
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Encapsulation of short-chain bioactive peptides (BAPs) for gastrointestinal delivery: a review
Yoni
Atma
ab,
Brent S.
Murray
 a,
Amin
Sadeghpour
a and
Francisco M.
Goycoolea
*ac
a,
Amin
Sadeghpour
a and
Francisco M.
Goycoolea
*ac
aSchool of Food Science and Nutrition, University of Leeds, Leeds, LS2 9JT, United Kingdom. E-mail: f.m.goycoolea@leeds.ac.uk
bDepartment of Food Science and Technology, Universitas Trilogi, Jakarta, 12760, Indonesia
cDepartment of Cell Biology and Histology, Faculty of Biology, University of Murcia, Campus de Espinardo, 30100, Murcia, Spain
First published on 25th March 2024
Abstract
The majority of known peptides with high bioactivity (BAPs) such as antihypertensive, antidiabetic, antioxidant, hypocholesterolemic, anti-inflammatory and antimicrobial actions, are short-chain sequences of less than ten amino acids. These short-chain BAPs of varying natural and synthetic origin must be bioaccessible to be capable of being adsorbed systemically upon oral administration to show their full range of bioactivity. However, in general, in vitro and in vivo studies have shown that gastrointestinal digestion reduces BAPs bioactivity unless they are protected from degradation by encapsulation. This review gives a critical analysis of short-chain BAP encapsulation and performance with regard to the oral delivery route. In particular, it focuses on short-chain BAPs with antihypertensive and antidiabetic activity and encapsulation methods via nanoparticles and microparticles. Also addressed are the different wall materials used to form these particles and their associated payloads and release kinetics, along with the current challenges and a perspective of the future applications of these systems.
1 Introduction
Short-chain bioactive peptides (BAPs) are protein fragments composed of several amino acids (di-, tri-, tetra-, or oligopeptides) and have the potential to prevent diseases and promote human health. Over 60 therapeutic BAPs have been approved in the United States, Europe, and Japan. Over 170 are in active clinical development, and an additional 260 have been tested in human clinical trials.1,2 Foods are the most promising source of BAPs and are usually 2 to 20 amino acid residues long. They are obtained as a result of chemical synthesis, in vitro enzymatic hydrolysis or microbial fermentation, or generated in vivo by human gastrointestinal enzymes.3 Studies of the origin, identification, isolation, and purification of BAPs and their mechanisms of action and application have increased significantly in the last decade. In vitro and in vivo studies have shown that BAPs can display antihypertensive, antidiabetic, antioxidant, hypocholesterolemic, anti-inflammatory, and antimicrobial actions.4 Antihypertensive and antidiabetic activities include demonstration of the capacity to inhibit angiotensin converting enzyme (ACE) and dipeptidyl peptidase IV (DPP-IV). When used alongside synthetic drugs, the anti-ACE and DPP-IV BAPs have also a shown synergistic effect, further enhancing their therapeutic potential.5A detailed analysis, in which by “high” bioactivity, we consider an IC50 (standard half-maximal inhibitory concentration) <10 μM for ACE inhibition and <50 μM for DPP-IV inhibition, is shown in Fig. 1. This is based on a review of BAPs from various food sources such as milk,6–16 fish,17–32 animals,33–38 plants,39–46 microalgae,47–50 and mushrooms.51
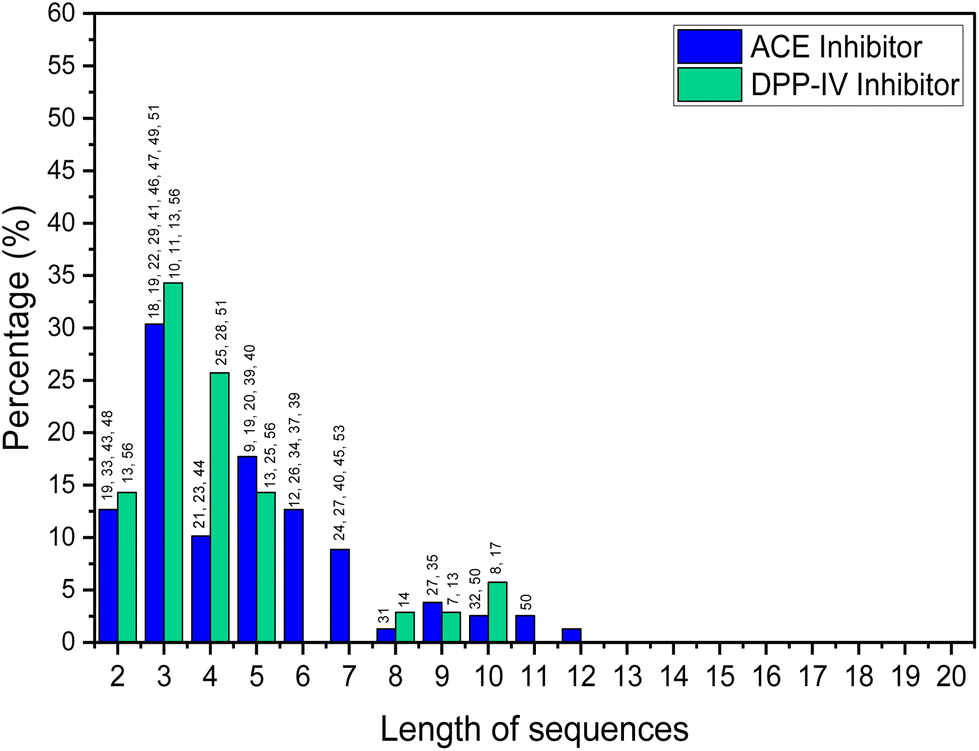 | ||
| Fig. 1 Percentage of food-derived BAPs based on high bioactivities (as ACE and DPP-IV inhibitors). | ||
The analysis shows that the greatest activity is displayed by BAPs of three amino acids. Skrzypczak et al.52 considered peptides of 2–10 amino acids as short-chain BAPs, while other workers only consider those containing 2–4 amino acid residues as short-chain BAPs.53 In general, peptide adsorption is most efficient for single amino acids or di- or tripeptides anyway.54,55 Certainly, the vast majority of food-based BAPs that have high bioactivities seem to be shorter than 10 amino acids residues.
Several recent reviews published over the last five years have delved into the encapsulation of bioactive peptides. For instance, Mirzapour-Kouhdasht et al.'s56 review focuses on enhancing the palatability of BAPs through encapsulation. Similarly, Berraquero-García et al.57 discuss the encapsulation of BAPs using spray-drying and electrospraying techniques. Additionally, Aguilar-Toala et al.'s58 provides an overview of strategies for encapsulating BAPs and presents a comprehensive analysis of the various methods for protection of the bioactivity and encapsulation of bioactive both proteins and peptides. They include a table summarizing encapsulation techniques for bioactive proteins and peptides. Furthermore, McClement's59 review addresses nanoparticle and microparticle systems for encapsulating proteins and peptides, exploring challenges in oral delivery. These reviews, despite being comprehensive and timely, have kept the focus on proteins or protein hydrolysates (peptides) with a molecular weight (Mw) exceeding ∼3 kDa. In the present review, the emphasis is on short BAPs, namely 10 to 50-fold smaller (2 to 20 amino acids) than the large therapeutic peptides and proteins which have been extensively studied and, in many cases, are available as approved drugs (generically known as “biologics”). BAPs have been discovered to exhibit antihypertensive, antidiabetic, anti-inflammatory, antimicrobial and antioxidant, among other activities. Features such as Mw, amino acid sequence, hydrophobicity and polarity, net charge, and ionisation state, dictate their association efficiency upon encapsulation in different type of carriers, as well as their release, bioaccessibility, and bioavailability. The case of low Mw and hydrophilic peptides entails unique challenges associated to their loading/encapsulation in different type of carriers, as further discussed in Section 4.1, but a simple case suffices to this well at the start. O'Neill et al.60 encapsulated a dipeptide and a pentapeptide in the same material but showed that the dipeptide had 3 times lower encapsulation efficiency and 10 times higher release rate. While the amino acid tryptophan (W) exhibits an encapsulation efficiency approximately 1.25 to 1.5 times lower and releases approximately 1.3 times more compared to the dipeptide alanine-tryptophan (AW), both amino acid and dipeptide maintain a consistent trend in charge across varying pH levels. Thus the Mw of BAPs has profound effects, as further discussed in this review.
Gastrointestinal stability is a crucial prerequisite of BAPs with regard to their full realisation of their potential as ingredients in functional foods, nutraceuticals, and food supplements. However, chemical and enzymatic digestion via proteases in the stomach, intestinal lumen, and intestinal brush border leads to degradation of their intact sequences and overall decrease of their oral bioaccesibility and bioavailability, as observed in both in vitro or in vivo studies.35,61–63 To overcome these shortcomings, encapsulation helps to preserve their structural integrity until they reach the desired site of adsorption in the intestinal epithelium. Fig. 2 depicts the major features and layers of factors to account for BAPs and their encapsulation to develop innovative functional food ingredients and supplements. The breadth of this review is on critical analysis of the recently published literature on BAPs, while identifying existing gaps of knowledge and the key research aspects that underpin their potential innovative applications in foods.
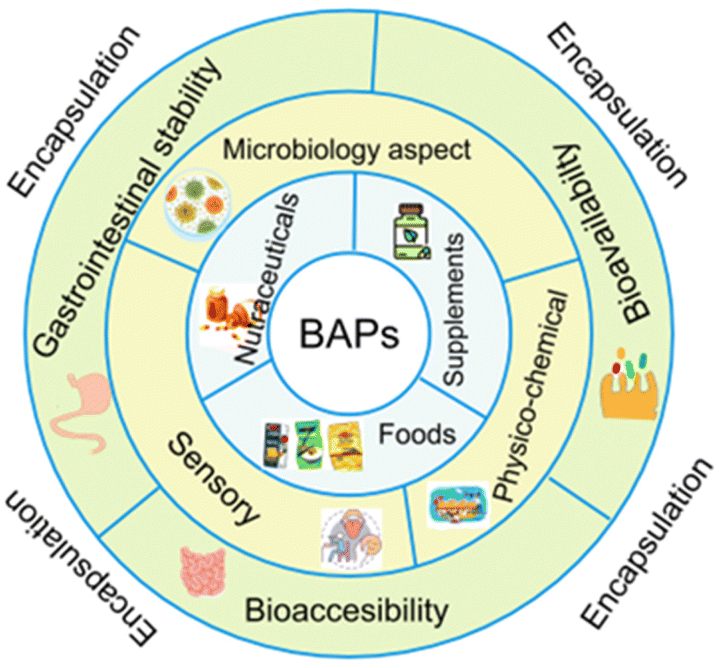 | ||
| Fig. 2 Multilevel pie chart illustrating some substantial aspects of the development of encapsulated BAPs as nutraceuticals, food supplements, and functional food ingredients. | ||
2 Types of short-chain BAPs
Since 2010, only limited studies (ca. 13) have addressed BAP encapsulation for gastrointestinal delivery and our review therefore focuses on this time period. These were focused on di-, tri-, tetra- and up to undecapeptides of molecular weight (Mw) ranging from 273.5 to 1493.7 g mol−1. Encapsulation of BAPs from varying sources with documented bioactivities such as antihypertensive, anti-inflammatory, antioxidant, antiproliferative, hypocholesterolemic, anxiolytic, anticancer and absorption permeation enhancers has been documented. Table 1 summarizes studies conducted on encapsulated BAPs of varying Mw, sequence, and their bioactivities.| Type BAPs | Sequencea | Molar mass (g mol−1) | Bioactivitiesb | Ref. |
|---|---|---|---|---|
| a Amino acids single letter code key: A = alanine; C = cysteine; E = glutamic acid; F = phenylalanine; G = glycine; I = isoleucine; K = lysine; L = leucine; M = methionine; N = asparagine; P = proline; Q = glutamine; R = arginine; S = serine; T = threonine; V = valine; W = tryptophan; Y = tyrosine. b NR = not reported. | ||||
| Dipeptide | AW | 275.3 | NR | 75 |
| FW | 351.4 | NR | 60 and 76 | |
| Tripeptide | IPP | 352.4 | Antihypertensive ACE inhibitor | 77 |
| LKP | 356.5 | Antihypertensive ACE inhibitor | 77 | |
| ECG | 307.3 | Antioxidant | 78 | |
| KPV | 342.4 | Anti-inflammatory | 79 and 80 | |
| YKT | 410.5 | Anticancer | 81 | |
| Tetrapeptide | LQPE | 485.5 | Hypocholesterole-mic | 68 |
| Pentapeptide | LWMRF | 751.9 | NR | 60 and 76 |
| VLPVP | 523.6 | Antihypertensive ACE inhibitor | 82 | |
| Hexapeptide | RLSFNP | 732.8 | Antihypertensive ACE inhibitor | 83 |
| Octapeptide | FCFWKTCT | 1035.2 | Antiproliferative | 71 |
| Decapeptide | YLGYLEQLLR | 1267.4 | Anxiolytic | 84 |
| Undecapeptide | GRKKRRQRRRP | 1493.7 | Permeation enhancer | 85 |
The studies in Table 1 are based on both synthetic and naturally-derived BAPs. Most model studies have used purified synthetic compounds, but some of these were also obtained from food sources. Tripeptide IPP and hexapeptide RLSFNP are each derived from casein64 and lactoglobulin,65 respectively; tripeptide LKP was sourced from bonito fish,66 and pentapeptide VLPVP was expressed in milk by engineered recombinant E. coli.67 Tetrapeptide LQPE was obtained from milk casein hydrolysed using Neutrase®,68 a commercialized brand mixture of proteases. Decapeptide YLGYLEQLLR (α-casozepine) is obtained from whey protein hydrolysis by bovine trypsin.69 Glutathione (γ-glutamyl-cysteinyl glycine, GSH, or peptide ECG) is produced by bioprocessing with Saccharomyces cerevisiae and Candida utilis.70 The tripeptide KPV is naturally produced in the human body, as alpha-melanocyte-stimulating hormone. Meanwhile, the octapeptide octreotide FCFWKTCT is a synthetic somatostatin with a cyclic structure.71 In spite of the demonstrated potent bioactivity of many BAPs, their absolute bioavailability, when administered orally, is very low (≲1%) due to the degradation and fast elimination.72,73 To improve their pharmacokinetics, colloidal carriers have been used.74 Of note, only a relatively small amount of small peptides (<20) have been selected for encapsulation studies for gastrointestinal delivery. These span a wide diversity in terms of their biological sources, methods of production, size and structure, as discussed further on.
