
Recent advances in methods for live-cell RNA imaging
Tien G.
Pham
 and
Jiahui
Wu
*
and
Jiahui
Wu
*
Department of Chemistry, University of Massachusetts, Amherst, Massachusetts 01003, USA. E-mail: jiahuiwu@umass.edu
First published on 21st February 2024
Abstract
As one of the most fundamental building blocks of life, RNA plays critical roles in diverse biological processes, from X chromosome inactivation, genome stability maintenance, to embryo development. Being able to visualize the localization and dynamics of RNA can provide critical insights into these fundamental processes. In this review, we provide an overview of current methods for live-cell RNA imaging with a focus on methods for visualizing RNA in living mammalian cells with single-molecule resolution.
 Tien G. Pham | Tien G. Pham received her Bachelor of Science of Chemistry from the University of Delaware, United States in 2023. She is currently a Ph.D. student in the Department of Chemistry at the University of Massachusetts, Amherst under the supervision of Prof. Jiahui (Chris) Wu. Her current research interest focuses on developing and applying RNA imaging technology to understand the function of non-coding RNA. |
 Jiahui Wu | Dr Jiahui (Chris) Wu is an Assistant Professor in the Department of Chemistry at the University of Massachusetts, Amherst. He received his B.Eng. in bioengineering from Guangdong University of Technology in 2009, and his Ph.D. in chemistry with Prof. Robert E. Campbell at the University of Alberta in 2014. Dr Wu then pursued his postdoctoral research in the laboratory of Prof. Samie R. Jaffrey at Weill Cornell Medicine of Cornell University. Dr Wu's research focuses on developing cutting-edge technologies to decipher the functional roles of RNA in both health and disease. |
1. Introduction
RNA is one of the most fundamental components in the regulation of gene expression. Small nuclear RNAs can assemble with proteins to form spliceosomes to generate mature mRNAs, mRNAs carry genetic information from the nucleus to different cellular locations for protein expression, and microRNAs can bind to Argonaute proteins to fine-tune protein expression levels.1,2 Besides RNA that codes for protein, genomics research has shown that the majority of the RNAs transcribed from the human genome are non-coding RNA (ncRNA), which are indispensable for maintaining cellular function.3,4RNA function is highly linked to its spatiotemporal distribution in the cell. Recent research has started to unravel the function of ncRNAs and have shown that they reside in specific subcellular locations to regulate critical biological processes, including X chromosome inactivation, genome stability maintenance, and embryo development.5–7 Importantly, dysregulation of RNA localization is highly associated with diseases, including amyotrophic lateral sclerosis, fragile X syndrome, and epithelial mesenchymal transition in cancer.8–10 Therefore, being able to visualize the localization and dynamics of RNA can provide critical insights into numerous fundamental processes and disease pathogenesis.
Imaging RNA in living cells is a highly challenging task, primarily because it requires selectively conferring fluorescence signals to the RNA of interest but not to the surrounding areas. Additionally, the transient and rapid-moving nature of RNA has made its dynamic behavior more elusive. Here, we review recent developments in methods for imaging RNA in living mammalian cells. We also discuss critical factors to consider when choosing a method for RNA imaging.
2. RNA imaging methods
2.1 RNA hairpin methods
One of the earliest methods to track the localization and dynamics of RNAs in living cells makes use of a naturally occurring RNA hairpin and its binding protein to tether fluorophores to an RNA of interest.11 In this method, multiple repeats of a bacteriophage-derived RNA hairpin, called MS2, are fused to the 3′ untranslated region (UTR) of an mRNA of interest. Each MS2 can recruit fluorescent proteins (FPs) by specifically binding to the homodimeric MS2 coat protein (MCP), which is fused to these FPs (Fig. 1). In this way, an aggregated fluorescence signal from multiple FPs is conferred to the RNA of interest with the MS2 tag. Using this method, Singer and coworkers were able to image the localization and dynamics of ASH1 mRNA in Saccharomyces cerevisiae.11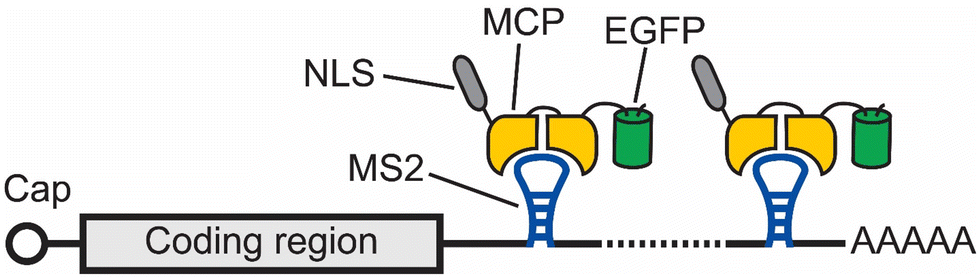 | ||
| Fig. 1 Schematic of the MS2 tag for imaging mRNA. In this method, multiple (usually 24) repeats of a bacteriophage-derived RNA hairpin, called MS2, are fused to the 3′ UTR of an mRNA of interest. Each MS2 can recruit an EGFP by specifically binding to the homodimeric MS2 coat protein (MCP). In this way, an aggregated fluorescence signal from multiple EGFPs is conferred to the RNA of interest with the MS2 tag. | ||
After its development, the MS2 method was applied to track RNAs with single-molecule resolution in living cells.12 To enable single RNA tracking, it is critical to tether enough FPs to the RNA of interest such that the fluorescence signals on each RNA is higher than the fluorescence signals from the unbound FPs. Therefore, an MS2 tag containing 24 copies of the MS2 hairpin is typically needed for single RNA imaging with each MS2 hairpin recruits two enhanced green fluorescent proteins (EGFPs).12 Even though MS2 binds to MCP with a high affinity (Kd = ∼0.2 nM), fluorescence fluctuation spectroscopy measurements showed that only about 60% of the MS2 hairpins in an MS2 tag are occupied by MCP.12,13
To further ensure a high fluorescence signal-to-noise ratio for detecting single RNA molecules, EGFP-MCP needs to be expressed at low levels, which is often achieved by inserting the EGFP-MCP gene into the host cell's genome. Additionally, each EGFP is fused to a nuclear localization sequence (NLS) so that the unbound MCP-EGFP is transported to the nucleus, thus decreasing the background fluorescence signals. It should be noted that having multiple NLS bound to the RNA of interest could alter the trafficking dynamics of this RNA.14 Another approach for decreasing background fluorescence signals involves using two orthogonal RNA hairpins to recruit the two halves of a split FP to the RNA of interest.15 The split FP is nonfluorescent unless both halves of this split FP are tethered to the RNA of interest. Besides these approaches, recent research has combined the SunTag technology16 with the RNA hairpin methods to further increase the signal-to-noise ratio for detecting single RNA molecules.17,18
