
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceBoron elemental and isotopic determination via the BF diatomic molecule using high-resolution continuum source graphite furnace molecular absorption spectrometry†
Maite
Aramendía
 ,
André L. M.
de Souza
,
Flávio V.
Nakadi
and
Martín
Resano
*
,
André L. M.
de Souza
,
Flávio V.
Nakadi
and
Martín
Resano
*
Department of Analytical Chemistry, Aragón Institute of Engineering Research (I3A), University of Zaragoza, Pedro Cerbuna 12, 50009 Zaragoza, Spain. E-mail: maiteam@unizar.es; andrelms@unizar.es; fvnakadi@unizar.es; mresano@unizar.es
First published on 24th January 2024
Abstract
Boron trace determination in biological materials is needed in different fields of application. Direct B determination by means of Graphite Furnace Atomic Absorption Spectrometry (SS-GFAAS) has been used in the past for this purpose, offering good detection limits hardly achievable by other techniques. However, such methods require the use of high atomization temperatures combined with large integration times to promote B atomization, which dramatically reduces the lifetime of the instrument's graphite parts. In this work, a new perspective for B determination by means of Graphite Furnace Molecular Absorption Spectrometry (GFMAS) is proposed. B was detected as the diatomic molecule BF (boron monofluoride), deploying a gas phase reaction with CH3F as fluorinating agent. Based on this strategy, a method for the direct determination of B in two biological certified reference materials (NIST SRM 1570a spinach leaves and NIST SRM 1573a tomato leaves) has been developed, providing similar detection capabilities to the GFAAS method (LOD of 0.24 ng) but requiring much milder furnace conditions. Moreover, the appearance of memory effects, very common in GFAAS methods, is also avoided with this method. Straightforward calibration with aqueous standard solutions was also found to be possible. To this end, a mixture of W (permanent), citric acid, and Ca as chemical modifiers was found to be essential for obtaining a reproducible and sufficiently sensitive signal for boron solutions, comparable to the signals obtained for the solid samples. With this method, accurate results were obtained for the direct analysis of both certified reference materials, provided that spectral interferences from the PO molecule were properly corrected. Precision values in the range of 15% RSD, as typically reported for direct solid sampling GFAAS, were found. Finally, and as an additional advantage of the GFMAS method, a large isotopic shift in the absorbance of the 10BF and 11BF molecules can be accurately monitored at a secondary transition for the BF molecule. This offers novel analytical possibilities for the method, which are also explored in this study. In this regard, control of the B concentration was found to be critical for obtaining accurate and precise isotope ratios for this element.
1. Introduction
Boron and boron compounds are extremely valuable in present-day societies, with many applications in different fields. Just to name a few examples, boron is used as a thermalizing agent in nuclear reactor materials1 or as a dopant in silicon wafers in the semiconductor industry.2 Boron compounds, on the other hand, are used for producing alloy fuels for propellants,3 ceramic structures such as borosilicate glass,4 dentistry materials,5 and synthetic herbicides and fertilizers, considering the key role that this element plays on plant growing and development.6 There are also other applications, yet to be fully exploited, such as the Boron Neutron Capture Therapy (BNCT),7 proposed for cancer treatment, where tumors are selectively destroyed by irradiation with slow neutrons after 10B administration to diseased tissues, or the production of boron-based semiconductors8 as an alternative to silicon-based materials.As a natural consequence of the above, analytical methods that are capable to perform B elemental and isotopic determinations at different concentration levels and in all sorts of samples for different purposes (e.g., industrial production control, food safety, BCNT-therapy administration, etc.) are needed. Among the existing analytical methods for B determination, reviewed elsewere,9,10 Inductively Coupled Plasma-Mass Spectrometry (ICP-MS) methods seem to be the most advantageous, providing the best limits of detection (LODs), and offering both elemental and isotopic information even at trace levels. However, when the target sample is solid, regardless of the analytical technique used, sample decomposition before analysis still remains a challenge for this element, worsening the LODs achievable. In fact, boron and its compounds are ubiquitous in nature, and some are rather volatile too, and hence, sample preparation is subjected to elevated risks of contamination and/or losses.11
One interesting way to address this issue is the use of direct methods of analysis, overcoming the necessity of sample pretreatment. In this regard, methods based on the use of the graphite furnace (i.e. Solid Sampling Electrothermal Vaporization-ICP-MS, SS-ETV-ICP-MS, and SS-Graphite Furnace-Atomic Absorption Spectrometry, SS-GFAAS), have proven useful in the past for B trace determination in biological solid samples.12,13 The use of ETV-ICP-MS, on the one hand, provides both elemental and isotopic information with low LODs. Unfortunately, and although it presents excellent analytical features,14 the use of this technique is at present very limited due to the lack of ETV instruments commercially available.
GF-Atomic Absorption methods, on the other hand, even though less sensitive in general terms, are still an interesting alternative to MS methods with lower running costs and renewed possibilities since the introduction of High-Resolution Continuum Source (HR CS) instruments.15,16 Among other features, these include the possibility to determine non-metals and to obtain isotopic information through the formation of transient molecules in the graphite furnace (most often diatomic) and the measurement of their absorption (HR CS GFMAS).17,18 This possibility could be exploited for B elemental and isotopic determinations, as a means to overcome the disadvantages related to GF-AAS methods for this element. And such disadvantages are not minor.
In fact, and as reviewed elsewhere,13 boron behavior in the graphite furnace is complicated when the goal is to promote the formation of B atoms and the measurement of their absorption. On the one hand, B easily forms volatile oxides that can be lost during the pyrolysis steps even at relatively low temperatures (600–900 °C) and are difficult to atomize at the temperatures of the GF, leading to low sensitivity and inaccuracies.19 On the other hand, B tends to form very refractory carbides, which makes necessary the use of high temperatures and long times for the atomization step. This fact reduces dramatically the lifetime of the graphite consumables, and still results in broad signals with a long tailing and important memory effects.20,21 To address these issues, a cocktail of chemical modifiers has been proposed in the literature with a double goal: (i) reducing the partial pressure of oxygen in the graphite furnace, thus hampering formation of boron oxides, and (ii) minimizing the formation of boron carbides. Still, sensitivity for B in GFAAS methods is poor (typical characteristic masses in the 0.2–0.3 ng range13) and the use of very harsh temperature programs is compulsory, resulting in premature degradation of the graphite parts. Additionally, the use of fluorinating agents for cleaning in between measurements seems mandatory for reducing memory effects, which also increases the total analysis time.21,22
This paper explores an alternative and potentially more advantageous method for B determination by means of HR CS GFMAS, based, precisely, in the mechanism of action of such fluorinating reagents typically added for cleaning purposes: the high tendency of B to form volatile fluoride species. This idea was previously exploited for B determination in ETV-ICP-MS,12 although with a critical difference, as only vaporization is needed in the ETV while ionization is carried out in the ICP-MS. For the HR CS GFMAS case, in contrast, the molecule formed must be stable for a certain amount of time in the graphite furnace such that its absorption can be monitored, which, as will be shown in this work, is not straightforward.
Promotion and measurement of the absorption of a boron fluoride molecular species would potentially have the advantage of softening the temperature program needed for analysis and reducing memory effects, thus circumventing the need for additional cleaning steps. Moreover, and as an additional advantage, the possibility to perform isotopic determinations would be also enhanced. In fact, it is well known that isotopic shifts in molecular spectra are much broader than for atomic ones, which is well documented for diatomic transient molecules.23 In the case of boron, for instance, and due to the relative wide mass differences of the two isotopes, small isotope effects have been observed in the atomic spectra (e.g. 2.5 pm displacement at the 208.9 nm maximum).24 These displacements are, however, difficult to monitor, leading to poor accuracy and precision values for the isotope ratio measurements. Such isotopic displacements have been used, for instance, for monitoring the isotope composition of alloyed steel reference materials by HR CS Flame AAS and were useful for identifying 10B enriched scrap, for which less rigorous requirements have to be met in terms of precision and accuracy.24 In contrast, isotopic displacements can reach tens to hundreds of picometers when monitoring diatomic molecules, depending on the transition monitored, such that measurement is much easier with the resolution available for HR CS GFAAS instruments. This idea, first applied to determine Cl isotope ratios by formation and measurement of the molecular absorption of the AlCl molecule,25 has already been tested for boron isotopes by formation of the BH molecule.22 However, and in spite of the good precision and accuracy values obtained with this method, the minimum B concentration needed for the method to be applied is 1 g L−1 on clean boric acid solutions, while intermediate cleaning steps are still needed for addressing memory effects. There is, hence, room for improvement also in this regard.
It is the goal of this work to develop a new analytical method for trace boron elemental and isotopic determinations in solid samples, based on the formation of the BF molecule and the monitoring of its absorption by HR CS GFMAS. Such method should allow for a more efficient analysis than methods based on boron atomic absorption spectrometry, reducing costs and total analysis time, and providing additional isotopic information. For proofing the concept, two botanical reference materials were targeted, considering the pivotal role that B has on plant growth and development. All features of the method, such as the formation of the molecule, calibration and overcoming of matrix effects, or evaluation of the possibilities for isotopic analysis, have been systematically considered and will be discussed in this paper.