Tripeptides IPP and LKP, pentapeptide VLPVP, and hexapeptide RLSFNP have been reported to mediate antihypertensive activity via inhibition of angiotensin-converting enzyme (ACE).77,82,83 Decapeptide YLGYLEQLLR (α-casozepine) has anxiolytic activity.84 Peptide ECG inhibits oxidative damage caused by free radicals from reactive oxygen species (ROS) circulating in the body.78 Octapeptide FCFWKTCT (octreotide) has antiproliferative activity via inhibition of pancreatic exocrine and endocrine secretion.71 In terms of their potential applications, even if the BAP does not need to circulate in the blood, in most cases, they must be absorbed systemically to display their bioactive effects fully. To this end, encapsulation is known to confer BAPs protection against degradation in the gastrointestinal environment, control their release in an intact form at the target sites, and promote their absorption, thus effectively, enhancing both their bioaccessibility and bioavailability. Other examples of encapsulated BAPs include hypocholesterolemic tetrapeptide LQPE and anti-inflammatory tripeptide KPV. LQPE demonstrated the ability to decrease micellar cholesterol solubility and affect the expression of proteins and enzymes related to cholesterol absorption in the small intestine epithelial cells.86 This peptide must be released in its intact form into the epithelial cells. KPV, in turn, has been reported as possessing anti-inflammatory activity via inhibition of proinflammatory cytokine synthesis and is naturally secreted into the colon. Thus, this tripeptide has been studied as a therapeutic peptide for treating inflammatory-related bowel diseases (IBD) in the large intestine.87,88
3 Types of particulate vehicles for encapsulation and their preparation
Colloidal nano- and microparticle carriers have been used to encapsulate therapeutic peptides and this has been comprehensively reviewed elsewhere.58,59,74 The various approaches to associating the peptides include complexation, conjugation, crystallization, or self-assembly through chemical or physical additions.89 Various types of particulate vehicles have been designed for encapsulation, protection and release of short-chain BAPs (hereafter simply referred to as ‘BAPs’). In general, there are two main ways of loading the BAPs into these structures: (i) during the particle formation, sometimes referred to as in situ loading or the internal method and (ii) loading after particle formation, sometimes referred to as pro-loading or the external method. Method (i) typically involves mixing the BAP with particle ingredients in some way possibly influencing the final size and structure of the vehicle. The BAP may simply diffuse into the structure. Method (ii) typically involves placing a suspension of the preformed vehicles in some sort of solution of the BAPs. The latter may also cause some change in the size and structure of the vehicle if it is subject to swelling or shrinkage in the BAP solution. In fact, the size of the vehicle is commonly used to classify different types of delivery vehicles.3.1 Nanoencapsulation
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) P octanolwater partition coefficient < 5.0).90 NPs can be comprised of polysaccharides such as chitosan (CS), alginate (ALG), hyaluronic acid, dextran, pectin, and arabinogalactans plus chemical derivatives of these, or by proteins such as zein, lysozyme, lactoferrin and gelatine, among others. NPs formed from natural biopolymers and macromolecules have been the focus of increasing attention, given the range of desirable properties they exhibit such as biocompatibility, biodegradability, stability and mucoactivity (mucoadhesion or mucodiffusion). In addition, they have the capacity to enhance the bioaccessibility and bioavailability of poorly absorbable payloads by promoting their epithelial absorption. Moreover, NPs based on these components can usually be produced under relatively mild conditions such as aqueous condition and room temperature, thus helping to preserve the bioactivity of delicate biological-type payloads such hormones, antibodies, vaccines, nucleic acids, as well as BAPs.91,92
P octanolwater partition coefficient < 5.0).90 NPs can be comprised of polysaccharides such as chitosan (CS), alginate (ALG), hyaluronic acid, dextran, pectin, and arabinogalactans plus chemical derivatives of these, or by proteins such as zein, lysozyme, lactoferrin and gelatine, among others. NPs formed from natural biopolymers and macromolecules have been the focus of increasing attention, given the range of desirable properties they exhibit such as biocompatibility, biodegradability, stability and mucoactivity (mucoadhesion or mucodiffusion). In addition, they have the capacity to enhance the bioaccessibility and bioavailability of poorly absorbable payloads by promoting their epithelial absorption. Moreover, NPs based on these components can usually be produced under relatively mild conditions such as aqueous condition and room temperature, thus helping to preserve the bioactivity of delicate biological-type payloads such hormones, antibodies, vaccines, nucleic acids, as well as BAPs.91,92
In foods, NPs based on polysaccharides and proteins are known to occur and form during traditional cooking processes of both animal- and plant-based foods, that have been referred to as “incidental NPs”.93 In addition, polymeric NPs may be deliberately manufactured and added to foods as various colloidal dispersions.94 NPs intended for oral delivery of BAPs have been largely prepared via two different protocols: (1) coacervation (ionic gelation),77,78,81,85 and (2) double emulsion and solvent evaporation.79,80 The preparation steps and main features of each of these methods are reviewed below.
With respect to the coacervation route, CS-based NP systems have been prepared from CS81 plus: sodium tripolyphosphate (TPP),77 cyclodextrin (CD),78 and the synthetic methyl acrylate based copolymers termed Eudragit.85 Under the double emulsion and solvent evaporation routes, CS/ALG79 and hyaluronic acid-CS80 NPs have been investigated.
Research conducted by Trapani et al.78 addressed the encapsulation of glutathione (peptide ECG) into CS NPs and CS/CD NPs using two types of CD (α-CD and sulphobutyl-ether-β-CD – termed SBE-β-CD). For the CS NPs, encapsulation was achieved by mixing ECG peptide and CS solutions then adding tripolyphosphate (TPP), to induce spontaneous NP formation via ionic cross-linking between the positive charges on the CS and the negative charges on the TPP. For encapsulation of tripeptides into CS/α-CD or CS/SBE-β-CD NPs, the tripeptide solution was first mixed with α-CD or SBE-β-CD solutions then CS solution added, with TPP added at the end. In some cases, NPs are formed and then further coated with another polymer. For example, Eudragit-coated CS NPs have been used to encapsulate a permeation enhancer undecapeptide alongside insulin (a 51 amino acid peptide). The undecapeptide was dissolved in acetic acid before being mixed with CS and then TPP crosslinking agent was added to form undecapeptide-insulin-CS NPs. Eudragit (dissolved in ethanol) was then slowly injected into the loaded NP dispersion and the ethanol was then removed by evaporation to end up coating the NPs.85 The antihypertensive tripeptides LKP and IPP have also been encapsulated into CS NP systems but a different order of addition of ingredients was used, i.e., the peptides were mixed with TPP first and then CS was added.77 These different orders of addition of ingredients (see Fig. 3) are likely to affect the size, structure, production yields and encapsulation efficiencies (EE) of the NPs.
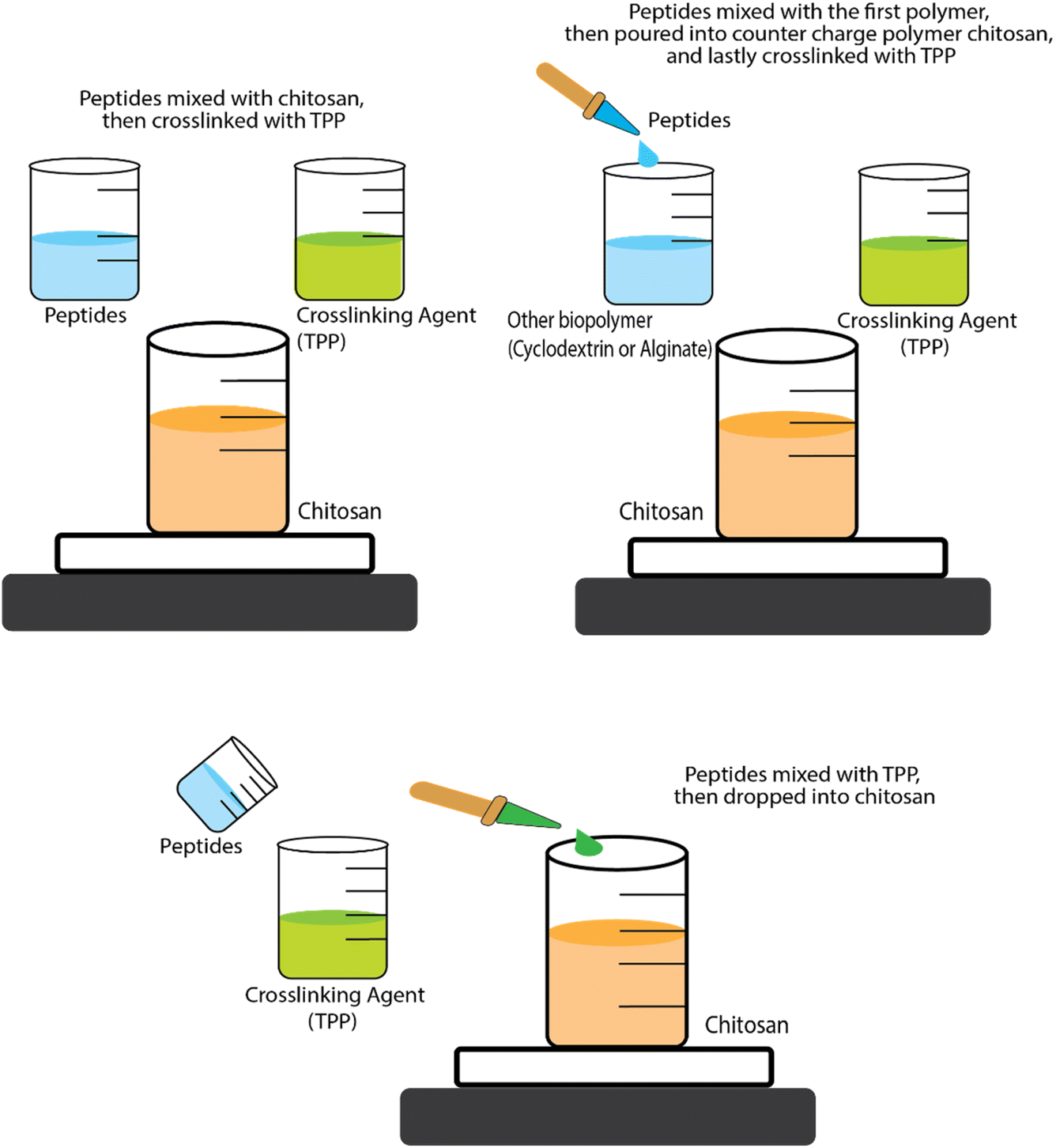 | ||
| Fig. 3 Schematic representation of the three approaches for encapsulation BAPs by ionic gelation/coacervation. | ||
Despite its important role in determining the properties of the formulation, the order of mixing of the components is often neglected. For instance, the size of CS-TPP NPs and the EE has been found to be sensitive to this. The strategic order of mixing might play a role in influencing the size of CS-TPP NPs and EE. Danish et al.77 demonstrated that dissolving the peptide into the TPP solution first, followed by the addition of CS, creating smaller-sized NPs and higher EE compared to the sequence proposed by Trapani et al.78 In the realm of CS-CD-TPP-NPs, no conclusive evidence seems to favour whether it is best to add first the ECG peptide into either CS or CD solution. Notably, despite SBE-β-CD having a molecular weight (Mw) twice as high as α-CD, CS/SBE-β-CD-TPP NPs exhibit a smaller size than both CS/α-CD-TPP and CS-TPP NPs. Additionally, the EE of ECG peptide into CS/SBE-β-CD-TPP NPs surpasses that of CS/α-CD-TPP or CS-TPP NPs by a noteworthy around fivefold. In fact, the accepted principle in ionic gelation is that the mixing order depends on the charge of each molecule. Dissolving the peptide into the phase with a similar charge before introducing the counter charged phase is generally preferred to prevent premature interactions and aggregation driven by full charge compensation and neutralisation leading to loss of electrostatic colloidal stabilisation.