Since its first invention, there have been multiple improvements on the MS2 tag. Because of its highly repetitive nature, the MS2 tag is prone to deletion and recombination during bacterial amplification and viral transduction.19,20 This can lead to MS2 tags with random deletion of the MS2 repeats and affect detection consistency of the tagged RNA. Singer and coworkers solved this problem by engineering a synonymous MS2 tag containing MS2 variants with different sequences while maintaining their MCP-binding ability.21 More recently, it has been shown that the high-affinity interaction between MS2 and MCP impairs mRNA degradation in Saccharomyces cerevisiae.22–25 Further improvements on the MS2 tag by decreasing the affinity between MS2 and MCP to a Kd of ∼2.5 nM have solved this problem (Table 1).26
Besides the MS2-MCP pairs, orthogonal RNA stem loop-protein pairs have been developed for imaging RNA.27,28 A widely used RNA imaging method is based on the PP7 RNA and its binding protein, called PP7 coat protein (PCP).27 Similar to MS2-MCP, PP7-PCP was derived from bacteriophage and showed a high binding affinity (Kd = ∼1.6 nM) (Table 1). Importantly, PP7 and MS2 can discriminately bind in favor of their coat protein by ∼1000 fold,27,29,30 which enabled simultaneous imaging two RNAs in the same cell at the same time.31 In addition, another RNA hairpin, called boxB, has been used for tethering FPs to RNA for live-cell RNA imaging (Table 1).28,32
2.2 Fluorogenic RNA aptamers
One limitation of the RNA hairpin methods is the high background fluorescence of the unbound fluorophores. To decrease background fluorescence, new methods were developed using fluorogenic RNA aptamers.33–46 Fluorogenic RNA aptamers are RNA sequences selected in vitro via systematic evolution of ligands by exponential enrichment (SELEX).47 These fluorogenic RNA aptamers can specifically bind and turn on the fluorescence signals of otherwise nonfluorescent small-molecule dyes (Fig. 2). When used for imaging RNA, an imaging tag containing multiple copies of fluorogenic RNA aptamers is fused to the 3′ UTR of an RNA of interest. Each fluorogenic RNA aptamer in this tag can bind and turn on the fluorescence of its cognate dye, thus conferring fluorescence to the RNA of interest in living cells (Fig. 2). | ||
| Fig. 2 Schematics of different fluorogenic RNA tags for imaging mRNA. In the fluorogenic RNA aptamer methods, an imaging tag containing multiple copies of fluorogenic RNA aptamers is fused to the 3′ UTR of an mRNA of interest. Each fluorogenic RNA aptamer in this tag can bind and turn on the fluorescence of otherwise nonfluorescent small-molecule dyes. Fluorescence turn-on can be achieved by (a) the fluorogenic RNA binding to the dye to stop the dye's cis–trans isomerization; (b) the fluorogenic RNA binding to the dye within a dye-quencher conjugate; (c) the fluorogenic RNA binding to the quencher within a dye-quencher conjugate. | ||
The first fluorogenic RNA aptamer, called Spinach, was selected in vitro to bind to a dye that mimics the GFP chromophore, called DFHBI.33 DFHBI exhibits minimal fluorescence in solution but shows a 2000-fold green fluorescence increase upon binding to Spinach with a Kd of 562 nM (Table 2 and Fig. 2a).33 Using Spinach, Jaffrey and coworkers were able to image the 5S ribosomal RNA in living mammalian cells.33 Further development of Spinach using systematic mutagenesis led to the development of Spinach2 (Table 2).34 Compared to Spinach, Spinach2 has higher folding efficiency and can track the dynamics of CGG trinucleotide repeat-containing RNA associated with Fragile X-associated tremor/ataxia syndrome.34 Subsequently, a strategy combining in vitro selection and in-cell fluorescence-based selection led to the development of a series of fluorogenic RNA aptamers with different colors, including Broccoli, Red Broccoli, Corn, Beetroot, and Squash (Table 2).35–38,48,49 Notably, Broccoli can bind an improved DFHBI derivative, called BI, and exhibits improved folding efficiency in mammalian cells.50 With BI, a Broccoli tag containing 24 copies of Broccoli enabled tracking of ACTB mRNA in living cells with single-molecule resolution.50
| Fluorophore | Ex/Ema (nm) | Extinction coefficient (M−1 cm−1) | Quantum yield | Brightness (×10−3 M−1 cm−1) | K d (nM) | Ref. |
|---|---|---|---|---|---|---|
| a Ex/Em: excitation/emission wavelength peak value. b Photophysical properties of ATTO 590. c N/A: not applicable. | ||||||
| Spinach/DFHBI | 469/501 | 24![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 300 300 |
0.72 | 17.50 | 562 | 33 |
| Spinach2/DFHBI-1T | 482/505 | 31000 |
0.94 | 29.14 | 560 | 34 |
| Broccoli/BI | 470/505 | 33600 |
0.67 | 22.51 | 51 | 50 |
| Broccoli/TBI | 485/527 | 35100 |
0.64 | 22.46 | 71 | 48 |
| Red Broccoli/OBI | 541/590 | 47300 |
0.67 | 31.69 | 23 | 49 |
| Corn/DFHO | 505/545 | 29000 |
0.25 | 7.25 | 70 | 36 |
| Beetroot/DFAME | 514/619 | 22500 |
0.17 | 3.83 | 460 | 37 |
| Squash/DFHBI-1T | 452/503 | 24200 |
0.71 | 17.18 | 45 | 38 |
| Squash/DFHO | 495/562 | 24600 |
0.60 | 14.76 | 54 | 38 |
| HBC-Pepper/HBC530 | 485/530 | 65300 |
0.66 | 43.10 | 3.5 | 39 |
| HBC-Pepper/HBC620 | 577/620 | 100000 |
0.58 | 58.00 | 6.1 | 39 |
| Clivia/NBSI574 | 490/574 | 23000 |
0.37 | 8.51 | 18 | 40 |
| Clivia/NBSI580 | 524/580 | 33000 |
0.48 | 15.84 | 55 | 40 |
| Clivia/NBSI595 | 492/595 | 25000 |
0.36 | 9.00 | 79 | 40 |
| Clivia/NBSI618 | 510/618 | 21000 |
0.17 | 3.57 | 25 | 40 |
| Chili/DMHBO+ | 456/592 | 22000 |
0.10 | 2.20 | 12 | 41 |
| o-Coral/Gemini-561 | 580/596 | 141000 |
0.58 | 81.78 | 73 | 42 |
| Mango II/TO1-Biotin | 510/535 | 77000 |
0.21 | 16.17 | 0.7 | 43 and 81 |
| RhoBAST/TMR-DN | 564/590 | 96000 |
0.57 | 54.72 | 15 | 44 |
| biRhoBAST/TMR2 | 564/590 | 165000 |
0.93 | 153.45 | 0.04 | 45 |
| Riboglow A/Cbl-5xPEG-ATTO 590 | 593/622b | 120000b |
0.31 | 37.20 | 34 | 46 |
| EGFP | 488/508 | 56000 |
0.67 | 37.52 | N/Ac | 82 |
| mNeonGreen | 504/517 | 113000 |
0.80 | 90.40 | N/Ac | 82 |
| mCherry | 586/610 | 85000 |
