
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Wear metal determination in lubricating oils by reversed-phase dispersive liquid–liquid microextraction and microwave induced plasma optical emission spectrometry†
Carmen
Sáez
 ab,
Dimitar
Stoitsov
c,
Miguel Ángel
Aguirre
*b,
Veselin
Kmetov‡
c,
Clara
Coscollà
d and
Antonio
Canals
*b
ab,
Dimitar
Stoitsov
c,
Miguel Ángel
Aguirre
*b,
Veselin
Kmetov‡
c,
Clara
Coscollà
d and
Antonio
Canals
*b
aPublic Health Laboratory of Alicante, 6, Plaza de España, Alicante, 03010, Spain
bDepartment of Analytical Chemistry and Food Science, University Institute of Materials, Faculty of Science, University of Alicante, P. O. Box 99, 03080, Alicante, Spain. E-mail: aguirre.pastor@ua.es; a.canals@ua.es
cDepartment of Analytical Chemistry and Computer Chemistry, University of Plovdiv Paisii Hilendarski, 24 Tzar Assen Str., Plovdiv, 4000, Bulgaria
dFoundation for the Promotion of Health and Biomedical Research in the Valencian Region, FISABIO-Public Health, 21, Avenida Catalunya, Valencia, 46020, Spain
First published on 15th December 2023
Abstract
A periodic study of engine oil can allow one to anticipate possible breakdowns that a vehicle could have. Due to the complexity of the matrix of this type of sample, a sample treatment prior to analysis is necessary. Analytical chemistry is constantly searching for simpler, more sensitive and environmentally friendly methods. The reversed phase dispersive liquid–liquid microextraction fulfills all the characteristics for the analysis in this type of sample by microwave induced plasma optical emission spectrometry (MIP OES). In this work, the extractant solvent used for the microextraction procedure is a diluted acid solution (i.e., 3 M HCl), which is a less-hazardous solvent, in comparison with other solvents used in microextraction procedures. Besides, it is perfectly compatible with MIP OES. The main experimental factors affecting the extraction of Cr, Cu, Mn, Mo, and Ni (i.e., amount of sample, extractant type, acid concentration, extractant volume, extractant time, and centrifugation time and speed) are optimized using a multivariate analysis consisting of two steps: a Plackett–Burman design followed by a circumscribed central composite design. Under optimum conditions (i.e., amount of sample: 5.9 g; extractant volume: 60 μL; extractant type: HCl; acid concentration: 3 M; extraction time: 3 min; centrifugation time: 3 min; centrifugation speed: 2000 rpm), the proposed analytical method is validated and employed to analyze different samples (i.e., used and unused engine oils). Two calibration methods have been evaluated since matrix effects have been observed in the used engine oil sample. These effects have been eliminated using standard addition calibration obtaining RSD and recovery values in the range of 4–12% and 94–106%, respectively, for samples spiked with 1 μg g−1 of Cr, Cu, Mn, Mo and Ni. Finally, the greenness of this method has been assessed by the Eco-Scale metrics.
1. Introduction
During standard engine operation, wearing is inevitable and mechanical erosion frequently introduces wear metals into the oil circulation.1 These wear products are composed of the same material as the metal surfaces from which they originated. Therefore, a periodic study of engine oil could allow one to anticipate possible breakdowns that the vehicle could have, replacing worn parts before their breakage or irreversible damage. This type of study is part of the maintenance plan of maritime transport, aviation, oil refineries, and mining or chemical plants.2 For instance, for some elements, there are limit values of concentration considered abnormally high (e.g., 4–28 mg kg−1 for Cr, 25–60 mg kg−1 for Cu, 1–3 mg kg−1 for Mn, 4–20 mg kg−1 for Mo, and 1–5 mg kg−1 for Ni) although these limits are very difficult to establish due to the wear metal content and depends on how much the oil has been used.3 However, engine oil analysis is often a difficult task due to its matrix complexity, viscosity and high organic content. Hence, analytical techniques with high sensitivity are needed, and efficient analytical methods have to be developed. Spectrometric techniques such as flame absorption atomic spectrometry (FAAS),1,4–6 electrothermal atomic absorption spectrometry (ETAAS),7–9 microwave induced plasma optical emission spectrometry (MIP OES),10–12 inductively coupled plasma mass spectrometry (ICP-MS),13,14 and inductively coupled plasma optical emission spectrometry (ICP OES),2,6,15–17 have been widely used for engine oil analysis. Among all analytical techniques mentioned above, MIP OES stands out for having advantages such as low acquisition and operating cost, multielemental determination capability and suitable detection limits, and has become a useful analysis tool with high stability and robustness for a variety of sample types.18 However, the lower plasma temperature compared with that of an ICP-based technique makes MIP OES more susceptible to matrix effects, especially with organic matrices.19 Whatever the analytical technique is employed, it is evident that the quantification of low analyte concentration in engine oils is a tough task and the difficulty rises when the concentration decreases. Hence, in engine oil analysis, the direct introduction of the sample in