
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
In situ biocatalytic ATP regulated, transient supramolecular polymerization†
Ananya
Mishra
 *ab,
Angshuman
Das
a and
Subi J.
George
*a
*ab,
Angshuman
Das
a and
Subi J.
George
*a
aSupramolecular Chemistry Laboratory, New Chemistry Unit and School of Advanced Materials (SAMat), Jawaharlal Nehru Centre for Advanced Scientific Research (JNCASR), Jakkur, Bangalore, 560064, India. E-mail: george@jncasr.ac.in
bCentre for Protolife Research, Centre for Organized Matter Chemistry, School of Chemistry, University of Bristol, Bristol BS81TS, UK. E-mail: ananya.mishra@bristol.ac.uk
First published on 19th August 2024
Abstract
Temporal control over self-assembly processes is a highly desirable attribute that is efficiently exhibited by biological systems, such as actin filaments. In nature, various proteins undergo enzymatically catalysed chemical reactions that kinetically govern their structural and functional properties. Consequently, any stimuli that can alter their reaction kinetics can lead to a change in their growth or decay profiles. This underscores the urgent need to investigate bioinspired, adaptable and controllable synthetic materials. Herein we intend to develop a general strategy for controlling the growth and decay of self-assembled systems via enzymatically coupled reactions. We achieve this by the coupling of enzymes phosphokinase/phosphatase with a bolaamphiphilic cationic chromophore (PDI) which selectively self-assembles with ATP and disassembles upon its enzymatic hydrolysis. The aggregation process is efficiently regulated by the controlled in situ generation of ATP, through enzymatic reactions. By carefully managing the ATP generating components, we realize precise control over the self-assembly process. Moreover, we also show self-assembled structures with programmed temporal decay profiles through coupled enzymatic reactions of ATP generation and hydrolysis, essentially rendering the process dissipative. This work introduces a novel strategy to generate a reaction-coupled one-dimensional nanostructure with controlled dimensions inspired by biological systems.
Introduction
Over the last few decades, research into the programmability and adaptivity of self-assembled polymers, with explicit structural and temporal control, is revolutionizing the field of supramolecular polymerization.1–3 Traditionally, this field has relied on the passive self-assembly process,4–8 but recent advances are steering it towards more active self-assembly techniques.9–11 This shift enables the creation of highly programmable self-assembled structures, which hold the potential of constructing kinetically and structurally controlled supramolecular polymers with predetermined length and dispersity.12,13 Additionally, these advanced techniques facilitate temporal programming resulting in supramolecular self-assembled structures achieved under non-equilibrium regimes.14–16 The exceptional precision in the self-assembly process found in naturally occurring proteins has inspired supramolecular chemists to translate these properties to the synthetic realms. Among the various strategies employed to achieve a temporal control over self-assembly, biocatalysis stands out as a particularly intriguing approach. Enzymatically controlled temporal self-assembly is ubiquitous in nature, directing essential processes such as cell motility, intracellular transport and maintaining the cell shape and structure.17–19 Biocatalysis is known to trigger in situ temporal changes in the self-assembly process, a property that has been exploited to mimic the self-assembly of biomacromolecules such as the formation of collagen fibrils.20 This has inspired researchers to investigate catalytic control over supramolecular polymerization which is crucial for combining structural and temporal control in synthetic systems.21Recent approaches have leveraged biocatalysis to obtain temporal control over supramolecular polymerization, which typically follows a nucleation growth mechanism to induce molecular order.22 In other examples, in situ hydrogel formation has also been explored through the integration of enzymatic networks with small molecules.23 In most of these examples, carefully designed peptide chains have been utilized, which undergo enzymatically triggered covalent bond cleaving or formation, initiating the self-assembly process. However, the next challenge in this field is to realize a universal approach for the predictive design of a controlled supramolecular polymerization process. Recent attempts have managed to achieve control over the structure24–30 and temporal31–34 aspects of the self-assembly process but often in separate systems. Drawing inspiration from biological programming, designing reaction-driven assemblies provides a straightforward strategy to achieve desirable control over supramolecular polymerization.35 With that idea, adenosine triphosphate (ATP) has recently been employed in multiple examples to provide a templating effect to the self-assembly process of supramolecular systems.36–39 While ATP has most commonly been used for templated transient assembly formation,40–42 there have been some reports towards the structural modulation of self-assembled systems with length dispersity control.43,44 Our recent work on the nucleation–elongation followed by seeded self-assembly facilitated by the structural modification of the self-assembling unit exhibits the first example of ATP triggered living supramolecular polymerization.43 However reaction regulated structural and temporal control similar to cytoskeletal self-assembly has so far not been achieved.
An interesting solution to this is to employ aggregating systems that can be controlled by biocatalysis, offering a more generalized strategy for constructing programmable supramolecular polymers. In a recent report from our group, we demonstrated the coupled nature of the enzymatic reaction with the growth of supramolecular polymers to realize structural control on the self-assembly.44 However, the coupling of antagonistic enzymatic reactions for controlled and transient supramolecular polymerization akin to the tandem functioning of cofilin and profilin in the treadmilling process of actin assembly45 by controlling ATP generation and decay rate remains unexplored. Herein we demonstrate a bioinspired strategy to achieve programmable supramolecular polymerization where an adenosine triphosphate (ATP) regulating, coupled enzymatic reaction network has been employed. We could successfully induce controlled supramolecular polymerization of a bolaamphiphilic dicationic molecule, perylene diimide functionalized with cationic trimethyl ammonium groups linked via ethylene spacers at the imide positions (PDI).46 This molecule is shown to undergo a time-dependent self-assembly when ATP was generated enzymatically from the precursor adenosine diphosphate (ADP) followed by a disassembly on enzymatic hydrolysis of ATP by employing the phosphokinase/phosphatase cycle. The resultant coupling of both enzymatic reactions resulted in the formation of transient assemblies of PDI. Taken together, this work establishes a facile method to create adaptive and active supramolecular systems driven by ATP regulation (Scheme 1).