0.3 | 25.50 | N/Ac | 82 |
While most of the fluorogenic aptamer-dye complex exhibits green to orange fluorescence emission, recent efforts further expanded the color palette of the fluorogenic aptamer-dye complex to cyan to red fluorescence emission.39,40 Yang and coworkers developed a fluorogenic RNA aptamer, called Pepper (we refer to this aptamer as HBC-Pepper to differentiate it from another Pepper aptamer that can bind and stabilize fluorogenic proteins described later in this review), that can bind and turn on a series of dyes derived from HBC with cyan to red fluorescence (Table 2).39 Notably, when expressed in mammalian cells, HBC-Pepper530 is about 10-fold brighter than the Broccoli/DFHBI-1T complex and the Corn/DFHO complex, respectively.39 Most recently, the Yang group reported another fluorogenic RNA aptamer, called Clivia, which can bind to a series of NBSI dyes and emit orange to red fluorescence with large Stokes shift (Table 2).40 Recent developments also led to other fluorogenic RNA-dye complexes including Chili/DMHBO+, o-Coral/Gemini-561, and Mango II/TO1-Biotin (Table 2).41,42,51
Since some of the fluorogenic aptamer-dye complexes have low photostability and fluorescence brightness,52 an alternative approach to image RNA involves using conventional small-molecule dyes with high photostability and brightness. A major challenge of using conventional dyes is their high background fluorescence even when they are not bound to the RNA of interest. To minimize this background fluorescence, these dyes are conjugated to a quenching moiety so that they only exhibit minimal fluorescence in solution. To selectively turn on the fluorescence of these dye-quencher conjugates, RNA aptamers were engineered to bind either the dye or the quencher to alleviate dye quenching.44,46
An early example of this approach from the Jäschke group involves engineering an RNA aptamer that binds sulforhodamine B and turns on the fluorescence of a sulforhodamine B-dinitroaniline conjugate (SR-DN) (Fig. 2b).53 Based on this concept, the same group reported another fluorogenic RNA aptamer called RhoBAST that can turn on the fluorescence of the tetramethylrhodamine-dinitroaniline conjugate (TMR-DN) (Table 2).44 Notably, due to its fast dye exchange kinetics, high photostability, and high brightness, the RhoBAST/TMR-DN complex enables super-resolution imaging of RNA in cells.44 A RhoBAST tag containing 16 copies of RhoBAST aptamers was used for imaging the FMR1 RNA containing excessive CGG repeats and revealed its architectural details in mammalian cells.44 More recently, Jäschke, Sunbul and coworkers exploited the avidity concept and developed a fluorogenic RNA aptamer, called biRhoBAST, with high molecular brightness and photostability for single mRNA imaging.45
Another elegant example of the dye-quencher approach involves using an RNA that binds to the quencher rather than the dye to achieve fluorescence turn on (Fig. 2c).46 Palmer and coworkers used cobalamin (Cbl) as the quencher and conjugated it to different dyes. The fluorescence of these Cbl-dye conjugates can be turned on by a Cbl-binding riboswitch (Table 2). This system, called Riboglow, enabled live-cell imaging of the small non-coding U1 RNA.46 Additionally, a Riboglow tag containing 12 copies of the Riboglow aptamers has been used for imaging mRNA with single-molecule resolution.54 Most recently, it has also been shown that the Riboglow system can also be used for fluorescence lifetime imaging microscopy.55
2.3 Fluorogenic proteins
Fluorogenic RNA aptamers require the addition of exogenous dyes, some of these dyes have high background fluorescence and exhibit limited permeability across cell membranes. To address these limitations, Jaffrey and coworkers developed a new method using genetically encoded fluorogenic proteins rather than exogenous dyes (Fig. 3).56 This method makes use of a protein degradation domain, called tDeg. tDeg is a 19-amino acid-long peptide containing an RNA-binding peptide from the bovine immunodeficiency virus and an Arg-Arg-Arg-Gly degron sequence. When fused to the C-terminus of an FP, tDeg can be recognized by the ubiquitin-proteasome system, causing protein degradation of this FP-tDeg fusion protein. However, an RNA aptamer, called Pepper, was designed to specifically bind to tDeg. When binding to tDeg, Pepper can block the Arg-Arg-Arg-Gly degron from being recognized by the ubiquitin-proteasome system, thus stabilizing the FP-tDeg fusion protein. In this way, tDeg converts an FP to an RNA-stabilized fluorogenic protein. | ||
| Fig. 3 Schematic of the Pepper tag for imaging mRNA. In this method, a Pepper tag containing multiple copies of Pepper RNA variants within the F30 RNA folding scaffold68 is fused to the 3′ UTR of an mRNA of interest. Each Pepper can bind and stabilize intrinsically unstable fluorogenic proteins, (mNeonGreen)4-tDeg. In this way, the fluorescence from the fluorogenic proteins is specially conferred to the RNA of interest tagged by the Pepper tag. | ||
Since the unbound fluorogenic proteins can be degraded, they do not require extra NLS or genomic incorporation to decrease background fluorescence levels. Additionally, this method does not require any exogenous dyes, thus making it fully genetically encoded. A Pepper tag consisting of 20 copies of Peppers enables tracking of single RNA molecules in living mammalian cells. Furthermore, this Pepper tag was used to reveal the trafficking dynamics of ACTB mRNA to stress granules when cells were under arsenite-induced stress.56 Additionally, recent research has shown the possibility of converting the Pepper-tDeg technology into fluorescent sensor for detecting cellular RNAs, such as survivin and MALAT-1.57
2.4 dCas-based methods
