3605 cm−1) reduces, while the broad signal at 3440 cm−1 (down shifted by 165 cm−1 from the previous maximum from hydrogen-bonded silanol nests) grows.
 | ||
| Fig. 1 (a) DRIFTS spectrum showing (a) the absorption of 100% ZSM-5 (black), a mixture of 75% ZSM-5 and 25% LDPE (red) after heating to 80 °C, both recorded under an inert (Argon) atmosphere, compared with a spectrum of pure LDPE (yellow), and (b) the influence of temperature on the spectrum of the 75% ZSM-5, 25% LDPE mixture. | ||
While the hydrogen-bonded silanol nests are responsible for the broad signal at ∼3450–3500 cm−1 in the empty zeolite,18,21 an interaction between the BAS and LDPE is responsible for the absorbance after combination with LDPE, which is enhanced at 80 °C (Fig. S4d, ESI†).18 Spoto et al. noted a downward shift of 389 cm−1 in the frequency of BAS due to hydrogen bonding of ethene, via the pi system,18 and Gakowski et al. recorded a shift of 174 cm−1 upon the adsorption of n-hexane.22 The shift observed here after LDPE mixing with H-ZSM-5 is most similar in magnitude to that recorded upon n-hexane adsorption which is reasonable given the fully saturated nature of LDPE. A shoulder at 2955 cm−1 is also seen, which is assigned to asymmetric CH3 stretching.23 CH2 groups in LDPE are also present, mixing by deformation bands at 1460 cm−1 (Fig. S4, ESI†).23,24 Overall, this highlights the interaction of LDPE and the ZSM-5 catalyst, suggesting these interactions are not limited to surface species, though primarily occur on the surface. We note there are only very subtle changes from the spectrum of pure LDPE (Fig. 1a and Fig. S4a, b, ESI†).
Increasing the temperature to 200 °C, showed little change in the CH stretch region, beyond the signals narrowing (Fig. 1b). Above 170 °C, the MS detects alkenes eluting from the reactor (Fig. S5, ESI†), the BAS signal (3605 cm−1) drops in intensity, and a new vibration at 1509 cm−1 emerges (Table S3, ESI†), in a region associated with hydrocarbons with multiple double bonds.25,26 The development of this band correlates with the erosion of BAS up to 200 °C, indicating that alkenes formed by initial LDPE cracking, have undergone further reaction with BAS and polymerised. MS data (Fig. S5, ESI†) indicates that by 170 and 200 °C, exclusively small alkenes are eluting from the reaction, as the cracking process is taking place, and these small alkenes can enter the pores to react with BAS. Taking the reaction above 200 °C (Fig. 2 and Fig. S6, ESI†) to 260 °C reveals subtle differences. The broad band at 3500 cm−1 (Fig. S6a, ESI†) related to hydrogen-bonded species decreases, which appeared when LDPE initially interacted with the zeolite. Similarly, we see a new signal at 3120 cm−1, attributed to sp2 hybridised C–H stretches of alkenes or aromatics (Fig. 2).27 Such species (particularly aromatics) are linked with pore blockage and catalytic deactivation. There is a pronounced increase in the 1509 cm−1 signal (Table S3 and Fig. S6b, ESI†), again linked to the formation of unsaturated carbocations, representing a conjugated sp2 C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C stretch. Therefore above 200 °C we begin to see the formation of alkene or aromatic species. Likely these reside within the pores, as they were not detected in the outlet MS data.
C stretch. Therefore above 200 °C we begin to see the formation of alkene or aromatic species. Likely these reside within the pores, as they were not detected in the outlet MS data.
