
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceConcomitant ion matrix effects in SCGD-OES enhanced with formic acid†
Yinchenxi
Zhang
,
Jaime
Orejas
 *,
Jorge
Pisonero
* and
Nerea
Bordel
*,
Jorge
Pisonero
* and
Nerea
Bordel
Grupo de Espectroscopía, Láseres y Plasmas (GELP), Department of Physics, University of Oviedo, C/ Gonzalo Gutiérrez Quirós S/N, Mieres 33600, Spain. E-mail: orejasjaime@uniovi.es; pisonerojorge@uniovi.es
First published on 25th January 2024
Abstract
Solution-Cathode Glow Discharge Optical Emission Spectrometry (SCGD-OES) is a cost-effective analytical technique due to its miniaturization potential and reduced consumption, useful for on-line and in situ aqueous solution elemental analysis, and to its adequate limits of detection, which are, on average, comparable with those of ICP-OES. The elemental response in SCGD-OES is frequently enhanced making use of low molecular weight compound additives, such as formic acid (HCOOH); however, this enhancement was shown to be affected by the presence of significant amounts of Na+ and Cl− ions, which are extensively present in a wide variety of samples. In this context, this work delves into the impact of HCOOH on analyte emission signals when analyzing solutions containing high concentrations of other cations (Na+, K+, Mg2+, and Ca2+) and anions (SO42−, Cl−, NO3−, HCO3− and Br−), which make up an ample variety of sample matrices, on SCGD-OES. In particular, the atomic emission intensity of certain analytes is observed to decline with increasing HCOOH additive concentration under certain sample matrix conditions. This intensity depression mainly appears when the sample matrix contains alkali cations, regardless of the anion type. Interestingly, elements with notable chemical vapor generation (CVG) efficiencies, such as Hg, Ag, Pb and In, maintain the positive HCOOH enhancement of emission intensity. HCOOH addition enhancement is also evaluated in a more complex sample, artificial seawater, showing similar results compared to those obtained with high NaCl concentration solutions due to the high proportion of these ions in seawater. Finally, the analytical performance of SCGD-OES is evaluated in terms of sensitivity and limits of detection, resulting that the addition of HCOOH for seawater analysis pays off when CVG-prone elements are targeted.
Introduction
Atmospheric pressure glow discharge generated using a solution electrode is increasingly utilized for spectrochemistry applications. The first application of this set-up in an analytical chemistry context dates back to 1993, when a direct current atmospheric pressure glow discharge in ambient air using an aqueous cathode was exploited as an excitation source for optical emission spectrometry (OES) by Cserfalvi and coworkers, successfully detecting alkali and alkali earth metals in water samples.1 Since then, the device has evolved towards simplified construction and reduced sample consumption. One of these approaches, the solution-cathode glow discharge (SCGD), was first described by Webb et al. in 2005,2 and gathers several attractive characteristics: low direct current power consumption (less than 100 W), no discharge gas requirements and analyte sampling directly achieved by plasma–liquid interaction. In particular, the required instrumentation does not include vacuum systems, pressurized gases, and radio-frequency powering nor nebulization systems, indispensable for other traditional elemental analysis techniques like inductively coupled plasma (ICP) coupled to OES or to mass spectrometry (MS). Accordingly, the SCGD is endowed with great potential for miniaturization and portabilization.3 Additionally, the SCGD provides competitive analytical performance with limits of detection (LODs) at the middle to low ppb3–5 levels and even lower for some alkali metals.6 Given the advantages of low cost, small size and simple operation, the SCGD was applied in various contexts and for different samples, such as seawater,3 waste water,4,7–9 saline solutions,10 brine,11 ores10,12 or stream sediments.9,13 Moreover, analysis of nanoparticles by SCGD was recently reported,14 as well as its application to analyze large biomolecules by coupling the SCGD to mass spectrometric detection.15The plasma–liquid interface in a running SCGD shows a complex behavior due to its rich chemical reactivity and the presence of different physical phenomena that, altogether, contribute to the transference of dissolved analyte ions in solution towards the plasma phase to be excited. In particular, this analyte solution-to-plasma transfer process comes from three main potential mechanisms. (a) Cathodic sputtering: being a glow discharge, the cathode solution in the SCGD is continuously bombarded with positive ions accelerated by the strong electric field in this region (of around 106 V m−1).16 As a result of this bombardment, dissolved ions are released into the bulk plasma phase in the form of solvated complexes. The ions are liberated through desolvation within the first 100 μm above the cathode surface, followed by atomization and excitation.17 (b) Droplet ejection from the plasma–liquid interface: thanks to the physical properties at the liquid–plasma boundary, droplets are formed, transferring the analytes from the sample into the discharge zone. These droplets can be formed through two different mechanisms: on one hand, the strong electric field reduces the liquid surface tension and forms Taylor cones capable of ejecting liquid droplets; on the other hand, the high temperature gradient between liquid and plasma phases can produce droplet ejection through a fast vaporization process followed by condensation.2,18–20 Several reports visualized the droplets arising from the solution surface by laser scattering and captured in short exposure images.16,21,22 (c) Chemical vapor generation (CVG): the liquid–plasma interface is a rich source of highly reactive species (e.g., solvated electrons, H˙, OH˙, H2O2 and UV radiation).23 These radicals facilitate redox chemical reactions with ions in the incoming solution, yielding volatile species capable of crossing the solution-plasma boundary to be excited.24,25
The incorporation of low weight organic compounds, e.g., formic acid (HCOOH), acetic acid (CH3COOH), methanol (CH3OH) and ethanol (CH3CH2OH), in the incoming solution is frequently employed to improve SCGD-OES analytical performance. For instance, previous studies showed that 0.5% MeOH promoted In emission 33 times;26 Hg emission was improved 15.5 times and 8.5 times by adding 4% CH3OH and 2% CH3CH2OH, respectively;7 1% CH3COOH led to Ag and Pb emission increases of 5.4 and 3.7 fold, respectively;18 a 23-fold Bi emission enhancement was achieved due to the addition of 3% HCOOH,27 while Pb emission was improved by a factor of 10 using 5% HCOOH.28 The correlation of the emission signal enhancements with changes in the electron density and temperature of the SCGD was investigated by Yu et al., concluding that the excitation conditions were not significantly modified by the presence of HCOOH.27 Therefore, emission signal enhancement was mainly related to a superior analyte transfer efficiency. The presence of additives altered the solution physical properties, e.g., boiling point, viscosity and surface tension, which might change the size of the droplets formed at the plasma–liquid interface or/and the droplet ejection rate, affecting the analyte solution-to-plasma transfer rate.21,29 In addition, additives are likely to be decomposed at the interface. Additive radicals, e.g., H˙, OH˙, and COO˙21 were shown to enhance the CVG process and increase sampling efficiency.29–32
In relation to the use of HCOOH (one of the most common additives), previous work from our research group showed that the influence of HCOOH on elemental emission is significantly affected in solutions containing high amounts of NaCl.33 Specifically, while the HCOOH additive improved the signals of Fe, In, Sr and Li when NaCl was not included in the incoming solution, this was not the case when introducing solutions with 3.5 g L−1 of the salt (one-tenth the total concentration of seawater), In being the exception among the tested elements. Considering that Na+ and Cl− are extremely common ions present in many kinds of matrices, understanding how these and other representative ions may alter the influence of HCOOH on the SCGD-OES performance is crucial since it could provide a useful guideline on whether to use it as a sensitivity enhancer in actual analytical methodologies. Therefore, this work aims at systematically studying matrix effects due to the presence of concomitant ions on the SCGD-OES analytical performance when HCOOH is included in the incoming solution. Given that the positive influence of HCOOH on analyte response can be interfered by the presence of Na+ and Cl−, experiments are carried out to infer the contribution of other common cations (K+, Mg2+ and Ca2+) and anions (NO3−, HCO3−, Br−, and SO42−) on this sensitivity enhancement to a set of selected analytes (Fe, Sr, Li and In). Additionally, the particular effect of Na+ and Mg2+, present in solution together with Cl−, NO3−, Br− or SO42−, on elements (Zn, Cd, Hg, Ag and Pb) known to be efficient in CVG, is studied with more detail. Finally, the analytical features of SCGD-OES with and without HCOOH in solution are evaluated in ultrapure water and artificial seawater.
