
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAsymmetric copper-catalyzed alkynylallylic dimethylamination†
Si-Yuan
Luo
abc,
Guo-Qiang
Lin
ab and
Zhi-Tao
He
 *cd
*cd
aState Key Laboratory of Chemical Biology, Shanghai Institute of Organic Chemistry, University of Chinese Academy of Sciences, Shanghai, 200032, China. E-mail: hezt@sioc.ac.cn
bSchool of Physical Science and Technology, ShanghaiTech University, Shanghai, 201210, China
cState Key Laboratory of Organometallic Chemistry, Shanghai Institute of Organic Chemistry, University of Chinese Academy of Sciences, Shanghai, 200032, China
dSchool of Chemistry and Materials Science, Hangzhou Institute for Advanced Study, University of Chinese Academy of Sciences, Hangzhou 310024, China
First published on 5th December 2023
Abstract
A feasible protocol for Cu-catalyzed asymmetric alkynylallylic dimethylamination is developed with the discovery of tetramethyldiaminomethane as a new, stable and convenient surrogate of dimethylamine. A series of enantioenriched 1,4-enynes are constructed in reasonable yields, high regioselectivities and moderate to good enantioselectivities. Mechanistic experiments show that the tertiary amine works as a nucleophile directly followed by the release of the expected dialkylamine unit, different from the conventional primary and secondary amines with the cleavage of a proton as the nucleophile. DFT calculations elucidate the origin of regio- and enantioselectivity for the present transformation.
Introduction
Transition metal-catalyzed η3-substitution reactions have been widely used for the construction of different stereogenic centers.1–6 In particular, since the pioneering work of van Maarseveen7 and Nishibayashi8 groups, Cu-catalyzed propargylic substitution has emerged as a reliable strategy to introduce C–N,9–16 C–O,17–20 C–C21–29 and C–S30,31 bonds vicinal to an alkyne unit stereoselectively. In this case, a propargylic leaving group is required to be preprepared for the following generation of a critical Cu-allenylidene intermediate. However, when a leaving group is located at a remote position to the alkyne unit, such a motif is generally not considered a potential substrate for propargylic substitution. Recently, Fang,32 Xu33 and our group34,35 independently demonstrated the concept of Cu-catalyzed remote propargylic substitution reactions. In these studies, an olefin32–34 or aryl unit35 was used to link the alkyne unit and the γ-leaving group, and the reactive alkene-tethered or dearomatization-induced Cu-allenylidene intermediate was smoothly formed to induce the expected substitution. In particular, our group34 and Xu33 sequentially described the first asymmetric alkynylallylic substitution processes, in which both SN2′ and in situ substitution models were shown to be feasible to form chiral 1,4-enyne products and spirocycles, respectively. However, the substrate scope and reaction models are heavily limited to the use of common nucleophiles. Thus, the discovery of new applications and the establishment of efficient catalytic systems suitable for the less developed remote propargylic substitution reactions are highly desired.The chiral dimethylamino unit exists widely in bioactive molecules, such as rivastigmine,36 which is used to treat Alzheimer's disease, and the tetracycline family including tetracycline, doxycycline and tigecycline, a series of broad-spectrum antibiotics37 (Scheme 1a). Based on our previous work,34 we envisioned that with dimethylamine as the nucleophile, Cu-catalyzed asymmetric alkynylallylic substitution might provide a novel and valuable route to construct chiral 1,4-enynes bearing a dimethylamino stereocenter. The vicinal olefin and alkyne units could further be transformed into a series of functional groups, thus largely broadening the application potential of this methodology. However, dimethylamine is gaseous at room temperature and usually stored in special solvents, which limits the convenient use of this reagent and also influences the efficiency of the corresponding transformation (Scheme 1b). Therefore, the identification of different and efficient surrogates of the dimethylamine nucleophile is critical to realize the expected alkynylallylic dimethylamination reaction. Here, we discovered that tetramethyldiaminomethane38 worked as a novel and reliable precursor of dimethylamine and enabled enantioselective Cu-catalyzed alkynylallylic dimethylaminations to construct valuable chiral 1,4-enynes39 (Scheme 1b). A variety of 1,4-enynes bearing a dimethylamino stereocenter were constructed in good yields and moderate to good enantioselectivities. Mechanistic experiments and DFT calculations elucidated the origin of regio- and enantioselectivity and revealed that tetramethyldiaminomethane first participated in the reaction as a neutral tertiary amine nucleophile and then released the dimethylamino unit from the ammonium intermediate.