Moreover, the role of the biopolymer structural characteristics such as the degree of acetylation (DA) and pattern of acetylation (e.g. CS), degree of esterification (e.g. pectin), block composition (e.g. ALG), as well as the Mw and polydispersity (Đ) of the various biopolymer families, on the size, surface charge, morphology and peptides' EE (Section 4.2) of the different type of NPs, has been widely neglected.
Double emulsions and solvent evaporation have been used to encapsulate KPV into CS/ALG NPs and hyaluronic acid-CS NPs.79,80 In fact, many hydrophilic drugs have been encapsulated via water-in-oil-in-water (w/o/w) emulsions where the middle non-aqueous phase is a mixture of polymer + solvent/oil mixture. The solvent is finally evaporated away to leave a hardened polymer layer encasing the inner aqueous phase and its bioactive payload. For BAPs, the polymer used is often poly(lactic-acid) (PLA) or poly(lactide-co-glycolide-acid) (PLGA) dissolved in dichloromethane and a final stabilizing layer polyvinyl alcohol (PVA) is used to stabilize the dispersion in water. Such systems are often freeze-dried and then re-dispersed in CS and/or ALG solutions before use. One general disadvantage of the double emulsion route is the lack of natural, biocompatible water-in-oil (w/o) emulsifiers.
Ultimately, the utilization of nanotechnology in food applications necessitates a careful consideration of potential risks to consumers’ health, as emphasized by the European Food Safety Authority (EFSA) Committee. The EFSA guidance documents outline specific criteria for nano-specific risk assessment, particularly focusing on particles smaller than ∼500 nm, with a distribution exceeding 10% below 250 nm.95,96 Key elements in this assessment encompass the physicochemical properties, biokinetics, potential cellular uptake, physiological fate, and distribution of nanoparticles (NPs). The guidance underscores the importance of evaluating the persistence of particulate forms, such as solubility and dissolution rate, along with factors like dispersion, agglomeration, and biological relevance of particle size after reaching the gastrointestinal tract.97,98 Notably, for food composed of natural polymers at the nanoscale, which undergo digestion and assimilation without translocation outside the gastrointestinal tract, and exhibit no adverse effects, a risk assessment similar to conventional food products is deemed appropriate.
In the case of BAPs, liposomes have been studied and loaded with hypocholesterolemic tetrapeptides, antihypertensive hexapeptides, and antiproliferative octapeptides. The BAP-loaded liposomes were further modified by encapsulating with Lactobacillus surface (S-layer) protein to modify their adhesion.68 Alternatively, MLVs can be used to give extra protective layers to the payload.100
BAPs have been loaded into liposomes using both in situ and pro-loading methods.101In situ loading obviously follows the same steps for the liposome preparation but with the peptides present in the aqueous phase during liposome formation. In this way Zhang et al.83 encapsulated RLSFNP in soy lecithin + cholesterol liposomes using ethyl ether as solvent, while Jiang et al.68 encapsulated the LQPE via 1,2-dipalmitoyl-sn-glycero-3-phosphocholine + cholesterol + octadecylamine liposomes, using trichloromethane as solvent.
Parmentier et al.71 encapsulated the synthetic cyclic octreotide (Table 1) into tetraether lipid liposomes using a pro-loading approach. Egg phosphatidylcholine (EPC) dipalmitoyl phosphatidylcholine (DPPC), tetraether lipids from Archaea spp. and cholesterol were used as the lipid materials. After evaporation of the solvent and rehydration to MLVs, the latter were extruded through 400 and 200 nm filter membranes to create the (so-called) liposomes. Loading with octreotide was achieved by mixing with the liposome suspension, followed by freeze-drying and subsequent rehydration.
3.2 Microencapsulation
Microparticles (MPs) may be defined as particles that typically have average diameter sizes from one to several tens of μm. In other words, this class of particles is somewhat larger in size than NPs. Although these larger sizes may give rise to greater problems of sedimentation and colloidal stability, various types have been explored for BAP encapsulation.Another method of producing microgel MPs is via membrane emulsification and extrusion.82 These have been used to encapsulate the ACE inhibitor VLPVP in SA-coated O-carboxymethyl chitosan (O-CMCS) into a w/o emulsion. The SA and peptide in acetate formed the aqueous phase and paraffin oil containing petroleum ether and hexaglycerin penta ester surfactant formed the oil phase. After forcing the water phase to the oil phase through the pores in the membrane to form the first w/o emulsion, a second w/o emulsion containing CaCl2 was then formed and mixed with the first emulsion. The CaCl2 produced transfer of Ca2+ ions to cross-link the O-CMCS in the first emulsion and gel the water droplets. The peptide-containing microgel MPs obtained could then be washed and freeze dried. (The authors also tried extruding SA solution into CaCl2via a thin nozzle then coating the particles with O-CMCS.) Fig. 3 and 4 illustrate the ionic gelation/coacervation and double emulsion/solvent evaporation approaches.
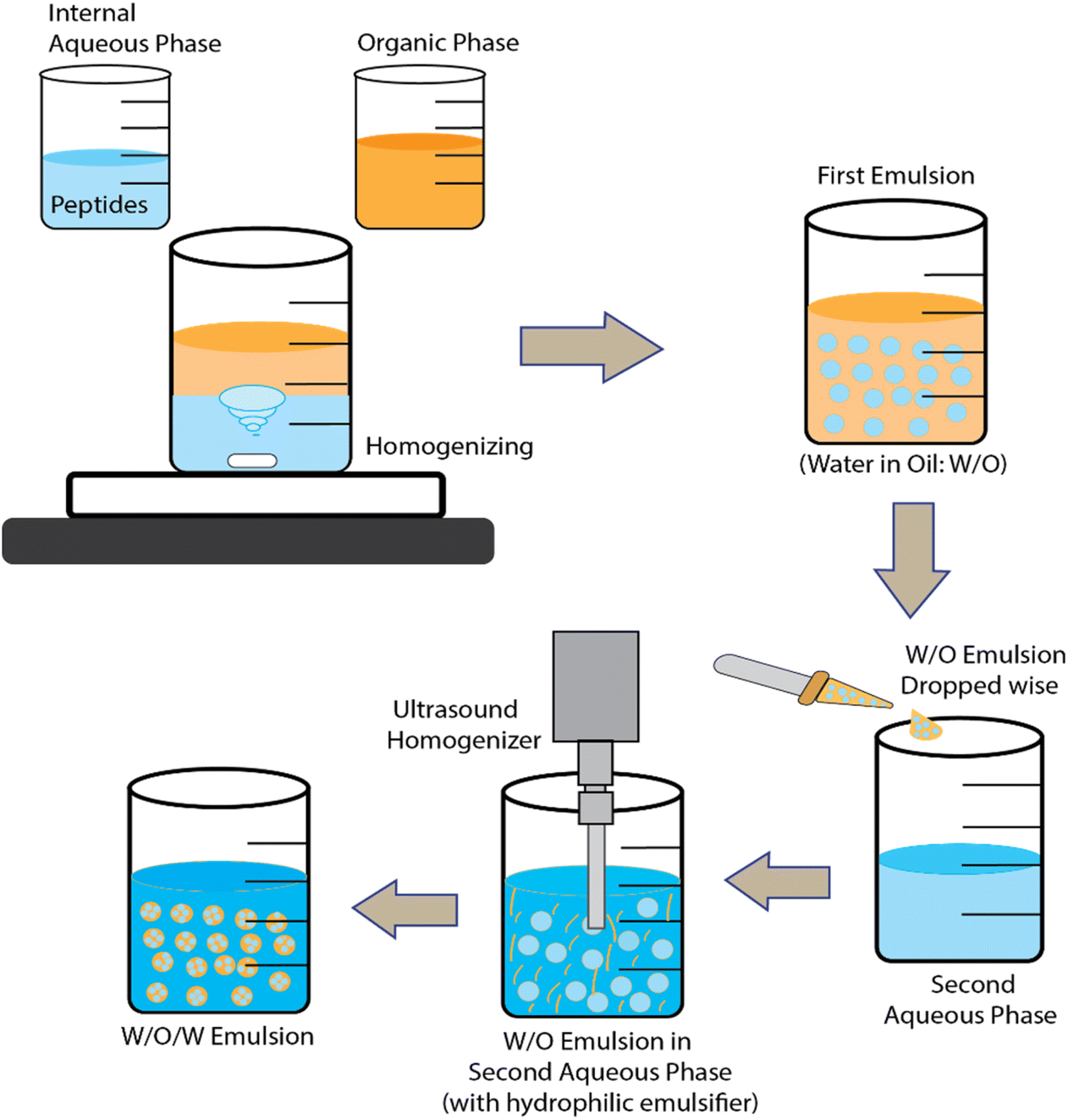 | ||
| Fig. 4 Schematic representation of the methods for encapsulation BAPs via double emulsions and solvent evaporation. | ||
However, among the above techniques of encapsulation of BAPs especially by ionic gelation/coacervation and double emulsion solvent evaporation, there were no studies on gastrointestinal stability of the vehicle or encapsulant, both loaded and unloaded. In a study by Laroui et al.79 BAPs were encapsulated within a double emulsion prepared via the solvent evaporation technique, after which the loaded NPs were embedded in a chitosan (CS)-alginate (ALG) hydrogel. But this group investigated the gastrointestinal stability of cross-linked CS-ALG hydrogel before loading with NPs. The CS-ALG mixed hydrogel, ionically cross-linked solution via Ca2+ and SO42− ions, was assessed for its kinetics of swelling as a measure of its stability in simulated gastric fluid pH 1 to 3 (2 M buffer acetate, 50 μg mL−1 pepsin, 5 μg mL−1 trypsin) and intestinal fluid pH 4 until 6 (0.1 M buffer phosphate, 5 μg mL−1 pepsin, 30 μg mL−1 trypsin). As a result, they concluded that the composite CS-ALG hydrogel was stable at gastric pH 1, 2, and at 3 for 24 h, but shrank at pH 4 and finally collapsed to form a dispersion at pH 5 and 6 after 24 h.
Apart from encapsulation of BAPs, the CS-TPP nanoparticles produced through ionic gelation were assessed for stability in simulated salivary fluid at pH 6.8. Comparative analysis with ALG-NP and pectin-NP (Pec-NP) revealed CS-NP to be the least stable, evidenced by an increase in polydispersity index (PDI) and size (hydrodynamic diameter), accompanied by a reduction in zeta-potential. Prior to integration with artificial saliva, CS-NP exhibited a PDI below 0.2, escalating to 0.4 post-mixing and exceeding 0.6 after a 20-min incubation. The size of the CS-NPs continued to grow progressively after 40 minutes of incubation. Although the zeta-potential remained stable throughout the testing period (0 to 120 minutes), it decreased from +25 mV (pre-mixing) to below +10 mV (post-blending with artificial salivary fluid). These changes were attributed to an increase in pH, leading to CS deprotonation (i.e. pKo ∼ 6.1), and the presence of sulphates, phosphates, and carbonates in the buffer solution, which could bind to chitosan's charged groups, causing particle bridging, reduced stability, and the formation of aggregates and precipitates.102
In one of our studies,103 we explored the stability of CS-TPP-ALG NPs loaded with insulin using the ionic gelation method and its potential efficacy for intranasal delivery of insulin. Remarkably, the colloidal particles exhibited robust stability for an extended period, enduring up to 80 minutes of incubation in acetic buffer with a pH of 4.3. This was evidenced by the absence of any discernible alterations in their hydrodynamic radius, as determined via dynamic light scattering (DLS). At lower pH levels, CS undergoes increased protonation, fostering stronger ionic interactions between CS and its cross linkers such as TPP, as well as oppositely charged polyanionic macromolecules. The heightened protonation of the cationic CS contributes to the reinforcement of these ionic linkages. It is noteworthy that the stability of NPs formed via ionic gelation, such as CS-TPP NPs, is not solely contingent on the ionic strength of the medium; rather, it is intricately influenced by the molar ratio between the involved molecules.
In relation to emulsion-based encapsulation systems, such as water-in-oil-in-water (w/o/w) double emulsions, in terms of water being the dispersed or continuous phase, and situated on the outermost part of the system, this technique shares similarities with the single oil-in-water (o/w) emulsion. The w/o/w encapsulation system is generally employed to address the inefficient encapsulation of highly hydrophilic bioactive compounds that tend to diffuse during formulation. The stability of this system in the gastrointestinal tract is influenced by the type and concentration of stabilizers used in the system.104 Proteins and surfactants typically utilized as stabilizers for o/w emulsion formation have limitations due to the unwanted flocculation and coalescence of oil droplets, possibly resulting from pepsinolysis and the ionic environment during digestion.105
A study conducted by Torres et al.105 analysed the stability of the o/w emulsion-based encapsulation system during in vitro gastric and intestinal digestion using whey protein isolate (WPI) as a stabilizer. In this study, the emulsion system was exposed to simulated gastric fluid (SGF) at pH 3 containing pepsin, simulating the fasted condition of the stomach. Subsequently, it was subjected to simulated intestinal fluid (SIF) at pH 6.8, consisting of bile salt and pancreatin solution. The findings indicated that the emulsion experienced flocculation and coalescence in the SGF, resulting in a slightly more stable emulsion droplet distribution in the SIF, with sizes ranging between approximately 1 to 50 μm, lasting longer than the emulsion mixed with SIF without in vitro gastric pre-digestion. In the latter case, the emulsion destabilized within 30 min (becoming very polydisperse) with a prominent peak in the size range 100 to 1000 μm. During intestinal conditions, bile salts can potentially displace the WPI at the interface. (The size measurements in this study were obtained via confocal laser scanning microscopy – CLSM.)