Experimental setup
Fig. 1 illustrates the SCGD configuration applied in this work, which was described in detail elsewhere.20,33 The discharge zone is composed of a vertically oriented sharpened tungsten electrode (Saf-Fro, 3.2 mm diameter) and a co-axially situated borosilicate glass capillary tube (Hirschmann ringcaps, 20 μL) used for sample introduction, which is surrounded by grounded graphite. A direct current high voltage power supply (Kepco Inc., BHK 2000-01, New York, USA) sustains the discharge in current limited mode (80 mA), providing positive potential to the tungsten electrode and negative (grounded) potential to the graphite. The tungsten electrode can be precisely situated in X, Y and Z directions with the aid of micrometer screws (Newport, M-460A series) and its distance from the edge of the capillary is set at 3 mm for every experiment described here. Under these conditions, applied potentials to the discharge range between 650 and 750 V, depending on the composition of the incoming solution. The electrical circuit is closed when the liquid conductive sample overflows the capillary tip and reaches the graphite. A peristaltic pump (Reglo ICC, Ismatec, Wertheim, Germany) drives and controls the flow rate (3.0 mL min−1) of the sample solution into the discharge area. A section of the tubing (Ismatec, inner diameter 2.06 mm) is filled with glass beads (Merck, diameter 2 mm) to reduce the flow rate fluctuation caused by the peristaltic pump. The discharge construction coaxially stands in a 20 mL home-made 3D printed polyether ether ketone base that serves as a residue reservoir including an exit tube for residue disposal. | ||
| Fig. 1 Schematic diagram of the SCGD system. | ||
A pair of lenses (focal length of 300 mm, diameter 50.8 mm, LB4710, Thorlabs, Germany) are located on a 3D platform (Newport, M-460A series) between the spectrometer and the discharge zone for focusing the radiation from the plasma onto the entrance slit of the spectrograph, resulting in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 magnification. A diaphragm (diameter 50.8 mm, Thorlabs, Germany) is placed between these two lenses to reduce spherical aberration in spatially resolved measurements. Detection of the optical emission is wavelength-dispersed using a Czerny-Turner spectrograph (SpectraPro 2500i, Teledyne Princeton Instruments, USA), equipped with two diffraction gratings of 3600 lines per mm and 2400 lines per mm, effective in the 200–550 nm and 200–750 nm spectral ranges, respectively. The emission spectrum is detected by an intensified charge coupled device (PI-MAX 1024RB, Teledyne Princeton Instruments, USA). The entrance slit is set at 50 μm, leading to a wavelength resolution of 0.02 nm (grating 3600 lines per mm) and 0.04 nm (grating 2400 lines per mm), respectively. Winspec/32 (Teledyne Princeton Instruments, USA) is used to control the parameters of the spectrograph and the detector.
:1 magnification. A diaphragm (diameter 50.8 mm, Thorlabs, Germany) is placed between these two lenses to reduce spherical aberration in spatially resolved measurements. Detection of the optical emission is wavelength-dispersed using a Czerny-Turner spectrograph (SpectraPro 2500i, Teledyne Princeton Instruments, USA), equipped with two diffraction gratings of 3600 lines per mm and 2400 lines per mm, effective in the 200–550 nm and 200–750 nm spectral ranges, respectively. The emission spectrum is detected by an intensified charge coupled device (PI-MAX 1024RB, Teledyne Princeton Instruments, USA). The entrance slit is set at 50 μm, leading to a wavelength resolution of 0.02 nm (grating 3600 lines per mm) and 0.04 nm (grating 2400 lines per mm), respectively. Winspec/32 (Teledyne Princeton Instruments, USA) is used to control the parameters of the spectrograph and the detector.
The selected elements and wavelengths employed in this work are listed in Table 1. The emission intensity corresponds to the integrated peak area once the baseline has been subtracted. Unless otherwise stated, these intensity values are calculated as the mean of five repetitions, and the corresponding standard deviation as an estimate of the statistical error. For each simulated matrix solution, the emission intensity measured for a particular emission line at different HCOOH concentrations is normalized to the corresponding emission intensity obtained for 0% HCOOH concentration solution. The normalized intensity deviation results from the following equation:

| Element | Compound | Work concentration (mg L−1) | Wavelength (nm) |
|---|---|---|---|
| a Sigma-Aldrich. b VWR Chemicals. c Alfa-Aesar. d Merck. e Labkem. | |||
| Zn I | ZnCl2a | 1.00 | 213.8 |
| Cd I | Cd(NO3)2·4H2Oc | 1.00 | 228.8 |
| Fe I | Fe(NO3)3·9H2Ob | 10.0 | 252.3 |
| Hg I | Hg(NO3)2d | 10.0 | 253.7 |
| Ag I | AgNO3e | 0.100 | 338.3 |
| Pb I | Pb(NO3)2b | 10.0 | 368.3 |
| In I | In(NO3)3·xH2Oc | 1.00 | 451.1 |
| Sr I | Sr(NO3)2a | 10.0 | 460.7 |
| Li I | LiNO3c | 1.00 | 670.8 |
LODs are estimated from the 3σ/S criterion, where S (sensitivity) is calculated from the net analyte emission intensity of the measurement of a single solution of the analyte divided by the corresponding added concentration of the target element and σ is obtained from the standard deviation of twenty consecutive measurements of a blank sample.