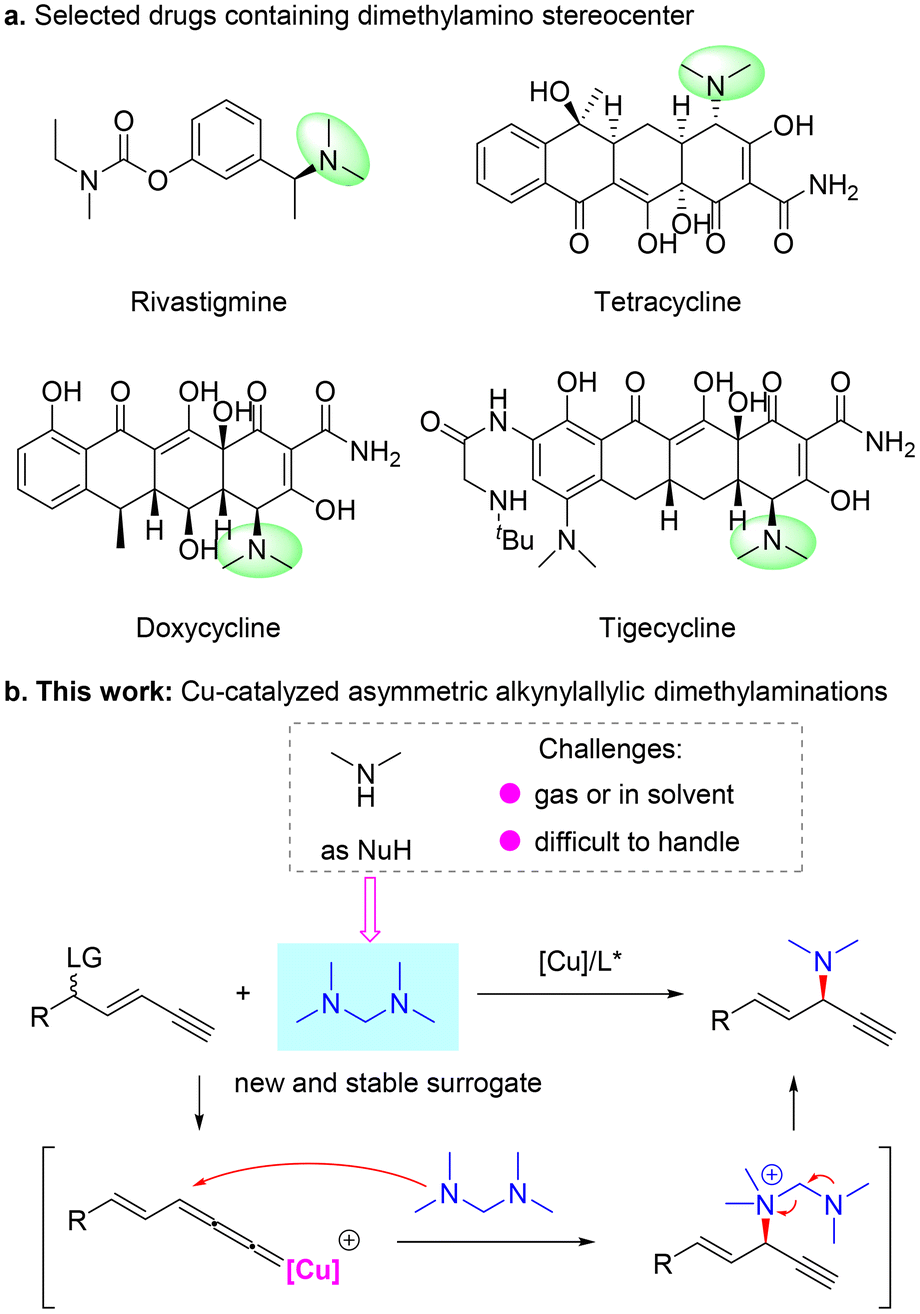 | ||
| Scheme 1 Representative drugs with dimethylamino stereocenters and our design. | ||
Results and discussion
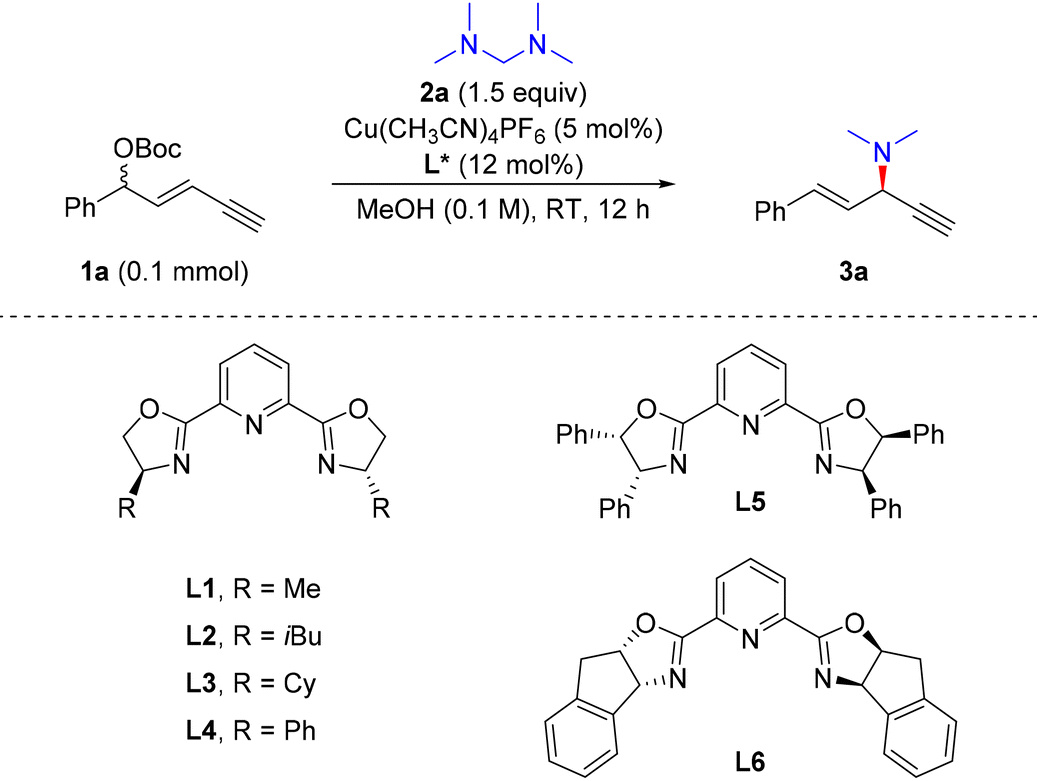
We initiated the reaction with racemic 1,3-enyne 1a as the electrophile and tetramethyldiaminomethane (2a) as a new dimethylamine source under copper(I) catalysis (Table 1). A set of PyBox ligands which were proven to be efficient for stereocontrol in the previous alkynylallylic substitutions32–34 were evaluated first. When L1 was used, the reaction proceeded smoothly, providing 3a in a total 42% yield, 75![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :25 er and 2.5:1 rr (entry 1). Other PyBox ligands showed similar reactivity and stereocontrol effects to L1 as the ligand (entries 2–5). However, when L6 was used as the ligand, 3a was obtained in a high yield and good stereoselectivity, i.e., 94% yield, 19:1 rr and 91:9 er (entry 6). When the solvent was changed to EtOH or DCM, obvious erosion of the yield and enantioselectivity of 3a was observed (entries 7 and 8). Reducing the loading of the ligand led to a slight increase in the enantioselectivity, reaching 93:7 er (entry 9). The evaluations of reaction temperature were unfavourable to the regio- and enantioselectivity of 3a (entries 10–12). In addition, when OBoc as the leaving group in 1a was changed to OAc, the alkynylallylic amination proceeded with lowered efficiency and stereocontrol (entry 13). Thus, the optimal conditions for alkynylallylic substitution were identified as the combination of 1,3-enyne 1a (1 equiv.), nucleophile 2a (1.5 equiv.), Cu(MeCN)4PF6 (5 mol%) and L6 (10 mol%) in MeOH (0.1 M) at room temperature for 12 h. In this context, when dimethylamine in methanol solution was used directly as the nucleophile, a heavily decreased yield, regioselectivity and enantioselectivity of 3a were observed (entry 14).