2. Experimental
2.1. Instrumentation
All absorption measurements were carried out using a HR CS atomic absorption spectrometer ContrAA 800 G (Analytik Jena AG, Germany). This instrument is equipped with a transversally heated graphite tube atomizer and both automated solid sampling and liquid sampling accessories (Analytik Jena AG). Pyrolytic graphite tubes and platforms were used throughout the study.Moreover, an inductively coupled plasma mass spectrometer NexION 300X (PerkinElmer, USA) was used to determine the boron isotopic composition of the standards that were subjected to analysis by HR CS GFMAS. The ICP-MS parameters used for this purpose were: 15 L min−1 plasma Ar gas flow, 1.1 L min−1 nebulizer Ar gas flow, 1.2 L min−1 auxiliary Ar gas flow and 1450 W RF power. The nuclides monitored were 10B+ and 11B+ and the dwell time for each nuclide was 50 ms.
2.2. Reagents and materials
All reagents used in the work were of analytical purity grade. Deionized water (18 MΩ cm) obtained from a Milli-Q water system (Millipore, France) was used for dilutions throughout the work. A 1000 mg L−1 B elemental standard (boric acid in water, Merck, Germany) was used as stock solution to prepare natural isotope composition boron solutions. For B isotope measurements, two isotopic reference standard solutions (boric acid in water) enriched in 10B and 11B, respectively, were obtained from Inorganic Ventures (USA). The concentration of the standards was approximately 10 mg L−1 (10.157 ± 0.080 mg L−1 for the one enriched in 10B, and 10.516 ± 0.083 mg L−1 for the one enriched in 11B) and their isotopic composition was 0.9954 10B mole fraction and 0.9990 11B mole fraction, respectively. A boron isotopic standard of 0.97 11B/10B theoretical ratio was prepared by mixing appropriate amounts of these isotopic standards.For preparation of the chemical modifiers tested, 1000 mg L−1 mono elemental standard solutions of Ca (as nitrate) and W (as ammonium tungstate (VI)) were obtained from Merck. A 15 g L−1 citric acid solution was prepared by dissolving the solid reagent (Merck) in deionized water.
For acquisition of the model spectra for correction of the spectral interference from the PO molecule, a 1000 mg L−1 mono elemental P standard solution (H3PO4 in water) was purchased from Merck.
As fluorinating agents, two options were tested and are compared in the work. The first option consisted of the addition of NH4F·HF in aqueous solution. A 10% m/m solution was prepared by diluting the solid in deionized water (99.9% ammonium bifluoride, Sigma Aldrich, USA). As a second option, a 1% v/v methyl fluoride–argon mixture (Nippon Gases, Spain), which will be denoted as Ar/CH3F throughout the manuscript, was added in gas phase during the graphite furnace program (described in Section 2.3).
Two biological standard reference materials (SRMs) were analyzed for optimization of the analytical method and accuracy verification: spinach leaves 1570a and tomato leaves 1573a, both from the National Institute of Standards and Technology (NIST, USA).
2.3. Measurement protocol for elemental and isotopic determinations
Boron and boron isotopes were determined through formation of the BF molecule and monitorization of its absorbance. In particular, two vibronic transitions within the X1Σ → A1Π electronic transition were monitored for this molecule,26 and each one of them was used either for the elemental determination of B or for the determination of B isotopes.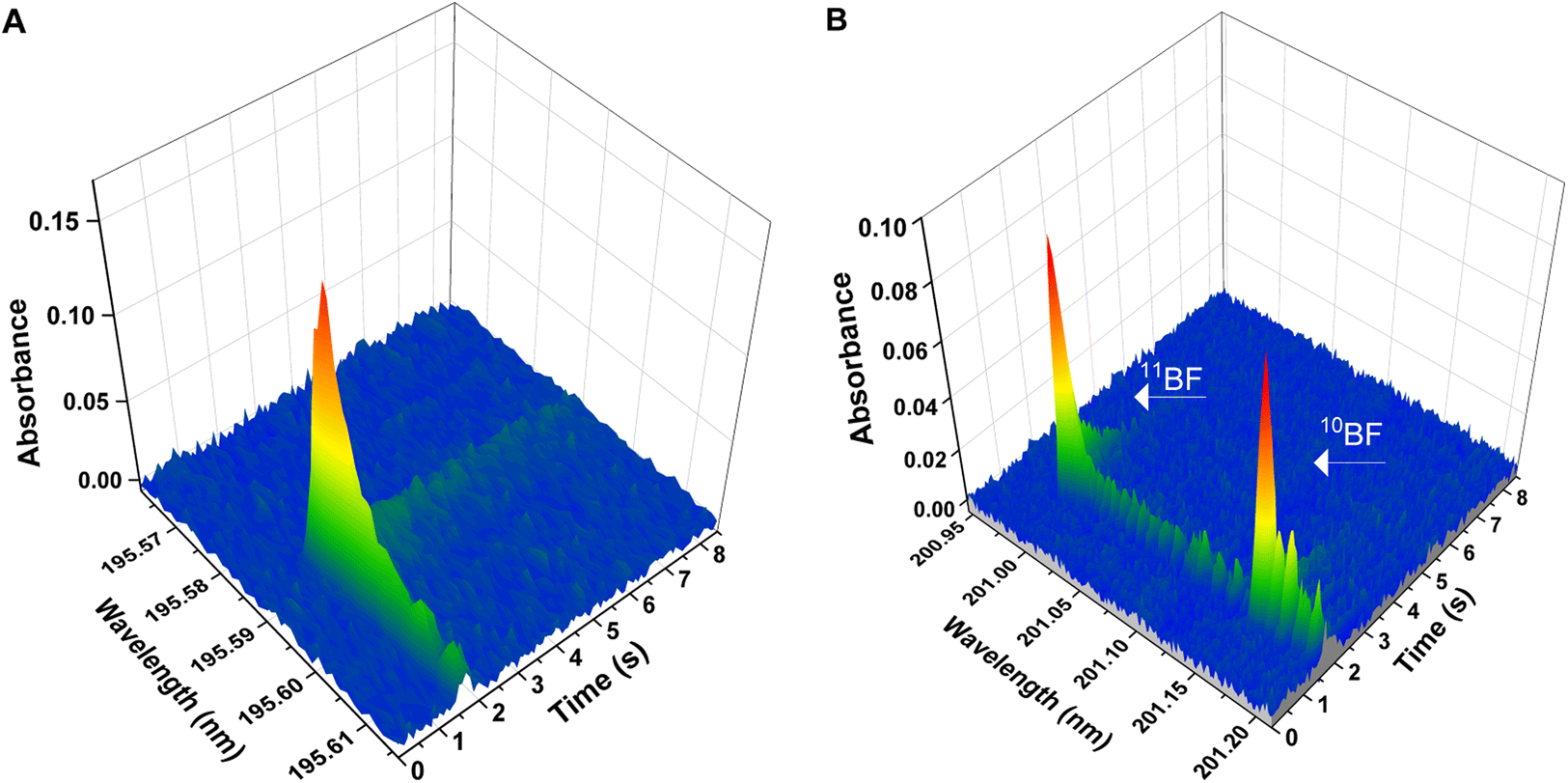 | ||
| Fig. 1 Wavelength- and time-resolved spectra for the two different BF transitions deployed in the work. Spectra were obtained with aqueous standard solutions and deploying the temperature program presented on Table 1 (chemical modifiers: citric acid 150 μg, Ca 10 μg, Wpermanent 250 μg). (A) Spectrum for the X1Σ → A1Π (0,0) transition generated using 10 ng of B elemental standard of natural isotopic composition. The central pixel was adjusted at 195.589 nm (detection pixel). (B) Spectrum for the X1Σ-A1Π (1,0) transition, showing two separated peaks for 10BF and 11BF. Spectrum generated using two isotopic B standards (approximately 50 ng of each boron isotope). The central pixel was set at 201.08 nm; 11BF maximum peak is found at 200.993 nm, and 10BF maximum at 201.160 nm. | ||
Solid samples were directly analyzed by HR CS GFMAS without any prior preparation step. The solid sampling device used allows for automatic weighing and transport of the samples, once deposited on the graphite platform, to the furnace. The empty platform (previously modified with 250 μg of tungsten as permanent modifier, see Section 3.2 for more details), was first transported to the built-in balance with a pair of tweezers for taring. Next, an appropriate amount of sample was loaded on the platform and weighed. The corresponding amount of liquid modifier (10 μL of a 15 mg L−1 citric acid solution and 10 μL of a 1 g L−1 Ca solution) was added afterwards. Finally, the platform was transferred into the graphite furnace and subsequently subjected to the temperature program. All these operations were fully controlled from the instrument software, except for the deposition of the sample and modifiers on the platform, which was carried out manually. The operating conditions are summarized in Table 1.