Reagents and solutions
All chemicals involved throughout this work are of analytical grade or purer. Concentrated HNO3 (65%, Labkem) is added to ultrapure water (18.2 MΩ cm) obtained from a water purification system (Merck Millipore, Germany) to reach a concentration of 1 vol% as a basis for further solution preparation. The solutions introduced into the SCGD are prepared by diluting concentrated stock solutions of analyte elements with each simulated matrix solution. Specifically, the concentration of stock solution for each element is 1000 μg mL−1, obtained by dissolving a corresponding amount of each compound listed in Table 1 individually in 1% HNO3 solution. On the other hand, the salts listed in Table 2 are used to prepare simulated matrix solutions. These salts are also dissolved in 1% HNO3 separately to obtain solutions containing 59.8 mM (consistent with the concentration of our previous work, 3.5 g per L NaCl33) of a cation (Na+, Mg2+ or Ca2+). K+ was an exception since a concentration of 59.8 mM led to unsustainable plasma conditions. Therefore, a K+ concentration of 49.8 mM (equal to 3.5 g per L KCl) is applied in the corresponding experiments. The analyte stock concentrated solutions, together with formic acid (HCOOH, 98% weight, VWR Chemicals), are added to each of these simulated matrix solutions to obtain the final mixture work solutions that are introduced into the SCGD for the experiments. HCOOH is included in the solutions at concentrations of 0–7% vol/vol.Artificial seawater was purchased from VWR Chemicals (VWRC79789LH). Several solutions are prepared using 1:10 diluted artificial seawater and acidified with HNO3 to reach 1% vol HNO3 and with HCOOH at different concentrations (0–7% vol). These solutions are spiked with the selected analytes (using the abovementioned concentrated analyte solutions) before introducing them into the SCGD. The seawater dilution degree is chosen to generate a stable glow discharge that allows sustained analyses.
Results and discussion
Cation influence on analyte signal enhancement by HCOOH addition
The analytical performance of SCGD-OES was recently investigated with solutions containing different concentrations of NaCl and HCOOH, demonstrating that the influence of this additive under high NaCl content conditions can be negative.33 To extend this study to other matrices, the co-presence of HCOOH and different cations and anions is considered. Fig. 2 shows the normalized atomic emission intensity of four analytes (Fe, In, Sr and Li) when introducing different simulated matrix solutions made up with different chloride salts, adding different HCOOH concentrations, in the SCGD.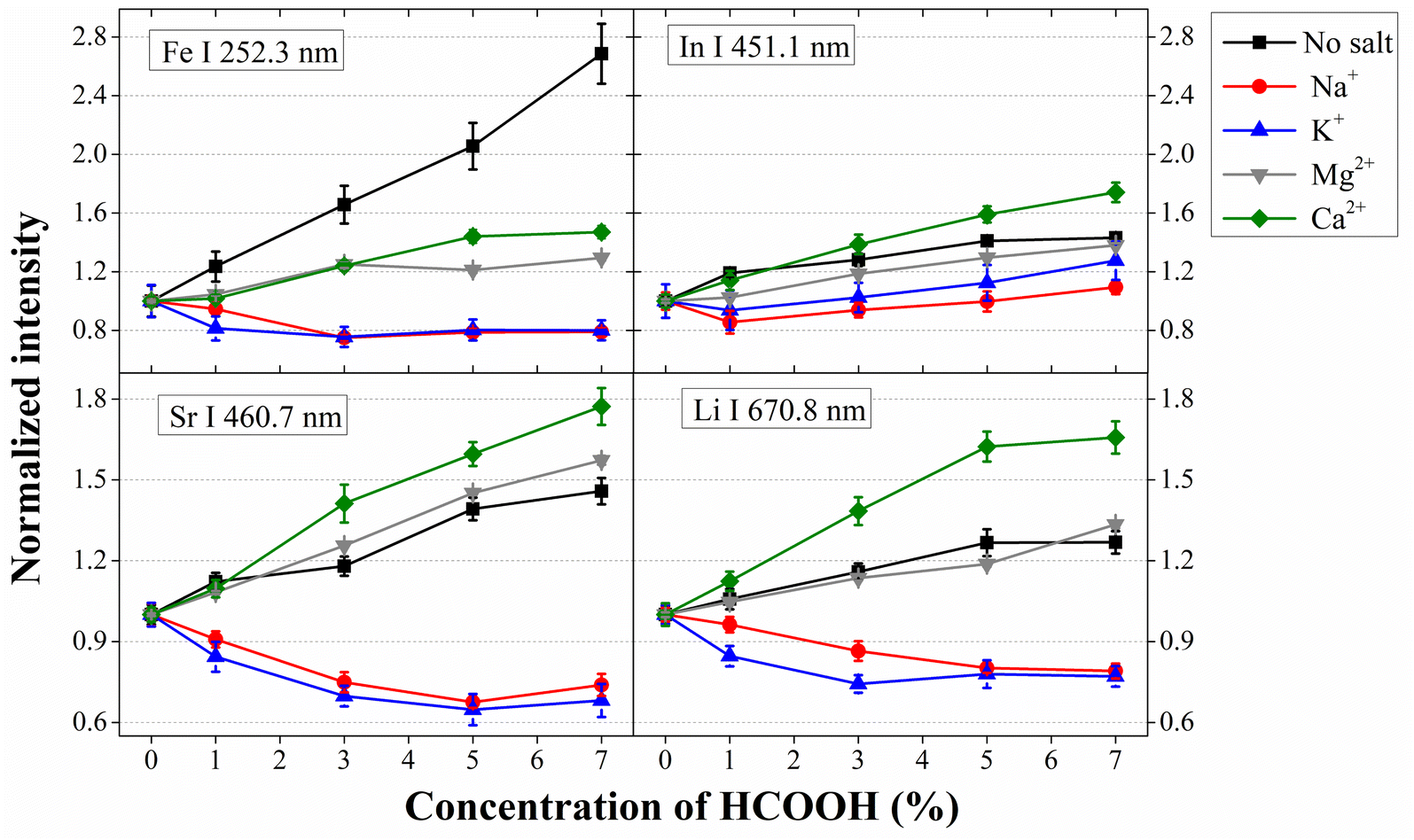 | ||
| Fig. 2 Normalized elemental emission evolutions vs. HCOOH concentration in the presence of different metal chlorides (cation concentrations: 59.8 mM for Na+, Mg2+ and Ca2+, and 49.8 mM for K+). | ||
The results show different trends for In, compared to Fe, Sr and Li. The In emission signal is not hampered by HCOOH addition, except at low HCOOH concentrations when alkali metals are present in solution, though the emission reduction is not significant. However, the influence of HCOOH addition on Fe, Sr and Li emission signals depends on whether the cation is an alkali or an alkali-earth metal. On one hand, in the presence of high amounts of an alkali metal, Na+ or K+, the addition of HCOOH depresses the emission intensity of the three mentioned analytes. This observation matches the result from Cai et al., showing that the positive HCOOH influence was hampered by Na+ presence, though CVG-prone elements suffered less.34 On the other hand, analyte response is still promoted by the addition of HCOOH even if there is a high concentration of alkali earth metals (Mg2+ and Ca2+) in the incoming solution. However, Mg2+ and Ca2+ decrease the HCOOH enhancement for Fe while Ca2+ promotes HCOOH enhancement for Sr and Li.