the analytical system is not recommended and a sample preparation must be performed.2 Several sample preparation procedures are proposed in the bibliography, including (i) engine oil dilution,2,15,16,20 (ii) engine oil emulsification,1,2,4,14,20 (iii) microwave digestion,2,21 and (iv) (micro)extraction.7The sample dilution with a miscible organic solvent is an attractive sample preparation procedure since it is quick and simple. Unfortunately, the problems associated with organic vapor loading into the plasma, such as carbon deposit generation and plasma cooling, are greatly aggravated with this procedure.20 Another popular sample preparation method is microwave-assisted acid digestion. Although this procedure completely removes the organic matrix of the sample, it is time-consuming and there is a high risk of sample contamination and volatile analyte losses.20 For engine oil emulsification, the most important drawbacks are the low stability of the emulsions and that the samples must be diluted for the preparation of emulsions, which is detrimental to the limits of detection and quantification.22
Regarding the microextraction procedure, it is an attractive procedure due to its simplicity, high enrichment factor and greenness. Nowadays, laboratories employ analytical methods that ensure compliance with the principles of green analytical chemistry,23 such as reversed phase dispersive liquid–liquid microextraction (RP-DLLME), where the extractant is an aqueous solution, a consolidated green sample preparation. In this mode, a microvolume of a diluted acidic aqueous solution allows analytes to be completely separated from the organic phase with a high enrichment factor.24
In this work, RP-DLLME was proposed as a green sample preparation method for the extraction of heavy metals from engine oils before MIP OES quantification. To the best of our knowledge, this is the first application combining RP-DLLME and MIP OES for the determination of metals in engine oils. Finally, the greenness of this method was confirmed by the Eco-Scale metrics.25
2. Experimental
2.1 Reagents and samples
Calibration standards for Cr, Cu, Mn, Mo, and Ni were prepared from a multielement solution (Conostan S-21, SCP Science, Baie D'Urfé, Canada) using petroleum ether as a solvent (Panreac, Barcelona, Spain). In this work two calibration protocols were used, external calibration for starting experiments and standard addition calibration to correct matrix effects. In both calibration protocols, 5 standards were prepared covering the range of 0.1–3.0 mg L−1.For multivariate optimization, a standard of 2 μg g−1 was used with all analytes and the extractant solvent was prepared from HNO3 65% (w/w) (Merck Pro Analysis, Darmstadt, Germany) or HCl 32% (w/w) (Merck) in MilliQ water (resistivity 18 MΩ cm).
Used engine oil was obtained from a local automobile workshop near the University of Alicante. The used lubricating oils were drained from an automobile engine during a routine service in a garage after use at a certain mileage (20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 km). It was stored in amber glass flasks at 4 °C. The engine oil was diluted gravimetrically with a dilution factor of 10 (DF = 10) with petroleum ether to decrease its viscosity.
000 km). It was stored in amber glass flasks at 4 °C. The engine oil was diluted gravimetrically with a dilution factor of 10 (DF = 10) with petroleum ether to decrease its viscosity.
2.2 RP-DLLME procedure
For analyte extraction, a sample amount of 5.9 g was placed in a 10 mL glass tube with 60 μL of extractant solvent (i.e., 3 M HCl). The mixture was shaken for 3 min with a vortex-type stirrer. Afterwards, the sample was centrifuged for 3 minutes at a speed of 2000 rpm. Finally, the organic phase was removed, and the aqueous extract (i.e., 50 μL) was introduced into the MIP OES (Fig. 1). | ||
| Fig. 1 Scheme of the analytical procedure for Cu, Ni, Mn, Mo, and Cr quantification. | ||
2.3 Instrumentation
An Agilent 4100 (Agilent, Melbourne, Australia) was used in this study. The MIP OES instrument is equipped with a double-pass cyclonic spray chamber (Glass Expansion, Victoria, Australia) and an inert OneNeb® nebulizer (Ingeniatrics, Seville, Spain). Additional details about instrumental parameters of MIP OES are shown in Table 1.| Instrumental parameter | Value |
|---|---|
| Nebulization gas flow rate (L min−1) | 0.70 |
| Liquid flow rate (μL min−1) | 100 |
| Integration time (s) | 1 |
| Replicates | 3 |
| Plasma observation position | 0 |
| Background correction | Automatic |
| Emission lines (nm) | Cr (425.433) |
| Cu (324.754) | |
| Mn (403.076) | |
| Mo (379.825) | |
| Ni (352.454) |
The sample introduction system of the MIP OES spectrometer was modified in order to introduce a low volume of aqueous extract (Fig. S1†). A pipette tip of 200 μL was inserted in the sample capillary of the OneNeb® nebulizer. By using a micropipette, a portion (50 μL) of the aqueous extract is transferred from the bottom of the sample tube to the tip. Afterwards, the spectrometer peristaltic pump transports the portion of the extract to the nebulizer. After the introduction of the aqueous extract, 200 μL of MilliQ water was sequentially introduced in order to rinse the sample introduction system and to prepare it for the next run.