4 Key characteristics of delivery vehicles
Table 2 lists various encapsulation vehicles, their sizes, methods of formation and encapsulation efficiency (EE) and release characteristics of various BAPs and Fig. 5 also illustrates the typical range of sizes and structures.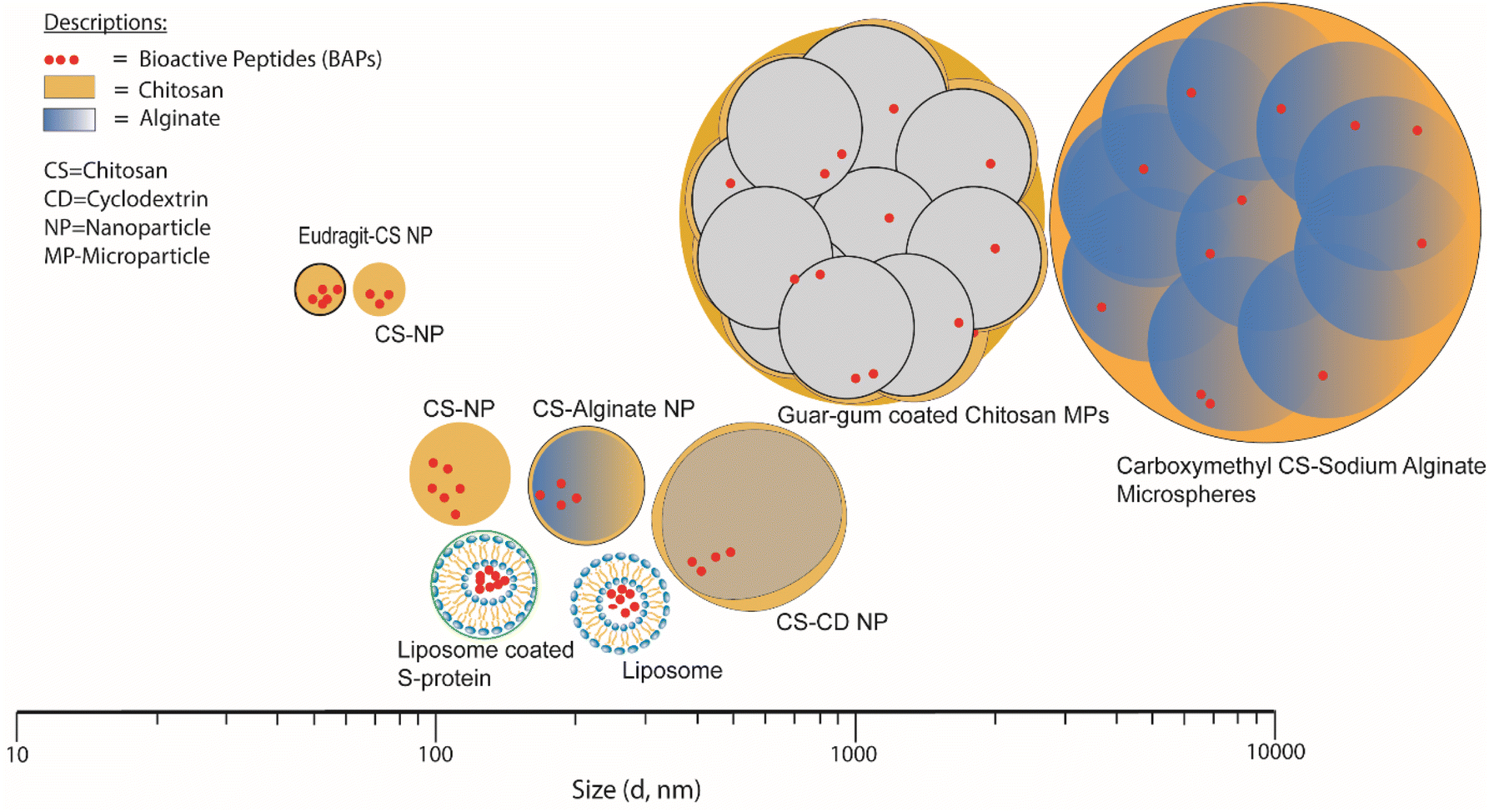 | ||
| Fig. 5 Size distribution of NPs and MPs for short-chain BAPs encapsulation in biopolymer-based systems. | ||
| BAPs payload | Encapsulation wall materials | Encapsulation methods | Encapsulation efficiency (EE %) | Diameter (nm) | Release | Ref. |
|---|---|---|---|---|---|---|
| NR = not reported. | ||||||
| AW | FSP microfibers | Electrospinning | NR | 100–700 and 1000 | 61.5% in SGF after 30 min, and max 71.8% in SIF after 15 min | 75 |
| FW | Whey microbeads | Cold set/ionic gelation | 32–56 | 56% (6 h), ∼80% (Vbead/Vaq < 0.2) | 60 and 76 | |
| IPP | Chitosan NPs (CS: Mw = 110 kDa, DA = 14%) | Ionic gelation/coacervation | 22.6–43.9 | 113–209 | ∼75% in SGF (120 min); >90% in SIF (240 min). Over 60% released within the first 2 h in acidic simulated fluid | 77 |
| LKP | Chitosan NPs (CS: Mw = 110 kDa, DA = 14%) | Ionic gelation/coacervation | 40.9–65.1 | 125–207 | ∼75% in SGF (120 min); ∼90% in SIF (240 min). Over 60% released within the first 2 h in acidic simulated fluid | 77 |
| ECG | Chitosan/CD NPs (CS: Mw = 110 kDa, DA = 14%) | Ionic gelation/coacervation | 7.1–25.1 | 190–500 | pH 1.2: CS/α-CD, 100% (<200 min); CS/SBE-β-CD, ∼25% (<200 min). pH 6.8: CS/α-CD, <60% (<200 min); CS/SBE-β-CD, 100% (<100 min) | 78 |
| KPV | Chitosan/ALG NPs, and hyaluronic acid (HA)-chitosan/ALG NPs (CS: Mw = NR, DA = NR; ALG: Mw = NR, M/G = NR; HA: Mw = 20 kDa) | Double emulsions and solvent evaporation | NR | 273–300 | 25% at pH 6.2 within 30 min. After 30 minutes, release kinetics is more linier about 35% within 30 h | 79 and 80 |
| YKT | Chitosan MPs (CS: Mw = 50–190 kDa, DA = NR) | Ionic gelation/coacervation | 35 | ∼57 | 26% release at first 8 h, 34% (24 h), 43% (48 h), and 97% (264 h/11th day) | 81 |
| LQPE | Liposome coated SLP | Liposome | 89 | 218.2 | liposome-LQPE: 60% after 24 h, SLP-L-LQPE: 40% after 24 h | 68 |
| LWMRF | Whey microbeads | Cold set/ionic gelation | 89–95.1 | NR | <5% (6 h), ∼10% (Vbead/Vaq < 0.2) | 60 and 76 |
| VLPVP | Carboxymethyl chitosan/Na-ALG microspheres (O-CMCS: Mw = NR, DS = 35%; ALG: Mw = NR, M/G = NR) | Membrane emulsification combined ionic gelation | 64.6–87.8 | 5390–43810 |
9.59% at pH 1.2 (2 h), and 87.63% at pH 6.8 (5 h) | 82 |
| RLSFNP | Liposome | Liposome | 67.50 | 199 | Diffuse through dialysis membrane: 75% (1 h), 98.4% (2 h). Cumulative release RLSFNP: 75% (12 h) and 94.92% (24 h). | 83 |
| FCFWKTCT | Tetraether lipid liposomes | Liposome | 13 | 130–207 | NR | 71 |
| YLGYLEQLLR | Guar gum Film-coated chitosan MPs (GG: Mw = NR, CS: Mw = 50–190 kDa, DA = 15–25%) | Ionic gelation/coacervation | 86 | 1810 | <2.5% (4 h) | 84 |
| GRKKRRQRRRP | Eudragit (ES)-coated chitosan NPs (CS: Mw = 100 kDa, DA = 5%) | Ionic gelation/coacervation | 50 | 50.6 | CS NPs: 85% (pH 1.2, 4 h), 40% (pH6.8, 1 h). ES-coated CS NPs: 40% (pH 6.8, 1 h), 50% (pH 6.8, 4 h), 70% (pH 7.4, 1 h), almost completely released (pH 7.4, 4 h) | 85 |
4.1 Association/entrapment efficiency (EE)
Generally, BAP association/entrapment/encapsulation efficiency (EE%) is defined as the % of BAPs added to a system that are successfully entrapped within the vehicles.68,77,78,81,83 EE is therefore determined either by measuring the amount of peptide lost to the delivery vehicle from the original BAP solution, or by extracting the amount of BAP contained within the vehicles post loading, e.g., by dialysis, centrifugation and dissolution of the vehicle. High performance liquid chromatography (HPLC) or reverse phase HPLC (RPHPLC) is by far the most common method to assay for BAP content. It is often difficult to attain EE values close to 100%, whilst it is equally important the vehicle delivers a sufficiently high dose of bioactive at the target site.106As mentioned earlier, loading BAPs in nano- and microparticle carriers deserves special considerations and challenges to identify the most optimal carrier system. Indeed, the EE is influenced by Mw, amino acid sequence, solubility, hydrophobicity, polarizability, net charge and ionisation state of the peptide, as it has been reviewed in a few comprehensive reviews focused on therapeutic peptides.74,107 On the one hand, during their formulation into matrix-type systems such as nanoprecipitates obtained by solvent displacement, they are not retained and are released into the aqueous phase. On the other, when loaded into self-assembled electrostatic polyelectrolyte complexes, the low Mw BAP interacting with one of the polyelectrolyte components is ejected from the system, given that the low charge density and lack of cooperativity of the BAP is outcompeted by the polyelectrolyte bearing the same charge. The loading efficiency of low Mw BAPs is bound to be determined by similar phenomena to those that dictate the association of small drug molecules. Previous studies in model drugs of low Mw (242 to 479 Da) have shown that the hydrophobicity and polarizability, dictated their association efficiency. The more hydrophilic compound with lowest polarizability showed the highest tendency to be displaced by chitosan and associated with efficiencies as low as 12%.108 As shown in Table 2, the BAPs associated in various types of carrier formulations, with efficiencies varying over a wide range 7 to 95%, thus reflecting the complexity of factors at play.
As already described, a relatively wide range of delivery vehicles has been developed, but no single BAP has been tested for EE and release across a wide range of different vehicles: at most, a maximum of two,68,85 and rarely three78 different types of vehicle seem to have been tested for the same BAP. At the same time, each type of vehicle has mostly only been tested with one BAP and occasionally two different BAPs.77 The most optimal type of carrier for a given low Mw peptide must be identified considering the intrinsic characteristics of both the peptide and the carrier components. This makes it hard to compare the efficiency of different systems and the effects that the BAP itself may have on the loaded vehicle size and structure. In almost all cases HPLC or RPHPLC have been used to assay for the BAP content to determine EE. One exception is KPV loading into w/o/w emulsions, where fluorescence spectroscopy was used.79 In general, in situ methods of loading different peptides into different types of vehicle seem to have been more widespread but EE values seem to be lower – exceptions being YLGYLEQLLR into CS + TPP + GG microparticles (86%)84 and LQPE into liposomes (89.0%).68 YLGYLEQLLR will have both a positive and negative charges at neutral pH whereas LQPE has one negative charge.
Nanoparticles formed by CS-TPP systems, where three different tripeptide BAPs (IPP, LKP and YKT) were encapsulated in situ, had EE values ranging between 23 and 65%, depending upon the TPP:CS ratio;77,81 the same vehicle gave only EE = 7% for encapsulation of ECG.78 The reasons for these differences with different peptides are not clear, although it should be noted that LKP and YKT will both have a positive charge at neutral pH whereas ECG will have a negative charge. Using the slightly different CS + α-CD + TPP and CS + β-CD + TPP systems78 for encapsulation of ECG, EE values were 6 and 25%, respectively. The BAP was mixed first with the CD before adding the CS, so that the difference here probably relates to the greater affinity of ECG for β-CD. Using the higher Mw GRKKRRQRRRP and the same general CS + TPP method84 EE = 51%, i.e., higher than most of the other CS + TPP systems tested, so this might point to higher retention of higher Mw peptides using the in situ method, but varying peptide hydrophobicity and charge must also be taken into account – GRKKRRQRRRP has a relatively high number of eight positive charges at neutral pH. The addition of Eudragit polymer to the last system, after the initial encapsulation, resulted in no change in EE.
Turning to liposomal nanoparticles where EE has been fully quantified,68in situ encapsulation gave EE values of 89%, for liposomes uncoated and coated with SLP. It seems obvious that dissolution of all the ingredients in a solvent which is then evaporated off before re-hydration will force intimate contact between the BAP and the encapsulating ingredients, so leading to high EE values. (However, this does not necessarily mean that retention will be high on rehydration – see next section that discusses the BAPs release.) NPs formed via the double emulsion and solvent evaporation route79 similarly gave high EE values of 70 to 71%. On the other hand, pro-loaded microgels have also been shown to give relatively high EE values. For example, WPI microbeads pro-loaded with FW and LWMRF using different volume ratios of microbead particles to BAP solution60,109 gave EE values up to 95.1%. EE increased as the ratio increased and at the same ratio the maximum EEs obtained were 56.3% for FW and 95.1% for LWMRF, the latter having a positive charge. SAL + O-CMC beads formed by the membrane emulsification method82 gave EE as high as 87.8% for VLPVP loading, though here loading was in situ. Interestingly, attempts to form the same types of particle by extrusion though ‘encapsulator’ nozzles and needles gave lower values of in situ EE (maximum values of 65 and 35%, respectively), pointing to the superiority of the emulsification method for this system.