 | ||
| Scheme 1 Biocatalytic ATP regulated temporal assembly of PDI. Molecular structure and schematic representation of (a) dicationic perylene diimide (PDI) and (b) enzymatic adenosine triphosphate (ATP) generation from adenosine diphosphate (ADP) in the presence of creatine phosphokinase (CPK) and phosphocreatine (PCr). This releases creatine (Cr) as a by-product. Enzymatic ATP hydrolysis to adenosine and PO43− is triggered in the presence of calf intestinal alkaline phosphatase (CIAP). (c) Schematic representation of the overall biocatalytic controlled growth and seeding of ATP bound PDI through in situ generation of ATP followed by transient characteristics achieved through coupled enzymatic reactions and ATP regulation. | ||
Results and discussion
ATP selective self-assembly of PDI
To realize bioinspired, enzymatically controlled supramolecular polymerization, we selected ATP as the enzyme-responsive template molecule capable of inducing integrative dynamic assembly. First, we investigated ATP selectivity in the supramolecular polymerization process of PDI. Due to its bolaamphiphilic nature, PDI (5 × 10−5 M) remained in a monomeric state in 100% DMSO. However, as the percentage of HEPES increased in the HEPES/DMSO mixture, PDI underwent aggregation, indicated by a reversal in the absorbance of vibronic bands (A492/A530 from 0.64 to 1.48 for 100% DMSO and 100% HEPES, respectively) and the appearance of a new aggregate band at 580 nm (Fig. S1, ESI†). Therefore, we selected a 50/50 HEPES/DMSO (v/v) mixture for further experiments where PDI formed small nano-aggregates due to randomly arranged molecules that self-assemble through electrostatic interaction with ATP (Fig. 1 and Fig. S2, ESI†). As the equivalents of ATP were increased in the 5 × 10−5 M PDI solution (50% HEPES/DMSO mixture), a gradual increase in the aggregate band at λ580nm, along with broadening of the absorption spectra, suggested ATP-templated self-assembly of PDI. Furthermore, the chiral induction to the assembly as evident in the CD spectra from the chiral ribose sugar and quenched emission corroborated this observation (Fig. S2–S5 and Table S1, ESI†). Detailed spectroscopic characterization of ATP-bound PDI indicated the formation of H-type aggregates resulting from π–π stacking of the chromophores. The titration plot obtained by monitoring the absorbance at 580 nm followed a sigmoidal profile that saturated at 2.0 equivalents of ATP, suggesting 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 binding of PDI with ATP with an association constant (Ka) of 4.21 × 105 M−1 (Fig. 1b and Fig. S2 and S3, ESI†). In contrast, titrating PDI with increasing equivalents of ADP showed negligible changes in the absorption spectra (at 580 nm), indicating ATP-selective aggregation of PDI (Fig. 1a and b and Fig. S2d–f, ESI†). However, the slight quenching in the corresponding emission spectra suggests the weak binding of ADP with PDI (Fig. S2e and f, ESI†). The ATP-selective self-assembly of PDI was further evident from the extent of chiral induction in the PDI-ATP/ADP stack (Fig. S4, ESI†). The CD titration plot showed a CD silent state up to 0.8 equivalents of ATP, beyond which the CD signal increased gradually, saturating around 2.0 equivalents of ATP due to allosteric regulation. Conversely, no chiral induction in the CD signal was observed with increasing equivalents of ADP. From these spectroscopic experiments, we concluded that PDI exhibits ATP selectivity in self-assembly. For further experiments, we used 2.0 equivalents of ATP to maximize the extent of PDI self-assembly.
:1 binding of PDI with ATP with an association constant (Ka) of 4.21 × 105 M−1 (Fig. 1b and Fig. S2 and S3, ESI†). In contrast, titrating PDI with increasing equivalents of ADP showed negligible changes in the absorption spectra (at 580 nm), indicating ATP-selective aggregation of PDI (Fig. 1a and b and Fig. S2d–f, ESI†). However, the slight quenching in the corresponding emission spectra suggests the weak binding of ADP with PDI (Fig. S2e and f, ESI†). The ATP-selective self-assembly of PDI was further evident from the extent of chiral induction in the PDI-ATP/ADP stack (Fig. S4, ESI†). The CD titration plot showed a CD silent state up to 0.8 equivalents of ATP, beyond which the CD signal increased gradually, saturating around 2.0 equivalents of ATP due to allosteric regulation. Conversely, no chiral induction in the CD signal was observed with increasing equivalents of ADP. From these spectroscopic experiments, we concluded that PDI exhibits ATP selectivity in self-assembly. For further experiments, we used 2.0 equivalents of ATP to maximize the extent of PDI self-assembly.
 | ||
| Fig. 1 ATP selective self-assembly of PDI. (a) Comparative absorption spectra of PDI (pink), PDI-ADP (purple) and PDI-ATP (orange). (b) Comparative titration curves of PDI obtained from absorption (λ580nm) with increasing equiv. of ATP (red curve) and ADP (purple curve). (c) TEM image (stained with uranyl acetate, 1 wt% in water) of PDI with 2 equiv. of ATP. (d) AFM height image along with a height profile of 2 equiv. of ATP bound PDI. The red line corresponds to the region from where the height profile has been extracted. (e) Schematic representation of the cross-section of ATP bound PDI tapes. ([PDI] = 5 × 10−5 M, H2O or HEPES/DMSO, 50/50, (v/v)). | ||
Furthermore, the ATP-selective self-assembly was confirmed by dynamic light scattering (DLS) experiments, which showed an increase in size for ATP compared to ADP (Fig. S6a, ESI†). Transmission electron microscopy (TEM) images revealed the presence of nanoparticle-like assemblies (∼100 ± 5 nm) of PDI in an H2O/DMSO 50/50 (v/v) mixture (Fig. S6b, ESI†). These assemblies transformed into 1D nano-tape-like structures with polydisperse lengths (>5 μm) and ∼60 (±10) nm breadth upon interaction with 2.0 equivalents of ATP (Fig. 1c and d and Fig. S6d, ESI†). Atomic force microscopy (AFM) height analysis showed the height of the tape to be ∼5 (±0.3) nm, obtained by tracing an individual tape across the breadth which matches the molecular dimensions of PDI bonded with ATP at both ends (Fig. 1e). This suggests that PDI molecules self-assemble through π–π stacking along the tape's length, with hydrophobic interactions between the chromophores along the breadth where ATP is bound to the trimethylammonium cation exposed to the hydrophilic environment. Combined spectroscopic and morphological experiments indicate effective binding and self-assembly of PDI in the presence of trivalent ATP, resulting in elongated and entangled tape-like structures. In contrast, divalent ADP does not induce self-assembly of PDI, even though it binds to the molecule (Fig. S2 and S6c, ESI†). The uncontrolled elongation of the nanotape was attributed to the instantaneous self-assembly of PDI upon binding with 2 equivalents of ATP, as observed in time-dependent spectroscopic changes (Fig. S7, ESI†). We noticed an immediate increase in absorbance (at λ580 nm) and emission (at λ630 nm), indicating the spontaneous formation of ATP-bound PDI assemblies (Fig. 2a red plot and Fig. S7a, ESI†). To induce controlled self-assembly of PDI, we employed the biocatalytic in situ generation of ATP and further investigated the enzymatic modulation of temporal self-assembly of PDI (vide infra).