Since all the above methods require inserting an RNA imaging tag to confer fluorescence to an RNA of interest, they are mostly used for imaging exogenously expressed RNAs. To image endogenous RNA without the need of an imaging tag, a new method was developed based on the CRISPR-Cas technology.58–62 In this method, a nuclease-deficient Cas protein (dCas) is fused to an EGFP (in some cases, multiple FPs). This dCas-EGFP fusion protein uses a guide RNA (gRNA) to bind to untagged endogenous RNAs of interest in a programmable manner, thus conferring fluorescence to these RNAs for imaging (Fig. 4). This method was first shown to be able to track endogenous ACTB, CCNA2, and TFRC mRNAs in cells using a dCas9 by Yeo and coworkers.58 After realizing the possibility of targeting RNA using a dCas protein, different types of dCas13 proteins have been used to track both coding and non-coding RNAs, respectively.59–62 Notably, some dCas13-based methods can image RNA with single-molecule resolution when this RNA contains highly repetitive sequences.60,62,63 These repetitive sequences can be a genetically inserted tag, such as 12× or 48× GCN4 repeats, or repeat regions from an endogenous gene, such as exon 2 of the MUC4 mRNA.60,62,63 | ||
| Fig. 4 Schematic of using dCas-EGFP for imaging mRNA. In this method, a nuclease-deficient Cas protein (dCas) is fused to an EGFP (in some cases, multiple FPs). This dCas-EGFP fusion protein uses a gRNA to bind to untagged endogenous RNAs of interest in a programmable manner, thus conferring fluorescence to these RNAs for imaging. | ||
3. Key factors to consider when choosing an RNA imaging method
3.1 Tag length and size
Detecting single RNA molecules in living cells requires conferring enough fluorescence signals to the RNA compared to the background. To achieve such signal-to-noise ratio, multiple copies of stem loops or fluorogenic RNAs are often required.26,50,56,64 For example, the MS2 tag (1660 nt) contains 24 copies of the MS2 stem loop, the Pepper tag (1812 nt) contains 20 copies of the Pepper RNA, and the Broccoli tag (1449 nt) contains 24 copies of the Broccoli aptamer (Table 3).26,50,56,64 However, recent studies have shown that long RNA tag can induce degradation of the tagged RNA via the nonsense-mediated mRNA decay (NMD) pathway.64 In this work by Singer and coworkers, an MS2 tag with 24 copies of the MS2 stem loop was shown to induce RNA degradation of the tagged RNA through NMD.64 However, this problem was alleviated in mammalian cells when an NMD-corrective protein, PABPC1(F142 V, F337 V) or eRF3, was fused to MCP-EGFP.64 It should be noted that, if tag length is the major determinant of NMD pathway activation, this problem might also occur with other imaging tags with similar or longer length compared to the MS2 tag.| Name | Number of RNA copies in the tag | Tag length (nt) | Fluorophore (molecular weight in kDa) | Tag sizea (kDa) | Fully genetically encoded | Reports on imaging transcription | Ref. |
|---|---|---|---|---|---|---|---|
| a Tag size is calculated assuming each hairpin or fluorogenic RNA aptamer is bound by its cognate fluorophore. | |||||||
| MS2 tag | 24 | 1660 | NLS-tandem dimer MCP-EGFP-eRF3 (112.4) | 3230 | Yes | Yes | 64 |
| PP7 tag | 24 | 1446 | NLS-tandem dimer PCP-EGFP (57.5) | 1846 | Yes | Yes | 13 |
| Broccoli tag | 24 | 1449 | BI (0.368) | 478 | No | No | 50 |
| Mango II tag | 24 | 1187 | TO1-Biotin (0.749) | 408 | No | Yes | 51 |
| biRhoBAST tag | 8 | 1242 | TMR2 (1.824) | 418 | No | No | 45 |
| Riboglow tag | 12 | 1367 | Cbl-4xGly-ATTO 590 (2.4) | 471 | No | No | 54 |
| Pepper tag | 20 | 1812 | (mNeonGreen)4-tDeg (112.2) | 2628 | Yes | No | 56 |
While tag length can affect RNA stability, another critical factor to consider is the size of the imaging tag. The size of an imaging tag includes the total molecular weight of the RNA tag sequence and all the fluorophores that are tethered to this imaging tag. Presumably, a larger tag size is more likely to affect the dynamics and trafficking behavior of the tagged RNA. Assuming each MS2 stem loop is occupied by one NLS-tandem dimer MCP-EGFP-eRF3, the latest version of the MS2 tag containing 24 copies of synonymous MS2 stem loops has a tag size of 3230 kDa, (Table 3).26,64 Compared to the MS2 tag, fluorogenic RNA tags usually have a smaller tag size (Table 3). Notably, the Mango II tag containing 24 copies of the Mango II fluorogenic RNA aptamer binding to its cognate dye, TO1-biotin, has a tag size of 408 kDa, which is the smallest among the fluorogenic RNA tags (Table 3).51 When using fluorogenic proteins to image RNA, a Pepper tag with 20 copies of Peppers with each Pepper binds to four copies of mNeonGreens gives a 2628 kDa tag size (Table 3).56 While the dCas method does not require insertion of any imaging tag, an NLS-dPspCas13b-3xEGFP-NLS protein has a size of 216.7 kDa.60 Therefore, the MS2 tag has the largest tag size, while the Mango II tag offers the smallest tag size for imaging RNA with single-molecule resolution.
3.2 Fluorophore maturation time
Maturation time of the fluorophore is a critical factor to consider when imaging RNA during transcription. Fluorescence of the fluorophores should be instantaneous upon binding to the nascent RNA. For fluorogenic aptamers, although the fluorogenic dyes are readily available upon addition to cells, it takes time for the fluorogenic RNA aptamers to fold before binding and turning on the fluorescence of the dyes. Thus, fast-folding fluorogenic RNA aptamers would shorten this delay of fluorescence onset. Among the fluorogenic RNA tags, the Mango II tag has been shown to image transcription.51 For fluorogenic proteins, nascent fluorogenic proteins are not instantaneous fluorescent as it usually takes 4–30 minutes for most fluorescent protein's chromophore to mature to be fluorescent. Therefore, fluorogenic proteins may not be able to confer instantaneous fluorescence to the nascent RNA. Using a fast-maturing fluorescent protein, such as mCerulean,65 mNeonGreen,65 and mScarlet-I3,66 could shorten this delay of fluorescence onset.Among all the imaging methods, the RNA hairpin method can bring instantaneous fluorescence to the nascent RNA because each RNA hairpin can bind and recruit fluorophores with constitutive fluorescence.31,67 Notably, the PP7-PCP method was used for real-time observation of transcription initiation, elongation, and termination in Saccharomyces cerevisiae.67 Similarly, the dCas method can also bring instantaneous fluorescence to the nascent RNA containing repetitive sequences that can be bound by the EGFP-dCas-gRNA complex.59 Therefore, for imaging transcription, the RNA hairpin method and the dCas method are advantageous because they can confer instantaneous fluorescence to the nascent transcripts.