Anion influence on analyte signal enhancement by HCOOH addition
A similar study is carried out to evaluate the effect of common anions such as SO42−, NO3− and Br−. In particular, analyte emission signals are measured introducing solutions with HCOOH and one of these salts: MgCl2, Mg(Br)2, Mg(NO3)2 and MgSO4. Only alkali earth metals are initially considered since they maintain the HCOOH-produced enhancement. Fig. 3a shows the normalized analyte emission intensity versus HCOOH concentration in the presence of a high concentration of Mg2+ and different anions. Although Li analyte signals are slightly decreased at 1% vol HCOOH, overall, analyte emission signals are still enhanced if more HCOOH is included in solution, each element to different degrees, also depending on the specific anions present in solutions. Similar results are obtained introducing solutions with Ca2+, including CaCl2 and Ca(NO3)2 salts. The HCOOH enhancement changes (see Fig. 3a) depending on the anions present in solution, suggesting that they are also involved in the HCOOH enhancement.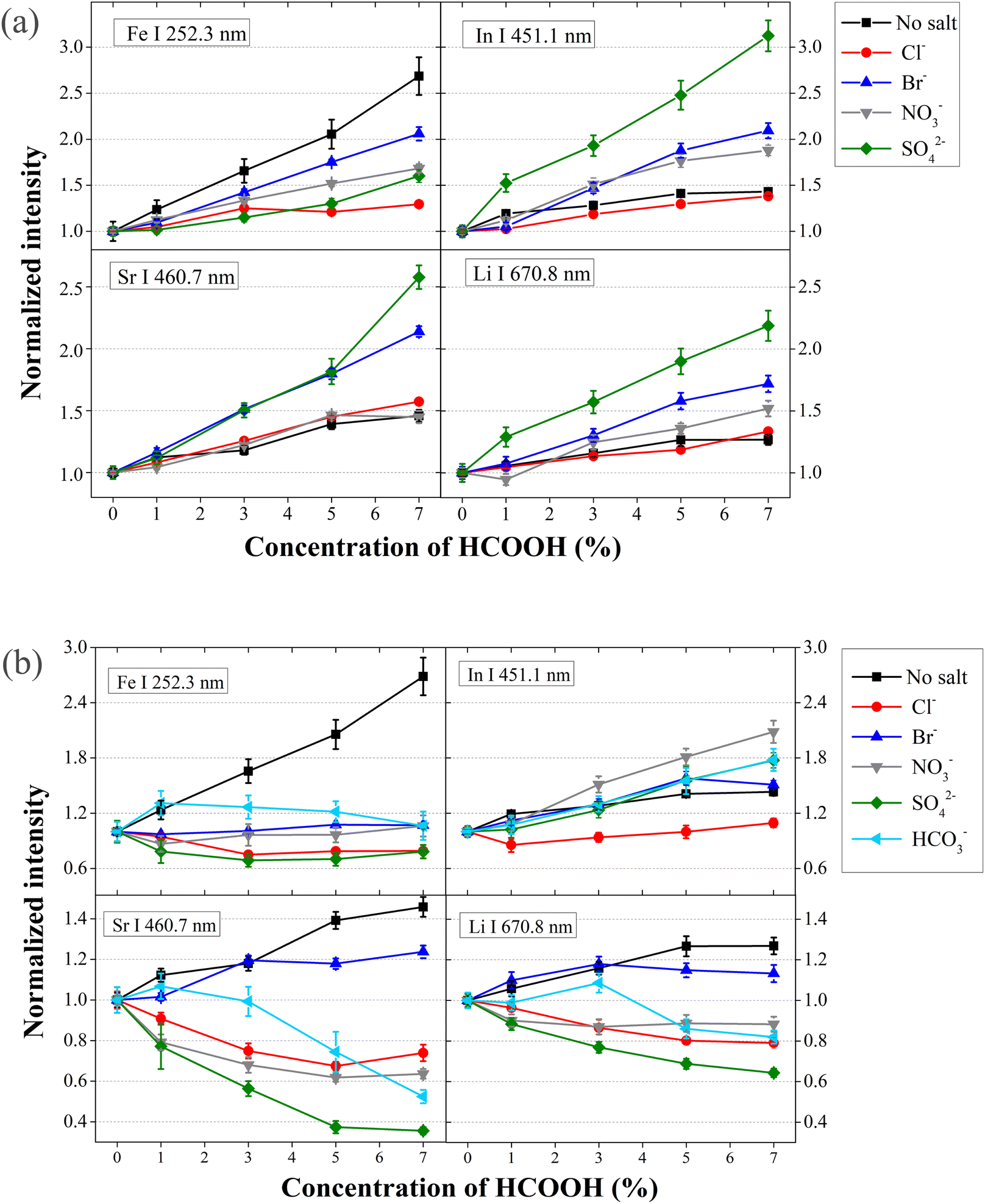 | ||
| Fig. 3 HCOOH influence on analyte emission in the presence of (a) 59.8 mM Mg2+ combined with various anions; (b) 59.8 mM Na+ combined with various anions. | ||
To further study the effect of anions (Cl−, SO42−, NO3−, Br− and HCO3−) on HCOOH-produced signal enhancement, analyte emission was monitored introducing solutions including different Na-based salts (Table 2). The results are shown in Fig. 3b. In this case, a more complex behavior is observed. Again, In seems to keep the enhancement, no matter which anion is present. For the rest of the elements, the emission enhancement caused by adding HCOOH disappears if HCO3−, NO3−, or SO42− is present although to different degrees. Br− deteriorates the HCOOH enhancement on Fe though not on Sr and Li. In general, it seems that each anion generates slightly different changes in the HCOOH enhancement, though the nature of the cation content is more critical, since the presence of alkali ions eliminates the enhancement in many cases.