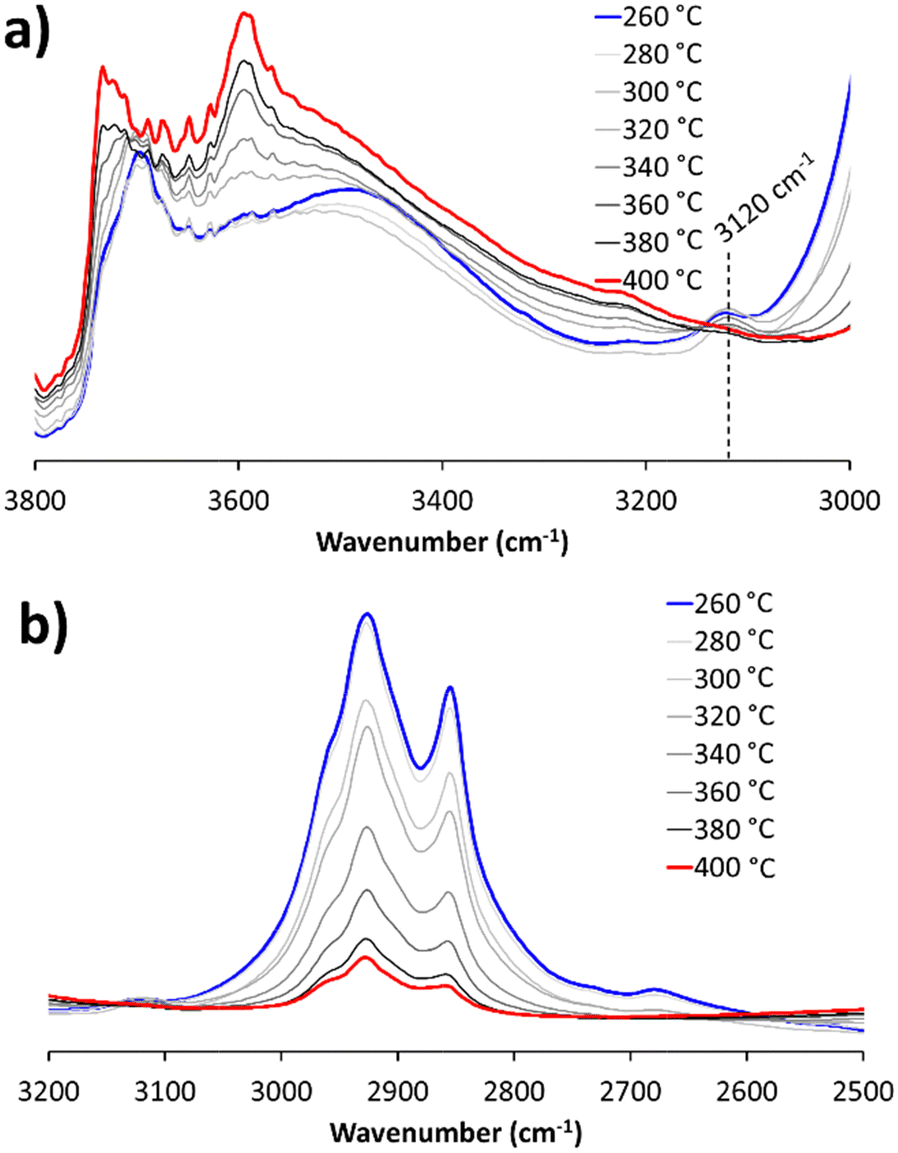 | ||
| Fig. 2 DRIFTS spectra of the LDPE/ZSM-5 mixture, as an evolution of temperature from 260 to 400 °C, focussing on the (a) 3800–3000 cm−1, and (b) 3200–2500 cm−1 regions. | ||
Increasing the temperature further shows that from 320 °C the BAS begin regenerating, as shown by the return of the signal at 3605 cm−1. This is due to the bound hydrocarbons (alkenes) desorbing from the catalyst, reforming the BAS. This coincides with the concentration of hydrocarbons decreasing above 300 °C, in the effluent gas stream (Fig. S5, ESI†). As the temperature approaches 400 °C, C–H stretches from 2800–2950 cm−1 continue to drop. The OH stretch of external silanol groups (3737 cm−1) recover as the hydrogen bonded hydrocarbons are removed from the zeolite surface (Fig. 2b).
The signal at 3120 cm−1 also declines at high temperatures as the unsaturated species on the surface from cracking or aromatic condensation, react further akin to the processes occurring in the methanol-to-olefin system.28 This shows the near-complete removal of hydrocarbon species, as the reaction approaches 400 °C, in line with our TGA findings (Fig. S3, ESI†), suggesting quantitative conversion had been achieved.