3.3 Molecular brightness and photostability of the fluorophores
Fluorescence brightness and photostability are major factors that can affect imaging duration, especially during imaging of RNA with single-molecule resolution. Tracking RNA dynamics often requires continuous imaging of the fluorescently tagged RNA. To achieve this, the fluorescence detector needs to collect enough photons from the RNA to differentiate its fluorescence signals from the background fluorescence. To ensure this, common strategies include using fluorophores with higher molecular brightness/photostability and recruiting more fluorophores to the tagged RNA. For example, to image RNA with single-molecule resolution, the MS2 tag generally recruits 24–48 copies of EGFPs, the Broccoli tag recruits 24 BI, and the Pepper tag recruits up to 80 copies of mNeonGreens.26,50,56 In addition, when using fluorogenic RNA aptamer tags, it is important to use the optimal concentration of the cognate dye as excess dye can interact with cellular proteins or DNA, thus generating high background fluorescence.It is worth noting that not necessarily every RNA aptamer on an RNA imaging tag is folded properly. Therefore, an RNA imaging tag might not be fully occupied by fluorophores. For example, it has been shown that the in-cell fluorophore occupancy of an MS2 tag is about 60%.12,13 In contrast, a PP7 tag has an in-cell fluorophore occupancy of close to 100%.13 Suboptimal in-cell RNA folding efficiency was also reported in fluorogenic RNA aptamers.50 To solve this problem, Jaffrey and coworkers developed dyes that can promote folding efficiency of the fluorogenic RNA aptamers.50 Notably, BI can promote cellular Broccoli folding and induce a more than 10-fold increase in fluorescence brightness than DFHBI-1T in Broccoli-expressing cells.50 Another approach to increase RNA folding efficiency involves the use of RNA folding scaffold.68 However, an increase of tag size and tag length will be a trade-off.
Continuous excitation of the fluorophores inevitably causes photobleaching, which in turn decreases the signal-to-noise ratio over time. Therefore, fluorophores with high photostability are critical to combat photobleaching during continuous RNA imaging. In fluorophores with similar photostability, the ones with a higher molecular brightness can also alleviate their susceptibility of photobleaching. Molecular brightness of a fluorophore is determined by how well it absorbs light (extinction coefficient) and how efficiently it converts the absorbed light to emitted photons (quantum yield). The amount of photons emitted from a single fluorophore is proportional to the amount of photons used for its excitation (i.e. excitation power). Therefore, to emit the same amount of photons, a fluorophore with high molecular brightness would require less photon input than a fluorophore with low molecular brightness would, thus decreasing its susceptibility to photobleaching.
FPs, such as EGFP and mCherry, are often used for live-cell imaging of RNA and have shown versatility in imaging different types of RNAs.26,31 The latest development of FPs with high molecular brightness and photostability, such as mStayGold69,70 and mScarlet3,66 should further extend the duration of single RNA tracking in living cells. Additionally, chemogenetic fluorophores, such as the HaloTag-binding Janelia Fluor dyes,71–73 also offer greatly improved molecular brightness and photostability compared to FPs for live-cell RNA imaging.
3.4 Multicolor imaging
Being able to visualize the interactions between different RNAs in living cells can provide valuable insights into their roles in critical cellular processes such as transcription, splicing, and translation. An early example of two-color RNA imaging used two orthogonal RNA hairpin methods.31 In this study by Singer and coworkers, MDN1 from two different alleles was tagged with the MS2 tag and the PP7 tag, respectively. Coexpression of PCP-2yEGFP and MCP-mCherry enabled simultaneous tracking of these two RNAs.31 More recently, a dCas13-based system, called CRISPRpalette, was developed to achieve three-color RNA imaging in living cells.62 Unlike conventional dCas methods, which fuse FPs directly to the dCas protein for imaging, CRISPRpalette uses MS2-MCP, PP7-PCP, and Pepper-tDeg to tether fluorophores to different gRNAs to confer fluorescence to the target RNAs of interest.62 In addition, a new method, called seqFRIES, utilizes orthogonal fluorogenic RNA aptamers to image up to four RNAs in the same cell.74 In this method, multiplexed imaging is achieved by sequential rounds of addition and withdrawal of cognate dyes of different fluorogenic RNA aptamers.4. Conclusions and future perspectives
In this review, we have discussed recent progress in imaging methods for tracking RNA in living mammalian cells. As pioneering technologies for live-cell RNA imaging, the RNA hairpin methods, such as MS2-MCP and PP7-PCP, remain the gold standard for single-molecule resolution tracking of RNA in living cells. In the past 15 years, there have been tremendous developments in innovative methods for live-cell tracking of RNA, including the use of fluorogenic aptamers, fluorogenic proteins, and dCas13. These methods have enabled a major leap towards understanding the role of RNA as a critical regulator for gene expression and numerous cellular processes. Looking forward, there are exciting opportunities to further expand the toolbox for RNA imaging.Being able to image the dynamic behavior of endogenous RNAs with single-molecule resolution is critical for understanding their function. While the dCas method enables imaging of untagged endogenous RNA in living cells with single-molecule resolution, it is limited to MUC4 which contains highly repetitive sequences for multiple EGFP-dCas-gRNA complexes to bind. New research has shown the possibility of combining the state-of-the-art fluorescence signal amplification technologies, such as SunTag16 and MoonTag,75 to the dCas methods to increase signal-to-noise ratio for imaging RNA.76 This represents an exciting area towards tracking untagged endogenous RNAs in living cells. Another possibility to image endogenous RNA is to utilize methods with high signal-to-noise ratios, such as fluorogenic aptamers and fluorogenic proteins. An RNA imaging tag can be genetically inserted to the locus of an endogenous gene of interest. When this endogenous gene of interest is transcribed, the corresponding transcripts will be tagged with the imaging tag. By incubating with fluorogenic dyes or coexpressing fluorogenic proteins, these tagged endogenous RNA transcripts can be tracked in living cells.