Statistical software (NemrodW® version 2007, LPRAI, Marseille, France) was used for generation of the experimental design matrices and for data processing. Furthermore, the Statgraphics statistical computer package “Statgraphics Centurion XVI” (Warrenton, VA, USA) was used to analyze the desirability function.
3. Results and discussion
3.1 Optimization of RP-DLLME experimental parameters
A multivariate approach was employed to optimize the main experimental factors affecting metal extraction. In order to identify the most important experimental factors affecting the RP-DLLME procedure, a previous screening study (Plackett–Burman design) was carried out. After that, significant factors were optimized by means of a circumscribed central composite design (CCCD). In both cases, the emission signal of each analyte obtained from MIP OES analysis of the resulting extractions was used as the response.A standard containing 2 μg g−1 of the analytes was used as a model sample in all the screening experiments. After the RP-DLLME procedure, the resulting analyte-enriched extract was analyzed by MIP OES, evaluating the emission signal obtained of each analyte.
Fig. S2† shows the Pareto charts obtained from the screening experiments. The length of the bars is proportional to the significance of the factors that affect the emission intensities of the analytes. The bars which extend beyond the dashed vertical line indicate statistically significant factors at 95% probability. Also, the direction of the bar is related to the sign of the effect produced by the corresponding factor. For example, rightward bars indicate a positive effect on the response when a factor value increases, while leftward bars indicate a negative effect during the increase in the same factor value.
In this study, the factors significantly affecting the emission signals at the different analyte emission lines were the type of extractant, sample weight and extractant volume. During the screening step, it was found that when HNO3 is used as the extractant, significantly lower emission signals were achieved for each of the investigated analyte emission lines compared to the emission signals obtained with HCl used as the extracting phase. For this reason, HCl was chosen to be used as the extractant in the next step of the optimization study. Meanwhile, the quantitative factors, sample weight and extractant volume, both significantly affect the responses of Cu, Mn, Mo, and Ni but in opposite directions. Increasing sample weight leads to an increase in the emission signals of the analytes, while increasing the extractant volume causes a decrease in the analyte responses. In order to make the RP-DLLME/MIP OES method possible to be used for multielement determination of Cr, Cu, Mn, Mo, and Ni in lubricating oils, all of the analytes must be determined under a set of the same experimental conditions. For this reason, both the factors, the sample weight and extractant volume, must be optimized simultaneously and finally if it is needed, compromised common optimized conditions must be accepted for Cr, Cu, Mn, Mo, and Ni. As a result, a circumscribed central composite design was used for the optimization of sample weight and extractant volume. Finally, the other factors (i.e., concentration of the extraction phase, extraction time, centrifugation time, and centrifugation speed) were fixed at the most convenient level. The concentration of the extraction phase, extraction time, and the centrifugation time were fixed at their high levels (i.e., HCl 3 M, 3 min, and 3 min, respectively), while the centrifugation speed was fixed at its low level (i.e., 2000 rpm).
Because different optimized values were obtained for the different analytes (Table S5†), a desirability function was employed. This function allows the simultaneous optimization of more than one response, finding the best compromise for all of them.26 The overall desirability was calculated by determining the geometric mean of individual desirabilities, represented on a dimensionless scale (i.e., from zero to one). In this graphic, one represents the wanted response and zero is the undesirable response. Fig. 2 shows the desirability function and its contour plot. As a result, the desirable values for simultaneous extraction of all the analytes were 5.9 g (i.e., sample weight) and 60 μL (i.e., extractant volume). In summary, optimal RP-DLLME conditions were 5.9 g of sample weight, 3 M HCl as the extractant phase, 60 μL of extractant volume, 3 min of extraction and centrifugation time, and 2000 rpm of centrifugation speed.