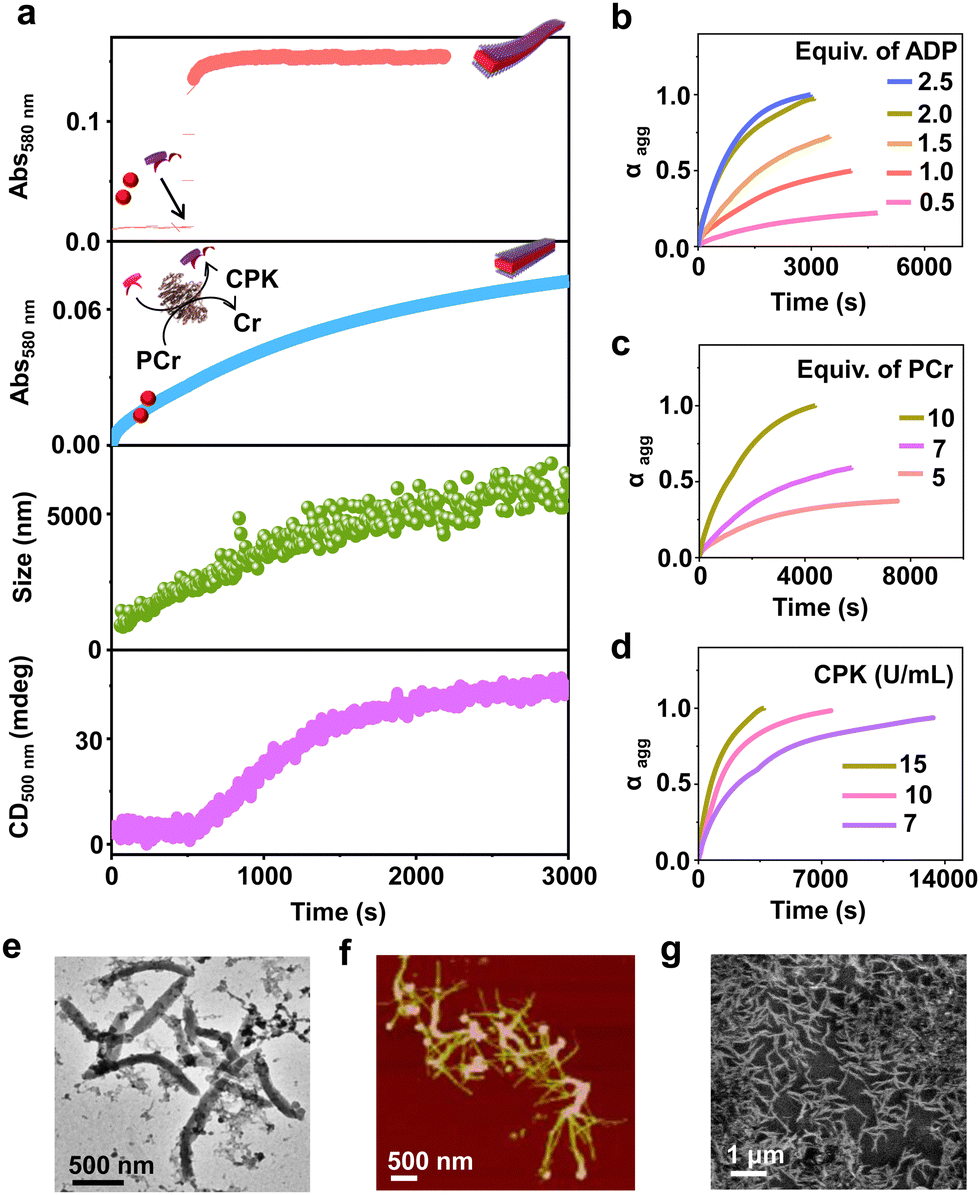 | ||
| Fig. 2 Biocatalytic generation of ATP driven temporally controlled self-assembly of PDI. (a) Comparative growth kinetics showing instantaneous self-assembly of PDI on addition of 2 equiv. of ATP from time dependent absorption changes (λ580nm, red) and time dependent spectroscopic evolution of ATP-driven self-assembly of PDI as ADP enzymatically converts to ATP in the presence of CPK monitored by absorption (λ580nm, blue) and scattering from DLS (green) and CD (λ500nm, magenta). Schematic representation of ATP triggered and enzymatically generated ATP driven PDI self-assembly has also been shown. Time dependent absorption changes monitored at λ580nm for enzymatically grown ATP bound PDI (b) for varying concentrations of ADP (CPK = 15 U mL−1, PCr = 10 equiv.), (c) for varying concentrations of PCr (ADP = 2 equiv., CPK = 15 U mL−1) and (d) for varying concentrations of CPK (ADP = 2 equiv., PCr = 10 equiv.). (e) TEM, (f) AFM and (g) FESEM images of enzymatically grown tapes of PDI ([PDI] = 5 × 10−5 M, ADP = 2 equiv., CPK = 15 U mL−1, PCr = 10 equiv., H2O or HEPES/DMSO, 50/50, (v/v), 25 °C). | ||
Enzymatic in situ generation of ATP for controlled self-assembly of PDI
Having observed the uncontrolled growth of PDI with the direct addition of ATP, we next controlled the elongation process by in situ generation of ATP from ADP in the presence of phosphocreatine (PCr) as a substrate, using the creatine phosphokinase (CPK) enzyme (Scheme 1). The ATP selectivity facilitated the successful use of this enzymatic reaction for controlling supramolecular polymerization, where the CPK enzyme transfers the PO43− group from PCr to ADP, generating ATP and releasing creatine (Cr) as a by-product. Thereby the ATP produced over time slowly induced the growth of PDI-ATP tapes. To a 5 × 10−5 M solution of PDI in DMSO/HEPES 50/50 (v/v) containing 15.0 U mL−1 CPK and 10.0 equivalents of PCr, 2.0 equivalents of ADP were injected to initiate the biocatalytic reaction, generating ATP in situ. As a result of controlled ATP generation by CPK, kinetically controlled growth was observed, monitored at the 580 nm band in UV-vis spectroscopy, which saturated at 3000 s (blue plot, Fig. 2a), whereas emission showed quenching over time monitored at λ630nm (Fig. S8, ESI†). This kinetic profile contrasts with the sudden jump observed for the direct addition of ATP to the PDI solution (Fig. S7a, ESI†), though the extent of aggregation for enzymatically produced 2.0 equivalents of ATP (from an equivalent amount of ADP) was less compared to the direct addition of 2.0 equivalents of ATP (Fig. S9, ESI†).A similar kinetic profile was observed in DLS, showing an increase in scattering over time due to increased aggregation (green plot, Fig. 2a). However, the corresponding time-dependent change in CD signal followed sigmoidal kinetics with a 500 s lag phase (monitored at 500 nm, magenta plot, Fig. 2a), differing from the absorbance kinetics (blue plot, Fig. 2a). This suggests that supramolecular chirality induction sets in only after certain extent of growth of self-assembly. Interestingly, the enzymatically grown ATP-bound PDI assembly showed a narrower size distribution compared to PDI assemblies grown instantaneously, highlighting the importance of the controlled self-assembly process regulated by the enzymatic reaction (Fig. S8e, ESI†).