Another exciting area is to track the dynamic behavior of RNA in primary cells and in vivo, which can provide unparalleled insight into how changes in RNA localization and dynamics are translated to changes in cellular and ultimately animal behavior. While there are examples of imaging RNA in primary neurons using the MS2-MCP method,77–79 major challenges remain as this method requires GFP-MCP to be expressed at low levels. This involves generating genetically modified organisms, which can be highly laborious. Conceivably, imaging RNA in primary cells or in vivo could be achieved by exogenously expressing a fluorescently tagged RNA via the fluorogenic aptamer method. While the in vivo toxicity and permeability of the fluorogenic dyes have not been rigorously characterized, recent examples of using chemogenetic dyes in vivo80 hold promise for future application of fluorogenic dyes in vivo. Another way to achieve imaging of exogenously expressed RNA in primary cells and in vivo may be through the use of fluorogenic proteins. Since fluorogenic proteins are fully genetically encoded, and the excess fluorogenic proteins should be degraded in cells, this method also has potential for in vivo applications.
Author contributions
T. G. Pham and J. Wu wrote the manuscript.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
This work was supported by the University of Massachusetts Amherst startup funds (T. G. P. and J. W.). We thank Dr. Yi Shen for useful comments and suggestions.References
- A. G. Matera and Z. Wang, Nat. Rev. Mol. Cell Biol., 2014, 15, 108–121 CrossRef CAS PubMed.
- H. Kobayashi and Y. Tomari, Biochim. Biophys. Acta, 2016, 1859, 71–81 CrossRef CAS PubMed.
- E. Pennisi, Science, 2012, 337, 1159–1161 CrossRef CAS PubMed.
- M. de Hoon, J. W. Shin and P. Carninci, Mamm. Genome, 2015, 26, 391–402 CrossRef CAS PubMed.
- J. M. Engreitz, A. Pandya-Jones, P. McDonel, A. Shishkin, K. Sirokman, C. Surka, S. Kadri, J. Xing, A. Goren, E. S. Lander, K. Plath and M. Guttman, Science, 2013, 341, 1237973 CrossRef PubMed.
- S. Lee, F. Kopp, T.-C. Chang, A. Sataluri, B. Chen, S. Sivakumar, H. Yu, Y. Xie and J. T. Mendell, Cell, 2016, 164, 69–80 CrossRef CAS PubMed.
- J. Rosenbluh, D. Nijhawan, Z. Chen, K.-K. Wong, K. Masutomi and W. C. Hahn, PLoS One, 2011, 6, e26270 CrossRef CAS PubMed.
- O. J. Ziff, J. Harley, Y. Wang, J. Neeves, G. Tyzack, F. Ibrahim, M. Skehel, A. M. Chakrabarti, G. Kelly and R. Patani, Neuron, 2023, 111, 3011–3027 CrossRef CAS PubMed.
- D.-I. Kao, G. M. Aldridge, I. J. Weiler and W. T. Greenough, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 15601–15606 CrossRef CAS PubMed.
- R. Mitra, X. Chen, E. J. Greenawalt, U. Maulik, W. Jiang, Z. Zhao and C. M. Eischen, Nat. Commun., 2017, 8, 1604 CrossRef PubMed.
- E. Bertrand, P. Chartrand, M. Schaefer, S. M. Shenoy, R. H. Singer and R. M. Long, Mol. Cell, 1998, 2, 437–445 CrossRef CAS PubMed.
- D. Fusco, N. Accornero, B. Lavoie, S. M. Shenoy, J.-M. Blanchard, R. H. Singer and E. Bertrand, Curr. Biol., 2003, 13, 161–167 CrossRef CAS PubMed.
- B. Wu, J. A. Chao and R. H. Singer, Biophys. J., 2012, 102, 2936–2944 CrossRef CAS PubMed.
- S. Tyagi, Nat. Methods, 2009, 6, 331–338 CrossRef CAS PubMed.
- B. Wu, J. Chen and R. H. Singer, Sci. Rep., 2014, 4, 3615 CrossRef PubMed.
- M. E. Tanenbaum, L. A. Gilbert, L. S. Qi, J. S. Weissman and R. D. Vale, Cell, 2014, 159, 635–646 CrossRef CAS PubMed.
- Y. Guo and R. E. C. Lee, Cells Rep. Methods, 2022, 2, 100226 CrossRef CAS PubMed.
- Y. Hu, J. Xu, E. Gao, X. Fan, J. Wei, B. Ye, S. Xu and W. Ma, eLife, 2023, 12, e82178 CrossRef PubMed.
- K. A. Delviks and V. K. Pathak, J. Virol., 1999, 73, 7923–7932 CrossRef CAS PubMed.
- W. An and A. Telesnitsky, J. Virol., 2002, 76, 7897–7902 CrossRef CAS PubMed.
- B. Wu, V. Miskolci, H. Sato, E. Tutucci, C. A. Kenworthy, S. K. Donnelly, Y. J. Yoon, D. Cox, R. H. Singer and L. Hodgson, Genes Dev., 2015, 29, 876–886 CrossRef CAS PubMed.
- J. F. Garcia and R. Parker, RNA, 2015, 21, 1393–1395 CrossRef CAS PubMed.
- S. Heinrich, C. L. Sidler, C. M. Azzalin and K. Weis, RNA, 2017, 23, 134–141 CrossRef CAS PubMed.
- J. F. Garcia and R. Parker, RNA, 2016, 22, 657–659 CrossRef CAS PubMed.
- G. Haimovich, D. Zabezhinsky, B. Haas, B. Slobodin, P. Purushothaman, L. Fan, J. Z. Levin, C. Nusbaum and J. E. Gerst, RNA, 2016, 22, 660–666 CrossRef CAS PubMed.
- E. Tutucci, M. Vera, J. Biswas, J. Garcia, R. Parker and R. H. Singer, Nat. Methods, 2018, 15, 81–89 CrossRef CAS PubMed.
- J. A. Chao, Y. Patskovsky, S. C. Almo and R. H. Singer, Nat. Struct. Mol. Biol., 2008, 15, 103–105 CrossRef CAS PubMed.
- N. Daigle and J. Ellenberg, Nat. Methods, 2007, 4, 633–636 CrossRef CAS PubMed.
- J. Carey, V. Cameron, P. L. de Haseth and O. C. Uhlenbeck, Biochemistry, 1983, 22, 2601–2610 CrossRef CAS PubMed.