CVG-prone analyte signal enhancement by HCOOH addition
The SCGD can induce chemical vapor generation (CVG) due to the redox reactions at the liquid–plasma interface, which represents an important mechanism contributing to the analyte solution-to-plasma transfer. For instance, Fig. 3 shows that the emission intensity from In, a CVG-prone element, is systematically enhanced by HCOOH addition in the presence of different ions. In order to verify whether emission signals from CVG-prone elements are enhanced by the addition of HCOOH even if other concomitant ions are present, the study is extended to other elements such as Zn, Cd, Hg, Ag and Pb, which are notable CVG elements.In particular, solutions containing 1.00 mg per L Zn, 1.00 mg per L Cd, 10.0 mg per L Hg, 0.100 mg per L Ag and 10.0 mg per L Pb, respectively, are introduced into the SCGD to register the analyte emission intensity in different simulated matrix solutions. Fig. 4a shows the normalized analyte emission signals at increasing concentration of HCOOH measured considering solutions prepared without and with salt (NaCl and MgCl2). It is observed that Hg I, Ag I and Pb I emission signals achieve a sustained rise at increasing HCOOH with the three solutions employed. Notably, 1% HCOOH provides the maximum improvement of the Hg emission signal, maintained at higher concentrations of the additive, when no salt is present in the solution, which agrees with previously reported results.7,35 It should be remarked that solutions containing Cl− result in the formation of AgCl precipitates, but simply shaking the solution prior to its introduction in the sample line, a suspension is formed leading to a sustainable Ag emission signal in the SCGD-OES system. In contrast to these analytes' behavior, emission intensity from Zn decreases and emission intensity from Cd levels off, when HCOOH concentration increases in the solution containing Na+, while both are still enhanced in solutions containing Mg2+. Nevertheless, all the HCOOH enhancement levels for CVG-prone elements, compared to the case without salt, encounter reduction when the incoming solution contains concomitant ions, though the discharge containing Mg2+ is less affected.
 | ||
| Fig. 4 Variation of Zn, Cd, Hg, Ag and Pb emission signals at increasing HCOOH concentration in presence of (a) salts composed of Cl− and various cations and (b) salts composed of Na+ and various anions. | ||
Another experiment is conducted to evaluate how anions affect the HCOOH performance on CVG-prone elements. In particular, the analyte emission response is recorded including NaCl, NaBr, NaNO3 and Na2SO4 in solution separately, at increasing HCOOH concentrations. Normalized analyte emission intensities versus HCOOH concentration are presented in Fig. 4b. Similarly, HCOOH improves the emission of Hg, Ag and Pb to different degrees and influences Zn and Cd differently, depending on the anion present in solution. Specifically, HCOOH promotes the emission signal of Zn and Cd when high amounts of SO42− or NO3− are present in the solution. However, the presence of Cl− and HCOOH depresses the emission of Zn and does not enhance Cd emission.
Spatial distribution of analyte emission signals in the presence of HCOOH and alkali and alkali-earth ions
Elemental spatial emission profiles at selected wavelengths of Fe I, In I, Sr I and Li I at several HCOOH concentrations in solutions containing NaCl or MgCl2 are compared with the case without salt. In this study, plasma emission is acquired without any CCD pixel binning, allowing correlation of the height of each CCD row to a height in the axis of the SCGD plasma. The profiles depict the whole plasma, as well as the region 0.5 mm above the tungsten and 0.5 mm below the solution surface. Fig. 5 and 6 show the spatial profiles obtained with NaCl and MgCl2 solutions, respectively, together with those obtained without adding any salt in solution, prepared with and without HCOOH. | ||
| Fig. 5 HCOOH influence on Fe, In, Sr and Li emission intensity spatial distribution with and without Na+ in solution. | ||
 | ||
| Fig. 6 HCOOH influence on Fe, In, Sr and Li emission intensity spatial distribution with and without Mg2+ in solution. | ||
Fe, Sr and Li share similarities in their emission pattern when the solution contains no salt, showing strong emission within 2–3 mm from the anode and another emission zone in the area of 0–2 mm from the anode. In only presents an emission maximum close to the cathode (2–3 mm from the anode). With the addition of Na+, every element emission near the cathode solution surface falls drastically. In this situation, the main contributing region to the total optical emission lies in the 0–2 mm, except for In. Interestingly, the emission in this region actually increases for Sr, Li and In. Moreover, emission around 0 mm shifts beyond the anode for every element, although each one present different behavior. The Fe intensity drops, the Sr intensity barely changes, and the In and Li intensities increase. From the description presented here, it is clear that the addition of Na+ modifies the emission pattern notably. The strong emission depression in the 2–3 mm region produced by the addition of Na+ salt can be ascribed to a localized drop in the excitation temperature.20
The addition of Mg2+ in solution produces more subtle changes, without notable changes in the emission spatial pattern. The 0–2 mm region emission intensity increases, without the shift observed in the Na+ case. The 2–3 mm region suffers element dependent changes: Fe and In emission increases, while Sr and Li emission drops.
When no concomitant ion is present in solution, the addition of HCOOH promotes the emission along the whole plasma column, though to different degrees depending on the specific analyte (see Fig. 5 and 6), and without noticeable changes in the emission spatial distribution. If Mg2+ is present in the solution, together with HCOOH, minor changes are found: the overall emission increases without changing its spatial distribution. When comparing the elemental emission pattern in the presence of Na+, with and without HCOOH, different changes along the plasma column can be observed. Specifically, the emission region closer to the cathode (2–3 mm) increases their intensity when HCOOH is present in solution while the emission maxima situated closer to the anode, in contrast, reduce their intensity, except for In, that barely changes its emission in this region. For Fe, Sr and Li, this generalized emission drop in the 0–2 mm range, which is the main contributor to the overall emission, explains the loss in sensitivity, which is not compensated for by the emission rise in the 2–3 mm region.
The spatial emission profiles of CVG-prone analytes along the SCGD plasma axis are plotted in Fig. S1.† Similarly, the elemental spatial emission patterns are not affected by the addition of Mg2+ alone. When solutions contain Na+, the 0–1 mm emission intensity is shifted to the anode, and the emission at 0.2–3 mm drops. The sole addition of HCOOH does not significantly change the spatial emission profiles, except for an increase in the general intensity. Na+ presence in the incoming solution does not hamper HCOOH enhancement along the whole plasma spatial intensity of Hg, Ag and Pb, although the region close to electrodes benefit more. Zn and Cd emission between 0 and 1 mm has different behavior: Zn emission drops and Cd emission barely changes; the rest behave similarly: 1–2 mm drops and 2–3 mm increases.