| Electronic transition | X1Σ-A1Π |
|---|---|
| Wavelengths | 195.589 nm (total BF) |
| 200.993 nm (11BF) | |
| 201.160 nm (10BF) | |
| Number of detection pixels summed per line | 3 in area (total B determination) |
| 1 in height (11BF/10BF isotope ratio determination) | |
| Pixels for baseline setting (isotope ratio determination only) | 32 and 89 (11BF); 149 and 174 (10BF) |
| Chemical modifiers in aqueous solution | Citric acid: 10 μL, 15 g L−1 |
| Ca: 10 μL, 1000 mg L−1 | |
| Permanent modifier | W: 250 μL, 1000 mg L−1 |
| Aqueous sample volume (isotope ratio determination) | 10 μL |
| Solid sample mass (total B determination) | 0.2–0.4 mg |
| Measurement time | 8.5 s |
| Measurement delay | 4.5 s |
| Spectral interferences correction | TAP (NIST 1570a) |
| None (NIST 1573a) |
| Temperature program | |||||
|---|---|---|---|---|---|
| Step | Temperature (°C) | Ramp (°C s−1) | Hold (s) | Gas flow (L min−1) | |
| Ar | Ar/CH3F | ||||
| Drying | 80 | 6 | 20 | 2.0 | 0 |
| Drying | 110 | 5 | 40 | 2.0 | 0 |
| Pyrolysis | 850 | 300 | 20 | 2.0 | 0 |
| Gas adaptation | 850 | 0 | 10 | 0 | 0.5 |
| Vaporization | 2500 | 1800 | 7 | 0 | 0 |
| Cleaning | 2700 | 3000 | 5 | 2.0 | 0 |
The calibration was performed using 10 μL of a B aqueous solution of the appropriate concentration, added with a micropipette onto the sampling platform instead of the solid sample. Five points plus the blank were used to build a calibration curve between 5 and 25 ng B. Each point of the calibration curve was measured in quintuplicate.
Seven replicate measurements were carried out for each solid sample analysis. For analysis of NIST 1570a spinach leaves, the TAP correction method, was used for obtaining accurate results in accordance to the NIST certificate. This method, described elsewere,27 is based on the fact that the time-absorbance profile (TAP) of any species, measured under the same instrumental conditions using a GF as the atomizer, should be the same at every wavelength. Therefore, using a TAP normalized spectrum of the interfering species should be sufficient to subtract it from the normalized absorbance of the atomic line, leaving only the analytical signal of the analyte. For the calculation of the wet content, the approach recommended by NIST was used. An amount (approximately 0.2 g) of every sample was weighed and dried in a desiccator at room temperature (22 °C) for 120 h and the loss of water was quantified by differential weighing (0.09% for 1570a spinach leaves and 0.07% for 1573a tomato leaves moisture contents, respectively). The moisture content was taken into account for the final calculation of the HR CS GFMAS results, which were reported on a dry-mass basis.
3. Results and discussion
3.1. Target molecule formation and wavelength selection
As discussed in the introduction, the goal of the work was to explore the formation of a volatile boron fluoride molecule that could soften the temperature program needed for boron determination in the graphite furnace, while also enabling the measurement of boron isotope ratios. For this purpose, and as in most HR CS GFMAS works, the formation of a diatomic molecule (BF in the current case) was targeted. During element atomization processes at high temperatures (such as those occurring in a graphite furnace), atomic recombination processes occur that result in the formation of diatomic transient molecules. Many spectral characteristics of such molecules, including isotopic effects, are well documented in the literature,23,26 which can serve as a starting point for program optimization. According to the data available, we selected the X1Σ → A1Π (0,0) vibronic transition at 195.74 nm26 for optimizing the formation method for the BF molecule, which, at least in theory, should be the most sensitive transition available and measurable with our spectrometer.The BF molecule shows a high bond dissociation energy (732 kJ mol−1),28 so that, once formed, it should be fairly stable in the graphite furnace, which favors sensitivity for absorption measurements. Besides, and considering that fluorine is monoisotopic, the potential isotopic shift to be monitored for the BF molecule should only obey to B isotopes, which is an additional advantage. However, and as already hinted at in the introduction, formation of such molecule in the graphite furnace in the most conventional way, i.e., starting from the dissolved reagents, is challenging.
In fact, and as has been shown with ETV-ICP-MS measurements,12 B tends to form BF3 in the presence of dissolved fluorinating agents (such as NH4F·HF or NaF), a stable but extremely volatile molecule (boiling point −101 °C),29 which would be lost well before the BF diatomic molecule could be formed in the furnace after elemental atomization-recombination. This is why fluorinating agents in solution have mostly been used in GF-AAS methods for B determination as cleaning agents in-between sample runs, in order to cope with the memory effects caused by the formation of refractory boron carbides in the furnace.21 It is not surprising then that our attempts to promote the BF molecule by using fluorinating agents in solution were unsuccessful. In particular, we tested ammonium bifluoride solutions because of its relatively high boiling point (240 °C),29 which would result in a lower loss of the forming reagent during the drying step. For these experiments, a considerable high amount of B (10 μL of a 1000 mg L−1 B standard solution) was introduced into the graphite furnace to maximize the probability to obtain an analytical signal. Additionally, 10 μL of a 10% m/m NH4F·HF solution were pipetted and placed on the platform together with the B standard. The graphite furnace was programmed with a low pyrolysis temperature (150 °C) and a relatively slow heating ramp (300 °C s−1) up to 2700 °C, the maximum temperature achievable by the instrument. This strategy allowed us to scan a broad range of vaporization temperatures, although no signal for the BF molecule could be recorded at any of the temperatures tested.
Next, we tried an alternative formation protocol with a fluorinating agent in the gas phase (a 1% v/v CH3F in Ar mixture) added before the atomization step. This strategy had provided good results in a previous work of the group for promoting the SrF molecule and performing Sr isotope ratio determinations, although this approach also requires careful optimization.30 Briefly, the protocol for fluorination in the gas phase consists of filling the graphite tube with the reactive gas (Ar/CH3F in this case) at the end of the pyrolysis step, then stopping the inner gas flow in the furnace for reducing the diffusion rate of gases out of the furnace and subsequently heating the system to vaporize/atomize the analyte in an atmosphere rich in the reactive gas, where the target fluoride molecule can be formed. For the particular case of boron, the presence of the fluorinating agent in the gas phase during this step should promote B vaporization from the sampling platform at a lower temperature than in its absence (probably as a fluoride species), even if very refractory carbides would have been formed. This has already been shown in other works for coping with memory effects with carbide-forming elements in the graphite furnace.22,31,32 On the other hand, the high temperatures at which this vaporization occurs should permit the rapid atomization of both the analyte and the forming reagent, that would then be able to recombine and form the BF molecule.
To test if these assumptions were true, a preliminary temperature program based on the results shown in ref. 30 was tested. Again, 10 μL of a 1000 mg L−1 B standard solution were introduced in the graphite furnace, to maximize the probability to obtain an analytical signal. After drying, a pyrolysis step at low temperature (150 °C) was programmed, at the end of which the graphite furnace was filled with the Ar/CH3F mixture, introduced at a rate of 0.5 L min−1 for 6 seconds. Next, the flow was stopped, the temperature was raised to 2700 °C (at a fast rate of 3000 °C s−1), and the absorption in the range of 195.74 nm was monitored to record the X1Σ → A1Π (0,0) vibronic transition of BF. With this strategy, an absorption band with its maximum at 195.5897 nm could be finally observed (see Fig. 1A). As a result, the protocol for fluorination in the gas phase was the only considered for all further experiments.
3.2. Optimization of the furnace program for direct B determination in solid samples. Temperatures and chemical modifiers
To this end, optimization experiments were conducted with a much lower amount of B, (10 ng of B) as it was clear from first experiments that the sensitivity achievable by monitoring the BF molecule in the 195 nm region was not far from that reported for the monitoring of the main atomic line for B at 249.8 nm. Taking into consideration our previous experience with this element, a mixture of W as permanent modifier and citric acid in solution was used. The exact mechanism by which this combination of modifiers acts is unclear (different hypothesis exist), but it offered the best results for reducing signal tailing and improving sensitivity in B determination by GFAAS when compared to other approaches and was then also implemented in this work.13
The method optimized in ref. 13 was used with some adaptations, which mostly concerned W deposition. Soaking the graphite parts in a W solution before use has been described as the most efficient way to deposit this permanent modifier. However, this approach gave problems for the well-functioning of the ContrAA 800 G instrument (different error messages were displayed, as the conductivity of the graphite parts seems to be affected) and could not be used. Instead, only the graphite platforms were covered with W before use, following the protocol described by Lima et al.33 This method consists of four sequential depositions of 50 μL of a 1 g L−1 W solution onto the graphite platform (250 μg W, in total), followed by a furnace program with different steps at different temperatures. With this protocol, no error messages were given by the spectrometer. A detailed description of this method is included in Table S1 of the ESI.† Additionally, 10 μL of a 15 g L−1 citric acid aqueous solution were also added at the beginning of the temperature program.