:25 er and 2.5:1 rr (entry 1). Other PyBox ligands showed similar reactivity and stereocontrol effects to L1 as the ligand (entries 2–5). However, when L6 was used as the ligand, 3a was obtained in a high yield and good stereoselectivity, i.e., 94% yield, 19:1 rr and 91:9 er (entry 6). When the solvent was changed to EtOH or DCM, obvious erosion of the yield and enantioselectivity of 3a was observed (entries 7 and 8). Reducing the loading of the ligand led to a slight increase in the enantioselectivity, reaching 93:7 er (entry 9). The evaluations of reaction temperature were unfavourable to the regio- and enantioselectivity of 3a (entries 10–12). In addition, when OBoc as the leaving group in 1a was changed to OAc, the alkynylallylic amination proceeded with lowered efficiency and stereocontrol (entry 13). Thus, the optimal conditions for alkynylallylic substitution were identified as the combination of 1,3-enyne 1a (1 equiv.), nucleophile 2a (1.5 equiv.), Cu(MeCN)4PF6 (5 mol%) and L6 (10 mol%) in MeOH (0.1 M) at room temperature for 12 h. In this context, when dimethylamine in methanol solution was used directly as the nucleophile, a heavily decreased yield, regioselectivity and enantioselectivity of 3a were observed (entry 14).
|
|
||||
|---|---|---|---|---|
| Entry | L* | Yield (%) | rr | er |
| a The reactions were carried out on a 0.1 mmol scale. The yields and rr values were determined by crude 1H NMR. The er values were determined by HPLC analysis. b EtOH as the solvent. c DCM as the solvent. d L6 (10 mol%) was added. Isolated yield. e The reaction temperature was −20 °C. f The reaction temperature was −10 °C. g The reaction temperature was 60 °C. h OAc was used instead of OBoc as the leaving group. i Dimethylamine was used as the nucleophile. | ||||
| 1 | L1 | 42 | 2.5:1 |
75:25 |
| 2 | L2 | 39 | 1:1 |
77:23 |
| 3 | L3 | 34 | 1.2:1 |
81:19 |
| 4 | L4 | 58 | 15:1 |
73:27 |
| 5 | L5 | 45 | 1.9:1 |
54:46 |
| 6 | L6 | 94 | 19:1 |
91:9 |
| 7b | L6 | 66 | 13:1 |
86:14 |
| 8c | L6 | 21 | >20:1 |
56:44 |
| 9d | L6 | 90 | >20:1 |
93:7 |
| 10d,e | L6 | 72 | 10:1 |
91:9 |
| 11d,f | L6 | 79 | >20:1 |
90:10 |
| 12d,g | L6 | 55 | 11:1 |
87:13 |
| 13d,h | L6 | 55 | >20:1 |
90:10 |
| 14d,i | L6 | 68 | 3.8:1 |
79:21 |
With the optimal conditions in hand, we investigated the scope of 1,3-enyne electrophiles for this remote propargylic dimethylamination reaction and the results are summarized in Scheme 2. Various electron-rich and electron-deficient functional groups at the para- or meta-positions of aryl-substituted 1,3-enynes 1 showed high compatibility with the substitution. For example, alkoxyl, nitryl, halide, and ester moieties were well tolerated in the reaction, providing the corresponding dimethylamination products in 64–93% yields with generally >20:1 rr and >90:10 er (3b–3j and 3m). Notably, substrates that are prone to cross-coupling reactions also yielded amination products smoothly (3f and 3g). Multi-substituted aryl enyne was also a suitable electrophile for the transformation (3k). In addition, 1,3-enyne bearing an ortho-methoxy unit afforded 3l in 86% yield, >20:1 rr and 90:10 er. However, alkyl-derived enyne as the electrophile failed to undergo the alkynylallylic dimethylamination reaction (3n).