Mechanistic insights into the HCOOH-concomitant ion crossed effect
Because the SCGD is a non-uniform plasma, with different plasma conditions (electron densities or temperatures) at different positions, the composition of the incoming solution can determine the excitation conditions (e.g., electron density and temperatures of varied species) of each discharge region. These modifications should lead then to changes in the optical emission distribution. Indeed, the addition of high Na+ amounts in solution produces localized variations in electron density and excitation temperatures.20 In this sense, similar changes in the spatial emission distribution are observed here for all the studied elements when Na+ is presented in the solution. Though to a lesser degree, Mg2+ presence in solution produces similar localized emission depression for Sr and Li. In this sense, Mg2+ may result in alike changes in the plasma parameters, e.g., worse excitation conditions. In addition, the analyte transfer rates might be affected by the presence of these ions as well and can also contribute to the matrix effects leading to sensitivity losses.In contrast, the SCGD excitation conditions seem to be barely affected by HCOOH (without the strong presence of dissolved ions). Additionally, even if there is a strong presence of concomitant ions, the emission spatial distribution patterns are barely modified with the addition of HCOOH. This suggests that only the concentration of the analyte inside the plasma is changed, and the excitation conditions are maintained. In this sense, the effect of HCOOH on the SCGD performance seems to be more related to an enhanced analyte transfer rate that, in the presence of high alkali metal ions, quenches. Some other results aim at this situation. For instance, Webb et al. did not observe any difference between the plasma structures before and after adding 3% HCOOH in the discharge;21 and Yu et al. observed that the excitation temperature and electron density showed relatively constant values with increasing HCOOH concentrations.27
The analyte transfer process is related to three mechanisms: cathodic sputtering, droplet ejection and CVG. The first two mechanisms are quite likely less element dependent than CVG, which is clearly element dependent through the specific volatile species that are formed by the interaction between the reducing radicals and elements. In this sense, the reduction capacity of the plasma–liquid interface is critical for the analyte transfer through this means, which can be promoted by the presence of HCOOH and its decomposed products. In detail, UV radiation and reactive species at the interface where the plasma is in contact with the solution containing this additive can produce reducing radicals through pathways illustrated in Fig. 7.18,27,36,37 These radicals, together with HCOO−, can convert analytes into their volatile species, e.g., neutral atoms, hydrides, and organic-metal compounds, which drive analytes into the plasma to be atomized and excited.
 | ||
| Fig. 7 Probable radical formation pathways at the interface where the plasma is in contact with solution containing HCOOH. | ||
Since the surface tension of the liquid continues to decrease due to HCOOH addition,38 analyte transfer through droplet formation or cathodic sputtering would be improved, even in the presence of NaCl, together with the analyte response. Nevertheless, the results shown here disagree. The presence of high alkali ion concentrations likely deteriorates the sampling efficiency by hampering these CVG processes, for example by preventing the reduction of analytes with low reduction potentials. The reduction potentials for the dissolved elemental ions to be converted into neutral atoms are collected in Table S1.†39 According to them, CVG processes for Li or Sr are quite difficult to achieve and are probably transferred into the plasma by different alternative means. Zn and Cd have higher reduction potentials. Thus, their CVG process can be promoted and its sensitivity is enhanced by adding HCOOH, whenever the ion content is not high. Hg, Ag and Pb have even higher reduction potentials, and their CVG can be promoted by HCOOH addition, even in the presence of considerable amounts of alkali ions in solution. In addition, some reports have proven that the specific volatile species that is formed is highly element dependent, e.g., Hg is usually formed as neutral atoms, while Zn, Cd or Pb volatile species are also in the form of hydrides, which can be marked by the available H.36,40,41 Therefore, it is likely that the reactive species are varied hampering the production of analyte volatile species due to the presence of alkali metal ions, either through reduction or a different pathway. In contrast, the presence of alkaline-earth metals does not have a strong impact here. An exception here should be pointed out for Fe, which, being in the Fe3+ form, should be more prone to CVG than, for example, Pb, according to their reduction potentials. Nevertheless, partial reduction to its Fe2+ form, with quite high reduction potential, might be happening. Once in the Fe2+ form, the reduction potential is lower (−0.45 V). This two-step process might be concerned and Fe signal promotion via HCOOH addition can be concerned, and probably other mechanisms than CVG might be relevant here.
As for the rest of the solution-to-plasma transfer mechanisms (sputtering or droplet ejection), they can also be promoted with the addition of HCOOH. But when added in high ion content solutions, its enhancement disappears for elements less prone to CVG (Fe, Sr or Li), indicating that the efficiency of these mechanisms might be negatively affected, which should also be partially responsible for the less enhancement factors in CVG-prone elements. Of course, further experiments more dedicated to these mechanisms are required to confirm these hypotheses, since the excitation conditions are also affected by the presence of concomitant ions.20
Artificial seawater analysis
SCGD-OES enhancement with HCOOH is tested in terms of the resulting analytical features using a more complex matrix such as artificial seawater solutions (1:10 diluted to attain stable discharge conditions) spiked with certain analyte concentrations (Zn at 1.00 mg L−1, Cd at 1.00 mg L−1, Fe at 10.0 mg L−1, Hg at 10.0 mg L−1, Pb at 10.0 mg L−1, In at 1.00 mg L−1, Sr at 10.0 mg L−1 and Li at 1.00 mg L−1 and Ag at 0.100 mg L−1). Fig. 8 shows the normalized emission intensities of different analytes when these solutions are introduced into the SCGD-OES set-up. Their emission intensity behaviors at increasing HCOOH concentration can be classified into three groups: Fe, Sr and Li intensities present a generalized drop; Zn and Cd intensities level off; and In, Ag, Pb and Hg emission intensities increase (though In and Ag intensity increase is minor). Overall, the variation in the elemental emission caused by HCOOH in artificial seawater resembles the results obtained in solutions containing 59.8 mM Na+ (and Cl−), probably due to the high content of these ions in seawater (45 mM of both Na+ and Cl− in 1:10 dilution seawater and make up 69% of the overall ion content of seawater).
 | ||
| Fig. 8 Normalized emission intensity of several analytes versus concentration of HCOOH added in 1:10 diluted artificial sea water: (a) Fe, Sr and Li; (b) Zn and Cd; (c) In, Ag, Pb and Hg. | ||
In addition, LODs for the tracked elements are estimated in acidified ultrapure water and 1:10 diluted artificial seawater, respectively, including three different concentrations of HCOOH (0, 3 and 7% vol). The results are listed in Tables 3 and 4, respectively. Uncertainties were obtained from the standard deviation of five measurements. In acidified ultrapure water, HCOOH improves the sensitivity and reduces the noise, improving the LODs within 2–10 times. The situation changes in diluted artificial seawater as not all the LODs are improved. In particular, the LODs of analytes with high CVG efficiency are still improved with the addition of HCOOH, though to a lesser degree than in ultrapure water. Also interestingly, the Li LOD improves in diluted seawater, despite the slightly lower sensitivity.