Pyrolysis and vaporization curves were first carried out and are shown in Fig. 2 (red trace). For this experiment, each temperature point was obtained as the average of 5 consecutive measurements. First, the vaporization temperature was optimized using a low pyrolysis temperature at which no B losses should occur (150 °C). Well defined signals with almost no tailing could be obtained at temperatures as low as 2200 °C, which represents an important advantage in terms of thermal preservation for the graphite parts if compared to the program needed for measuring B atomic absorption, where long atomization steps (20 s) at very high temperatures are mandatory. This is illustrated in Fig. 3. For BF vaporization, possible from 2200 °C (although Fig. 3B was obtained at 2500 °C), the signal returned to the baseline in less than 4 seconds, while for B atomization (Fig. 3A) 15 s–20 s at 2600 °C were needed for the complete atomization of B. As typically observed for this element in GFAAS, higher vaporization temperatures also lead to higher sensitivities for BF, which seems to indicate that B pre-atomization is needed for the efficient formation of the molecule. For further experiments however, and in order to preserve the graphite parts from thermal degradation, we selected 2500 °C as a compromise vaporization temperature, which provided intermediate integrated absorbance values with good precision (repeatability of 5 consecutive measurements was typically of about 5% RSD).
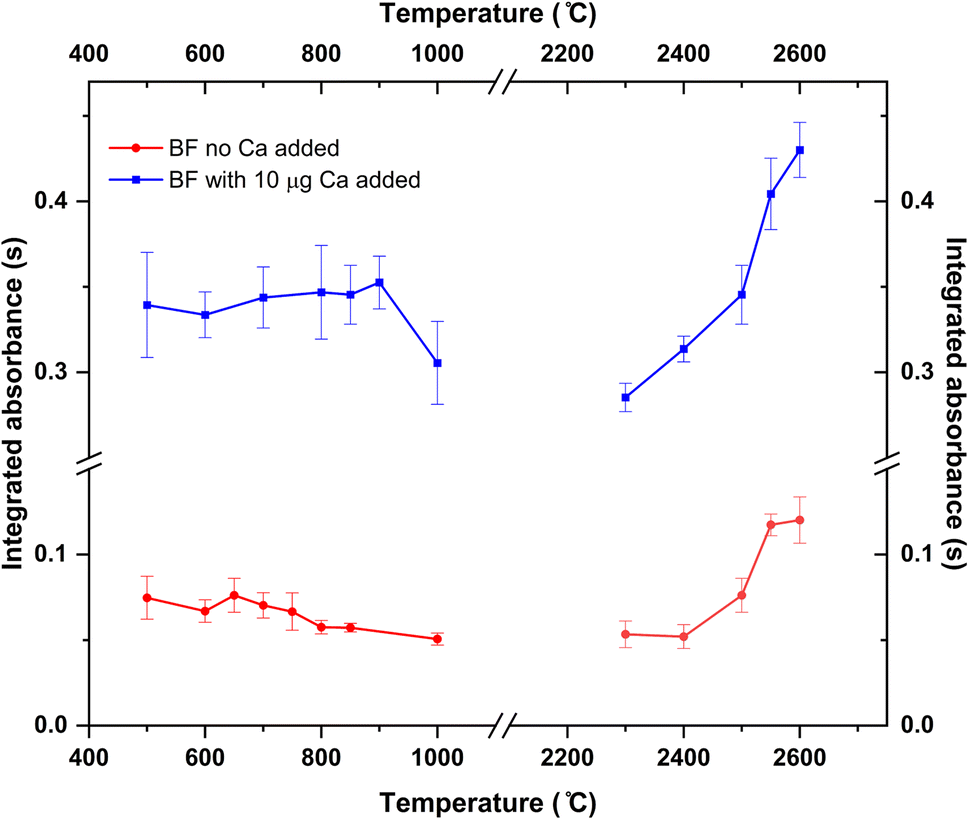 | ||
| Fig. 2 Pyrolysis and vaporization curves for the formation and monitoring of the BF molecule obtained for aqueous standard solutions with (blue trace) and without (red trace) the addition of 10 μg Ca as modifier. 10 ng of B were used in all cases. Error bars represent one standard deviation of five consecutive replicates. | ||
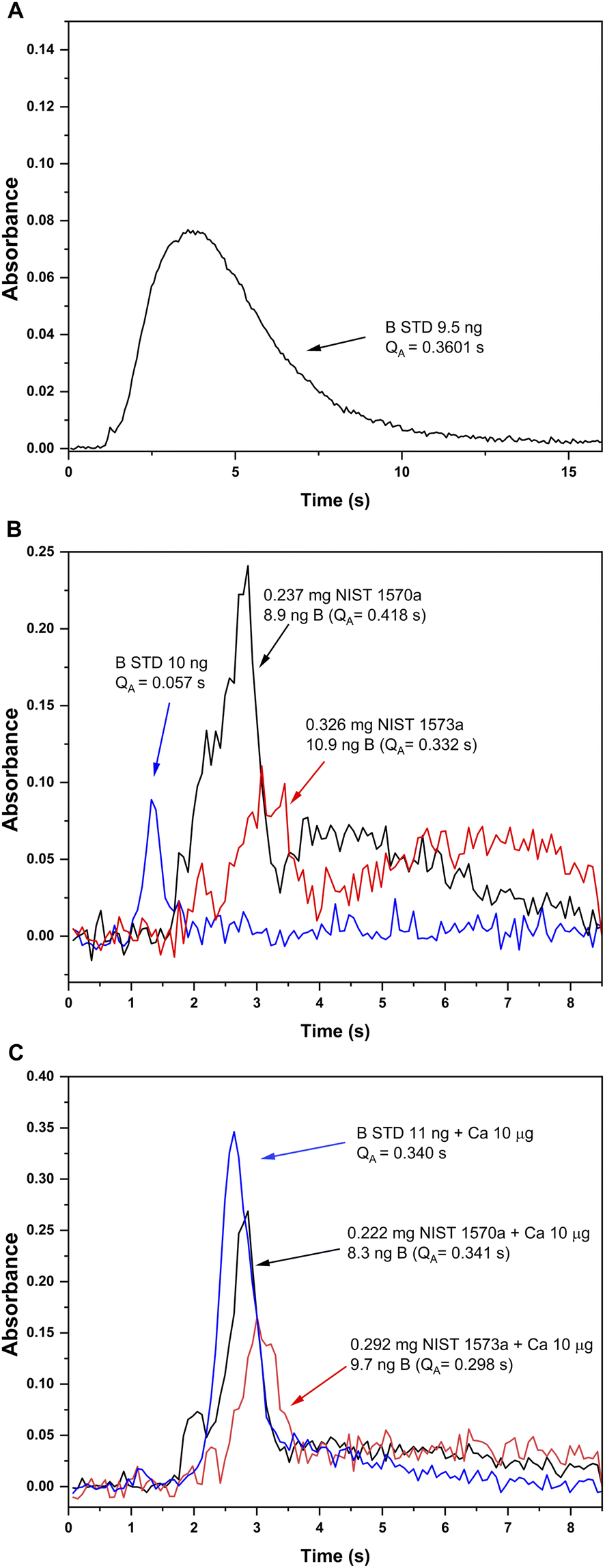 | ||
| Fig. 3 Absorbance-time profiles obtained for B aqueous standard solutions and solid samples under different furnace conditions. (A) Atomic absorption for a B aqueous standard solution monitored at 249.8 nm (CP ± 1) with the method developed in ref. 13 (chemical modifiers: W-permanent, and citric acid; pyrolysis 1200 °C, atomization 2600 °C). (B) and (C) Absorption profiles for the BF molecule monitored at 195.5897 nm (CP ± 1) and vaporized from a B aqueous standard solution (blue), solid NIST1570a spinach leaves (red) and solid NIST 1573a tomato leaves (black). Measurement conditions for (B) and (C) are those presented on Table 1 except for the addition of 10 μg Ca as modifier, which was only carried out for measurements presented in (C). | ||
As for the pyrolysis temperature, and as seen from Fig. 2, temperatures up to 900 °C could be used without significant analyte losses, which is a much lower temperature than that reported in the literature for the W + citric acid mixture (2100 °C). This maximum pyrolysis temperature reduction could be related to the procedure used for W deposition.13 In any case, and concerning matrix elimination, a pyrolysis at 800–900 °C should be sufficient for eliminating most organic components of the matrix, although it might not be enough for certain inorganic components. Self-evidently, this is something that should be checked for each particular sample under investigation.
The same operation conditions that were found to be optimum for B standard solutions in the previous sections were applied to the direct analysis of the solid samples, introducing around 0.3 mg of the sample in the graphite platform (corresponding to about 10 ng B). First experiments with NIST SRM 1570a proved that the pyrolysis temperature used (850 °C) was enough to eliminate the matrix, as no residues were observed at the end of the program. B interaction with the different modifiers seemed to be also achieved with no further measures, as promotion of the BF molecule was observed. However, three important issues were identified.
(i) In the first place, the absorbance-time profile obtained for the solids was bimodal, with a first peak appearing slightly delayed if compared to that observed for the aqueous standard solution (maximum at about 3 s) and a second peak appearing even later (maximum at about 4.5 s). This is shown in Fig. 3B.