An analogous INS study was also performed, which focuses on wavenumbers below 1500 cm−1. With the lack of selection rules for INS, signal intensity is not dependent on absorption coefficients and different features can be probed. As expected, due to its low hydrogen content, the ZSM-5 barely contributes to the INS spectra, with no discernible features seen in hydroxyl region (3500–4000 cm−1; Fig. S7A, ESI†). The INS spectra of our LDPE/ZSM-5 system (Fig. 3 and Fig. S7B, ESI†), are in excellent agreement with previous INS studies on LDPE.29 Assignments for the main features are found in Table S4 of the ESI.† Given LDPE's melting temperature (105–115 °C), the similarity of these two spectra suggests no noticeable changes during melting and recrystallisation. Further the MS data of the samples (Fig. S8, ESI†) shows that despite cracking occurring beyond 120 °C, with significant cracking occurring at 200 °C, there is minimal noticeable change on heating. This suggests there are no significant changes to bulk LDPE at these temperatures, with no notable evidence of confined LDPE. This supports the DRIFTS findings that initial contact and activation occur on the surface of the catalyst, such that the initial cracking of LDPE is not shape-selective. This does not preclude smaller cracked hydrocarbons later entering the pores, as per the schematic given in Fig. S9 (ESI†).
 | ||
| Fig. 3 INS spectra showing the scaled LDPE/ZSM-5 mix after being exposed to different temperatures, spectra have been artificially separated by a factor of 0.0015 for ease of comparison. | ||
Through a combination of operando DRIFTS and INS spectroscopy we gained valuable insights into a promising plastic recycling process; acid-catalysed LDPE cracking. DRIFTS provided evidence of internal BAS interacting with LDPE chains below 100 °C, but mainly with external silanol groups. At higher temperatures the intensity of signals from BAS decrease, as smaller olefins and paraffins molecules form. This suggests that there is limited interaction between the internal sites and the LDPE chains during the initial cracking, however, these sites do catalyse further reactions with smaller hydrocarbons, formed by the initial cracking. This hypothesis is also supported by the INS study. In both cases there were minimal changes in the bands associated with the chemical structure of the LDPE, suggesting the polymer breakdown is highly systematic. Overall, the novel combination of operando DRIFTS and INS for the acid catalysed LDPE degradation has given insight into the mechanism, and warrants further exploration, understanding, and application to other solid acid catalysts.
Data availability
INS data in this proposal was obtained on the TOSCA instrument, at the ISIS Neutron and Muon Source under experiment RB2220645. The DOI for this is: https://doi.org/10.5286/ISIS.E.RB2220645.Conflicts of interest
There are no conflicts to declare.Notes and references
- https://www.oecd.org/en/about/news/press-releases/2022/06/global-plastic-waste-set-to-almost-triple-by-2060.html .
- L. D. Ellis, N. A. Rorrer, K. P. Sullivan, M. Otto, J. E. McGeehan, Y. Román-Leshkov, N. Wierckx and G. T. Beckham, Nat. Catal., 2021, 4, 539 CrossRef CAS.
- I. Vollmer, M. J. F. Jenks, M. C. P. Roelands, R. J. White, T. van Harmelen, P. de Wild, G. P. van der Laan, F. Meirer, J. T. F. Keurentjes and B. M. Weckhuysen, Angew. Chem., Int. Ed., 2020, 59, 15402 CrossRef CAS PubMed.
- A. Schade, M. Melzer, S. Zimmermann, T. Schwarz, K. Stoewe and H. Kuhn, ACS Sustainable Chem. Eng., 2024, 12, 12270 CrossRef CAS.
- S. Tsubota, S. Kokuryo, K. Tamura, K. Miyake, Y. Uchida, A. Mizusawa, T. Kubo and N. Nishiyama, Catal. Sci. Technol., 2024, 14, 1369 RSC.
- A. Maity, S. Chaudhari, J. J. Titman and V. Polshettiwar, Nat. Commun., 2020, 11, 3828 CrossRef PubMed.
- K. Pyra, K. A. Tarach, E. Janiszewska, D. Majda and K. Gora-Marek, Molecules, 2020, 25, 926 Search PubMed.
- X. Tian, Z. Zeng, Z. Liu, L. Dai, J. Xu, X. Yang, L. Yue, Y. Liu, R. Ruan and Y. Wang, J. Cleaner Prod., 2022, 358, 131989 CrossRef CAS.
- J. P. Hittinger and D. F. Shantz, Microporous Mesoporous Mater., 2022, 343, 112170 CrossRef CAS.