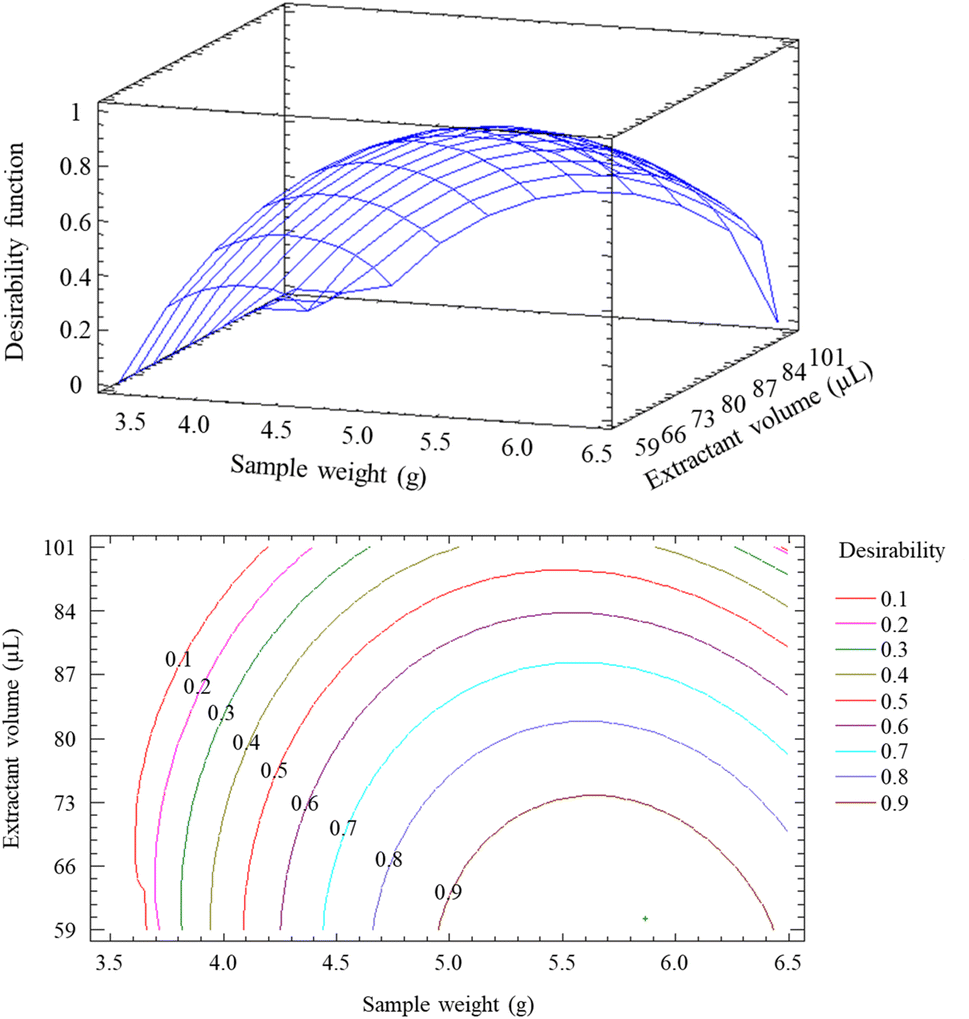 | ||
| Fig. 2 The desirability function and its contour plot. | ||
3.2 Validation of the proposed method
In preliminary experiments, the effect of different matrices (i.e., petroleum ether and real-world matrices) on the analyte extraction was studied. In these experiments, negative matrix effects were confirmed when analyzing different real-world samples (results not given). As a consequence, standard addition calibration was employed to evaluate the analytical figures of merit and assess the applicability of the proposed analytical method under optimized extraction conditions.Table 2 presents the analytical figures of merit of the proposed method using standard addition calibration. First, ten blanks containing the solvent (petroleum ether) and unused lubricating oil (i.e., the first point of the standard addition calibration) were measured after the RP-DLLME procedure and the limits of detection (LOD) and quantitation (LOQ) were estimated. The obtained LOD and LOQ values were estimated in the range of ng g−1 levels. The lowest LOD and LOQ values were calculated for Cu and the highest were obtained for Mo. Also, the highest sensitivity was observed for Cu in comparison with the other analytes (i.e., Cr, Mn, Mo, and Ni). Moreover, the repeatability of the method was tested by measuring two standard solutions containing different analyte concentrations (1 and 2 μg g−1, respectively). An average RSD under 10% was obtained for the developed vortex-assisted RP-DLLME/MIP OES method.
| Analyte | Linear range (μg g−1) | Sensitivitya (cps g μg−1) | r | LODc (ng g−1) | LOQc (ng g−1) | RSDd (%) | RSDe (%) |
|---|---|---|---|---|---|---|---|
| a Slope ± standard error. The calibration standards are five. b Correlation coefficient. The calibration standards are five. c 10 blanks (petroleum ether) measured with an MIP OES after the RP-DLLME procedure. d RSD obtained with 5 standard solutions containing 1 μg g−1 of the analytes. e RSD obtained with 5 standard solutions containing 2 μg g−1 of the analytes. | |||||||
| Cr | 0–3 | 40800 ± 900 |
0.9990 | 21 | 71 | 11 | 5 |
| Cu | 0–3 | 195000 ± 4000 |
0.9991 | 3 | 9 | 10 | 6 |
| Mn | 0–3 | 39200 ± 900 |
0.9990 | 34 | 114 | 7 | 5 |
| Mo | 0–3 | 62600 ± 1400 |
0.9991 | 45 | 149 | 10 | 9 |
| Ni | 0–3 | 31500 ± 400 |
0.9994 | 34 | 112 | 6 | 3 |
The optimized proposed method was applied for the determination of Cr, Cu, Mn, Mo, and Ni in two types of lubricating oils (i.e., unused and used oils) using external calibration (i.e., without matrix effect correction) and using standard addition calibration. For method validation, the following criteria for RSD and recovery values were accepted: RSD < 16% (Horwitz criterion)27 and acceptable recovery percentages (80–110%).28 As can be seen from the results in Table 3, the trueness of the proposed method, expressed as the recovery value, is higher for the determination of all analytes in unused lubricating oil than in the case of used lubricating oil using external calibration. The difference between the obtained recovery values for these analytes, determined, respectively, in unused and used lubricating oil, is proof for an existing difference between the matrix composition of both types of lubricating oils. In this case, the lower recovery values obtained for the analytes in used lubricating oil (Table 3), can be explained by the fact that the dispersion of the extractant phase could be different in both solvents, and this was confirmed by visual inspection. In addition, it must be noted that the concentrations of the analytes found in the original unused lubricating oil were below the LOQ, except for Mo. The higher concentrations of heavy metals found in the original used lubricating oil can be explained by taking into account the wear of metal parts of machines during their operational life when elements such as Cu, Ni, Mo, Mn and Cr can be accumulated in lubricating oil. It is obvious from Table 4 that, in the case of standard addition calibration, higher recovery values were obtained for Cr, Cu, Mo, and Ni in comparison with the external calibration (Table 3). The higher recovery values were a sign of the correction of the matrix effect of the proposed method for Cr, Cu, Mo, and Ni in unused and used lubricating oils. Only the recovery value of Mn was not affected by the matrix effect of the unused lubrication oil matrix if a comparison is made with the external calibration method. In comparison with external calibration, it appears that standard addition calibration can be chosen as a more appropriate calibration strategy for the determination of wear metals in lubricating oils.