(ii) Next, the sensitivity obtained for BF in the solids was clearly higher (about 5 times, see Fig. 3B) than that obtained for the aqueous standard solution. Self-evidently, this would lead to results biased high for both reference materials if external calibration with aqueous standard solutions is performed, which is always the intention for a straightforward procedure.
(iii) Finally, the spectrum obtained for the solid samples showed additional lines around the BF transition at 195.59 nm, corresponding to additional molecules and/or atoms coming from the sample matrices. This is shown in Fig. 4 for the two solid sample materials. The software interference library of the instrument suggested PO as potential interference in both samples, as well as the presence of two secondary atomic lines for Fe at around 195.6 nm and 196.7 nm, respectively. These additional lines did not seem to interfere with the main BF transition for NIST SRM1573a, although for NIST SRM 1570a a clear interference was observed, as will be commented below.
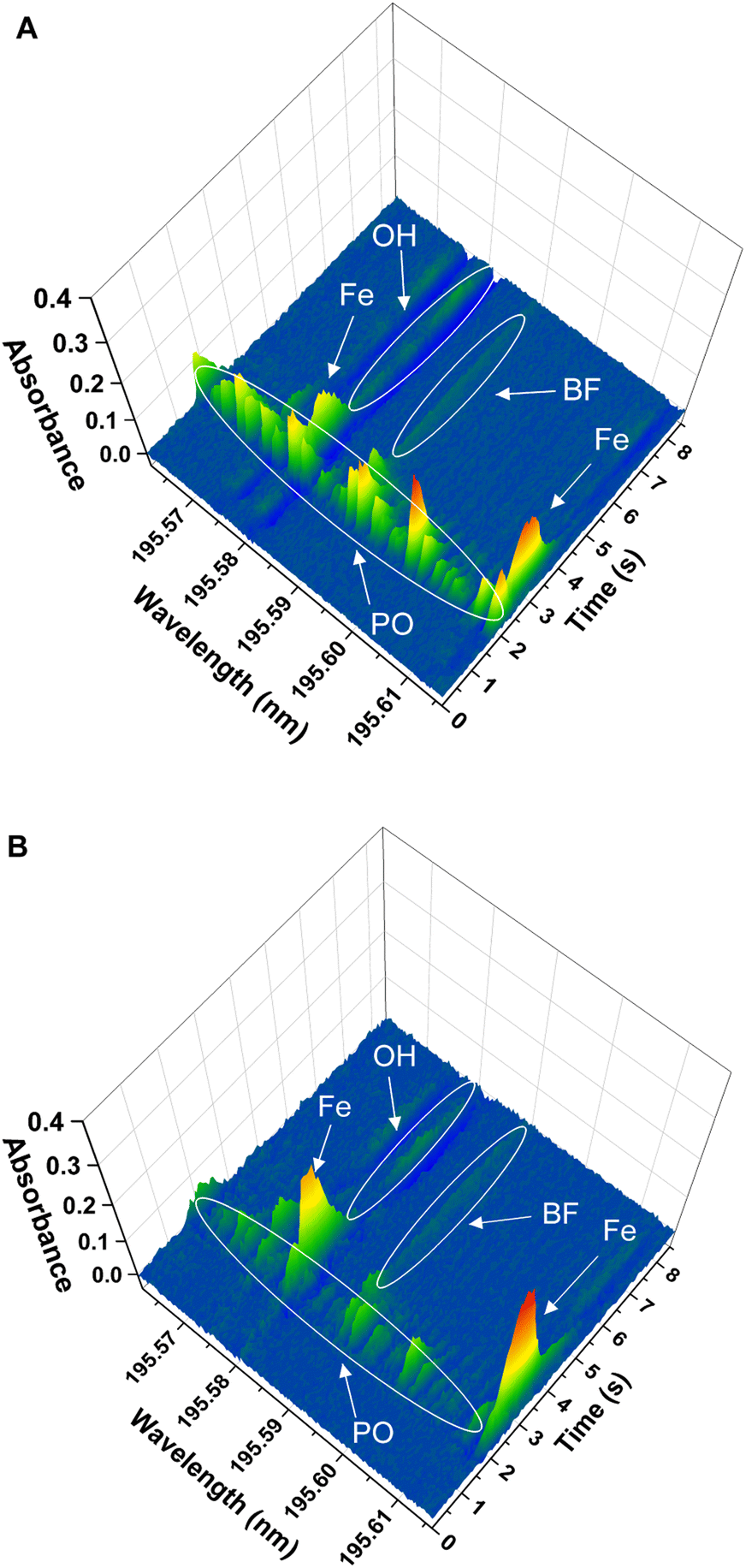 | ||
| Fig. 4 Spectra in the 195.59 nm region obtained for the direct analysis of the two solid samples analyzed, obtained using the furnace program presented in Table 1. Peak identification included in the figures was performed by comparison with model spectra from the literature and further experimental confirmation with P and Fe standards. (A) Spectrum obtained for analysis of 0.222 mg of the NIST SRM 1570a spinach leaves. (B) Spectrum obtained for analysis of 0.292 mg of the NIST SRM 1573a tomato leaves. | ||
Therefore, additional measures had to be developed in order to obtain accurate results for the direct analysis of the solid samples, particularly regarding the second issue. For this purpose, the certified composition of the target samples was examined, revealing a high Ca content in both of them (1.527% and 5.05% mass fraction, respectively, according to the corresponding certificates of analysis). Calcium salts (nitrates and/or chlorides) have been proposed in the literature as chemical modifiers for B determination with GFAAS.20,34 Again, the mechanism of action for these modifiers is unclear from existing literature, although it has been suggested that it might be related to the chemical stabilization of B as refractory borides and/or borates.13,29 Thus, it was hypothesized that the natural presence of Ca in the solid samples could be behind the higher sensitivity obtained for the solid samples, and addition of Ca as chemical modifier was next studied.
To this end, 10 μL of a 1 g L−1 Ca solution (10 μg Ca) were added together with the citric acid solution at the beginning of the furnace program. Ca addition to the B aqueous standard solutions resulted in broader peaks for the absorbance-time profile of the BF transition, also slightly delayed if compared to the peaks obtained without Ca as shown in Fig. 3C. Most importantly, the addition of this modifier seemed to correct for the difference in sensitivity observed for the aqueous standard solution and the solid samples. Pyrolysis and vaporization curves for B aqueous standard solutions with the addition of Ca were constructed next and are also shown in Fig. 2 (blue trace). As seen from this figure, the addition of Ca did not significantly change the general trends already observed in the absence of Ca in terms of the optimum operation temperatures recommended, although it provides roughly a five-fold increase in sensitivity.
The addition of the Ca modifier also affected the absorbance-time profile obtained for the solid samples, although the sensitivity was not affected significantly. As shown in Fig. 3C, even though not exactly coincident, the time profiles obtained for the standard and the solid samples with the addition of 10 μg Ca are much more similar, and integrated absorbances are comparable. As a consequence, the addition of this modifier was implemented in the analytical procedure. The exact mechanism by which Ca addition produces these effects still needs to be elucidated. It seems feasible that the Ca naturally present in the solid samples aids stabilizing boron before atomization, preventing the formation of other species such as oxides and/or carbides. However, it is clear from our experiments that the Ca added externally has an additional effect on the mechanism of BF promotion. There are many potential explanations (e.g., fluorine stabilization by Ca leading to a more efficient fluorine atomization) but, in any case, specific experiments should be conducted to clarify this issue. This, however, was out of the scope of this work and such study was not carried out.
To continue with program optimization, the fact that the absorbance–time profiles obtained for the solids were broader was easily solved by simply enlarging the duration of the vaporization step up to 8.5 s, to allow the signal to practically return to the baseline. This measure would affect the lifetime of the graphite parts, although to a much lower extent than the time and temperature needed for measuring B atomic absorption (15–20 s at 2600 °C).
Finally, the effect of the potential PO interference on the BF signal obtained for the solid samples was investigated. To confirm the identity of the PO interference, a model PO spectrum was experimentally obtained. To this end, 10 μL of a 10 mg L−1 P standard solution were introduced in the graphite furnace and were subjected to the same furnace program used for BF monitoring (see Table 1), including the addition of the Ca modifier. Visual comparison of the spectrum obtained for the P standard with those obtained for the solid samples confirmed the identity of the PO molecule. As previously indicated and as shown in Fig. 4 and 5, this PO interference seemed to only significantly affect the BF transition at 195.59 nm for NIST 1570a (Fig. 4A and 5A), but not that for NIST 1573a (Fig. 4B and 5B), which is consistent with the lower P amount present in the latter sample according to the corresponding certificates of analysis (0.518% and 0.216% mass fraction, respectively). Due to this interference, results biased high were obtained for NIST 1570a if no interference correction was carried out.