- A. Zachariou, A. P. Hawkins, R. F. Howe, J. M. S. Skakle, N. Barrow, P. Collier, D. W. Nye, R. I. Smith, G. B. G. Stenning, S. F. Parker and D. Lennon, ACS Phys. Chem. Au, 2023, 3, 74 CrossRef CAS PubMed.
- V. Daligaux, R. Richard and M.-H. Manero, Catalysts, 2021, 11, 770 CrossRef CAS.
- I. L. C. Buurmans, F. Soulimani, J. Ruiz-Martínez, H. E. van der Bij and B. M. Weckhuysen, Microporous Mesoporous Mater., 2013, 166, 86 CrossRef CAS.
- L. Maggiulli, V. L. Sushkevich, O. Kröcher, J. A. van Bokhoven and D. Ferri, ACS Catal., 2024, 14, 11477 CrossRef CAS.
- M. E. Potter, H. Cavaye, J. J. M. Le Brocq, L. L. Daemen and Y. Cheng, Phys. Chem. Chem. Phys., 2024, 26, 25969 RSC.
- A. J. O'Malley, S. F. Parker and C. R. A. Catlow, Chem. Commun., 2017, 53, 12164 Search PubMed.
- Z. Cen, X. Han, L. Lin, S. Yang, W. Han, W. Wen, W. Yuan, M. Dong, Z. Ma, F. Li, Y. Ke, J. Dong, J. Zhang, S. Liu, J. Li, Q. Li, N. Wu, J. Xiang, H. Wu, L. Cai, Y. Hou, Y. Cheng, L. L. Daemen, A. J. Ramirez-Cuesta, P. Ferrer, D. C. Grinter, G. Held, Y. Liu and B. Han, Nat. Chem., 2024, 16, 871 CrossRef CAS PubMed.
- S. Bordiga, I. Roggero, P. Ugliengo, A. Zecchina, V. Bolis, G. Artioli, R. Buzzoni, G. Marra, F. Rivetti, G. Spanò and C. Lamberti, J. Chem. Soc., Dalton Trans., 2000, 3921 RSC.
- G. Spoto, S. Bordiga, G. Ricchiardi, D. Scarano, A. Zecchina and E. Borello, J. Chem. Soc., Faraday Trans., 1994, 90, 2827 RSC.
- M. Trombetta, T. Armaroli, A. D. Gutièrrez Alejandre, J. Ramirez Solis and G. Busca, Appl. Catal., A, 2000, 192, 125 CrossRef CAS.
- M. Bjørgen, K.-P. Lillerud, U. Olsbye, S. Bordiga and A. Zecchina, J. Phys. Chem. B, 2004, 108, 7862 CrossRef.
- A. Zecchina, S. Bordiga, G. Spoto, D. Scarano, G. Spanò and F. Geobaldo, J. Chem. Soc., Faraday Trans., 1996, 92, 4863 RSC.
- M. Gackowski, J. Podobiński and M. Hunger, Microporous Mesoporous Mater., 2019, 273, 67 CrossRef CAS.
- S. Krimm, C. Y. Liang and G. B. B. M. Sutherland, J. Chem. Phys., 1956, 25, 549 Search PubMed.
- M. Milosevic and S. L. Berets, Appl. Spectrosc. Rev., 2002, 37, 347 CrossRef CAS.
- C. Pazè, B. Sazak, A. Zecchina and J. Dwyer, J. Phys. Chem. B, 1999, 103, 9978 CrossRef.
- D. A. Long, J. Raman Spectrosc., 2004, 35, 905 CrossRef.
- L. Palumbo, F. Bonino, P. Beato, M. Bjørgen, A. Zecchina and S. Bordiga, J. Phys. Chem. C, 2008, 112, 9710 CrossRef CAS.
- E. Borodina, F. Meirer, I. Lezcano-González, M. Mokhtar, A. M. Asiri, S. A. Al-Thabaiti, S. N. Basahel, J. Ruiz-Martinez and B. M. Weckhuysen, ACS Catal., 2015, 5, 992 CrossRef CAS.
- S. F. Parker, J. Chem. Soc., Faraday Trans., 1996, 92, 1941 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Including further information on experimental methods, additional data on catalyst characterisation, mass-spectrometry data, and further data and assignments of infrared and inelastic neutron scattering signals. See DOI: https://doi.org/10.1039/d4cc06209d |
| This journal is © The Royal Society of Chemistry 2025 |