- F. Lim, T. P. Downey and D. S. Peabody, J. Biol. Chem., 2001, 276, 22507–22513 CrossRef CAS PubMed.
- S. Hocine, P. Raymond, D. Zenklusen, J. A. Chao and R. H. Singer, Nat. Methods, 2013, 10, 119–121 CrossRef CAS PubMed.
- R. J. Austin, T. Xia, J. Ren, T. T. Takahashi and R. W. Roberts, J. Am. Chem. Soc., 2002, 124, 10966–10967 CrossRef CAS.
- J. S. Paige, K. Y. Wu and S. R. Jaffrey, Science, 2011, 333, 642–646 CrossRef CAS PubMed.
- R. L. Strack, M. D. Disney and S. R. Jaffrey, Nat. Methods, 2013, 10, 1219–1224 CrossRef CAS PubMed.
- G. S. Filonov, J. D. Moon, N. Svensen and S. R. Jaffrey, J. Am. Chem. Soc., 2014, 136, 16299–16308 CrossRef CAS PubMed.
- W. Song, G. S. Filonov, H. Kim, M. Hirsch, X. Li, J. D. Moon and S. R. Jaffrey, Nat. Chem. Biol., 2017, 13, 1187–1194 CrossRef CAS PubMed.
- J. Wu, N. Svensen, W. Song, H. Kim, S. Zhang, X. Li and S. R. Jaffrey, J. Am. Chem. Soc., 2022, 144, 5471–5477 CrossRef CAS PubMed.
- S. K. Dey, G. S. Filonov, A. O. Olarerin-George, B. T. Jackson, L. W. S. Finley and S. R. Jaffrey, Nat. Chem. Biol., 2022, 18, 180–190 CrossRef CAS PubMed.
- X. Chen, D. Zhang, N. Su, B. Bao, X. Xie, F. Zuo, L. Yang, H. Wang, L. Jiang, Q. Lin, M. Fang, N. Li, X. Hua, Z. Chen, C. Bao, J. Xu, W. Du, L. Zhang, Y. Zhao, L. Zhu, J. Loscalzo and Y. Yang, Nat. Biotechnol., 2019, 37, 1287–1293 CrossRef CAS PubMed.
- L. Jiang, X. Xie, N. Su, D. Zhang, X. Chen, X. Xu, B. Zhang, K. Huang, J. Yu, M. Fang, B. Bao, F. Zuo, L. Yang, R. Zhang, H. Li, X. Huang, Z. Chen, Q. Zeng, R. Liu, Q. Lin, Y. Zhao, A. Ren, L. Zhu and Y. Yang, Nat. Methods, 2023, 20, 1563–1572 CrossRef CAS PubMed.
- C. Steinmetzger, N. Palanisamy, K. R. Gore and C. Höbartner, Chemistry, 2019, 25, 1931–1935 CrossRef CAS PubMed.
- F. Bouhedda, K. T. Fam, M. Collot, A. Autour, S. Marzi, A. Klymchenko and M. Ryckelynck, Nat. Chem. Biol., 2020, 16, 69–76 CrossRef CAS PubMed.
- E. V. Dolgosheina, S. C. Y. Jeng, S. S. S. Panchapakesan, R. Cojocaru, P. S. K. Chen, P. D. Wilson, N. Hawkins, P. A. Wiggins and P. J. Unrau, ACS Chem. Biol., 2014, 9, 2412–2420 CrossRef CAS PubMed.
- M. Sunbul, J. Lackner, A. Martin, D. Englert, B. Hacene, F. Grün, K. Nienhaus, G. U. Nienhaus and A. Jäschke, Nat. Biotechnol., 2021, 39, 686–690 CrossRef CAS PubMed.
- B. Bühler, J. Schokolowski, A. Benderoth, D. Englert, F. Grün, A. Jäschke and M. Sunbul, Nat. Chem. Biol., 2023, 19, 478–487 CrossRef PubMed.
- E. Braselmann, A. J. Wierzba, J. T. Polaski, M. Chromiński, Z. E. Holmes, S.-T. Hung, D. Batan, J. R. Wheeler, R. Parker, R. Jimenez, D. Gryko, R. T. Batey and A. E. Palmer, Nat. Chem. Biol., 2018, 14, 964–971 CrossRef CAS PubMed.
- C. Tuerk and L. Gold, Science, 1990, 249, 505–510 CrossRef CAS PubMed.
- X. Li, J. Wu and S. Jaffrey, Angew. Chem., Int. Ed. Engl., 2021, 60, 24153–24161 CrossRef CAS PubMed.
- X. Li, L. Mo, J. L. Litke, S. K. Dey, S. R. Suter and S. R. Jaffrey, J. Am. Chem. Soc., 2020, 142, 14117–14124 CrossRef CAS PubMed.
- X. Li, H. Kim, J. L. Litke, J. Wu and S. R. Jaffrey, Angew. Chem., Int. Ed., 2020, 59, 4511–4518 CrossRef CAS PubMed.
- A. D. Cawte, P. J. Unrau and D. S. Rueda, Nat. Commun., 2020, 11, 1283 CrossRef CAS PubMed.
- K. Y. Han, B. J. Leslie, J. Fei, J. Zhang and T. Ha, J. Am. Chem. Soc., 2013, 135, 19033–19038 CrossRef CAS PubMed.
- M. Sunbul and A. Jäschke, Angew. Chem., Int. Ed., 2013, 52, 13401–13404 CrossRef CAS PubMed.
- E. Braselmann, T. J. Stasevich, K. Lyon, R. T. Batey and A. E. Palmer, bioRxiv, 2019 DOI:10.1101/701649v1.
- N. Sarfraz, E. Moscoso, T. Oertel, H. J. Lee, S. Ranjit and E. Braselmann, Nat. Commun., 2023, 14, 867 CrossRef CAS PubMed.
- J. Wu, S. Zaccara, D. Khuperkar, H. Kim, M. E. Tanenbaum and S. R. Jaffrey, Nat. Methods, 2019, 16, 862–865 CrossRef CAS PubMed.
- W.-J. Zhou, H. Li, K.-K. Zhang, F. Wang, X. Chu and J.-H. Jiang, J. Am. Chem. Soc., 2021, 143, 14394–14401 CrossRef CAS PubMed.
- D. A. Nelles, M. Y. Fang, M. R. O'Connell, J. L. Xu, S. J. Markmiller, J. A. Doudna and G. W. Yeo, Cell, 2016, 165, 488–496 CrossRef CAS PubMed.