Further evidence of reaction-controlled growth was observed when the growth was influenced by various components present in the enzymatic reaction, such as ADP, PCr, and CPK (Fig. 2b–d and Fig. S10–S12, ESI†). First, we varied the concentrations of ADP and monitored the changes spectroscopically. Since ADP enzymatically generates equivalent and quantitative amounts of ATP, increasing the concentration of ADP (0.5–2.5 equivalents) resulted in an increased self-assembly process. This was evidenced by a decrease in t50 values from 2700 (±400) s to 1436 (±150) s and an increase in νmax from 1.01 (±0.5) to 5.41 (±0.2) s−1, monitored through absorbance changes at 580 nm, due to faster production and higher concentration of ATP in solution (Fig. S11a, ESI† and Fig. 2b). Analysis through DLS showed a gradual increase in size from 290 (±75) nm to 2115 (±94) nm with increasing equivalents of ADP, with a linear increase characteristic of controlled supramolecular polymerization which saturated at 2 equiv. of ADP (Fig. S11, ESI†).
To further confirm the effects of enzymatically produced ATP on the self-assembly kinetics of PDI, we varied the concentrations of PCr and CPK and carefully monitored their effects on the resulting elongation kinetics of ATP-bound PDI. With increasing PCr concentrations from 5.0 to 10.0 equivalents at fixed ADP (2.0 equivalents) and CPK (15.0 U mL−1) in the PDI solution (5 × 10−5 M), an increase in the growth rate (νmax) was observed from 1.01 (±0.3) s−1 to 5.12 (±0.12) s−1 (Fig. 2c). Since PCr is the PO43−-generating substrate that enzymatically converts ADP to ATP, higher equivalents of PCr generate higher concentrations of PO43− quickly, thereby increasing the growth rate. These results are corroborated by the decrease in t50 from 3750 (±250) s to 1501 (±150) s (Fig. S12a, ESI†). Similarly, with increasing concentrations of CPK, a decrease in the total time, required for the completion of the growth process from, 14000 s to 3000 s was observed, although the extent of growth remained similar (Fig. 2d). These observations were further reflected in the derived time parameter t50 values from 6667 (±450) s to 1359 (±150) s and rate of elongation νmax values from 0.97 (±0.2) to 5.01 (±0.3) s−1 (Fig. S12b, ESI†). All these outcomes confirm that the rate of in situ ATP generation directly governs the kinetics of PDI elongation. The enzymatically grown ATP-bound PDI assemblies were further characterized in detail using various morphological techniques. TEM, FESEM, and AFM images of the enzymatically generated PDI-ATP assemblies showed the formation of 1-D nanotapes with uniform dimensions (length ∼1 ± 0.1 μm) with a dispersity value of 0.02 obtained directly from DLS (Fig. 2e–g). Next, the enzymatic reaction-controlled generation of ATP was utilized to control the length of the PDI-ATP supramolecular polymer through a seeded approach, which is a characteristic feature of actin assembly (vide infra).