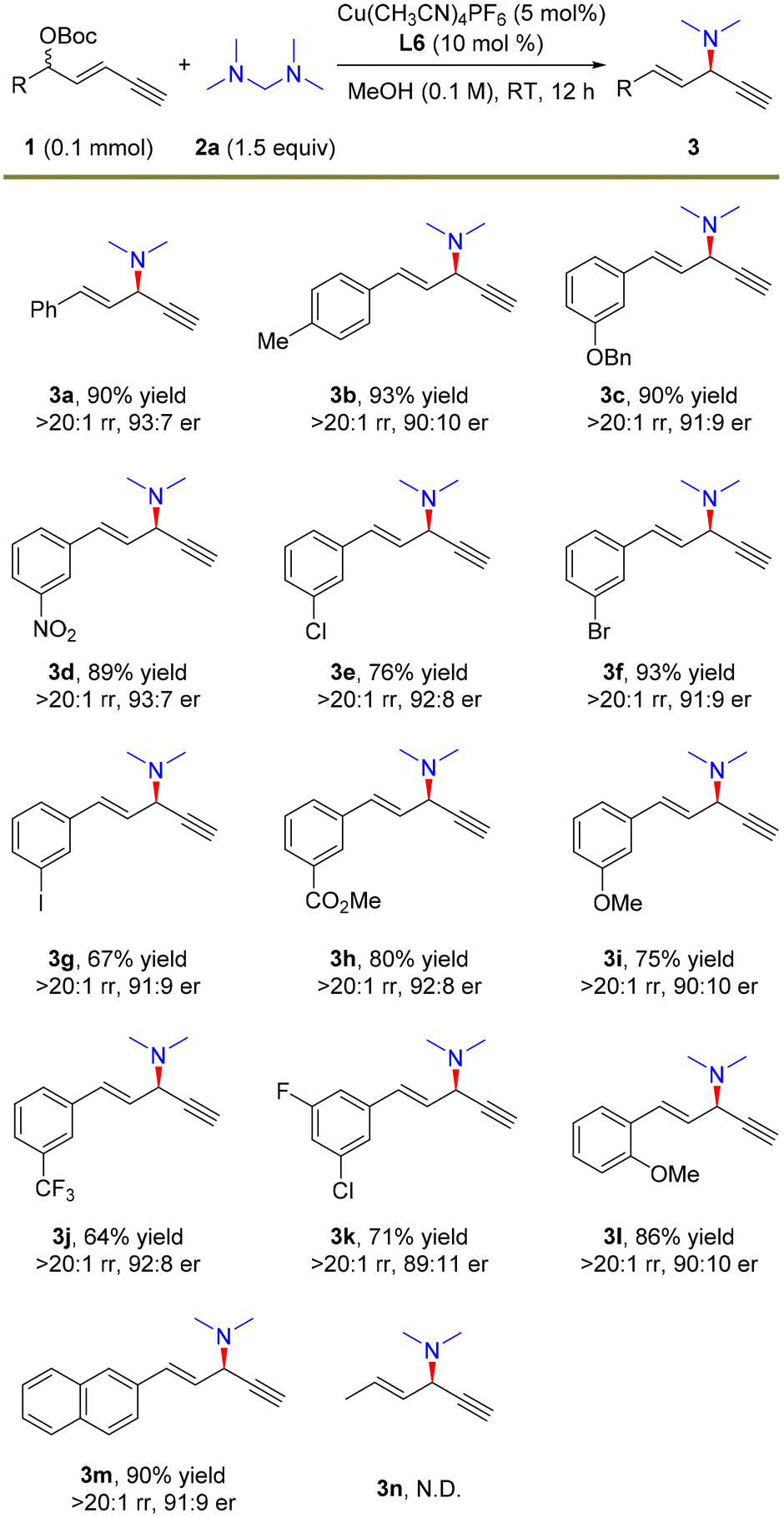 | ||
| Scheme 2 The scope of electrophiles. The reactions were carried out on a 0.1 mmol scale. Isolated yields. The rr values were determined by 1H NMR. The er values were determined by HPLC analysis. | ||
After demonstrating the feasibility of the remote propargylic dimethylamination with this novel dimethylamine source, we further explored the application potential of this protocol with other amine nucleophiles and the results are summarized in Scheme 3. A set of secondary amine sources as the surrogates of dimethylamine, pyrrolidine and morpholine were suitable for the alkynylallylic substitution, furnishing the corresponding products 4a–4c in high yields, >20:1 regioselectivity and up to 94:6 er. This strategy was also applicable to the introduction of different diarylmethyl amines, and the corresponding products 4d and 4e were obtained in 95:5 and 96:4 er, respectively. The absolute configuration of 4d was determined to be (R)-4d based on the same reported compound.34a However, the introduction of the diarylamine moiety via this route did not work (4f).
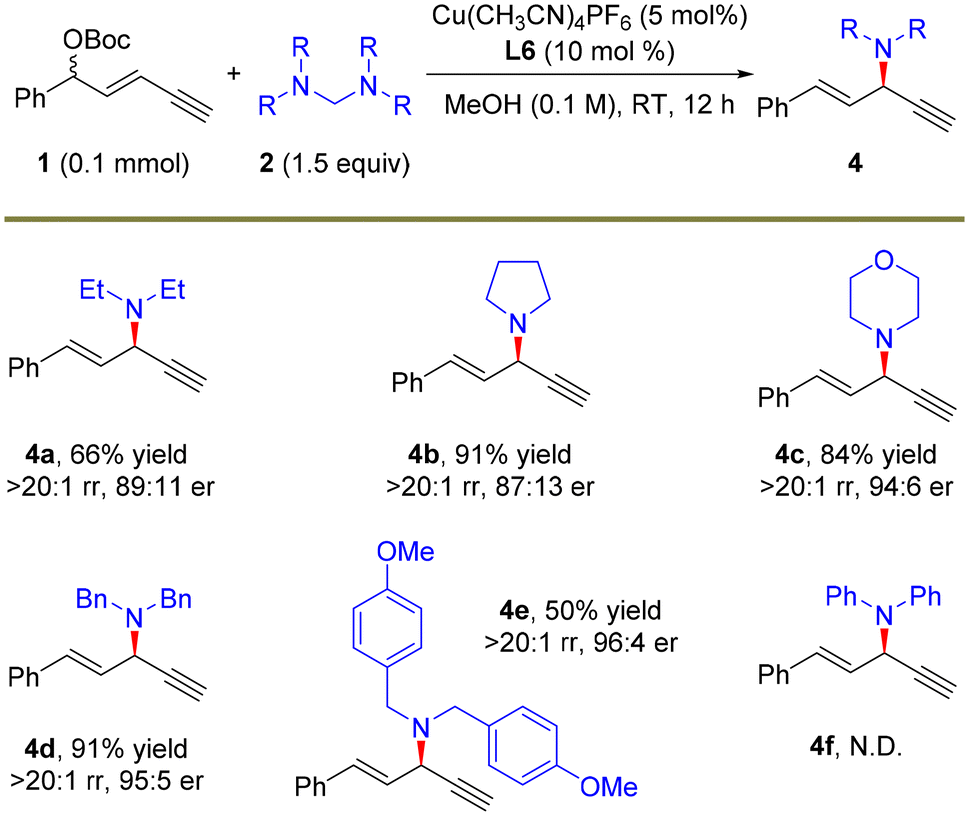 | ||
| Scheme 3 The scope of amine precursors. The reactions were carried out on a 0.1 mmol scale. Isolated yields. The rr values were determined by 1H NMR. The er values were determined by HPLC analysis. | ||
To figure out the potential reaction mechanism, a set of mechanistic studies were conducted (Scheme 4). As it was unclear whether the nucleophile 2a underwent the substitution via the in situ release of dimethylamine or serving as the tertiary amine nucleophile first and then releasing the dimethylamine unit, several control experiments were carried out with 2e as the substrate (Scheme 4a). When the reaction was conducted under standard conditions