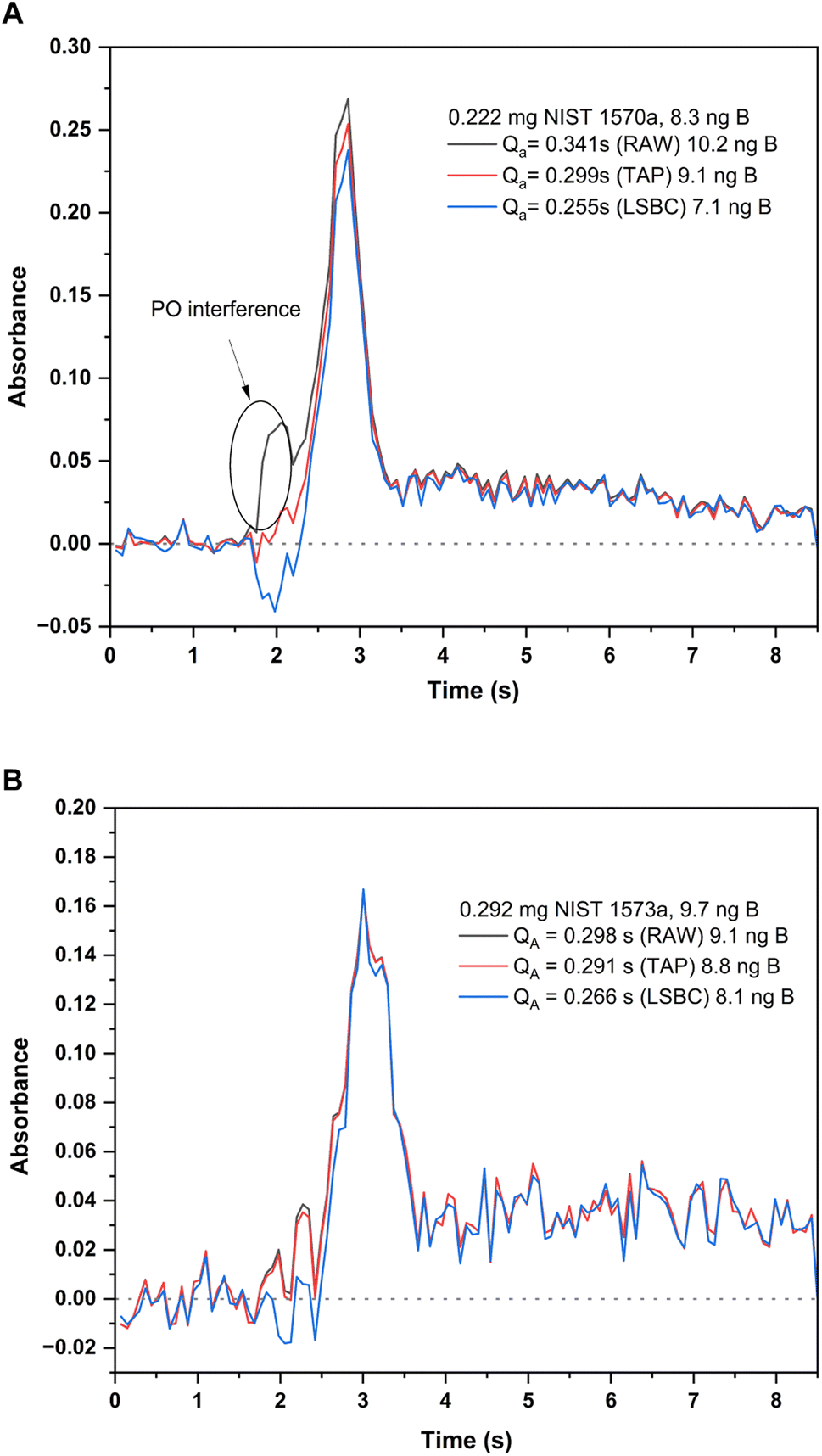 | ||
| Fig. 5 Absorbance-time profiles measured for the BF molecule at 195.5897 nm (CP ± 1) for the two solid samples analyzed and with different interference correction methods: original profile (black trace), TAP correction method (red trace) and LSBC (blue trace). Integrated absorbances and the corresponding B contents interpolated with each of these methods is indicated in the figure. Measurement conditions are those included in Table 1. (A) Profiles for the analysis of 0.222 mg of NIST SRM 1570a spinach leaves. (B) Profiles for the analysis of 0.292 mg of SRM 1573a tomato leaves. | ||
To tackle this issue, the Least Squares Background Correction (LSBC) method implemented in the instrument's software was first attempted. Application of this method, however, was not entirely successful. As described in the literature, one of the possible issues that hampers the successful application of LSBC is the difference in the absorbance–time profile for the reference when compared to that actually obtained for the sample.27 If this condition is not perfectly fulfilled, the interference correction might not work for all or some of the transitions recorded, and this was the case for NIST 1570a spinach leaves. This effect is shown in Fig. 5A. As seen from this figure, application of the LSBC method leads to a clear overcorrection of the interference in the BF transition monitored, therefore leading to results biased low for this sample with this correction method. To cope with this issue, the TAP correction algorithm developed in our lab was applied.27 The correction in this case was better achieved, leading to quantitative results in accordance with the NIST certificate of analysis for the 1570a material, as illustrated in Fig. 5A. As already discussed, the application of these algorithms did not change the results obtained for NIST 1573a, as the interference was not significant in this case, which is illustrated in Fig. 5B.
Finally, it is worth mentioning that the two Fe atomic lines monitored at 195.6 nm and 195.7 nm did not cause any overlap on the BF transition. The line at 195.6 nm is hardly usable as it overlaps with the intense OH absorption, which causes a clear overcorrection in this region of the spectrum (this can be observed in both Fig. 4A and B: the baseline signal falls below zero in the area where OH absorbs, which may be better appreciated when looking at the rear wavelength axes of such figures). However, the line at 195.7 nm could in principle be used for Fe and B simultaneous determination, if desired.
3.3. Direct solid sampling analysis for total B determination and method figures of merit
Analysis of the two reference materials targeted was finally carried out following the optimized program described in Section 2.3.1 and summarized in Table 1. A five-point calibration curve was measured at the beginning of each working session covering the range between 0 and 25 ng B, although linearity of the method was checked up to 50 ng. It is important to indicate at this point that, when higher B contents were introduced into the graphite tube the signal obtained showed a longer tailing, so that it did not return to the baseline within the 8.5 s programmed and linearity was lost. The use of a longer vaporization time could solve this problem, so that samples with higher B contents could be directly analyzed. Performing this study would certainly reduce the lifetime of the graphite consumables, so that we decided not to carry it out as we considered it to be out of the scope of the work.Next, analysis of both NIST samples was carried out, introducing a sample mass in the range 0.2–0.4 mg for each determination, ensuring measurements within the linearity range. Samples were analyzed without any previous treatment. Each sample measurement took about 7 min (considering everything, including sample weighing). Results for these determinations were finally corrected for the moisture content, (see Section 2.3.1. for details) and are included on Table 2. As seen from this table, results show a good agreement with the certified values. Precision in the range of 11–20% RSD was obtained for these determinations, which is typical for this technique and should be adequate for control laboratories, especially taking into account that sample pretreatment is avoided. For calculation of the limit of detection (LOD) and quantification (LOQ), also included in Table 2, 10 replicate measurements of the blank (10 μL of deionized water) were measured following the same working program described in Table 1. These absolute LOD and LOQ correspond to a B concentration in the solid sample of about 0.6 mg kg−1 and 1.8 mg kg−1, respectively (considering 0.4 mg of sample introduced in the furnace). These figures of merit can be considered as similar to those previously published for the measurement of boron atomic absorption,13 even if they are a little bit less competitive. However, the logistic advantages of this method cannot be disregarded. In the first place, addition of the fluorinating agent in gas phase as a forming agent has a clear effect on matrix digestion. This results in a more straightforward program, without the need for pre-digesting modifiers, as well as the elimination of memory effects, which avoids the need for cleaning steps in between measurements. Both effects simplify the protocol and reduce the total time required for analysis. Second, the temperature program required is milder (lower vaporization temperatures maintained for a shorter period of time), which reflects on a longer lifetime for the graphite parts. In this sense, it was estimated that the graphite platform and tube could be used without problems for the determination of at least 90 solid samples with this method. Finally, and as will be covered in the next section, the use of this method opens the possibility to perform isotopic analyses, so that a wider range of experiments, potentially providing information of better quality, could be developed (e.g., tracer experiments).
| Sample | Reference (mg kg−1) | SS HR GF MAS (mg kg−1) |
|---|---|---|
| NIST 1570a spinach leaves | 37.6 ± 1.0 | 35.6 ± 9.4 |
| Correction method: TAP | ||
| NIST 1573a tomato leaves | 33.3 ± 0.7 | 28.5 ± 6.8 |
| Correction method: None | ||
| Limit of detection | 0.6 mg kg−1 | |
| Limit of quantification | 1.8 mg kg−1 | |
3.4. Determination of boron isotopes
As previously indicated, one of the potential advantages of performing molecular absorption measurements (instead of atomic) is the fact that isotopic information becomes more accessible. In fact, the wavelength isotopic shifts observed for the absorption of diatomic molecules generated in the graphite furnace are expected to be much larger than those observed for atomic absorption spectra,18 to a point where it is possible to separately detect the absorption of different isotopes for certain transitions with modern, high-resolution continuum source spectrometers.22,25,30,35,36To explore this possibility, expected isotopic shifts for a given molecule can be calculated a priori with reasonable accuracy, as they respond to the theoretical equation proposed by Herzberg (originally available in page 162 of ref. 23; reproduced and commented in ref. 25 and on the ESI†) and the constants needed for calculations are available for many of these molecules. From this theoretical equation, it follows that transitions starting from higher vibrational levels will also show larger isotopic shifts. However, these transitions normally offer a lower sensitivity, since, at the temperatures reached by graphite atomizers, the fundamental level is still the most populated one. Therefore, when optimizing the working conditions for obtaining isotopic information, there is always a tradeoff between achieving sufficient sensitivity and large enough isotopic shifts, such that a suitable balance needs to be found experimentally. Moreover, and depending on the spectrum complexity for each transition monitored, the signals for the different isotopes can be mixed up,35 so that it is important to perform measurements with (pure) enriched isotopic standards to verify that the transitions measured respond to a particular isotope only.