| Analyte | Spiked value (μg g−1) | Unused lubricating oil | Used lubricating oil | ||||
|---|---|---|---|---|---|---|---|
| Found concentrationa (μg g−1) | Recovery (%) | Original concentration (μg g−1) | Found concentrationa (μg g−1) | Recovery (%) | Original concentration (μg g−1) | ||
| a DF = 10. | |||||||
| Cr | <LOQ | — | <LOQ | 0.121 ± 0.002 | — | 1.21 ± 0.02 | |
| 1.05 | 0.98 ± 0.02 | 93 ± 2 | 9.8 ± 0.2 | 1.030 ± 0.014 | 86.3 ± 1.1 | 10.30 ± 0.14 | |
| Cu | <LOQ | — | <LOQ | 1.25 ± 0.11 | — | 12.5 ± 1.1 | |
| 1.05 | 0.883 ± 0.010 | 84.1 ± 1.1 | 8.83 ± 0.10 | 1.92 ± 0.05 | 64 ± 12 | 19.2 ± 0.5 | |
| Mn | <LOQ | — | <LOQ | 0.550 ± 0.013 | — | 5.50 ± 0.13 | |
| 1.05 | 0.99 ± 0.03 | 94 ± 3 | 9.9 ± 0.3 | 1.412 ± 0.014 | 81.8 ± 1.0 | 14.12 ± 0.14 | |
| Mo | 0.160 ± 0.008 | — | 1.60 ± 0.08 | 0.99 ± 0.04 | — | 9.9 ± 0.4 | |
| 1.05 | 1.073 ± 0.011 | 86.9 ± 0.2 | 10.73 ± 0.11 | 1.58 ± 0.03 | 57 ± 6 | 15.8 ± 0.3 | |
| Ni | <LOQ | — | <LOQ | 0.121 ± 0.008 | — | 1.21 ± 0.08 | |
| 1.05 | 0.90 ± 0.03 | 86 ± 3 | 9.0 ± 0.3 | 0.97 ± 0.03 | 81 ± 3 | 9.7 ± 0.3 | |
| Analyte | Spiked value (μg g−1) | Unused lubricating oil | Used lubricating oil | ||||
|---|---|---|---|---|---|---|---|
| Found concentrationa (μg g−1) | Recovery (%) | Original concentration (μg g−1) | Found concentrationa (μg g−1) | Recovery (%) | Original concentration (μg g−1) | ||
| a DF = 10. | |||||||
| Cr | <LOQ | — | <LOQ | 0.15 ± 0.03 | — | 1.5 ± 0.3 | |
| 1.05 | 1.02 ± 0.11 | 97 ± 10 | 10.2 ± 1.1 | 1.21 ± 0.08 | 101 ± 9 | 12.1 ± 0.8 | |
| Cu | <LOQ | — | <LOQ | 1.46 ± 0.12 | — | 14.6 ± 1.2 | |
| 1.05 | 1.02 ± 0.12 | 96 ± 12 | 10.2 ± 1.2 | 2.57 ± 0.09 | 106 ± 9 | 25.7 ± 0.9 | |
| Mn | <LOQ | — | <LOQ | 0.81 ± 0.08 | — | 8.1 ± 0.8 | |
| 1.05 | 0.99 ± 0.10 | 94 ± 10 | 9.9 ± 1.0 | 1.92 ± 0.04 | 106 ± 9 | 19.2 ± 0.4 | |
| Mo | 0.16 ± 0.07 | — | 1.6 ± 0.7 | 1.62 ± 0.12 | — | 16.2 ± 1.2 | |
| 1.05 | 1.23 ± 0.10 | 102 ± 9 | 12.3 ± 1.0 | 2.66 ± 0.10 | 99 ± 10 | 26.6 ± 1.0 | |
| Ni | <LOQ | — | <LOQ | 0.28 ± 0.04 | — | 2.8 ± 0.4 | |
| 1.05 | 1.11 ± 0.12 | 106 ± 12 | 11.1 ± 1.2 | 1.371 ± 0.004 | 103 ± 4 | 13.71 ± 0.04 | |
3.3 Application of the Eco-Scale
In order to assess the greenness of the new RP-DLLME/MIP OES method, the Eco-Scale metrics25 was used to calculate the penalty points of the whole analytical process, the results of which are shown in Table S6.† The result obtained in the Eco-Scale metrics was >75, representing an excellent green analysis.3.4 Comparison with other analytical methods
A summary of different analytical methods employed for the analysis of wear metals in lubricating oils can be seen in Table 5. The most commonly used sample treatment is emulsification with a surfactant agent such as Triton. In the case of He et al.14 and Leite et al.,29 the LOD values obtained were higher in comparison with those of the proposed analytical method, even with the use of more sensitive and more expensive analytical instrumentation (i.e., ICP-MS and ETAAS, respectively). In the emulsion procedure proposed by Azcarate et al.,10 the detection technique was the same as the proposed analytical method (i.e., MIP OES), providing higher LOD values. Alternatively, Carballo et al.8 suggested another type of emulsion in which a surfactant is not needed, but a cosolvent is used, creating a three-component