4.2 Size, surface properties and shape (morphology)
Morphology, size, surface charge and shape of NPs and MPs are other key aspects can affect EE and release, as well as the mechanisms of interaction with biological interfaces such as mucosa, epithelia, skin and therefore cellular uptake and intracellular trafficking. These aspects can be controlled to some extent by the overall composition and method of preparation.In terms of size, it is known that only lipid NPs of size ∼30 nm are able to cross the cell membrane by passive diffusion.110 Larger particles rely on active, energy-dependent processes.111 The main mechanism of cellular particle uptake in eukaryotic cells is endocytosis, which is commonly receptor-mediated pinocytosis and phagocytosis. In general, nanoparticles ≲100 nm can be taken up pinocytosis, whilst ∼150–200 nm particles are internalised by receptor-mediated endocytosis. Particles ranging from ∼250 nm to 3 μm are taken up by phagocytic mechanisms.112 Other studies have shown that particles have to be under ∼200 nm in order to diffuse easily through the through the intestinal mucus layers to the epithelial cells.113,114 In terms of shape, rod-like NPs with a higher aspect ratio were shown to be more efficient in penetrating into human cell line (HeLa) and colorectal adenocarcinoma-2 (Caco-2) cells.115
Examples of NPs and MPs that have been more thoroughly investigated in terms of their size distribution include Eudragit-coated CS NPs and CS NPs, whose mean diameters were reported as 51 and 57 nm, respectively81,85 whilst the largest size reported for this type of NP was 500 nm, for CS/CD NPs.78 It is found that in the context of producing CS-based NPs consistently, it is advisable to consider manipulating the initial concentration and the solvent environment as controllable factors.116 The liposomes loaded with BAPs have been reported as having average diameters ranging from ∼130 to 218 nm.68,71,83 Guar film-coated CS MPs loaded with BAPs were ∼1.8 μm in diameter84 and carboxymethylated gum/sodium ALG microspheres loaded with BAPs varied in size from ∼5.4 to 43.8 μm.82 Excluding carboxymethylated gum/sodium ALG microspheres characterised by laser diffraction using equipment such as a Mastersizer (Malvern Panalytical Ltd) to measure size distribution, all the vehicle sizes mentioned were measured by DLS, more suitable for particles of smaller size (<1 μm), to determine the distribution of the hydrodynamic radius (Rh). DLS relies on the measurement of the diffusion coefficient of particles under Brownian motion in a solution in which small particles move rapidly, while bigger particles move slowly. This method is usually applied for particles less than 10 μm. By assuming the particles shapes are spherical or round, the apparent particle diameter is inversely related to the coefficient of diffusion of particles according to the Stokes–Einstein equation.117,118 Therefore, temperature and viscosity of buffer are critical to obtain appropriate results.119 Study conducted by Luque-Alcaraz et al.92 on CS NPs via nanoprecipitation found that the Rh depended on the suspension medium for measurement (methanol, water, or acetone). Furthermore, polydispersity aspects are limiting factors that affect the accuracy and precision of this method.120
As already mentioned, NP and MP surface properties are also known to significantly impact the interaction with the biological interfaces and, therefore cellular uptake.121 Detailed topology of soft surfaces and particle shape can be characterised for fixed or frozen samples via transmission electron microscopy (TEM) and cryo-scanning electron microscopy (SEM),122 provided one is aware of the artefacts that these techniques can introduce.81 Some workers68,78,79,83 have found fine correspondence between the results of DLS and such electron microscopy (EM) techniques. However, it should be noted that DLS averages the distribution as intensity (even though mathematically, can also provide volume or number distributions), and assumes all particles are spherical, whereas in analysing EM images, a number average is obtained, as well as information on particle shape. In principle, atomic force microscopy (AFM) can examine the surface of native particles in an aqueous environment, subject to the limitations of particle immobilisation on a solid surface and the softness of the surface structure versus the AFM cantilever stiffness.123,124 Small-angle X-ray scattering (SAXS) and neutron scattering (SANS) can reveal the external internal structure of particles,125 also hydrodynamic thickness of NPs MPs layers can be investigated118 related to stability and release where these aspects are discussed hereafter. Surface charge is most commonly inferred from electrophoretic mobility and zeta potential measurements.
NPs and MPs loaded with BAPs have been reported to exhibit a regular spherical morphology for liposomes coated with SLP, sodium ALG-O-CMCS microspheres and Eudragit-coated CS NPs.68,82,85 Huang et al.82 studied VLPVP-loaded sodium ALG-O-CMCS microspheres produced via membrane emulsification. They noticed that surface of the microspheres was rough but became smoother as the membrane pore size increased to 5 μm or higher.
4.3 Stability, release, absorption/permeability, and bioactivity of encapsulated BAPs
The bioaccessibility of BAPs, reflecting the proportion of peptides released from their delivery system during digestion for absorption in the gastrointestinal tract to manifest their bioactivities, underscores the critical importance of both BAPs’ stability and the efficacy of the encapsulating agent. By enhancing their stability and bioaccessibility, encapsulation also aims to increase their bioavailability, since many studies have found that the biological activity of non-encapsulated BAPs is significantly reduced after gastrointestinal digestion and in vivo analysis.126–128 In the pharmaceutical sector, the target is to increase the bioavailability of therapeutic peptides to 30–50%.129 This section addresses the intricate interplay surrounding BAPs and their bioaccessibility within the gastrointestinal milieu. Table 2 and Fig. 6 summarise the various encapsulation methods that have been sought to enhance the bioaccessibility of BAPs in the gastrointestinal tract.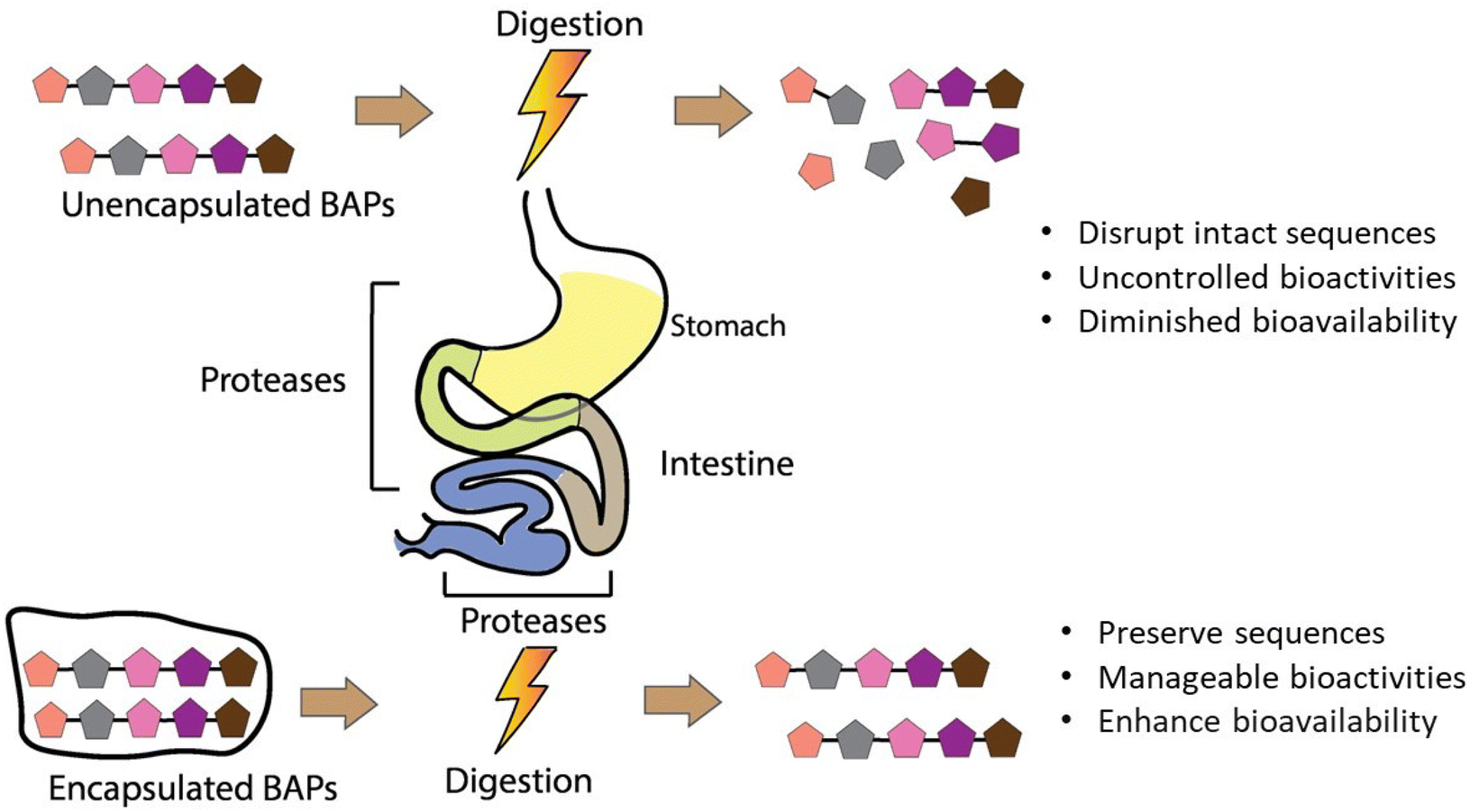 | ||
| Fig. 6 The effect of encapsulation on peptide digestion. | ||
Other notable results include VLPVP encapsulated within O-carboxymethyl CS/SA microspheres, where only 9.6% was released after 2 h at pH 1.2 but 87.6% after 5 h at pH 6.8,82 indicating a system potentially highly suited to gastric protection but intestinal release. Guar film-coated CS MPs also successfully protected YLGYLEQLLR for 4 h in artificial saliva (pH 6.8), giving <2.5% release after 4 h.84 However, all these results also indicate the need for a consistent approach where systems are studied under the same set of physiological conditions.
Yet an alternative approach has focused on the release of small peptides of varying Mw (3.4 to 20 kDa) conjugated in poly(ethylene glycol) diacrylate (PEGDA)-peptide hydrogels.132 To this end, the degradation by matrix metalloproteinase-2 (MMP-2) proteases to drive the release of peptides was harnessed. It was found that the peptidés Mw had a direct effect on the hydrogel mesh size that itself influenced the penetration of the MMP-2 protease into the gel, and the optimal release of the bioactive peptide was for the PEGDA-10 kDa. Further research is needed to investigate these phenomena in matrix-type nano- and microparticles where the peptide is loaded, not as a polymer conjugate, but by virtue of other interactions, where the peptide release is mediated by the degradation of the particles by proteases, such as protein-polysaccharide systems.
Based on the published contradicting and inconsistent results, we cannot conclude firmly about roles of the BAPs sequences, type of encapsulation agents, and physiological medium to their release kinetic and behaviour. Only further systematic studies using identical standardised protocols will help to elucidate in greater detail the role of each these factors.
The apparent permeability coefficient of liposomal RLSFNP liposome was at least 2× higher than free RLSFNP.83 Similarly, the permeability of YLGYLEQLLR loaded into CS MPs across a human adenocarcinoma Caco-2/HT29 colorectal cell line was 4-fold higher than the free decapeptide.84 The results of this study are consistent with the notion that encapsulation under different strategies contributes to enhance the bioavailability of BAPs. More mechanistic studies are needed to elucidate the optimal encapsulation systems for given BAPs, so as to identify the most optimal systems to enhance their absorption.
The assessment of the bioactivity of the tripeptides LKP and IPP subsequent to encapsulation involved measuring the loaded-NPs capacity to inhibit angiotensin-converting enzyme (ACE) during the conversion of the substrate hippuryl-L-histidyl-L-leucine (HHL) to the product hippuric acid (HPA). This methodology entailed quantifying the total HPA concentration in the reaction solution via HPLC. A diminished concentration of HPA in the reaction solution signified elevated ACE inhibitory activity. In turn, the assessment of LKP and IPP activity post-release entailed sonication of the loaded-NPs, followed by centrifugation. The quantification of BAPs in the supernatant was conducted using HPLC, and the BAPs were subsequently introduced into a reaction solution containing ACE enzyme and HHL. The total HA content in the reaction solution was analysed using HPLC-UV/Vis.77
In the study conducted by Huang et al.82 encapsulating antihypertensive BAPs into microspheres, freeze-dried loaded microspheres were added into borate buffer, followed by sonication for one hour. The quantification of peptide release was achieved through HPLC, and ACE inhibitory activity was determined using spectrophotometry, following the method outlined by Cushman and Cheung (1971).133
4.4 Cytotoxicity
Establishing any cytotoxicity of NPs/MPs is of paramount if particular encapsulating vehicles are to be used in practice. Caco-2, HT29-MTX, and IEC6 cell lines have been used to mimic gastrointestinal cells, and HepG2 cells were applied to mimic liver exposure. There are three commonly used assays to determine the cell viability of NPs/MPs (with or without loaded BAPs), namely 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl-2H-tetrazolium bromide (MTT) or 3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium (MTS), cell counting kit-8 (CCK-8), and water-soluble tetrazolium salt (WST-1) assay.CS-NPs loaded with IPP and LKP (at 1–10 mM) showed no effects on cell viability (Caco-2 and HepG2).77 Similarly, KPV encapsulated into (HPA)-CS/SA NPs did not affect cellular death rates of both Caco2-BBE and IEC6 cell lines over 72–80 h exposure.79,80 Membrane integrity of Caco-2 cells was not significantly affected after treatment with Eudragit-CS NPs loaded with GRKKRRQRRRP.85 Liposome loaded hexapeptide RLSFNP also displayed no cytotoxicity effect on Caco-2 cells.83
MPs loaded with decapeptide YLGYLEQLLR showed no cytotoxicity effects either, with a cell viability of about 75–99% after incubation with 0.1–5 μg mL−1 of peptides on MPs solution.84 Thus there seems very little evidence that these types of biopolymer-based NPs and MPs themselves have strong cytotoxic effects, pointing again to the advantage of basing delivery vehicles based (mostly) on food-derived materials.