| 1% vol HNO3 and MiliQ water | |||||||
|---|---|---|---|---|---|---|---|
| Element | Analyte conc. (μg mL−1) | 0% vol HCOOH | 3% vol HCOOH | 7% vol HCOOH | |||
| Sensitivity (×104 counts (mL μg−1)) | LOD (μg mL−1) | Sensitivity (×104 counts (mL μg−1)) | LOD (μg mL−1) | Sensitivity (×104 counts (mL μg−1)) | LOD (μg mL−1) | ||
| Zn I | 0.500 | 110 ± 4 | 0.08 | 145 ± 4 | 0.026 | 194 ± 3 | 0.013 |
| Cd I | 0.500 | 81 ± 4 | 0.034 | 135 ± 0.7 | 0.01 | 206 ± 4 | 0.008 |
| Fe I | 5.00 | 3.82 ± 0.12 | 0.75 | 6.44 ± 0.11 | 0.26 | 9.56 ± 0.15 | 0.23 |
| Hg I | 5.00 | 7.0 ± 0.3 | 0.26 | 48.0 ± 0.5 | 0.05 | 49.3 ± 0.8 | 0.03 |
| Ag I | 0.100 | 104 ± 7 | 0.008 | 737 ± 20 | 0.002 | 953 ± 30 | 0.0007 |
| Pb I | 5.00 | 20.7 ± 0.4 | 0.32 | 137.4 ± 0.6 | 0.042 | 179.7 ± 0.8 | 0.018 |
| In I | 0.500 | 905 ± 20 | 0.005 | 1332 ± 7 | 0.005 | 1710 ± 9 | 0.004 |
| Sr I | 5.00 | 48.3 ± 0.4 | 0.05 | 63.6 ± 0.4 | 0.033 | 84.7 ± 1.2 | 0.053 |
| Li I | 0.500 | 230 ± 7 | 0.005 | 321 ± 5 | 0.004 | 359 ± 8 | 0.003 |
:10 diluted seawater and different HCOOH concentrations
| 1% vol HNO3 and 1:10 seawater |
|||||||
|---|---|---|---|---|---|---|---|
| Element | Analyte conc. (μg mL−1) | 0% vol HCOOH | 3% vol HCOOH | 7% vol HCOOH | |||
| Sensitivity (×104 counts (mL μg−1)) | LOD (μg mL−1) | Sensitivity (×104 counts (mL μg−1)) | LOD (μg mL−1) | Sensitivity (×104 counts (mL μg−1)) | LOD (μg mL−1) | ||
| Zn I | 1.00 | 32.6 ± 1.4 | 0.14 | 33.4 ± 0.9 | 0.13 | 26.8 ± 1.6 | 0.037 |
| Cd I | 1.00 | 42.9 ± 1.0 | 0.046 | 37.2 ± 1.2 | 0.031 | 28.3 ± 1.1 | 0.038 |
| Fe I | 10.0 | 1.4 ± 0.3 | 1.2 | 1.18 ± 0.12 | 0.89 | 0.69 ± 0.03 | 2.2 |
| Hg I | 10.0 | 3.75 ± 0.13 | 0.54 | 14.3 ± 0.6 | 0.073 | 12.1 ± 0.5 | 0.11 |
| Ag I | 0.200 | 48.1 ± 1.0 | 0.016 | 55.6 ± 3 | 0.013 | 59 ± 4 | 0.034 |
| Pb I | 10.0 | 8.1 ± 0.5 | 0.9 | 27.1 ± 1.2 | 0.21 | 41.8 ± 0.8 | 0.13 |
| In I | 1.00 | 257 ± 20 | 0.056 | 234 ± 7 | 0.036 | 377 ± 9 | 0.11 |
| Sr I | 10.0 | 50.8 ± 0.9 | 0.11 | 26.9 ± 0.9 | 0.19 | 14.5 ± 0.9 | 1.9 |
| Li I | 1.00 | 274 ± 7 | 0.003 | 244 ± 7 | 0.008 | 255 ± 8 | 0.009 |
Conclusions
Though the addition of HCOOH is widely considered as an efficient way to improve elemental emission signals in SCGD-OES and similar techniques, its effect on SCGD analytical performance strongly depends on the sample matrix and the targeted analyte. HCOOH depresses some analyte emission signals if the solution has a high alkali metal ion content. Some exceptions appear for elements with high reduction potentials (e.g., Hg, Ag, Pb and In), where solution-to-plasma transfer is dominated by CVG. Differently, alkaline-earth metal ion content does not remove the HCOOH enhancement. Additionally, matrix-effect induced by anion content, though present, is less relevant than the nature of the cation in the HCOOH enhancement.Elemental spatial resolved patterns are varied when alkali metals are present in the solution, experiencing emission depression close to the cathode, resulting in the area close to the anode to be the main emission contributor. Similarly, the presence of Mg2+ ions in solution is observed to depress Sr and Li emission close to the cathode, though to a lesser degree. The reported lower excitation conditions in this region, in the presence of Na+, should be responsible for this result, and something similar might be happening in the presence of Mg2+. On the other hand, spatial emission patterns are barely altered by the presence of HCOOH alone, except for an overall increase in the emission intensity. This situation remains unchanged when introducing the solution containing Mg2+. In this sense, the elemental emission enhancements are ascribed to the improved analyte transfer at the plasma–solution interface. However, when Na+ is present in solution, together with HCOOH, the anode elemental emission signal drops for the elements not prone to CVG, while an overall spatial emission increase along the plasma axis is observed for CVG-prone elements. This anode emission signal deterioration in the co-presence of Na+ and HCOOH probably arises from less efficient analyte transfer at the solution–plasma interface, where notable CVG elements suffer less. In this sense, further dedicated experiments studying the analyte abundance in the plasma column are needed. Finally, in analytical terms, adding HCOOH for artificial seawater analysis resembles that in solutions containing a large amount of alkali metals, due, probably, to the high proportion of Na+ in seawater. In general, HCOOH addition seems to be convenient if little alkali ion content is present, and/or if elements prone to CVG are targeted. Otherwise, the enhancement it provides can be concerned and lead to poorer sensitivities.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge funding from the project MCI-21-PID2020-113951GB-I00 and project MCINN-22-TED2021-131619B-I00, funded by the Spanish Ministerio de Ciencia e Innovación and the Agencia Estatal de Investigación. Yinchenxi Zhang appreciates the fellowship support from the China Scholarship Council (CSC).References
- T. Cserfalvi, P. Mezei and P. Apai, J. Phys. D: Appl. Phys., 1993, 26, 2184–2188 CrossRef CAS.
- M. R. Webb, F. J. Andrade, G. Gamez, R. McCrindle and G. M. Hieftje, J. Anal. At. Spectrom., 2005, 20, 1218–1225 RSC.
- X. Peng, X. Guo, F. Ge and Z. Wang, J. Anal. At. Spectrom., 2019, 34, 394–400 RSC.
- J. Yu, S. Yang, D. Sun, Q. Lu, J. Zheng, X. Zhang and X. Wang, Microchem. J., 2016, 128, 325–330 CrossRef CAS.
- J. Yu, X. Zhang, Q. Lu, L. Yin, F. Feng, H. Luo and Y. Kang, ACS Omega, 2020, 5, 19541–19547 CrossRef CAS PubMed.
- J. Wang, P. Tang, P. Zheng and X. Zhai, J. Anal. At. Spectrom., 2017, 32, 1925–1931 RSC.