without a Cu catalyst or ligand or Cu/ligand, no product 4d was formed in these cases. In particular, dibenzylamine 5 was not detected. When electrophile 1a was not used under the standard conditions, no dibenzylamine 5 was observed similarly. These facts indicated that the in situ generation of 5 from 2e might be unfavourable under the present reaction system, and tertiary amine 2e might directly work as the nucleophile.38
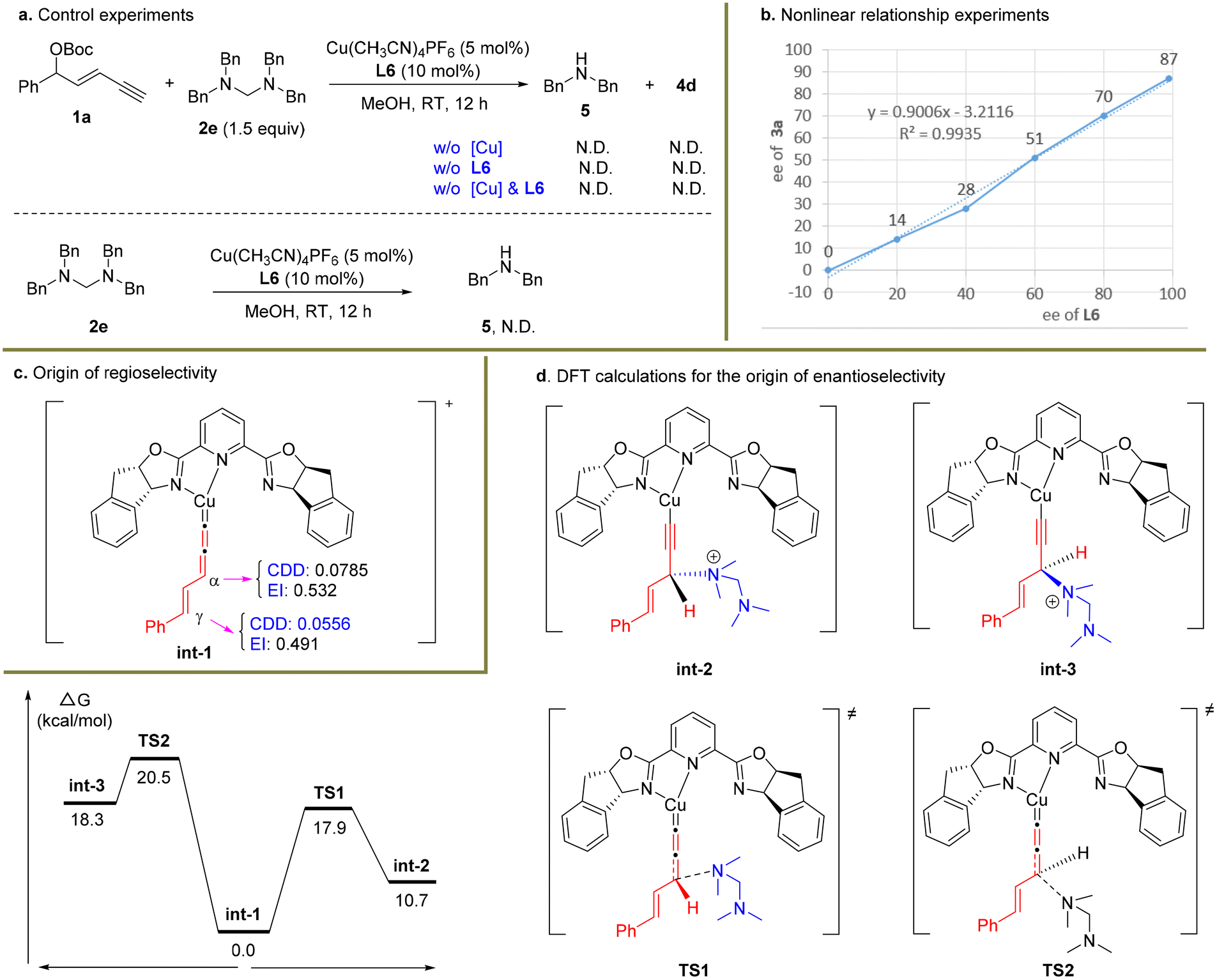 | ||
| Scheme 4 Preliminary mechanistic studies. BP86/def2tzvp/SMD(methanol)//B3LYP-D3(BJ)/6-31g*/SDD was used for DFT calculations. | ||
Next, a nonlinear relationship experiment showed a linear curve between the enantiopurity of chiral ligand L6 and that of the product 3a (Scheme 4b). This result suggested that monocopper/L6 as the catalyst might be involved in the enantio-determining step. Based on this fact, DFT calculations were conducted to uncover the origin of regio- and enantioselectivity for the alkynylallylic amination reaction. Both the condensed dual descriptor (CDD) and electrophilicity index (EI) were determined for the optimized structure of int-1 (Scheme 4c).40,41 The relatively larger CDD and EI data for the α-carbon center than those for the γ-carbon center showed that the α-carbon center is more electrophilic than the γ-carbon center. This is consistent with the observed preferred nucleophilic attack at the α-carbon position. Finally, calculations were conducted to uncover the energy profile of enantioselective substitution processes (Scheme 4d). It was obvious that the Re-face nucleophilic attack occurred viaTS1 with only 17.9 kcal mol−1 while the Si-face nucleophilic attack viaTS2 was 20.5 kcal mol−1. Therefore, the (R)-configuration for the formed stereocenter was observed in the present transformation.