5 Summary – gastrointestinal delivery vehicles for BAPs: challenges and opportunities
Overall, BAP release from the various NPs and MPs studied so far seems to be in the range 60–75% in simulated gastric fluid (SGF) but higher in (e.g., 90%) in simulated intestinal fluid (SIF). Obviously, these rates could be advantageous or disadvantageous depending on the actual desired site of action, but a general positive aspect is that most vehicles seem to give sustained (not burst) release in both SGF and SIF, e.g., typically 2 h in SGF and 4 h in SIF for full release. Food in the human digestive system needs around 2–6 h for full digestion and adsorption. Since more than half of encapsulated BAPs are released under gastric conditions, this might give a chance for pepsin to hydrolyse them and change their original amino sequence before reaching intestinal brush border. Pepsin is an enzyme that hydrolyses all peptide bonds between aromatic amino acid residues.134 Moreover, achieving even 90% release of BAPs in the intestine does not guarantee their successful arrival at target sites, because they encounter further challenges from other proteases such as trypsin, chymotrypsin, carboxypeptidase A, elastase and carboxypeptidase B. Trypsin predominantly cleaves peptide bonds involving the carboxyl groups of arginine (R) or lysine (K).135 Chymotrypsin targets peptide bonds formed by the carboxyl groups of Tyr (Y), Phe (F), Trp (W), and Leu (L).136 Carboxypeptidase A selectively cleaves C-terminal peptides, with increased hydrolysis rates observed when the side-chain R is aromatic or branched aliphatic, such as +Phe (F), +Tyr (Y), +Trp (W), +Leu (L), or + Ile (I).137 Furthermore, additional proteases are secreted by the brush border of the small intestine, including aminopeptidases and dipeptidases,54 potentially hydrolysing BAPs further. Based on this review, CS and CS-ALG NPs seem to give a prolonged release of BAPs under gastrointestinal conditions (see Table 2) no matter whether or not the BAPs encapsulated might be susceptible to any of the above enzymes. This biopolymer combination is therefore not particularly disrupted by human gastric and intestinal enzymes and of course inherently is not susceptible to protease action. One of core concept of encapsulation for food application is protecting bioactives during gastric digestion and release them on intestine at certain specific rate (fast/sustained/control) to maximize bioavailability.138A second critical challenge is creating encapsulation vehicles in the appropriate size range. The pore size of the mucus layer covering the epithelial cells in the intestine is ∼150–200 nm, so sizes smaller than this are required to enable vehicles to freely penetrate this layer. It seems challenging to create biopolymer-based vehicles that have diameters less than ∼200 nm that possess at least two or three biopolymer layers surrounding the BAPs, which of course is partly a reflection of the dimensions of the biopolymer molecules themselves. On the other hand, partly synthetic Eudragit-CS NPs can have sizes less than 60 nm,85 whereas purely biopolymer-based NPs seem to have the advantage of low toxicity. Thus, a balance may have to be reached between the use of synthetic versus natural biopolymers with regard to controlling and minimizing NP size and toxicity. NPs that are able to reach the intestine and penetrate through the epithelial cell membrane must undergo the nano-specific risk assessment stages as discussed earlier. Mucosa-adhesive carriers, such as CS, ALG, or their composite on the vehicle surfaces, not only augment the absorption and delivery of particles within the mucosa but also present attributes of biodegradability and minimal toxicity.103,139
There seems to have been a limited number of studies that have fully characterized BAP stability during gastrointestinal simulation fluids and the consequent effects on the bioavailability and bioactivity of any encapsulated BAPs, so that much more work is required in this area. In particular, attention needs to be paid to the internal structure of the vehicles and the location of the BAPs, plus the effects that this has on BAP bioactivity. In this respect, there are opportunities for further exploration of structures based on more than two biopolymer types, for instance, a third biopolymer (or other) type of coating layer. For example, possible ALG/CS/ALG or CS/ALG/CS combinations. Another potentially promising scenario is using a lipid to protect the BAPs inside the biopolymer layers.59 This creates steric hindrance so that the peptides are prevented from diffusing into the outer aqueous biopolymer layers.
Most of previous studies used buffers during in vitro gastrointestinal simulation without added proteases nor other attributes (e.g., bile salts, lipases) to mimic human gastrointestinal digestion. Only a few studies have applied in vivo models. However, given the elevated costs of in vivo digestion, in vitro approaches are preferred. However, they are complicated to set up and recapture the physiological context. Numerous studies have documented the use of in vitro static and dynamic digestion models. For examples, Chen et al.140 developed a simple model device to imitate dynamic physiological processes within gastrointestinal tract in vitro. They examined the influence of gastric juice ratio, pH, fluid flow, and pepsin concentration on peanut digestion. By this model, the parameters that influence gastric digestion like digestion time, enzyme concentration, food to gastric juice ratio, shearing and hydrodynamic flow were controllable and adjustable easily. Kozu et al.141 developed the Gastric Digestion Simulator (GDS) for in vitro gastric digestion, focusing on replicating peristalsis effects. This device observes food disintegration and gastric content changes. Using two tofu types as food models, it unravelled stomach digestion mechanisms. Egger et al.130 aimed to standardize in vitro simulation protocols, emphasizing enzyme presence. In vitro models, despite human studies being gold standards, offer advantages like speed, cost-efficiency, ethics, and scalability, especially for screening. They replicate oral, gastric, small intestinal phases, and sometimes large intestinal fermentation, controlling factors like enzymes, pH, duration, and salts.131 Some computer models simulate digestion dynamically,142 but many remain static.143 These approaches can investigate bioactive compounds (e.g., BAPs) in particles or functional food models.
All in all, much further work is required in order to optimize encapsulation vehicles for BAPs that provide them with adequate protection during gastrointestinal digestion yet, provide adequate release characteristics that ensure the required activity of the BAPs (Table 3).
| BAPs payloads | Encapsulation materials | Remarks on essential characteristics | Ref. |
|---|---|---|---|
| AW | FSP microfibers | Stability: microfiber degraded slowly by low activity SGF until 24 h (compared to after 30 min with higher amount of pepsin); microfiber has low activity in SIF, only 50% (after 24 h) (compared 100% in the higher amount of pancreatin) | 75 |
| IPP | Chitosan nanoparticles | Cytotoxicity: IPP loaded CS NPs were tested on Caco-2 and HepG2 cell lines, and no significant decrease of cell viability was found compared to negative control analysed by MTS viability assay. Bioactivity: 84% (after encapsulation); 82% (controlled release) | 77 |
| LKP | Chitosan nanoparticles | Cytotoxicity: LKP loaded CS NPs were tested on Caco-2 and HepG2 cell lines, and no significant decrease of cell viability was found compared to negative control analysed by MTS viability assay. Bioactivity: 85% (after encapsulation); 83% (controlled release) | 77 |
| ECG | Chitosan/CD nanoparticles | Absorption/Permeability: CS/α-CD: 1.42 times higher (on proximal segment) and 1.3 times higher (on distal segment). CS/SBE-β-CD: 1.2 times higher (on proximal segment) and 1.8 higher (on distal segment). All of these compared to unencapsulated tripeptide ECG | 78 |
| KPV | Chitosan/ALG nanoparticles, and hyaluronic acid (HA)-chitosan/ALG nanoparticles | Absorption/Permeability: qualitative data shown that surface functionalization with HA can promote the cellular uptake efficiency of NPs by targeting cells. Bioactivity: HA-KPV-CS NPs can enhance the healing of the wounded colonic epithelial layer. Cytotoxicity: concentration 1 mg mL−1 do not cause cellular death of Caco2-BBE and IEC6 cell line over 72–80 h. Cell treated with HA-KPV-NPs had significantly lower TNF-a mRNA expression levels compared with those treated with KPV-NPs | 79 and 80 |
| YKT | Chitosan nanoparticles | Cytotoxicity: viability (control). 100%. (1) 0.1–2.5 mg mL−1: 62.72–79% (after 24 h); (2) 0.1–1.0 mg mL−1: 34–38% (after 48 h). | 81 |
| LQPE | Liposome coated Lactobacillus S-layer protein (SLP) | Absorption/Permeability: all of the transhipment significantly increased. Compared L-LQPE, L-LQPE coated by SLP significantly increases the concentration of LQPE on the Basolateral side with initial concentration of 2 and 6 mmol L−1. Stability: LQPE Residue. Intestinal juice: 74.5% (SLP-L-LQPE), 55% (L-QPE). Gastric juice: 68% (SLP-L-LQPE), 50% (L-LQPE). | 86 |
| VLPVP | Carboxymethylated gum/sodium ALG microspheres | Bioactivity: 48.2% (before encapsulation); 45.2% (after encapsulation) | 82 |
| RLSFNP | Liposome | Absorption/Permeability: both RLSFNP and RKSFNP liposomes exhibited red fluorescence, indicating cellular uptake. The transport of the hexapeptide RLSFNP in liposome increased significantly in comparison to the peptide alone. Cytotoxicity: MTT Results indicated that the RLSFNP display no cytotoxicity in Caco-2 Cells | 83 |
| FCFWKTCT | Tetraether lipid liposomes | Bioavailability:>750 pg mL−1 in plasma, after 100 μg dosage | 71 |
| YLGYLEQLLR | Guar film-coated chitosan MPs | Absorption/Permeability: MPs loaded peptide in Caco-2/HT29-MTX: permeability >70% (120 min) and ∼50% (60 min). Peptide alone <60% (120 min) and <30% (60 min). Cytotoxicity: cell viability 75–99% (MTT assay) (0–5 μg mL−1) | 84 |
| GRKKRRQRRRP | Eudragit (ET)-coated chitosan nanoparticles | Cytotoxicity: membrane integrity of Caco-2 was not destroyed after treatment with ES-Tat-cNPs and ET-cNPs. There were not toxic toward Caco-2 cell using CCK8 assay (100% of cell viability after treatment) | 85 |
Abbreviations
| ACE | Angiotensin converting enzyme |
| AFM | Atomic force microscopy |
| ALG | Alginate |
| AP | Apical |
| BAPs | Bioactive peptides |
| BL | Basolateral |
| CCK-8 | Cell counting kit-8 |
| CD | Cyclodextrin |
| CS | Chitosan |
| DA | Degree of acetylation |
| DLS | Dynamic light scattering |
| DPP-IV | Dipeptidyl peptidase IV |
| DPPC | Dipalmitoyl phosphatidylcholine |
| EE | Encapsulation efficiency |
| EFSA | European Food Safety Authority |
| EPC | Egg phosphatidylcholine |
| ET | Eudragit |
| GDS | Gastric digestion simulator |
| GG | Guar gum |
| HA | Hyaluronic acid |
| HHL | Hippuryl-L-histidyl-L-leucine |
| HPA | Hippuric acid |
| MLVs | Multilamellar vesicles |
| MPs | Microparticles |
| MTS | 3-(4,5-Dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium |
| MTT | 3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyl-2H-tetrazolium bromide |
| NPs | Nanoparticles |
| O-CMCS | O-carboxymethyl chitosan |
| o/w | Oil-in-water emulsion |
| PDI | Polydispersity index |
| PEGDA | Poly(ethylene glycol) diacrylate |
| PLA | Poly(lactic acid) |
| PLGA | Poly(lactide-co-glycolic acid) |
| PVA | Polyvinyl alcohol |
| SA | Sodium alginate |
| SANS | Small angle neutron scattering |
| SAXS | Small angle X-ray scattering |
| SBE-β-CD | Sulphobutyl ether-β-cyclodextrin |
| SEM | Scanning electron microscopy |
| SLP | Lactobacillus S-layer protein |
| SGF | Simulated gastric fluid |
| SIF | Simulated intestinal fluid |
| TEM | Transmission electron microscopy |
| TPP | Tripolyphosphate |
| w/o | Water-in-oil emulsion |
| WPI | Whey protein isolate |
| WST-1 | Water-soluble tetrazolium |
Author contributions
YA: conceptualization, writing – original draft, visualization; BS: conceptualization, writing – review and editing, supervision; AS: writing – review and editing, supervision; FG: conceptualization, writing – review and editing, supervision.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors are grateful for the funding provided by the Center for Higher Education Funding (BPPT), the Ministry of Education, Culture, Research and Technology of the Republic of Indonesia, Ref. Number 1145/PLPP.1/BPI.LG/III/2023 and Indonesian Endowment Fund for Education (LPDP) in the form of a full scholarship for the PhD study of Yoni Atma.References
- L. Wang, N. Wang, W. Zhang, X. Cheng, Z. Yan, G. Shao, X. Wang, R. Wang and C. Fu, Therapeutic peptides: current applications and future directions, Signal Transduction Targeted Ther., 2022, 7, 48 CrossRef CAS PubMed.