- H. Wang, M. Nakamura, T. R. Abbott, D. Zhao, K. Luo, C. Yu, C. M. Nguyen, A. Lo, T. P. Daley, M. La Russa, Y. Liu and L. S. Qi, Science, 2019, 365, 1301–1305 CrossRef CAS PubMed.
- L.-Z. Yang, Y. Wang, S.-Q. Li, R.-W. Yao, P.-F. Luan, H. Wu, G. G. Carmichael and L.-L. Chen, Mol. Cell, 2019, 76, 981–997 CrossRef CAS PubMed , e7.
- O. O. Abudayyeh, J. S. Gootenberg, P. Essletzbichler, S. Han, J. Joung, J. J. Belanto, V. Verdine, D. B. T. Cox, M. J. Kellner, A. Regev, E. S. Lander, D. F. Voytas, A. Y. Ting and F. Zhang, Nature, 2017, 550, 280–284 CrossRef PubMed.
- L.-Z. Yang, B.-Q. Gao, Y. Huang, Y. Wang, L. Yang and L.-L. Chen, Cell Insights, 2022, 1, 100044 CrossRef PubMed.
- Y. Huang, B.-Q. Gao, Q. Meng, L.-Z. Yang, X.-K. Ma, H. Wu, Y.-H. Pan, L. Yang, D. Li and L.-L. Chen, Genome Biol., 2023, 24, 15 CrossRef CAS PubMed.
- W. Li, A. Maekiniemi, H. Sato, C. Osman and R. H. Singer, Nat. Methods, 2022, 19, 1558–1562 CrossRef CAS PubMed.
- E. Balleza, J. M. Kim and P. Cluzel, Nat. Methods, 2018, 15, 47–51 CrossRef CAS PubMed.
- T. W. J. Gadella Jr., L. van Weeren, J. Stouthamer, M. A. Hink, A. H. G. Wolters, B. N. G. Giepmans, S. Aumonier, J. Dupuy and A. Royant, Nat. Methods, 2023, 20, 541–545 CrossRef CAS PubMed.
- D. R. Larson, D. Zenklusen, B. Wu, J. A. Chao and R. H. Singer, Science, 2011, 332, 475–478 CrossRef CAS PubMed.
- G. S. Filonov, C. W. Kam, W. Song and S. R. Jaffrey, Chem. Biol., 2015, 22, 649–660 CrossRef CAS PubMed.
- R. Ando, S. Shimozono, H. Ago, M. Takagi, M. Sugiyama, H. Kurokawa, M. Hirano, Y. Niino, G. Ueno, F. Ishidate, T. Fujiwara, Y. Okada, M. Yamamoto and A. Miyawaki, Nat. Methods, 2023 DOI:10.1038/s41592-023-02085-6.
- A. Crow, E. Ivorra-Molla, D. Akhuli, M. McAndrew, L. Kumar, W. Scott, M. Mishima, S. Palani and M. Balasubramanian, Res. Sq., 2023 DOI:10.1038/s41587-023-02018-w.
- J. B. Grimm, A. K. Muthusamy, Y. Liang, T. A. Brown, W. C. Lemon, R. Patel, R. Lu, J. J. Macklin, P. J. Keller, N. Ji and L. D. Lavis, Nat. Methods, 2017, 14, 987–994 CrossRef CAS.
- J. B. Grimm, A. N. Tkachuk, L. Xie, H. Choi, B. Mohar, N. Falco, K. Schaefer, R. Patel, Q. Zheng, Z. Liu, J. Lippincott-Schwartz, T. A. Brown and L. D. Lavis, Nat. Methods, 2020, 17, 815–821 CrossRef CAS PubMed.
- J. B. Grimm, B. P. English, J. Chen, J. P. Slaughter, Z. Zhang, A. Revyakin, R. Patel, J. J. Macklin, D. Normanno, R. H. Singer, T. Lionnet and L. D. Lavis, Nat. Methods, 2015, 12, 244–250 CrossRef CAS PubMed.
- R. Zheng, R. Wu, Y. Liu, Z. Sun, Y. Bagheri, Z. Xue, L. Mi, Q. Tian, R. Pho, S. Siddiqui, K. Ren and M. You, bioRxiv, 2023 DOI:10.1101/2023.04.20.537750.
- S. Boersma, D. Khuperkar, B. M. P. Verhagen, S. Sonneveld, J. B. Grimm, L. D. Lavis and M. E. Tanenbaum, Cell, 2019, 178, 458–472 CrossRef CAS PubMed.
- N.-H. Sun, D.-Y. Chen, L.-P. Ye, G. Sheng, J.-J. Gong, B.-H. Chen, Y.-M. Lu and F. Han, Theranostics, 2020, 10, 10993–11012 CrossRef CAS PubMed.
- S. Das, H. C. Moon, R. H. Singer and H. Y. Park, Sci. Adv., 2018, 4, eaar3448 CrossRef PubMed.
- H. Y. Park, H. Lim, Y. J. Yoon, A. Follenzi, C. Nwokafor, M. Lopez-Jones, X. Meng and R. H. Singer, Science, 2014, 343, 422–424 CrossRef CAS.
- A. R. Buxbaum, B. Wu and R. H. Singer, Science, 2014, 343, 419–422 CrossRef CAS.
- A. S. Abdelfattah, T. Kawashima, A. Singh, O. Novak, H. Liu, Y. Shuai, Y.-C. Huang, L. Campagnola, S. C. Seeman, J. Yu, J. Zheng, J. B. Grimm, R. Patel, J. Friedrich, B. D. Mensh, L. Paninski, J. J. Macklin, G. J. Murphy, K. Podgorski, B.-J. Lin, T.-W. Chen, G. C. Turner, Z. Liu, M. Koyama, K. Svoboda, M. B. Ahrens, L. D. Lavis and E. R. Schreiter, Science, 2019, 365, 699–704 CrossRef CAS PubMed.
- R. J. Trachman 3rd, A. Abdolahzadeh, A. Andreoni, R. Cojocaru, J. R. Knutson, M. Ryckelynck, P. J. Unrau and A. R. Ferré-D'Amaré, Biochemistry, 2018, 57, 3544–3548 CrossRef PubMed.
- P. J. Cranfill, B. R. Sell, M. A. Baird, J. R. Allen, Z. Lavagnino, H. M. de Gruiter, G.-J. Kremers, M. W. Davidson, A. Ustione and D. W. Piston, Nat. Methods, 2016, 13, 557–562 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2024 |