Based on the above experimental and computational results, the proposed catalytic cycle is described in Scheme 5. The enyne substrate 1a first reacted with the copper catalyst in the presence of a base to give alkynyl copper intermediate int-4, which sequentially generated the Cu-allenylidene intermediate int-1 and tautomers int-5 and int-6via the cleavage of the leaving group. Then an outer-sphere nucleophilic attack by tertiary amine 2a occurred to provide ammonium salt int-7, which then released product 3a and regenerated the Cu catalyst. The released dimethylaminium might be further captured by potential nucleophiles in the reaction systems, such as methoxide.38,42
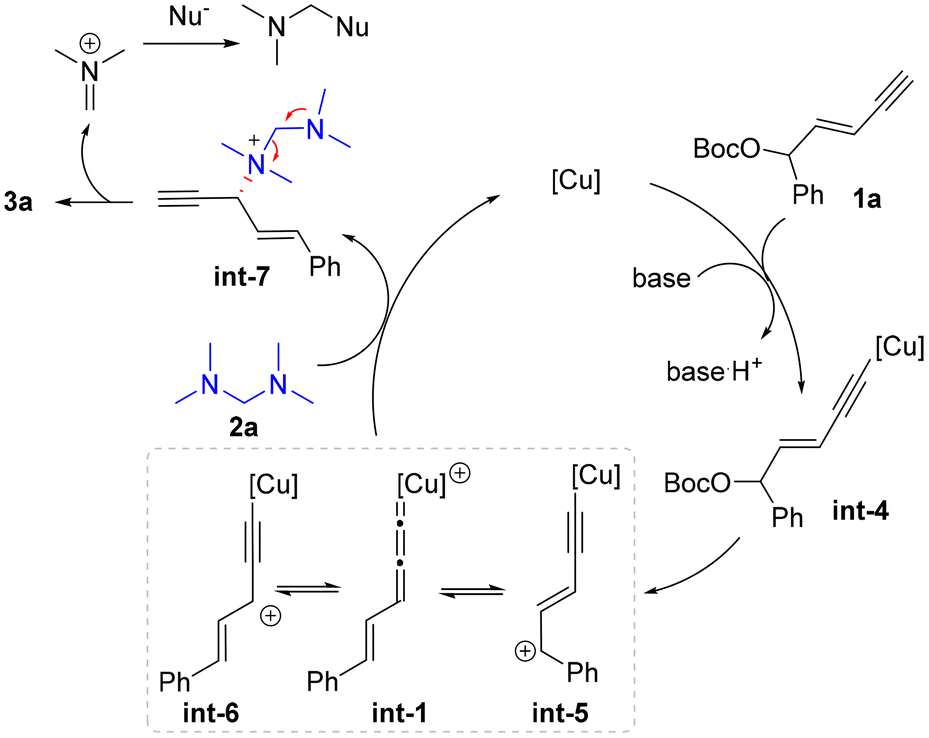 | ||
| Scheme 5 Proposed mechanism. | ||
Conclusions
In summary, we have established a feasible protocol for an asymmetric alkynylallylic dimethylamination reaction with the use of tetramethylmethanediamine as a new dimethylamine source. The novel protocol features reasonable functional group compatibility, high regioselectivity and moderate to good enantioselectivity. Different from the commonly used primary and secondary amines as nucleophiles with the cleavage of a proton, mechanistic experiments support the involvement of the tertiary amine as a neutral nucleophile directly followed by the release of dialkylamine. DFT calculations further elucidate the origin of regio- and enantioselectivity for the alkynylallylic substitution reaction.Author contributions
Z.-T. H. conceived the project. S.-Y. L. conducted the experiments and calculations. Z.-T. H. and G.-Q. L. supervised the project. Z.-T. H. and S.-Y. L. co-wrote the manuscript with feedback from all authors. Yuan-Xiang Yang is acknowledged for checking some results.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge the National Natural Science Foundation of China (22371292 and 22071262), the Natural Science Foundation of Ningbo (2023J036), the Shanghai Municipal Committee of Science and Technology (22ZR1475200), and the Shanghai Institute of Organic Chemistry for financial support.References
- L. Süsse and B. M. Stoltz, Enantioselective formation of quaternary centers by allylic alkylation with first-row transition-metal catalysts, Chem. Rev., 2021, 121, 4084–4099 CrossRef PubMed.
- G. Li, X. Huo, X. Jiang and W. Zhang, Asymmetric synthesis of allylic compounds via hydrofunctionalisation and difunctionalisation of dienes, allenes, and alkynes, Chem. Soc. Rev., 2020, 49, 2060–2118 RSC.
- R. J. Detz, H. Hiemstra and J. H. Van, Maarseveen, Catalyzed propargylic substitution, Eur. J. Org. Chem., 2009, 6263–6376 CrossRef.
- Y. Miyake, S. Uemura and Y. Nishibayashi, Catalytic propargylic substitution reactions, ChemCatChem, 2009, 1, 342–356 CrossRef.