Seeding via stepwise addition of ADP
Having demonstrated the enzymatic reaction-driven controlled formation of ATP-bound PDI nanotapes, we proceeded to control the degree of polymerization through a seeded assembly akin to actin polymerization (Fig. 3). In the previous section, we observed that the extent of tape growth could be controlled by the concentration of the substrate, ADP in the solution. Herein, we strategized the seeding process by stepwise addition of ADP into the same solution, differing from the conventional seeding process where a fresh batch of monomer is added each time into the seed solution. As a result, the supramolecular polymer formed by the first batch of ADP addition acts as a seed for the second batch of ADP addition into the same PDI solution containing fixed CPK and PCr concentrations (Fig. 3a). | ||
| Fig. 3 Seeded self-assembly of PDI by batch-wise addition of ADP. (a) Schematic representation of stepwise addition of ADP giving rise to enzymatically driven 1-D seeding on tapes where initially 0.5 equiv. of ADP is introduced to grow PDI-ATP followed by a second batch of 0.5, 1.0 or 1.5 equiv. of ADP addition. (b) Absorbance changes monitored at λ580nm for multi-batch addition of 0.4 equiv. of ADP. (c) Absorbance changes monitored at λ580nm for stepwise addition of ADP keeping the initial batch concentration of ADP at 0.5 equiv. and varying the second batch addition (numbers indicate the equiv. of ADP added in each batch). (d) Size changes obtained from DLS (pink) and dispersity from DLS for seed and seeded enzymatic generation of ATP leading to elongation of PDI by stepwise addition of ADP. Error bars are obtained from triplicate experiments. (e) TEM images obtained for two seeded growths of PDI-ATP through enzymatic generation of ATP (left: 0.5 + 0.5 equiv. of ADP, right: 0.5 + 1.5 equiv. of ADP) where ADP was introduced batchwise. Arrows indicate the addition of ADP into the solution ([PDI] = 5 × 10−5 M, CPK = 15 U mL−1, PCr = 10 equiv., HEPES/DMSO, 50/50 (v/v), 25 °C). | ||
Since all binding sites of PDI (5 × 10−5 M) were saturated with 2.0 equivalents of ADP, we added ADP in two sequential batches, keeping the total concentration of ADP fixed at 2.0 equivalents while maintaining a fixed 15.0 U mL−1 CPK and 10.0 equivalents of PCr (Fig. S13, ESI†). The seeded assembly was evident from the increased growth rate of PDI in the presence of the second batch of ADP (1.0 equivalent) compared to the first batch (1.0 equivalent), as monitored by absorbance at λ580nm (Fig. S13c, ESI†). This was further supported by the increase in the extent of aggregation after each batch of ADP addition and the corresponding increase in size in the DLS. Although the nucleation step of PDI-ATP was not evident from the absorption changes, as indicated by the absence of a lag phase in the initial growth of the seed, the seeded assembly from seed termini was evident from the increased growth rate for the second batch of ADP addition and the nonexistence of a bimodal distribution in DLS (Fig. S13b and c, ESI†).
We believe that the difference in the degree of polymerization during subsequent additions is a manifestation of in situ ATP generation being uniquely linked to seeded elongation characteristics. Since the subsequent additions of ADP are carried out in the presence of a preformed PDI-ATP seed, a faster elongation was observed due to the rapid generation of ATP in the solution. It has been previously reported by us that ATP-bound stacks tend to attract CPK, which can further facilitate positive feedback in the overall ATP generation and PDI stack seeded elongation process.44 The efficiency of the seeded assembly was further demonstrated by multicycle seeding, performed by adding 0.4 equivalents of ADP five times (multiple batch additions), keeping the total concentration of ADP fixed at 2.0 equivalents where we noticed seeded behaviour when monitored by absorbance at λ580nm (Fig. 3b).
A similar phenomenon was observed when the second batch of ADP addition was varied from 0.5 to 1.5 equivalents, keeping the initial batch of ADP constant at 0.5 equivalents (Fig. 3c). Interestingly, we noticed an increase in the growth rate along with an increase in the extent of aggregation due to the increased ADP equivalents in the second batch, indicating the seeded assembly of enzymatically controlled ATP-bound PDI (Fig. 3c). The increase in size from a seed to a seeded assembly was observed from scattering measurements for stepwise addition of ADP with varied equivalents (0.5, 1.0, and 1.5 equivalents) at the second step. This reiterates the seeded characteristic of PDI, which follows a linear fit with a good quality correlation curve (Fig. 3d, pink line and Fig. S14, ESI†). Moreover, the dispersity trends obtained directly from DLS indicate a controlled supramolecular polymerization process, as the values remain constant during the seeding experiments (Fig. 3d, green line). Although PDI-ATP structures are not spherical and the exact values obtained from DLS may not be accurate, they depict a qualitative trend in size increase during seeding experiments.
An increase in size was also observed when the seeded structures were visualized under an electron microscope (TEM), where the length of the PDI-ATP stack increased from 250 (±50) nm to 1100 (±160) nm with the increased ADP amount in subsequent batches with a polydispersity index close to 1.0 (Fig. 3e, S15 and S16, ESI†). This enzymatic reaction-driven, in situ generated ATP-triggered seeded supramolecular polymerization strategy provided controlled stack lengths, as evident from the low and consistent dispersity values, analogous to recently reported living supramolecular polymerization processes.43
Enzymatic-reaction controlled transient assembly
In addition to the reaction-controlled seeded assembly, another crucial aspect of the natural self-assembly process of actin is the temporal regulation of the structure, which governs various cellular functions. With an aim to mimic this, we formed a transient self-assembly of PDI using two antagonistic enzymes: CPK, which controls the growth by producing ATP in solution, and calf intestinal alkaline phosphatase (CIAP), which hydrolyses ATP to adenosine and phosphate, working in tandem (Fig. 4a). As shown previously, the self-assembly of PDI is selective to ATP. We envision that controlled enzymatic hydrolysis of ATP to phosphate will induce depolymerization of the tapes. First, we carried out control experiments where CIAP was introduced into a pre-grown ATP-bound PDI assembly, and we monitored the changes over time both spectroscopically and through scattering. We observed a decrease in the aggregation band over time, as indicated by the absorbance at 580 nm as well as the temporal changes in size obtained from DLS (Fig. S17, ESI†). Interestingly, the supramolecular polymer disassembled at a faster rate with increasing CIAP concentration (Fig. S17a, ESI†). This indicates that enzymatic hydrolysis of ATP induces passive disassembly of PDI tapes. | ||
| Fig. 4 Enzyme-coupled transient supramolecular polymerisation of PDI-ATP. (a) Schematic representation of active self-assembly of ATP bound PDI where coupling of an ATP generating (CPK) and hydrolyzing (CIAP) enzyme regulates ATP in solution, rendering a transient self-assembly. (b) Transient assembly of PDI by coupling of CPK (constant) and CIAP (varying) as evident from the time-dependent absorbance changes monitored at λ580nm. (c) Co-stacked absorbance (λ580nm) and scattering changes (CIAP = 0.4 U mL−1) showing multiple transient assembly cycles indicating growth followed by disassembly of ATP bound PDI on refuelling ADP into the solution. Arrows indicate the addition of ADP into the solution. TEM images showing the transient assembly of PDI-ATP at (d) t1h (steady state) and (e) t4h (after disassembly) (CIAP = 0.2 U mL−1) ([PDI] = 5 × 10−5 M, ADP = 2 equiv., CPK = 20 U mL−1, PCr = 10 equiv., HEPES/DMSO, 50/50, v/v, 25 °C). | ||
To extend this passive enzyme response of the ATP-bound PDI assembly to active regimes, a careful standardization of the concentrations of both ATP-generating enzyme (CPK) and ATP-hydrolyzing enzyme (CIAP) is required to achieve significant growth of the assembly before the disassembly process dominates. This can be achieved by increasing the CPK concentration to 20.0 U mL−1, where a fast in situ generation of ATP would accelerate the growth process. Thus, the assembly reaches its maximum before disassembly begins due to enzymatic hydrolysis of ATP. By coupling 20.0 U mL−1 of CPK with lower concentrations of CIAP (0.2–0.4 U mL−1), we could regulate the production and consumption of biofuel ATP in situ, rendering a transient self-assembly of PDI-ATP, as evidenced by the absorbance changes due to the differences in ATP generation and ATP hydrolysis rates. Interestingly, we noticed a decrease in the extent of growth, a shorter duration of the steady state, and faster disassembly when increasing the concentration of CIAP from 0.2 U mL−1 to 0.4 U mL−1 (Fig. 4b). The steady state could be due to the delay in disassembly of stable ATP-bound PDI assemblies due to the differential hydrolysis rate of free ATP in solution or the ATP bound to the PDI-ATP stack by CIAP enzyme. As a result, this prolongs the stability of PDI-ATP before initiating its disassembly (Fig. S18, ESI†).