In this case, an exploration of the different transitions theoretically available for the BF molecule in the region 190–207 nm (presented in Table S3 from the ESI†) was made using 10 μL of natural boron standards of different concentrations, and deploying the optimized method described in Section 2.3.2. Depending on the sensitivity for each transition monitored, amounts as high as 4 μg B had to be introduced in the furnace to observe an analytical signal. For confirmation of the theoretical isotopic shifts (Δλcalc), two different solutions enriched in one of the two existing boron stable isotopes were available at a 10 mg L−1 concentration. Self-evidently, and considering the concentration of these standards, confirmation of the potential isotopic shift was only possible for those transitions with sufficient sensitivity. In such cases, monitoring of the two enriched isotopic standards separately was conducted for confirmation and measurement of the shift. Theoretical and experimental values for these isotopic shifts are also included in Table S3.†
As expected, it was impossible to accurately measure the isotopic shift for the X1Σ-A1Π (0,0) transition at 195.589 nm (the most sensitive) with our instrumentation. The calculated shift (Δλcalc = 8.38 pm) would be reflected in a signal displacement of only 4–5 pixels (see Fig. 1A), resulting in the two isotopic peaks overlapped. Out of the other transitions monitored, the only transition that showed an isotopic shift measurable with the resolution of our spectrometer (Δλcalc = 173 pm), adequate sensitivity (roughly 10% of that observed for the main transition at 195.6 nm) and sufficiently simple spectra as to obtain a separate signal for each of the B stable isotopes, was the X1Σ-A1Π (1,0) transition around 201.08 nm. To be able to monitor the signals for the two B isotopes in the same spectral window (covering 266 pm in this region), the spectrometer was centered at 201.08 nm, as seen in Fig. 1B. With this configuration, the theoretical (Δλcalc = 173 pm) and experimental (Δλexp = 167 pm) shifts agreed reasonably well and the signals for both isotopes could be measured simultaneously (11BF at 200.9931 nm and 10BF at 201.1602 nm).
Next, experiments for exploring the possibilities for performing isotopic analyses at these wavelengths were conducted. First, the conditions for signal acquisition were optimized. The spectra obtained for a series of standard solutions enriched in 11B of increasing concentration showed that the flank of a secondary line for 11BF partially interfered the signal of 10BF. However, it was confirmed that this interference occurred at the flanks of the 10BF peak, but did not affect the 10BF signal at the central pixel (that of the maximum) at least up to 400 ng B, the maximum amount that could be measured with the B isotopic standards available in our lab (two consecutive injections of 20 μL of a 10 mg L−1 standard solution after addition of the modifiers). As a consequence, further signal evaluation was performed using only the central pixel for both transitions. Besides, and as is normally performed for isotopic measurements by means of GF MAS,25,30,35,36 signal evaluation was performed as the peak height. As the BF transitions for each isotope evolve from the head to longer wavelengths, the signal for 11BF at 200.9931 nm was free from interferences. Additionally, and to improve precision for the isotope ratio measurements, the baseline setting was carried out by selecting four specific pixels, one at each side of the 11BF and 10BF transitions, respectively, instead of using a dynamic method. In particular, pixels 32, 89, 149 and 174 were used for correction, while evaluation of the 11BF and 10BF signals was carried out at pixels 36 and 160, respectively.
Once the acquisition method was optimized, boron isotope ratios were determined for two reference solutions of different isotopic composition and at different boron contents. The goal of this experiment was to define the typical isotope ratio precision and accuracy directly achievable with this method, without any other correction measure. To this end, a first solution containing about 50% of each boron isotope was prepared using the two 10 mg L−1 enriched isotope standard solutions available (expected theoretical 11B/10B ratio of 0.97, as per the respective certificates of analysis). Additionally, a standard B solution of natural isotopic composition was also analyzed. Isotope ratios were simply calculated as the ratio of the absorbances measured as previously described, after blank correction. The standard B solution of natural isotopic composition showed a 11B/10B ratio of 4.314 ± 0.008 (95% confidence interval, n = 5), which was experimentally obtained by ICP-MS, where instrumental mass discrimination was corrected for by relying on external calibration using a prepared 11B/10B solution 80![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :20.
:20.
The results of this experiment are shown in Fig. 6. As seen from this figure, and as could be expected, the precision for the isotope ratios improves with increasing total B amounts for both solutions analyzed. For total B amounts below 100 ng, the signal to noise ratio is low, due to the relatively low sensitivity for the transition and the high instrumental noise at this region of the spectra. This results in RSD% values above 10% for the isotope ratios in both solutions. Precision improves with increasing boron contents but only down to values around 3–4%, which is on the same range as precision obtained for other isotope ratios when this technique is used25,30,35 (further improvement of these precision values might be possible by means of chemometrical approaches, such as partial least squares regression, as shown by Abad et al.22).
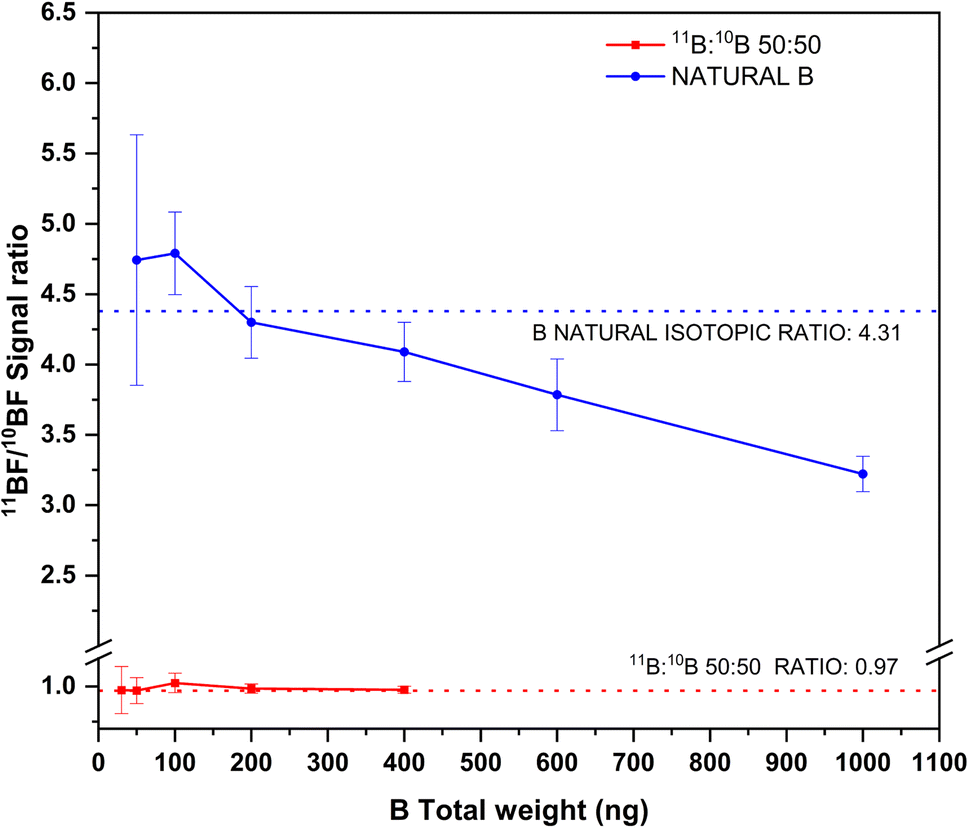 | ||
| Fig. 6 Experimental 11BF/10BF signal ratios obtained by HR CS GFMAS for B standard solutions of two different isotopic compositions and at different total B contents. The expected ratios are represented by the corresponding dashed lines. Measurement conditions are those indicated in Table 1. | ||
As for accuracy, results in accordance with the expected values are obtained for total boron amounts up to 400 ng. Beyond this value (which could only be measured for the boron standard of natural composition), 11B/10B ratios clearly biased low were obtained. This effect is probably due to the 11BF interference on the 10BF signal previously described, which will certainly become more important as the amount of 11B increases. To tackle this issue, mathematical models, as that developed by Abad et al.,22 could be used to compensate for this interference, thus providing improved accuracy and precision. Otherwise, total B contents should be strictly controlled if unbiased results are to be obtained by this method. The method as such, however, could be useful for performing tracer experiments or isotope dilution calibration while maintaining its simplicity.