- D.-Y. Zhang and X.-P. Hu, Recent advances in copper-catalyzed propargylic substitution, Tetrahedron Lett., 2015, 56, 283–295 CrossRef.
- K. Sakata and Y. Nishibayashi, Mechanism and reactivity of catalytic propargylic substitution reactions via metal–allenylidene intermediates: a theoretical perspective, Catal. Sci. Technol., 2018, 8, 12–25 RSC.
- R. J. Detz, M. M. Delville, H. Hiemstra and J. H. Van Maarseveen, Enantioselective copper-catalyzed propargylic amination, Angew. Chem., Int. Ed., 2008, 47, 3777–3780 CrossRef PubMed.
- G. Hattori, H. Matsuzawa, Y. Miyake and Y. Nishibayashi, Copper-catalyzed asymmetric propargylic substitution reactions of propargylic acetates with amines, Angew. Chem., Int. Ed., 2008, 47, 3781–3783 CrossRef.
- C. Zhang, Y.-H. Wang, X.-H. Hu, Z. Zheng, J. Xu and X.-P. Hu, Chiral tridentate P,N,N ligands for highly enantioselective copper-catalyzed propargylic amination with both primary and secondary amines as nucleophiles, Adv. Synth. Catal., 2012, 354, 2854–2858 CrossRef.
- G. Hattori, K. Sakata, H. Matsuzawa, Y. Tanabe, Y. Miyake and Y. Nishibayashi, Copper-catalyzed enantioselective propargylic amination of propargylic esters with amines: copper-allenylidene complexes as key intermediates, J. Am. Chem. Soc., 2010, 132, 10592–10608 CrossRef.
- R. J. Detz, Z. Abiri, R. l. Griel, H. Hiemstra and J. H. Van Maarseveen, Enantioselective copper-catalysed propargylic substitution: synthetic scope study and application in formal total syntheses of (+)-anisomycin and (-)-cytoxazone, Chem. – Eur. J., 2011, 17, 5921–5930 CrossRef PubMed.
- T. Mino, H. Taguchi, M. Hashimoto and M. Sakamoto, Copper-catalyzed asymmetric propargylic amination of propargylic acetates with amines using BICMAP, Tetrahedron: Asymmetry, 2013, 24, 1520–1523 CrossRef.
- M. Shibata, K. Nakajima and Y. Nishibayashi, Enantioselective intramolecular propargylic amination using chiral copper–pybox complexes as catalysts, Chem. Commun., 2014, 50, 7874–7877 RSC.
- D.-Y. Zhang, L. Shao, J. Xu and X.-P. Hu, Copper-catalyzed asymmetric formal [3 + 2] cycloaddition of propargylic acetates with hydrazines: enantioselective synthesis of optically active 2-pyrazolines, ACS Catal., 2015, 5, 5026–5030 CrossRef.
- Z.-T. Liu, Y.-H. Wang, F.-L. Zhu and X.-P. Hu, Enantioselective copper-catalyzed formal [4 + 2] cycloaddition of o-aminophenol derivatives with propargylic esters for synthesis of optically active 3,4-dihydro-2H-1,4-benzoxazines, Org. Lett., 2016, 18, 1190–1193 CrossRef PubMed.
- W. Guo, L. Zuo, M. Cui, B. Yan and S. Ni, Propargylic amination enabled the access to enantioenriched acyclic α-quaternary α-amino ketones, J. Am. Chem. Soc., 2021, 143, 7629–7634 CrossRef.
- K. Nakajima, M. Shibata and Y. Nishibayashi, Copper-catalyzed enantioselective propargylic etherification of propargylic esters with alcohols, J. Am. Chem. Soc., 2015, 137, 2472–2475 CrossRef.
- R.-Z. Li, H. Tang, K.-R. Yang, L.-Q. Wan, X. Zhang, J. Liu, Z. Fu and D.-W. Niu, Enantioselective propargylation of polyols and desymmetrization of meso 1,2-diols by copper/borinic acid dual catalysis, Angew. Chem., Int. Ed., 2017, 56, 7213–7217 CrossRef.
- R.-Z. Li, D.-Q. Liu and D.-W. Niu, Asymmetric O-propargylation of secondary aliphatic alcohols, Nat. Catal., 2020, 3, 672–680 CrossRef.
- Y. Hou, Z. Zhang, X. Sun, Z. Yang, Y.-X. Luan and P. Tang, Copper-catalyzed enantioselective trifluoromethoxylation of propargyl sulfonates, Angew. Chem., Int. Ed., 2023, 62, e202218919 CrossRef.
- P. Fang and X.-L. Hou, Asymmetric copper-catalyzed propargylic substitution reaction of propargylic acetates with enamines, Org. Lett., 2009, 11, 4612–4615 CrossRef PubMed.
- F.-L. Zhu, Y. Zou, D.-Y. Zhang, Y.-H. Wang, X.-H. Hu, S. Chen, J. Xu and X.-P. Hu, Enantioselective copper-catalyzed decarboxylative propargylic alkylation of propargyl beta-ketoesters with a chiral ketimine P,N,N-ligand, Angew. Chem., Int. Ed., 2014, 53, 1410–1414 CrossRef.
- W. Shao, H. Li, C. Liu, C. J. Liu and S.-L. You, Copper-catalyzed intermolecular asymmetric propargylic dearomatization of indoles, Angew. Chem., Int. Ed., 2015, 54, 7684–7687 CrossRef PubMed.
- K. Tsuchida, Y. Senda, K. Nakajima and Y. Nishibayashi, Construction of chiral tri- and tetra-arylmethanes bearing quaternary carbon centers: copper-catalyzed enantioselective propargylation of indoles with propargylic esters, Angew. Chem., Int. Ed., 2016, 55, 9728–9732 CrossRef.