solution. After vortex agitation, it is directly introduced in the ETAAS and the LOD obtained is in the range of μg g−1, which is higher than those obtained in the proposed analytical method (i.e., ng g−1). Tekie et al.2 proponed different analytical methods using ICP OES as a detection technique, obtaining the lowest LOD values using microwave digestion mineralization, which is a time-consuming procedure. In summary, the application of the proposed analytical method can enhance the sensitivity of the MIP OES technique obtaining LOD values slightly lower in comparison to those obtained with ICP-MS. Furthermore, the proposed method avoids the use of time-consuming procedures such as mineralization by microwave digestion or emulsification and it solves all problems related with an organic matrix in MIP OES since wear metals were extracted from a complex matrix (i.e., lubricating oil) to a more compatible aqueous solution.| Analytes | Sample preparation | Detection technique | LOD | Reference |
|---|---|---|---|---|
| Cr, Cu, Mn, Mo, and Ni | RP-DLLME vortex assisted with HCl 3 M | MIP OES | 3–45 ng g−1 | This work |
| Ag, Al, Ba, Ca, Cd, Cr, Cu, K, Mg, Mn, Mo, Ni, Pb, Si, Sn, Ti, V, and Zn | Emulsion with xylene and Triton X-114 | MIP OES | 0.46–2.09 μg g−1 | 10 |
| Mg, Cu, Ni, Cu, and Pb | Extraction induced by emulsion breaking with Triton X-114 | ICP-MS | 9–126 ng mL−1 | 14 |
| Ag, Ba, Cu, Mn, and Ni | Microwave digestion with nitric acid | ICP OES | 1.07–18 ng g−1 | 2 |
| Fe, Cr, and Cu | Emulsion with kerosene, Triton X-100, HNO3 and n-propanol | ETAAS | 0.04–1.85 μg g−1 | 29 |
| V, Ni, Cu, Cr, Pb, Mo, and Ag | Emulsion with MIBK, HNO3, HCl and n-propanol | ETAAS | 0.14–0.70 μg g−1 | 8 |
4. Conclusions
For the first time ever, the vortex-assisted RP-DLLME/MIP OES method was proposed for the determination of heavy metals (Cr, Cu, Mn, Mo, and Ni) in used and unused lubricating oils. The extraction method conditions were optimized by a two step experimental design and its analytical figures of merit were obtained and presented. With the optimized proposed method, instrumental limits of detection and quantitation can be achieved in the range of ng g−1 levels for Cr, Cu, Mn, Mo, and Ni. In comparison with the concentrations found for Cr, Cu, Mn, Mo, and Ni in μg g−1 levels in used lubricating oils, the concentrations of these elements found in unused lubricating oil are below the LOQ. The higher metal content in used lubricating oils is a result of the wear of metal parts of machines during their operational life, when accumulation of these elements in lubricating oils can occur. Also, due to matrix effects, better recovery values were obtained for Cr, Cu, Mn, Mo, and Ni in lubricating oils with the standard addition calibration method. Hence, the standard addition calibration method can be chosen as a more appropriate calibration strategy for the analysis of heavy metals in lubricating oils than external calibration.Finally, given the importance of developing environmentally friendly analytical methods, it is noteworthy that this novel and promising method is classified as excellent on the Eco-Scale metrics. Besides, the use of relatively cheap instrumentation such as an MIP OES makes the proposed analytical method affordable for any laboratory.
Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
The authors would like to thank the Spanish Ministry of Science and Innovation (PID2021-126155OB-I00), the Regional Government of Valencia (Spain) (CIPROM/2021/062), and the University of Alicante (UAIND21-03C) for the financial support. 15% of this study is financed by the European Union-NextGenerationEU, through the National Recovery and Resilience Plan of the Republic of Bulgaria, project number BG-RRP-2.004-0001-C01. This article is based upon work from the Sample Preparation Study Group and Network, supported by the Division of Analytical Chemistry of the European Chemical Society. Finally, the authors would also like to thank Agilent Technologies Inc. for the loan of the MIP OES spectrometer and Ingeniatrics for the OneNeb® Series 2 provided. This work is part of the PhD degree of C. S.References
- R. Q. Aucélio, R. M. de Souza, R. C. de Campos, N. Miekeley and C. L. P. da Silveira, The Determination of Trace Metals in Lubricating Oils by Atomic Spectrometry, Spectrochim. Acta, Part B, 2007, 62, 952–961, DOI:10.1016/j.sab.2007.05.003
.
- H. A. Tekie, R. I. McCrindle, P. J. J. G. Marais and A. A. Ambushe, Evaluation of Six Sample Preparation Methods for Determination of Trace Metals in Lubricating Oils Using Inductively Coupled Plasma-Optical Emission Spectrometry, S. Afr. J. Chem., 2015, 68, 76–84, DOI:10.17159/0379-4350/2015/v68a12
- Limit Values for Lubricants, https://en.oelcheck.com/wiki/limit-values-for-lubricants/, accessed 2023-10-27 Search PubMed.
- I. M. Goncalves, M. Murillo and A. M. González, Determination of Metals in Used Lubricating Oils by AAS Using Emulsified Samples, Talanta, 1998, 47, 1033–1042, DOI:10.1016/S0039-9140(98)00186-6
- E. L. C. Silveira, R. C. Coelho, J. M. M. Neto, C. V. R. De Moura and E. M. De Moura, Determinação de Metais Em Óleos Lubrificantes, Provenientes de Motores de Ônibus Urbano, Utilizando a FAAS, Quim. Nova, 2010, 33, 1863–1867, DOI:10.1590/S0100-40422010000900008
- P. Vähäoja, I. Välimäki, K. Heino, P. Perämäki and T. Kuokkanen, Determination of Wear Metals in Lubrication Oils: A Comparison Study of ICP-OES and FAAS, Anal. Sci., 2005, 21, 1365–1369, DOI:10.2116/analsci.21.1365
- M. Á. Aguirre, A. Canals, I. López-García and M. Hernández-Córdoba, Determination of Cadmium in Used Engine Oil, Gasoline and Diesel by Electrothermal Atomic Absorption Spectrometry Using Magnetic Ionic Liquid-Based Dispersive Liquid-Liquid Microextraction, Talanta, 2020, 220, 121395, DOI:10.1016/j.talanta.2020.121395
- S. Carballo, J. Terán, R. M. Soto, A. Carlosena, J. M. Andrade and D. Prada, Green Approaches to Determine Metals in Lubricating Oils by Electrothermal Atomic Absorption Spectrometry (ETAAS), Microchem. J., 2013, 108, 74–80, DOI:10.1016/j.microc.2013.01.002
- J. M. Andrade, S. Carballo-Paradelo, J. Terán-Baamonde, A. Carlosena, R. M. Soto-Ferreiro and D. Prada-Rodríguez, Multivariate Calibration to Implement Green ETAAS Methods when Analysing Cu in Lubricating Oils, Anal. Methods, 2013, 5, 4039–4046, 10.1039/c3ay40410b
- S. M. Azcarate, L. P. Langhoff, J. M. Camiña and M. Savio, A Green Single-Tube Sample Preparation Method for Wear Metal Determination in Lubricating Oil by Microwave Induced Plasma with Optical Emission Spectrometry, Talanta, 2019, 195, 573–579, DOI:10.1016/j.talanta.2018.11.045
- J. Nelson, G. Gilleland, L. Poirier, D. Leong, P. Hajdu and F. Lopez-Linares, Elemental Analysis of Crude Oils Using Microwave Plasma Atomic Emission Spectroscopy, Energy Fuels, 2015, 29, 5587–5594, DOI:10.1021/acs.energyfuels.5b01026
- L. Poirier, J. Nelson, G. Gilleland, S. Wall, L. Berhane and F. Lopez-Linares, Comparison of Preparation Methods for the Determination of Metals in Petroleum Fractions (1000°F+) by Microwave Plasma Atomic Emission Spectroscopy, Energy Fuels, 2017, 31, 7809–7815, DOI:10.1021/acs.energyfuels.7b00654