We further analysed and quantified the assembly and disassembly rates (dA/dt) and the lifetimes of assembly, steady state, and disassembly from the spectroscopic traces obtained from absorption changes monitored at λ580 nm. We noticed that the assembly rate remains constant (∼3.2 × 10−4 min−1), but its lifetime decreases from 160 to 96 min with increasing concentrations of CIAP, whereas the disassembly rate increases from 7.97 × 10−5 to 12.5 × 10−5 min−1 along with its lifetime (Table S2, ESI†). These observations result from faster hydrolysis kinetics with higher concentrations of CIAP. Higher units of CIAP also decrease the extent of growth of ATP-bound PDI during the assembly process. Moreover, introducing ADP again into the solution led to a second transient cycle (red trace, Fig. 4c).
These changes were further corroborated by monitoring scattering changes, which indicate the temporal increase in size (elongation), followed by a steady state and then a decrease (disassembly) when monitored through DLS (blue trace, Fig. 4c). These results confirm the enzymatic regulation of ATP-driven transient self-assembly of PDI. Time-dependent spectra taken for the transient assembly of PDI showed the temporal assembly of ATP-bound PDI, as seen from the emergence of the aggregate band, whereas disassembly can be observed from the disappearance of the aggregate band over time (Fig. S19, ESI†). The initial spectra of assembly (tinitial, assembly) and the final spectra after the completion of disassembly (tfinal, disassembly) could be overlaid, indicating a return to the same state. Furthermore, the transient self-assembly of the PDI-ATP assemblies was visualised at different time points by TEM, exhibiting the presence of elongated tapes at the end of 1 hour of the transient cycle, corresponding to the beginning of the steady state, and the absence of any elongated tapes at the end of 5 hours (Fig. 4d and e). Thus, we could achieve transient states of PDI-ATP through temporal control over the self-assembly followed by disassembly by coupling the enzymes CPK and CIAP.
Conclusions
Herein, we present a bioinspired approach to achieve programmable supramolecular polymerization, mimicking reaction-controlled assembly, modulation of length through a seeded approach, and temporal control over the self-assembly process akin to biological systems. Drawing inspiration from the actin self-assembly process, we coupled ATP-generating and ATP-hydrolyzing enzymes, CPK and CIAP, respectively, within the same system. This coupling allowed for in situ ATP regulation, leading to the formation of transient assemblies. By adjusting the concentrations of molecular cues influencing the self-assembly process, we efficiently modulated the extent and kinetics of the assemblies. Additionally, we demonstrated the interdependency between the enzymatic reactions and the growth of the supramolecular polymer PDI-ATP. These enzymatic reaction-driven assemblies also exhibited the capability for seeded supramolecular polymerization, allowing for precise control over their length and dispersity. The enzymatic coupling of self-assembly and disassembly processes in PDI-ATP systems resulted in the formation of aggregates in out-of-equilibrium regimes. The development of these strategies holds the potential to significantly impact applications targeted by self-assembled structures, paving the way for the creation of next-generation materials with unprecedented control over their structural and functional properties.Materials and methods
General
All chemicals were purchased from commercial sources and were used as such. ATP, ADP, PCr, CPK and CIAP were purchased from Sigma Aldrich. Spectroscopic grade solvents were used for all optical measurements.Spectroscopic measurements
Electronic absorption spectra were recorded on a Perkin Elmer Lambda 900 UV-vis-NIR Spectrometer and emission spectra were recorded on a Perkin Elmer Ls 55 Luminescence Spectrometer. Circular dichroism measurements were performed on a Jasco J-815 spectrometer. UV-vis, emission and CD spectra were recorded in a 5 mm path length quartz cuvette. Corresponding temperature dependent measurements were performed with a CDF-426S/15 Peltier-type temperature controller.Dynamic light scattering (DLS) experiments
The measurements were carried out using a NanoZS (Malvern UK) employing a 635 nm laser at a backscattering angle of 173°. The samples were measured in a 10 mm glass cuvette.Transmission electron microscopy (TEM)
TEM measurements were performed on a JEOL, JEM 3010 operated at 300 kV and a JOEL JEM2100 PLUS operated at 200 kV. Samples were prepared by placing a drop of the solution on carbon coated copper grids followed by drying at room temperature. The images were recorded at operating voltages of 300 kV and 200 kV depending on the instrument used. In order to get a better contrast, the sample was stained with uranyl acetate (1 wt% in water) before the measurements. For TEM, water was used instead of aq. HEPES solution to avoid masking of nanostructures due to HEPES deposition upon drying.Atomic force microscopy (AFM)