- Q. Wang, T.-R. Li, L.-Q. Lu, M.-M. Li, K. Zhang and W.-J. Xiao, Catalytic asymmetric [4 + 1] annulation of sulfur ylides with copper-allenylidene intermediates, J. Am. Chem. Soc., 2016, 138, 8360–8363 CrossRef.
- K. Zhang, L.-Q. Lu, S. Yao, J.-R. Chen, D.-Q. Shi and W.-J. Xiao, Enantioconvergent copper catalysis: in situ generation of the chiral phosphorus ylide and its Wittig reactions, J. Am. Chem. Soc., 2017, 139, 12847–12854 CrossRef.
- J. Song, Z.-J. Zhang and L.-Z. Gong, Asymmetric [4 + 2] annulation of C1 ammonium enolates with copper-allenylidenes, Angew. Chem., Int. Ed., 2017, 56, 5212–5216 CrossRef PubMed.
- H.-D. Qian, Z.-H. Li, S. Deng, C. Yao, H.-M. Xiang, G. Xu, Z.-Q. Geng, Z. Wang, L. Chen, C. Liu, C. Zhu, X. Qi and H. Xu, Catalytic asymmetric vinylogous and bisvinylogous propargylic substitution, J. Am. Chem. Soc., 2022, 144, 15779–15785 CrossRef PubMed.
- F. Gong, X. Meng, S. Lan, J. Liu, S. Yang and X. Fang, Asymmetric semipinacol rearrangement enabled by copper-catalyzed propargylic alkylation, ACS Catal., 2022, 12, 12036–12044 CrossRef.
- J. E. Gomez, A. Cristofol and A. W. Kleij, Copper-catalyzed enantioselective construction of tertiary propargylic sulfones, Angew. Chem., Int. Ed., 2019, 58, 3903–3907 CrossRef CAS.
- X. Gao, Y.-L. Xiao, S. Zhang, J. Wu and X.-G. Zhang, Copper-catalyzed enantioselective trifluoromethylthiolation of secondary propargyl sulfonates, CCS Chem., 2021, 3, 1463–1471 CrossRef CAS.
- S. Niu, Y. Luo, C. Xu, J. Liu, S. Yang and X. Fang, Copper-catalyzed yne-allylic substitutions using stabilized nucleophiles, ACS Catal., 2022, 12, 6840–6850 CrossRef CAS.
- H.-H. Kong, C. Zhu, S. Deng, G. Xu, R. Zhao, C. Yao, H.-M. Xiang, C. Zhao, X. Qi and H. Xu, Remote enantioselective [4 + 1] annulation with copper-vinylvinylidene intermediates, J. Am. Chem. Soc., 2022, 144, 21347–21355 CrossRef CAS.
- (a) J.-S. Ma, H.-Y. Lu, Y.-W. Chen, W.-C. Zhao, Y.-Z. Sun, R.-P. Li, H.-X. Wang, G.-Q. Lin and Z.-T. He, Copper-catalysed convergent regio- and enantioselective alkynylallylic substitution, Nat. Synth., 2022, 2, 37–48 CrossRef; (b) J. H. van Maarseveen, Copper for alkynylallylic substitution, Nat. Synth., 2023, 2, 11–12 CrossRef.
- Y.-Z. Sun, Z.-Y. Ren, Y.-X. Yang, Y. Liu, G.-Q. Lin and Z.-T. He, Asymmetric Substitution by Alkynyl Copper Driven Dearomatization and Rearomatization, Angew. Chem., Int. Ed., 2023, 62, e202314517 CrossRef CAS.
- M. Rosler, R. Anand, A. C. Sain, S. Gauthier, Y. Agid, P. D. Bianco, H. B. Stahelin, R. Hartman and M. Gharabawi, Efficacy and safety of rivastigmine in patients with Alzheimer's disease: international randomized controlled trial, Br. Med. J., 1999, 318, 633–638 CrossRef CAS PubMed.
- (a) D. Schnappinger and W. Hillen, Tetracyclines: antibiotic action, uptake, and resistance mechanisms, Arch. Microbiol., 1996, 165, 359–369 CrossRef CAS PubMed; (b) A. N. Sapadin and R. Fleischmajer, Tetracyclines: nonantibiotic properties and their clinical implications, J. Am. Acad. Dermatol., 2006, 54, 258–265 CrossRef; (c) G. A. Pankey, Tigecycline, J. Antimicrob. Chemother., 2005, 56, 470–480 CrossRef CAS.
- Y. Zhou, Y. Xie, L. Yang, P. Xie and H.-M. Huang, Copper-catalyzed aerobic oxidative amination of arylboronic acid with aminal under base-free conditions, Tetrahedron Lett., 2013, 54, 2713–2716 CrossRef CAS.
- H.-Y. Lu and Z.-T. He, Catalytic asymmetric synthesis of 1,4- enynes, Chin. Chem. Lett., 2023, 34, 108105–108115 CrossRef CAS.
- T. Lu and F. Chen, Multiwfn: A Multifunctional Wavefunction Analyzer, J. Comput. Chem., 2012, 33, 580–592 CrossRef CAS PubMed.
- T. Lu and Q. Chen, Realization of conceptual density functional theory and information-theoretic approach in Multiwfn program, in Conceptual Density Functional Theory, ed. S. Liu, Wiley-VCH, 2022, pp. 631–647 Search PubMed.
- (a) B. E. Love, Facile synthesis of N-dialkylaminomethylsubstituted heterocycles, J. Org. Chem., 2007, 72, 630–632 CrossRef CAS PubMed; (b) R. R. Khairullina, B. F. Akmanov, R. V. Kunakova and A. G. Ibragimov, Synthesis of amino sulfides in the presence of rare-earth and transition metal catalysts, Russ. J. Org. Chem., 2012, 48, 902–907 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3qo01749d |
| This journal is © the Partner Organisations 2024 |