- Y. Al-Dalahmeh, H. M. Al-Swaidan and A. H. Al-Ghamdi, Combination of Ultrasonication and Induced Emulsion Breaking for Efficient Extraction of Wear Metals from Lubricating Oils with Inductively Coupled Plasma–Mass Spectrometry Determination, J. Anal. Chem., 2019, 74, 71–80, DOI:10.1134/S1061934819010039
- Y. M. He, F. F. Zhao, Y. Zhou, F. Ahmad and Z. X. Ling, Extraction Induced by Emulsion Breaking as a Tool for Simultaneous Multi-Element Determination in Used Lubricating Oils by ICP-MS, Anal. Methods, 2015, 7, 4493–4501, 10.1039/c4ay03024a
-
T. T. Nham and R. M. Bombelka, Determination of Metals in Lubricating Oil by ICP-OES, Agilent Technologies, 1991 Search PubMed
- T. Kuokkanen, P. Perämäki, I. Välimäki and H. Rönkkömäki, Determination of Heavy Metals in Waste Lubricating Oils by Inductively Coupled Plasma-Optical Emission Spectrometry, Int. J. Environ. Anal. Chem., 2001, 81, 89–100, DOI:10.1080/03067310108044347
- M. García, M. Á. Aguirre and A. Canals, A New Multinebulizer for Spectrochemical Analysis: Wear Metal Determination in Used Lubricating Oils by on-Line Standard Dilution Analysis (SDA) Using Inductively Coupled Plasma Optical Emission Spectrometry (ICP OES), J. Anal. At. Spectrom., 2020, 35, 265–272, 10.1039/c9ja00255c
- V. Balaram, Microwave Plasma Atomic Emission Spectrometry (MP-AES) and Its Applications – A Critical Review, Microchem. J., 2020, 159, 105483, DOI:10.1016/j.microc.2020.105483
- M. Y. Jung, J. H. Kang, Y. S. Choi, D. Y. Lee, J. Y. Lee and J. S. Park, Analytical Features of Microwave Plasma-Atomic Emission Spectrometry (MP-AES) for the Quantitation of Manganese (Mn) in Wild Grape (Vitis Coignetiae) Red Wines: Comparison with Inductively Coupled Plasma-Optical Emission Spectrometry (ICP-OES), Food Chem., 2019, 274, 20–25, DOI:10.1016/j.foodchem.2018.08.114
- P. Vähäoja, I. Välimäki, K. Roppola, T. Kuokkanen and S. Lahdelma, Wear Metal Analysis of Oils, Crit. Rev. Anal. Chem., 2008, 38, 67–83, DOI:10.1080/10408340701804434
- V. R. A. Filho and J. A. Gomes, Different Lubricating Oil Treatments for the Determination of Cu, Cr, Fe, Ni, Sb, Pb, and Zn by HR-CS FAAS, Anal. Lett., 2008, 41, 1555–1570, DOI:10.1080/00032710802122115
- P. O. Vicentino and R. J. Cassella, Novel Extraction Induced by Microemulsion Breaking: A Model Study for Hg Extraction from Brazilian Gasoline, Talanta, 2017, 162, 249–255, DOI:10.1016/j.talanta.2016.10.032
- A. Gałuszka, Z. Migaszewski and J. Namieśnik, The 12 Principles of Green Analytical Chemistry and the SIGNIFICANCE Mnemonic of Green Analytical Practices, TrAC, Trends Anal. Chem., 2013, 50, 78–84, DOI:10.1016/j.trac.2013.04.010
- E. Vidal, A. S. Lorenzetti, M. B. Álvarez, C. Domini, M. Á. Aguirre, L. Vidal and A. Canals, Reversed-Phase Dispersive Liquid–Liquid Microextraction for Elemental Analysis of Gasoline by Inductively Coupled Plasma Optical Emission Spectrometry, J. Anal. At. Spectrom., 2021, 36, 2338–2345, 10.1039/D1JA00259G
- A. Gałuszka, Z. M. Migaszewski, P. Konieczka and J. Namieśnik, Analytical Eco-Scale for Assessing the Greenness of Analytical Procedures, TrAC, Trends Anal. Chem., 2012, 37, 61–72, DOI:10.1016/j.trac.2012.03.013
- G. Derringer and R. Suich, Simultaneous Optimization of Several Response Variables, J. Qual. Technol., 1980, 12, 214–219, DOI:10.1080/00224065.1980.11980968
-
The Amazing Horwitz Function, AMC Technical Brief, ed. Thompson M., Royal Society of Chemistry, 2004, vol. 17 Search PubMed
- A. Gustavo González and M. Ángeles Herrador, A Practical Guide to Analytical Method Validation, Including Measurement Uncertainty and Accuracy Profiles, TrAC, Trends Anal. Chem., 2007, 26, 227–238, DOI:10.1016/j.trac.2007.01.009
- C. C. Leite, Determination of Fe, Cr and Cu in Used Lubricating Oils by ET AAS Using a Microemulsion Process for Sample Preparation, Anal. Methods, 2015, 7, 3363, 10.1039/c5ay00128e
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ja00319a |
| ‡ Deceased. |
| This journal is © The Royal Society of Chemistry 2024 |