AFM was analysed using a Bruker Dimension Icon and the imaging was carried out in peak force tapping mode. Samples were prepared by spin coating a 2 μL solution of the sample on silicon. Imaging was carried out under ambient conditions in tapping mode. The probe used for imaging was antimony doped silicon cantilever with a resonant frequency of 300 kHz and a spring constant of 40 N m−1. The software used for the analysis of the nanostructures was Nanoscope V7.30r1sr3. Water was used instead of aq. HEPES solution to avoid masking of nanostructures due to HEPES deposition upon drying.Field emission scanning electron microscopy (FESEM)
FESEM measurements were carried out on a ZEISS GeminiSEM 500. Samples were prepared by spin coating a 2 μL solution of the sample on silicon. Water was used instead of aq. HEPES solution to avoid masking of nanostructures due to HEPES deposition upon drying.PDI synthesis and characterization were carried out according to the literature procedure.47
Sample preparation for self-assembly experiments of PDI
All samples for spectroscopic measurements were prepared by injecting the required volume of PDI (stock solution 10−3 M in DMSO) into N-2-hydroxyethylpiperazine-N′-2-ethanesulphonic acid (HEPES) buffer solution (10 mM). The stock concentrations of ATP, ADP, and PCr were kept at 10−2 M by dissolving the measured solid in appropriate amounts of HEPES. Aliquots of stock solution of CPK (50 U mL−1) were used for every measurement.Measurements for enzymatically triggered temporal self-assembly of PDI
For spectroscopic measurements of enzymatically triggered self-assembly of PDI, a 5 × 10−5 M solution of PDI in DMSO/HEPES 50/50 (v/v) is taken in a cuvette, to which the required components (PCr and CPK) are added, mixed by shaking and the measurement is started within 30 seconds. ADP/ATP was introduced into the mixture in the end as the final component to trigger the temporal growth of PDI. All the measurements are carried out at 25 °C in cuvettes with a path length of 10 mm.Seeded self-assembly experiments of PDI
First, the seed solution was prepared by introducing x equiv. of ADP (x = 0.2, 0.5, 1.0 or 1.5) into the reaction mixture in a cuvette containing the required amounts of PDI (5 × 10−5 M), PCr (10 equiv.) and CPK (15 U mL−1). The initial growth kinetics of PDI was monitored through absorption changes at λ580nm. Subsequent batches of ADP (y = 0.2, 0.5, 1.0 or 1.5 equiv.) were introduced into the preformed seed solution and the time dependent changes were monitored.Enzymatic ATP regulation leading to transient assembly of PDI
Both the enzymes, CPK (20 U mL−1) and CIAP (0.2, 0.3 or 0.4 U mL−1), are introduced into a solution containing the required amounts of PDI (5 × 10−5 M). Measured volumes of the preformed mixture of PCr (10 equiv.) and ADP (2 equiv.) are introduced into the solution and the temporal measurements are initiated and monitored with absorbance at λ580nm or through scattering in DLS. Refuelling was carried out by introducing a fresh feed of PCr (10 equiv.) and ADP (2 equiv.) into the solution and the changes were continued to be monitored.Author contributions
A. M. and S. J. G. conceived the project. A. M. designed and performed the experiments. A. M., A. D. and S. J. G. discussed the data and wrote the manuscript. All authors have given approval to the final version of the manuscript.Data availability
The authors confirm that the data supporting the findings of this study are included within the manuscript and its ESI.† Additionally, the raw data can be made available upon reasonable request.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank JNCASR, Department of Science and Technology (DST), Government of India. The authors acknowledge SRISTI-BIRAC for funding provided through GYTI awards. A. M. thanks DST for the research fellowship. S. J. G. acknowledges the funding received from the Swarna Jayanti Fellowship award (DST/SJF/CSA01/2016-2017). A. D. thanks the CSIR for the fellowship. We thank SAMat research facilities and JNCASR for TEM experiments.Notes and references
- U. Alon, An Introduction to Systems Biology: Design Principles of Biological Circuits, Chapman and Hall Publishers, 2006 Search PubMed.
- R. F. Service, Science, 2005, 309, 95 CrossRef CAS PubMed.
- G. M. Whitesides and B. Grzybowski, Science, 2002, 295, 2418–2421 CrossRef CAS PubMed.
- L. Brunsveld, B. J. Folmer, E. W. Meijer and R. P. Sijbesma, Chem. Rev., 2001, 101, 4071–4098 CrossRef CAS PubMed.
- F. J. M. Hoeben, P. Jonkheijm, E. W. Meijer and A. P. H. J. Schenning, Chem. Rev., 2005, 105, 1491–1546 CrossRef CAS PubMed.
- T. Aida, E. W. Meijer and S. I. Stupp, Science, 2012, 335, 813–817 CrossRef CAS PubMed.
- T. F. A. de Greef, M. M. J. Smulders, M. Wolffs, A. P. H. J. Schenning, R. P. Sijbesma and E. W. Meijer, Chem. Rev., 2009, 109, 5687–5754 CrossRef CAS PubMed.
- E. E. Greciano, B. Matarranz and L. Sanchez, Angew. Chem., Int. Ed., 2018, 57, 4697–4701 CrossRef CAS PubMed.
- J.-M. Lehn, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 4763–4768 CrossRef CAS PubMed.
- J.-F. Lutz, J.-M. Lehn, E. W. Meijer and K. Matyjaszewski, Nat. Rev. Mater., 2016, 1, 16024–16038 CrossRef CAS.
- D. van der Zwaag, T. F. A. de Greef and E. W. Meijer, Angew. Chem., Int. Ed., 2015, 54, 8334–8336 CrossRef CAS PubMed.
- R. D. Mukhopadhyay and A. Ajayaghosh, Science, 2015, 349, 241–242 CrossRef CAS PubMed.
- X. Wang, G. Guerin, H. Wang, Y. Wang, I. Manners and M. A. Winnik, Science, 2007, 317, 644–647 CrossRef CAS PubMed.