- J. L. Lau and M. K. Dunn, Therapeutic peptides: historical perspectives, current development trends, and future directions, Bioorg. Med. Chem., 2018, 26, 2700–2707 CrossRef CAS PubMed.
- P. J. Patil, M. Usman, C. Zhang, A. Mehmood, M. Zhou, C. Teng and X. Li, An updated review on food-derived bioactive peptides: focus on the regulatory requirements, safety, and bioavailability, Compr. Rev. Food Sci. Food Saf., 2022, 21, 1732–1776 CrossRef PubMed.
- L. Jia, L. Wang, C. Liu, Y. Liang and Q. Lin, Bioactive peptides from foods: production, function, and application, Food Funct., 2021, 12, 7108–7125 RSC.
- R. Han, J. Maycock, B. S. Murray and C. Boesch, Identification of angiotensin converting enzyme and dipeptidyl peptidase-IV inhibitory peptides derived from oilseed proteins using two integrated bioinformatic approaches, Food Res. Int., 2019, 115, 283–291 CrossRef CAS PubMed.
- M. d. M. Contreras, R. Carrón, M. J. Montero, M. Ramos and I. Recio, Novel casein-derived peptides with antihypertensive activity, Int. Dairy J., 2009, 19, 566–573 CrossRef CAS.
- I. M. Lacroix and E. C. Li-Chan, Isolation and characterization of peptides with dipeptidyl peptidase-IV inhibitory activity from pepsin-treated bovine whey proteins, Peptides, 2014, 54, 39–48 CrossRef CAS PubMed.
- I. M. E. Lacroix, G. Meng, I. W. Y. Cheung and E. C. Y. Li-Chan, Do whey protein-derived peptides have dual dipeptidyl-peptidase IV and angiotensin I-converting enzyme inhibitory activities?, J. Funct. Foods, 2016, 21, 87–96 CrossRef CAS.
- X.-Y. Mao, J.-R. Ni, W.-L. Sun, P.-P. Hao and L. Fan, Value-added utilization of yak milk casein for the production of angiotensin-I-converting enzyme inhibitory peptides, Food Chem., 2007, 103, 1282–1287 CrossRef CAS.
- A. B. Nongonierma, C. Cadamuro, A. Le Gouic, P. Mudgil, S. Maqsood and R. J. FitzGerald, Dipeptidyl peptidase IV (DPP-IV) inhibitory properties of a camel whey protein enriched hydrolysate preparation, Food Chem., 2019, 279, 70–79 CrossRef CAS PubMed.
- A. B. Nongonierma and R. J. FitzGerald, Susceptibility of milk protein-derived peptides to dipeptidyl peptidase IV (DPP-IV) hydrolysis, Food Chem., 2014, 145, 845–852 CrossRef CAS PubMed.
- V. P. Shanmugam, S. Kapila, T. S. Kemgang, S. Reddi, R. Kapila, S. Muthukumar and D. Rajesh, Isolation and characterization of angiotensin converting enzyme inhibitory peptide from buffalo casein, Int. J. Pept. Res. Ther., 2021, 27, 1481–1491 CrossRef CAS.
- S. T. Silveira, D. Martinez-Maqueda, I. Recio and B. Hernandez-Ledesma, Dipeptidyl peptidase-IV inhibitory peptides generated by tryptic hydrolysis of a whey protein concentrate rich in beta-lactoglobulin, Food Chem., 2013, 141, 1072–1077 CrossRef CAS PubMed.
- H. Uenishi, T. Kabuki, Y. Seto, A. Serizawa and H. Nakajima, Isolation and identification of casein-derived dipeptidyl-peptidase 4 (DPP-4)-inhibitory peptide LPQNIPPL from gouda-type cheese and its effect on plasma glucose in rats, Int. Dairy J., 2012, 22, 24–30 CrossRef CAS.
- A. Yamada, T. Sakurai, D. Ochi, E. Mitsuyama, K. Yamauchi and F. Abe, Antihypertensive effect of the bovine casein-derived peptide Met-Lys-Pro, Food Chem., 2015, 172, 441–446 CrossRef CAS PubMed.
- J. Yan, J. Zhao, R. Yang and W. Zhao, Bioactive peptides with antidiabetic properties: a review, Int. J. Food Sci. Technol., 2019, 54, 1909–1919 CrossRef CAS.
- H. G. Byun and S. K. Kim, Purification and characterization of angiotensin I converting enzyme (ACE) inhibitory peptides from Alaska pollack (Theragra chalcogramma) skin, Process Biochem., 2001, 36, 1155–1162 CrossRef CAS.
- H. Fujita, K. Yokoyama and M. Yoshikawa, Classification and antihypertensive activity of angiotensin I-converting enzyme inhibitory peptides derived from food proteins, J. Food Sci., 2000, 65, 564–569 CrossRef CAS.
- R. Balti, A. Bougatef, A. Sila, D. Guillochon, P. Dhulster and N. Nedjar-Arroume, Nine novel angiotensin I-converting enzyme (ACE) inhibitory peptides from cuttlefish (Sepia officinalis) muscle protein hydrolysates and antihypertensive effect of the potent active peptide in spontaneously hypertensive rats, Food Chem., 2015, 170, 519–525 CrossRef CAS PubMed.
- R. Balti, N. Nedjar-Arroume, A. Bougatef, D. Guillochon and M. Nasri, Three novel angiotensin I-converting enzyme (ACE) inhibitory peptides from cuttlefish (Sepia officinalis) using digestive proteases, Food Res. Int., 2010, 43, 1136–1143 CrossRef CAS.
- A. Fahmi, S. Morimura, H. C. Guo, T. Shigematsu, K. Kida and Y. Uemura, Production of angiotensin I converting enzyme inhibitory peptides from sea bream scales, Process Biochem., 2004, 39, 1195–1200 CrossRef CAS.
- M. Ghassem, K. Arihara and A. S. Babji, Isolation, purification and characterisation of angiotensin I-converting enzyme-inhibitory peptides derived from catfish (Clarias batrachus) muscle protein thermolysin hydrolysates, Int. J. Food Sci. Technol., 2012, 47, 2444–2451 CrossRef CAS.
- M. Ghassem, K. Arihara, A. S. Babji, M. Said and S. Ibrahim, Purification and identification of ACE inhibitory peptides from Haruan (Channa striatus) myofibrillar protein hydrolysate using HPLC–ESI-TOF MS/MS, Food Chem., 2011, 129, 1770–1777 CrossRef CAS.
- P. A. Harnedy-Rothwell, C. M. McLaughlin, M. B. O'Keeffe, A. V. Le Gouic, P. J. Allsopp, E. M. McSorley, S. Sharkey, J. Whooley, B. McGovern, F. P. M. O'Harte and R. J. FitzGerald, Identification and characterisation of peptides from a boarfish (Capros aper) protein hydrolysate displaying in vitro dipeptidyl peptidase-IV (DPP-IV) inhibitory and insulinotropic activity, Food Res. Int., 2020, 131, 108989 CrossRef CAS PubMed.
- R. Intarasirisawat, S. Benjakul, J. Wu and W. Visessanguan, Isolation of antioxidative and ACE inhibitory peptides from protein hydrolysate of skipjack (Katsuwana pelamis) roe, J. Funct. Foods, 2013, 5, 1854–1862 CrossRef CAS.
- J. K. Lee, J.-K. Jeon and H.-G. Byun, Effect of angiotensin I converting enzyme inhibitory peptide purified from skate skin hydrolysate, Food Chem., 2011, 125, 495–499 CrossRef CAS.
- E. C. Y. Li-Chan, S. Hunag, C. L. Jao, K. P. Ho and K. C. Hsu, Peptides derived from atlantic salmon skin gelatin as dipeptidyl-peptidase IV inhibitors, J. Agric. Food Chem., 2012, 60, 973–978 CrossRef CAS PubMed.
- Y. H. Lin, C. A. Chen, J. S. Tsai and G. W. Chen, Preparation and identification of novel antihypertensive peptides from the in vitro gastrointestinal digestion of marine cobia skin hydrolysates, Nutrients, 2019, 11, 1351 CrossRef CAS PubMed.
- X. Liu, M. Zhang, Y. Shi, R. Qiao, W. Tang and Z. Sun, Production of the angiotensin I converting enzyme inhibitory peptides and isolation of four novel peptides from jellyfish (Rhopilema esculentum) protein hydrolysate, J. Sci. Food Agric., 2016, 96, 3240–3248 CrossRef CAS PubMed.
- D.-H. Ngo, T.-S. Vo, B. Ryu and S.-K. Kim, Angiotensin- I- converting enzyme (ACE) inhibitory peptides from Pacific cod skin gelatin using ultrafiltration membranes, Process Biochem., 2016, 51, 1622–1628 CrossRef CAS.
- P. B. So, P. Rubio, S. Lirio, A. P. Macabeo, H. Y. Huang, M. J. Corpuz and O. B. Villaflores, In vitro angiotensin I converting enzyme inhibition by a peptide isolated from Chiropsalmus quadrigatus Haeckel (box jellyfish) venom hydrolysate, Toxicon, 2016, 119, 77–83 CrossRef CAS PubMed.
- H. Wu, H.-L. He, X.-L. Chen, C.-Y. Sun, Y.-Z. Zhang and B.-C. Zhou, Purification and identification of novel angiotensin-I-converting enzyme inhibitory peptides from shark meat hydrolysate, Process Biochem., 2008, 43, 457–461 CrossRef CAS.
- S. Cao, Y. Wang, Y. Hao, W. Zhang and G. Zhou, Antihypertensive effects in vitro and in vivo of novel angiotensin-converting enzyme inhibitory peptides from bovine bone gelatin hydrolysate, J. Agric. Food Chem., 2020, 68, 759–768 CrossRef CAS PubMed.
- M. Miguel, M. A. Aleixandre, M. Ramos and R. Lopez-Fandino, Effect of simulated gastrointestinal digestion on the antihypertensive properties of ACE-inhibitory peptides derived from ovalbumin, J. Agric. Food Chem., 2006, 54, 726–731 CrossRef CAS PubMed.
- M. Miguel, A. Davalos, M. A. Manso, G. de la Pena, M. A. Lasuncion and R. Lopez-Fandino, Transepithelial transport across Caco-2 cell monolayers of antihypertensive egg-derived peptides. PepT1-mediated flux of Tyr-Pro-Ile, Mol. Nutr. Food Res., 2008, 52, 1507–1513 CrossRef CAS PubMed.
- P. Sangsawad, K. Choowongkomon, D. D. Kitts, X.-M. Chen, E. C. Y. Li-Chan and J. Yongsawatdigul, Transepithelial transport and structural changes of chicken angiotensin I-converting enzyme (ACE) inhibitory peptides through Caco-2 cell monolayers, J. Funct. Foods, 2018, 45, 401–408 CrossRef CAS.
- P. Sangsawad, S. Roytrakul, K. Choowongkomon, D. D. Kitts, X. M. Chen, G. Meng, E. C. Y. Li-Chan and J. Yongsawatdigul, Transepithelial transport across Caco-2 cell monolayers of angiotensin converting enzyme (ACE) inhibitory peptides derived from simulated in vitro gastrointestinal digestion of cooked chicken muscles, Food Chem., 2018, 251, 77–85 CrossRef CAS PubMed.
- P. Sangsawad, S. Roytrakul and J. Yongsawatdigul, Angiotensin converting enzyme (ACE) inhibitory peptides derived from the simulated in vitro gastrointestinal digestion of cooked chicken breast, J. Funct. Foods, 2017, 29, 77–83 CrossRef CAS.
- Y. Yang, E. D. Marczak, M. Yokoo, H. Usui and M. Yoshikawa, Isolation and antihypertensive effect of angiotensin I-converting Enzyme (ACE) inhibitory peptides from spinach rubisco, J. Agric. Food Chem., 2003, 51, 4897–4902 CrossRef CAS PubMed.
- J. Chen, S. Liu, R. Ye, G. Cai, B. Ji and Y. Wu, Angiotensin-I converting enzyme (ACE) inhibitory tripeptides from rice protein hydrolysate: purification and characterization, J. Funct. Foods, 2013, 5, 1684–1692 CrossRef CAS.
- Y. Gu and J. Wu, LC-MS/MS coupled with QSAR modeling in characterising of angiotensin I-converting enzyme inhibitory peptides from soybean proteins, Food Chem., 2013, 141, 2682–2690 CrossRef CAS PubMed.
- E. D. Marczak, H. Usui, H. Fujita, Y. Yang, M. Yokoo, A. W. Lipkowski and M. Yoshikawa, New antihypertensive peptides isolated from rapeseed, Peptides, 2003, 24, 791–798 CrossRef CAS PubMed.