4. Conclusion
This work proposes an alternative to the existing GFAAS methods for the direct determination of B in biological solid samples at the μg g−1 level based on the use of HR CS GF MAS. The method has been applied to botanical samples, for which the typical B contents relevant for plant growth and development falls within the μg g−1 level,6 but could be applied to other biological samples as well. The proposed method relays on the promotion of the BF molecule through the use of a fluorinating agent in the gas phase (CH3F-Ar mixture) and represents a much more efficient protocol in terms of time of analysis, consumption of graphite parts and method simplicity, as well as quality of the analytical information achieved, providing both elemental and isotopic information for this element.The use of the fluorinating agent for generating the analytical signal shows several advantages. First, it permits the use of a much milder temperature program than those used in AAS methods, which results in longer lifetime for the graphite parts. Additionally, it enablesan efficient digestion of the solid samples in the graphite furnace, eliminating the need for cumbersome and time-consuming pre-digestions, and minimizing the risk of analyte losses or contamination. Last but not least, memory effects are also absent with this strategy, which circumvents the use of cleaning steps in-between measurements. Self-evidently, all of these advantages greatly contribute to the method simplicity, which is complemented by the possibilities for calibration offered by the method. In this regard, straightforward calibration with aqueous standard solutions is possible for obtaining B elemental information directly from botanical solid samples, provided that the proper mixture of modifiers (W-permanent, citric acid, and Ca nitrate) is deployed at the beginning of the measurement protocol.
It needs to be highlighted at this point that the logistic advantages described are not detrimental to the method performance. In fact, detection capabilities for the method developed are similar to those achievable by GFAAS methods, but with the additional advantage of providing isotopic information also. This information is available by measuring an alternative BF transition, with lower but still acceptable sensitivity. The characteristic mass for BF main transition at 195.6 nm is approx. 0.13 ng, while the characteristic mass for BF secondary transition at 201.08 nm region is approx. 1.3 ng for each isotopic line. In this regard, and although precision and accuracy for the isotopic measurements cannot compete with those of mass spectrometry methods, the method offers the possibility to perform new experimental designs previously unavailable for atomic absorption spectrometry based on the use of such isotopic information, such as tracer experiments and/or isotope dilution calibration.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors are very grateful for the grant PID2021-122455NB-I00, funded by MCIN/AEI/10.13039/501100011033 and “FEDER. Una manera de hacer Europa”, and acknowledge the funding received by the Aragon Government (DGA, Construyendo Europa desde Aragón, Grupo E43_20R). André L. M. de Souza also acknowledges MCIN/AEI/10.13039/501100011033 for his predoctoral grant (PRE2019-091118).References
- I. Ursu, Physics and Technology of Nuclear Materials, Pergamon Press, Inc., 1985 Search PubMed.
- C. Wongchoosuk, Characteristics and Applications of Boron, IntechOpen, Rijeka, 2022 Search PubMed.
- D. Yang, R. Liu, W. Li and Q.-L. Yan, Fuel, 2023, 342, 127855 CrossRef CAS.
- W. Zhang, S. Yamashita and H. Kita, Adv. Appl. Ceram., 2019, 118, 222–239 CrossRef CAS.
- I. Mitruţ, I. R. Scorei, H. O. Manolea, A. Biţă, L. Mogoantă, J. Neamţu, L. E. Bejenaru, M. V. Ciocîlteu, C. Bejenaru, G. Rău and G. D. Mogoşanu, Rom. J. Morphol. Embryol., 2023, 63, 477–483 CrossRef PubMed.
- P. Chalk, C. J. Smith, D. Chen and J.-Z. He, Arch. Agron. Soil Sci., 2022, 68, 561–578 CrossRef CAS.
- M. Suzuki, Int. J. Clin. Oncol., 2020, 25, 43–50 CrossRef PubMed.
- J. Shin, G. A. Gamage, Z. Ding, K. Chen, F. Tian, X. Qian, J. Zhou, H. Lee, J. Zhou, L. Shi, T. Nguyen, F. Han, M. Li, D. Broido, A. Schmidt, Z. Ren and G. Chen, Science, 2022, 377, 437–440 CrossRef CAS PubMed.
- R. N. Sah and P. H. Brown, Microchem. J., 1997, 56, 285–304 CrossRef CAS.
- P. González, A. Sixto and M. Knochen, Talanta, 2017, 166, 399–404 CrossRef PubMed.
- A. S. Al-Ammar, E. Reitznerová and R. M. Barnes, J. Radioanal. Nucl. Chem., 2000, 244, 267–272 CrossRef CAS.
- M. Resano, M. Aramendía and F. Vanhaecke, J. Anal. At. Spectrom., 2006, 21, 1036–1039 RSC.
- M. Resano, J. Briceño, M. Aramendía and M. A. Belarra, Anal. Chim. Acta, 2007, 582, 214–222 CrossRef CAS PubMed.
- M. Aramendía, M. Resano and F. Vanhaecke, Anal. Chim. Acta, 2009, 648, 23–44 CrossRef PubMed.
- B. Welz, H. Becker-Ross, S. Florek, U. Heitmann and M. Vale, J. Braz. Chem. Soc., 2003, 14, 220–229 CrossRef CAS.
- M. Resano, E. García-Ruiz, M. Aramendía and M. A. Belarra, J. Anal. At. Spectrom., 2019, 34, 59–80 RSC.
- U. Heitmann, H. Becker-Ross, S. Florek, M. D. Huang and M. Okruss, J. Anal. At. Spectrom., 2006, 21, 1314–1320 RSC.
- M. Resano, M. Aramendía, F. V. Nakadi, E. García-Ruiz, C. Alvarez-Llamas, N. Bordel, J. Pisonero, E. Bolea-Fernández, T. Liu and F. Vanhaecke, TrAC, Trends Anal. Chem., 2020, 129, 115955 CrossRef CAS.
- J. Byrne, D. Grégoire, D. Goltz and C. Chakrabarti, Spectrochim. Acta, Part B, 1994, 49, 433–443 CrossRef.
- R. Nowka, K. Eichardt and B. Welz, Spectrochim. Acta, Part B, 2000, 55, 517–524 CrossRef.
- M. Burguera, J. L. Burguera, C. Rondón and P. Carrero, Spectrochim. Acta, Part B, 2001, 56, 1845–1857 CrossRef.
- C. Abad, S. Florek, H. Becker-Ross, M.-D. Huang, H.-J. Heinrich, S. Recknagel, J. Vogl, N. Jakubowski and U. Panne, Spectrochim. Acta, Part B, 2017, 136, 116–122 CrossRef CAS.
- G. Herzberg, Molecular Spectra and Molecule Structure I. Spectra of Diatomic Molecules, D. Van Nostrand Company, INC., Princeton, 2nd edn, 1950, p. 509 Search PubMed.
- H. Wiltsche, K. Prattes, M. Zischka and G. Knapp, Spectrochim. Acta, Part B, 2009, 64, 341–346 CrossRef.
- F. V. Nakadi, M. A. M. S. Da Veiga, M. Aramendía, E. García-Ruiz and M. Resano, J. Anal. At. Spectrom., 2015, 30, 1531–1540 RSC.
- R. W. B. Pearse and A. G. Gaydon, The Identification of Molecular Spectra, Chapman and Hall Ltd, London, 4th edn, 1976, p. 54 Search PubMed.
- F. V. Nakadi, M. C. García-Poyo, C. Pécheyran and M. Resano, J. Anal. At. Spectrom., 2021, 36, 2370–2382 RSC.
- Y.-R. Luo, Comprehensive Handbook of Chemical Bond Energies, CRC Press, 1st edn, 2007 Search PubMed.
- CRC Handbook of Chemistry and Physics, ed. D. R. Lide, CRC Press, Taylor and Francis, Boca Raton, FL, 89th edn, 2009 Search PubMed.
- A. Bazo, R. Garde, E. Garcia-Ruiz, M. Aramendía, F. V. Nakadi and M. Resano, J. Anal. At. Spectrom., 2022, 37, 2517–2528 RSC.
- H.-J. Heinrich and R. Matschat, Spectrochim. Acta, Part B, 2007, 62, 807–816 CrossRef.
- F. Börno, S. Richter, D. Deiting, N. Jakubowski and U. Panne, J. Anal. At. Spectrom., 2015, 30, 1064–1071 RSC.
- É. C. Lima, F. J. Krug and K. W. Jackson, Spectrochim. Acta, Part B, 1998, 53, 1791–1804 CrossRef.
- G. M. A. Botelho, A. J. Curtius and R. C. Campos, J. Anal. At. Spectrom., 1994, 9, 1263 RSC.
- F. V. Nakadi, M. A. M. S. Da Veiga, M. Aramendía, E. García-Ruiz and M. Resano, J. Anal. At. Spectrom., 2016, 31, 1381–1390 RSC.
- M. B. T. Zanatta, F. V. Nakadi, M. Resano and M. A. M. S. Da Veiga, J. Anal. At. Spectrom., 2019, 34, 2280–2287 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ja00420a |
| This journal is © The Royal Society of Chemistry 2024 |