- J. Yu, L. Yin, Q. Lu, F. Feng, Y. Kang and H. Luo, Anal. Chim. Acta, 2020, 1131, 25–34 CrossRef CAS PubMed.
- Q. Lu, H. Luo, J. Yu, Y. Kang, Z. Lu, J. Li and W. Yang, Microchem. J., 2020, 158, 105246–105253 CrossRef CAS.
- C. Huang, Q. Li, J. Mo and Z. Wang, Anal. Chem., 2016, 88, 11559–11567 CrossRef CAS PubMed.
- P. Zheng, X. Zhai, J. Wang and P. Tang, Anal. Lett., 2018, 51, 2304–2315 CrossRef CAS.
- C. Yang, L. Wang, Z. Zhu, L. Jin, H. Zheng, N. S. Belshaw and S. Hu, Talanta, 2016, 155, 314–320 CrossRef CAS PubMed.
- J. Yu, S. Yang, Q. Lu, D. Sun, J. Zheng, X. Zhang, X. Wang and W. Yang, Talanta, 2017, 164, 216–221 CrossRef CAS PubMed.
- J. Mo, L. Zhou, X. Li, Q. Li, L. Wang and Z. Wang, Microchem. J., 2017, 130, 353–359 CrossRef CAS.
- N. Hazel, J. Orejas and S. J. Ray, Spectrochim. Acta, Part B, 2021, 176, 106040–106054 CrossRef CAS.
- A. J. Schwartz, J. T. Shelley, C. L. Walton, K. L. Williams and G. M. Hieftje, Chem. Sci., 2016, 7, 6440–6449 RSC.
- A. J. Schwartz, S. J. Ray, E. Elish, A. P. Storey, A. A. Rubinshtein, G. C. Chan, K. P. Pfeuffer and G. M. Hieftje, Talanta, 2012, 102, 26–33 CrossRef CAS PubMed.
- T. Cserfalvi and P. Mezei, J. Anal. At. Spectrom., 2005, 20, 939–944 RSC.
- K. Swiderski, A. Dzimitrowicz, P. Jamroz and P. Pohl, J. Anal. At. Spectrom., 2018, 33, 437–451 RSC.
- P. Mezei, T. Cserfalvi and L. Csillag, J. Phys. D: Appl. Phys., 2005, 38, 2804–2811 CrossRef CAS.
- J. Orejas, Y. Zhang, C. Soto-Gancedo, L. J. Fernández-Menéndez, J. Pisonero and N. Bordel, Spectrochim. Acta, Part B, 2023, 209, 106786–106795 CrossRef CAS.
- D. E. Moon and M. R. Webb, J. Anal. At. Spectrom., 2020, 35, 1859–1867 RSC.
- J. Orejas, N. Hazel and S. J. Ray, Spectrochim. Acta, Part B, 2021, 181, 106209–106220 CrossRef CAS.
- P. J. Bruggeman, M. J. Kushner, B. R. Locke, J. G. E. Gardeniers, W. G. Graham, D. B. Graves, R. C. H. M. Hofman-Caris, D. Maric, J. P. Reid, E. Ceriani, D. Fernandez Rivas, J. E. Foster, S. C. Garrick, Y. Gorbanev, S. Hamaguchi, F. Iza, H. Jablonowski, E. Klimova, J. Kolb, F. Krcma, P. Lukes, Z. Machala, I. Marinov, D. Mariotti, S. Mededovic Thagard, D. Minakata, E. C. Neyts, J. Pawlat, Z. L. Petrovic, R. Pflieger, S. Reuter, D. C. Schram, S. Schröter, M. Shiraiwa, B. Tarabová, P. A. Tsai, J. R. R. Verlet, T. von Woedtke, K. R. Wilson, K. Yasui and G. Zvereva, Plasma Sources Sci. Technol., 2016, 25, 53002–53061 CrossRef.
- J. Hu, C. Li, Y. Zhen, H. Chen, J. He and X. Hou, TrAC, Trends Anal. Chem., 2022, 155, 116677 CrossRef CAS.
- P. Jamroz, K. Greda, A. Dzimitrowicz, K. Swiderski and P. Pohl, Anal. Chem., 2017, 89, 5729–5733 CrossRef CAS PubMed.
- K. Greda, S. Burhenn, P. Pohl and J. Franzke, Talanta, 2019, 204, 304–309 CrossRef CAS PubMed.
- J. Yu, Y. Kang, Q. Lu, H. Luo, Z. Lu, L. Cui and J. Li, Microchem. J., 2020, 159, 105507 CrossRef CAS.
- Y.-J. Zhou, J. Ma, F. Li, T. Xian, Q.-H. Yuan and Q.-F. Lu, Microchem. J., 2020, 158, 105224 CrossRef CAS.
- C. G. Decker and M. R. Webb, J. Anal. At. Spectrom., 2016, 31, 311–318 RSC.
- M. Gorska, K. Greda and P. Pohl, J. Anal. At. Spectrom., 2021, 36, 165–177 RSC.
- Q. Xiao, Z. Zhu, H. Zheng, H. He, C. Huang and S. Hu, Talanta, 2013, 106, 144–149 CrossRef CAS PubMed.
- Q. Lu, F.-f. Feng, J. Yu, L. Yin, Y.-j. Kang, H. Luo, D. Sun and W. Yang, Microchem. J., 2020, 152, 104308–104317 CrossRef CAS.
- Y. Zhang, J. Orejas, J. Fandiño, D. Blanco Fernández, J. Pisonero and N. Bordel, J. Anal. At. Spectrom., 2022, 37, 1150–1160 RSC.
- Z. Cai, Z. Ni, M. Gao and Z. Wang, Microchem. J., 2023, 195, 109501–109510 CrossRef CAS.
- R. Shekhar, Talanta, 2012, 93, 32–36 CrossRef CAS PubMed.
- Y. L. Xiaomin Pan, Y. Su, J. Yang, L. He, Y. Deng, X. Hou and C. Zheng, Anal. Chem., 2021, 93, 8257–8264 CrossRef PubMed.
- P. E. S. Jianli Yu, Kinet. Catal. React. Eng., 1998, 37, 2–10 Search PubMed.
- J. G. Pruyne, M.-T. Lee, C. Fábri, A. Beloqui Redondo, A. Kleibert, M. Ammann, M. A. Brown and M. J. Krisch, J. Phys. Chem. C, 2014, 118, 29350–29360 CrossRef CAS.
- Handbook of Chemistry and Physics, ed. W. M. Haynes, CRC press, 95th edn, 2014 Search PubMed.
- Q. T. Zhi-ang Li, X. Hou, K. Xu and C. Zheng, Anal. Chem., 2014, 86, 12093–12099 CrossRef PubMed.
- K. Greda, K. Swiderski, P. Jamroz and P. Pohl, Anal. Chem., 2016, 88, 8812–8820 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ja00372h |
| This journal is © The Royal Society of Chemistry 2024 |