- J. Boekhoven, A. M. Brizard, K. N. K. Kowlgi, G. J. M. Koper, R. Eelkema and J. H. van Esch, Angew. Chem., Int. Ed., 2010, 49, 4825–4828 CrossRef CAS PubMed.
- J. Boekhoven, W. E. Hendriksen, G. J. K. Koper, R. Eelkema and J. H. van Esch, Science, 2015, 349, 1075–1079 CrossRef CAS PubMed.
- C. G. Pappas, I. R. Sasselli and R. V. Ulijn, Angew. Chem., Int. Ed., 2015, 54, 8119–8123 CrossRef CAS PubMed.
- H. Lodish, Molecular Cell Biology, 6th edn, 2007, W. H. Freeman, New York Search PubMed.
- M. Kasai, S. Asakura and F. Oosawa, Biochim. Biophys. Acta, 1962, 57, 22–31 CrossRef CAS PubMed.
- L. A. Lowery and D. V. Vactor, Nat. Rev. Mol. Cell Biol., 2009, 10, 332–343 CrossRef CAS PubMed.
- E. Leikina, M. V. Mertts, N. Kuznetsova and S. Leikin, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 1314 CrossRef CAS PubMed.
- S. Dhiman and S. J. George, Bull. Chem. Soc. Jpn., 2018, 91, 687–699 CrossRef CAS.
- C. G. Pappas, I. R. Sasselli and R. V. Ulijn, Angew. Chem., Int. Ed., 2015, 54, 8119–8123 CrossRef CAS PubMed.
- N. Singh, B. Lainer, G. J. M. Formon, S. De Piccoli and T. M. Hermans, J. Am. Chem. Soc., 2020, 142, 4083–4087 CrossRef CAS PubMed.
- S. Ogi, K. Sugiyasu, S. Manna, S. Samitsu and M. Takeuchi, Nat. Chem., 2014, 6, 188–195 CrossRef CAS PubMed.
- T. Fukui, S. Kawai, S. Fujinuma, Y. Matsushita, T. Yasuda, T. Sakurai, S. Seki, M. Takeuchi and K. Sugiyasu, Nat. Chem., 2017, 9, 493–499 CrossRef CAS PubMed.
- J. Kang, D. Miyajima, T. Mori, Y. Inoue, Y. Itoh and T. Aida, Science, 2015, 347, 646–651 CrossRef CAS PubMed.
- M. Wehner and F. Würthner, Nat. Rev. Chem., 2020, 4, 38–53 CrossRef CAS.
- S. Patra, S. Chandrabhas, S. Dhiman and S. J. George, J. Am. Chem. Soc., 2024, 146, 12577–12586 CrossRef CAS PubMed.
- S. Sarkar, A. Sarkar, A. Som, S. S. Agasti and S. J. George, J. Am. Chem. Soc., 2021, 143, 11777–11787 CrossRef CAS PubMed.
- R. Laishram, S. Sarkar, I. Seth, N. Khatun, V. K. Aswal, U. Maitra and S. J. George, J. Am. Chem. Soc., 2022, 144, 11306–11315 CrossRef CAS PubMed.
- S. Toledano, R. J. Williams, V. Jayawarna and R. V. Ulijn, J. Am. Chem. Soc., 2006, 128, 1070–1071 CrossRef CAS PubMed.
- R. J. Williams, A. M. Smith, R. Collins, N. Hodson, A. K. Das and R. V. Ulijn, Nat. Nanotechnol., 2009, 4, 19–24 CrossRef CAS PubMed.
- H. He, W. Tan, J. Guo, M. Yi, A. N. Shy and B. Xu, Chem. Rev., 2020, 120, 9994–10078 CrossRef CAS PubMed.
- K. Jalani, A. D. Das, R. Sasmal, S. S. Agasti and S. J. George, Nat. Commun., 2020, 11, 3967 CrossRef CAS PubMed.
- S. Dhiman, A. Sarkar and S. J. George, RSC Adv., 2018, 8, 18913–18925 RSC.
- A. Mishra, S. Dhiman and S. J. George, Angew. Chem., Int. Ed., 2021, 133, 2772–2788 CrossRef.
- J. Deng and A. Walther, Adv. Mater., 2020, 32, 2002629 CrossRef CAS PubMed.
- J. Deng and A. Walther, Nat. Commun., 2020, 11, 3658 CrossRef CAS PubMed.
- M. Kumar, P. Brocorens, C. Tonnelé, D. Beljonne, M. Surin and S. J. George, Nat. Commun., 2014, 5, 5793 CrossRef CAS PubMed.
- S. Dhiman, A. Jain, M. Kumar and S. J. George, J. Am. Chem. Soc., 2017, 139, 16568–16575 CrossRef CAS PubMed.
- S. Dhiman, A. Jain and S. J. George, Angew. Chem., Int. Ed., 2017, 129, 1349–1353 CrossRef.
- S. Maiti, I. Fortunati, C. Ferrante, P. Scrimin and L. J. Prins, Nat. Chem., 2016, 8, 725–731 CrossRef CAS PubMed.
- A. Mishra, D. B. Korlepara, M. Kumar, A. Jain, N. Jonnalagadda, K. K. Bejagam, S. Balasubramanian and S. J. George, Nat. Commun., 2018, 9, 1295 CrossRef PubMed.
- A. Das, S. Ghosh, A. Mishra, A. Som, V. B. Banakar, S. S. Agasti and S. J. George, J. Am. Chem. Soc., 2024, 146, 14844–14855 CrossRef CAS PubMed.
- D. Didry, M.-F. Carlier and D. Pantaloni, J. Biol. Chem., 1998, 273, 25602–25611 CrossRef CAS PubMed.
- T. Ma, C. Li and G. Shi, Langmuir, 2008, 24, 43–48 CrossRef CAS PubMed.
- B. Russ, M. J. Robb, F. G. Brunetti, P. L. Miller, E. E. Perry, S. N. Patel, V. Ho, W. B. Chang, J. J. Urban, M. L. Chabinyc, C. J. Hawker and R. A. Segelman, Adv. Mater., 2014, 26, 3473–3477 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4tb01558d |
| This journal is © The Royal Society of Chemistry 2024 |