- V. S. Vallabha and P. K. Tiku, Antihypertensive peptides derived from soy protein by fermentation, Int. J. Pept. Res. Ther., 2013, 20, 161–168 CrossRef.
- F.-J. Wang, X.-Y. Yin, J. M. Regenstein and J.-Z. Wang, Separation and purification of angiotensin-I-converting enzyme (ACE) inhibitory peptides from walnuts (Juglans regia L.) meal, Eur. Food Res. Technol., 2015, 242, 911–918 CrossRef.
- J. Wu, R. E. Aluko and A. D. Muir, Purification of angiotensin I-converting enzyme-inhibitory peptides from the enzymatic hydrolysate of defatted canola meal, Food Chem., 2008, 111, 942–950 CrossRef CAS.
- R. Yang, Y. Zou, N. Yu and Z. Gu, Accumulation and identification of angiotensin-converting enzyme inhibitory peptides from wheat germ, J. Agric. Food Chem., 2011, 59, 3598–3605 CrossRef CAS PubMed.
- Y. H. Lin, G. W. Chen, C. H. Yeh, H. Song and J. S. Tsai, Purification and identification of angiotensin I-converting enzyme inhibitory peptides and the antihypertensive effect of Chlorella sorokiniana, protein hydrolysates, Nutrients, 2018, 10, 1397 CrossRef PubMed.
- J. Lu, Y. Sawano, T. Miyakawa, Y. L. Xue, M. Y. Cai, Y. Egashira, D. F. Ren and M. Tanokura, One-week antihypertensive effect of Ile-Gln-Pro in spontaneously hypertensive rats, J. Agric. Food Chem., 2011, 59, 559–563 CrossRef CAS PubMed.
- C. M. Montone, A. L. Capriotti, C. Cavaliere, G. La Barbera, S. Piovesana, R. Zenezini Chiozzi and A. Lagana, Peptidomic strategy for purification and identification of potential ACE-inhibitory and antioxidant peptides in Tetradesmus obliquus microalgae, Anal. Bioanal. Chem., 2018, 410, 3573–3586 CrossRef CAS PubMed.
- J. Xie, X. Chen, J. Wu, Y. Zhang, Y. Zhou, L. Zhang, Y. J. Tang and D. Wei, Antihypertensive effects, molecular docking study, and isothermal titration calorimetry assay of angiotensin I-converting enzyme inhibitory peptides from Chlorella vulgaris, J. Agric. Food Chem., 2018, 66, 1359–1368 CrossRef CAS PubMed.
- X. Geng, G. Tian, W. Zhang, Y. Zhao, L. Zhao, H. Wang and T. B. Ng, A Tricholoma matsutake peptide with angiotensin converting enzyme inhibitory and antioxidative activities and antihypertensive effects in spontaneously hypertensive rats, Sci. Rep., 2016, 6, 24130 CrossRef CAS PubMed.
- K. Skrzypczak, W. Gustaw, D. Szwajgier, E. Fornal and A. Wasko, kappa-Casein as a source of short-chain bioactive peptides generated by Lactobacillus helveticus, J. Food Sci. Technol., 2017, 54, 3679–3688 CrossRef CAS PubMed.
- A. Cerrato, S. E. Aita, A. L. Capriotti, C. Cavaliere, A. M. I. Montone, C. M. Montone and A. Lagana, Investigating the short peptidome profile of italian dry-cured ham at different processing times by high-resolution mass spectrometry and chemometrics, Int. J. Mol. Sci., 2022, 23, 3193 CrossRef CAS PubMed.
- Y. D. Bhutia and V. Ganapathy, in Physiology of the Gastrointestinal Tract, ed. H. M. Said, Academic Press, United Kingdom, 2018, vol. 6, ch. 47, pp. 1063–1086 Search PubMed.
- W. Shen and T. Matsui, Intestinal absorption of small peptides: a review, Int. J. Food Sci. Technol., 2019, 54, 1942–1948 CrossRef CAS.
- A. Mirzapour-Kouhdasht, D. J. McClements, M. S. Taghizadeh, A. Niazi and M. Garcia-Vaquero, Strategies for oral delivery of bioactive peptides with focus on debittering and masking, NPJ Sci. Food, 2023, 7, 22 CrossRef PubMed.
- C. Berraquero-García, R. Perez-Galvez, F. J. Espejo-Carpio, A. Guadix, E. M. Guadix and P. J. Garcia-Moreno, Encapsulation of bioactive peptides by spray-drying and electrospraying, Foods, 2023, 12, 2005 CrossRef PubMed.
- J. E. Aguilar-Toala, D. Quintanar-Guerrero, A. M. Liceaga and M. L. Zambrano-Zaragoza, Encapsulation of bioactive peptides: a strategy to improve the stability, protect the nutraceutical bioactivity and support their food applications, RSC Adv., 2022, 12, 6449–6458 RSC.
- D. J. McClements, Encapsulation, protection, and delivery of bioactive proteins and peptides using nanoparticle and microparticle systems: a review, Adv. Colloid Interface, 2018, 253, 1–22 CrossRef CAS PubMed.
- G. J. O’Neill, T. Egan, J. C. Jacquier, M. O’Sullivan and E. D. O’Riordan, Kinetics of immobilisation and release of tryptophan, riboflavin and peptides from whey protein microbeads, Food Chem., 2015, 180, 150–155 CrossRef PubMed.
- I. M. E. Lacroix, X. M. Chen, D. D. Kitts and E. C. Y. Li-Chan, Investigation into the bioavailability of milk protein-derived peptides with dipeptidyl-peptidase IV inhibitory activity using Caco-2 cell monolayers, Food Funct., 2017, 8, 701–709 RSC.
- L. Basirico, E. Catalani, P. Morera, S. Cattaneo, M. Stuknyte, U. Bernabucci, I. De Noni and A. Nardone, Release of angiotensin converting enzyme-inhibitor peptides during in vitro gastrointestinal digestion of Parmigiano Reggiano PDO cheese and their absorption through an in vitro model of intestinal epithelium, J. Dairy Sci., 2015, 98, 7595–7601 CrossRef CAS PubMed.
- R. Fernández-Musoles, J. B. Salom, M. Castelló-Ruiz, M. D. M. Contreras, I. Recio and P. Manzanares, Bioavailability of antihypertensive lactoferricin B-derived peptides: transepithelial transport and resistance to intestinal and plasma peptidases, Int. Dairy J., 2013, 32, 169–174 CrossRef.
- Y. Nakamura, N. Yamamoto, K. Sakai, A. Okubo, S. Yamazaki and T. Takano, Purification and characterization of angiotensin i-converting enzyme inhibitors from sour milk, J. Dairy Sci., 1995, 78, 777–783 CrossRef CAS PubMed.
- D. Pan and Y. Guo, Optimization of sour milk fermentation for the production of ACE-inhibitory peptides and purification of a novel peptide from whey protein hydrolysate, Int. Dairy J., 2010, 20, 472–479 CrossRef CAS.
- H. Fujita and M. Yoshikawa, LKPNM: a prodrug-type ACE-inhibitory peptide derived from fish protein, Immunopharmacology, 1999, 44, 123–127 CrossRef CAS PubMed.
- D. Liu, H. Sun, L. Zhang, S. Li and Z. Qin, High-Level expression of milk-derived antihypertensive peptide in Escherichia coli and its bioactivity, J. Agric. Food Chem., 2007, 55, 5109–5112 CrossRef CAS PubMed.
- X. X. Jiang, D. D. Pan, M. X. Tao, T. Zhang, X. Q. Zeng, Z. Wu and Y. X. Guo, New nanocarrier system for liposomes coated with Lactobacillus acidophilus S-layer protein to improve Leu-Gln-Pro-Glu absorption through the intestinal epithelium, J. Agric. Food Chem., 2021, 69, 7593–7602 CrossRef CAS PubMed.
- L. Miclo, E. Perrin, A. Driou, V. Papadopoulos, N. Boujrad, R. Vanderesse, J. F. Boudier, D. Desor, G. Linden and J. L. Gaillard, Characterization of alpha-casozepine, a tryptic peptide from bovine alpha(s1)-casein with benzodiazepine-like activity, FASEB J., 2001, 15, 1780–1782 CrossRef CAS PubMed.
- Y. Li, G. Wei and J. Chen, Glutathione: a review on biotechnological production, Appl. Microbiol. Biotechnol., 2004, 66, 233–242 CrossRef CAS PubMed.
- J. Parmentier, B. Thewes, F. Gropp and G. Fricker, Oral peptide delivery by tetraether lipid liposomes, Int. J. Pharm., 2011, 415, 150–157 CrossRef CAS PubMed.
- X. H. Zhou and A. L. W. Po, Peptide and protein drugs: II. non-parenteral routes of delivery, Int. J. Pharm., 1991, 75, 117–130 CrossRef CAS.
- A. Fjellestad-Paulsen, P. Hoglund, S. Lundin and O. Paulsen, Pharmacokinetics of 1-deamino-8-D-arginine vasopressin after various routes of administration in healthy volunteers, Clin. Endocrinol., 1993, 38, 177–182 CrossRef CAS PubMed.
- S. Martins, B. Sarmento, D. C. Ferreira and E. B. Souto, Lipid-based colloidal carriers for peptide and protein delivery – liposomes versus lipid nanoparticles, Int. J. Nanomed., 2007, 2, 595–607 CAS.
- K. Stephansen, I. S. Chronakis and F. Jessen, Bioactive electrospun fish sarcoplasmic proteins as a drug delivery system, Colloids Surf., B, 2014, 122, 158–165 CrossRef CAS PubMed.
- G. J. O'Neill, T. Egan, J. C. Jacquier, M. O'Sullivan and E. D. O'Riordan, Whey microbeads as a matrix for the encapsulation and immobilisation of riboflavin and peptides, Food Chem., 2015, 160, 45–52 Search PubMed.
- M. K. Danish, G. Vozza, H. J. Byrne, J. M. Frias and S. M. Ryan, Comparative study of the structural and physicochemical properties of two food derived antihypertensive tri-peptides, Isoleucine-Proline-Proline and Leucine-Lysine-Proline encapsulated into a chitosan based nanoparticle system, Innovative Food Sci. Emerging, 2017, 44, 139–148 CrossRef CAS.
- A. Trapani, A. Lopedota, M. Franco, N. Cioffi, E. Ieva, M. Garcia-Fuentes and M. J. Alonso, A comparative study of chitosan and chitosan/cyclodextrin nanoparticles as potential carriers for the oral delivery of small peptides, Eur. J. Pharm. Biopharm., 2010, 75, 26–32 CrossRef CAS PubMed.
- H. Laroui, G. Dalmasso, H. T. Nguyen, Y. Yan, S. V. Sitaraman and D. Merlin, Drug-loaded nanoparticles targeted to the colon with polysaccharide hydrogel reduce colitis in a mouse model, Gastroenterology, 2010, 138, 843–853 CrossRef CAS PubMed.
- B. Xiao, Z. Xu, E. Viennois, Y. Zhang, Z. Zhang, M. Zhang, M. K. Han, Y. Kang and D. Merlin, Orally targeted delivery of tripeptide KPV via hyaluronic acid-functionalized nanoparticles efficiently alleviates ulcerative colitis, Mol. Ther., 2017, 25, 1628–1640 CrossRef CAS PubMed.
- B. Bicak, Y. Budama-Kilinc, S. Kecel-Gunduz, T. Zorlud and G. Akman, Peptide based nano-drug candidate for cancer treatment: preparation, characterization, in vitro and in silico evaluation, J. Mol. Struct., 2021, 1240, 130573 CrossRef CAS.
- G. Q. Huang, J. X. Xiao, L. Q. Hao and J. Yang, Microencapsulation of an angiotensin i-converting enzyme inhibitory peptide VLPVP by membrane emulsification, Food Bioprocess Technol., 2017, 10, 2005–2012 CrossRef CAS.
- T. Zhang, M. Su, X. X. Jiang, Y. Q. Xue, J. X. Zhang, X. Q. Zeng, Z. Wu, Y. X. Guo and D. D. Pan, Transepithelial transport route and liposome encapsulation of milk-derived ACE-inhibitory peptide Arg-Leu-Ser-Phe-Asn-Pro, J. Agric. Food Chem., 2019, 67, 5544–5551 CrossRef CAS PubMed.
- P. Batista, P. M. Castro, A. R. Madureira, B. Sarmento and M. Pintado, Preparation, characterization and evaluation of guar films impregnated with relaxing peptide loaded into chitosan microparticles, Appl. Sci., 2021, 11, 9849 CrossRef CAS.
- S. X. Chen, F. Guo, T. T. Deng, S. Q. Zhu, W. Y. Liu, H. J. Zhong, H. Yu, R. Luo and Z. Y. Deng, Eudragit S100-coated chitosan nanoparticles co-loading Tat for enhanced oral colon absorption of insulin, AAPS PharmSciTech, 2017, 18, 1277–1287 CrossRef CAS PubMed.
- X. Jiang, D. Pan, T. Zhang, C. Liu, J. Zhang, M. Su, Z. Wu, X. Zeng, Y. Sun and Y. Guo, Novel milk casein-derived peptides decrease cholesterol micellar solubility and cholesterol intestinal absorption in Caco-2 cells, J. Dairy Sci., 2020, 103, 3924–3936 CrossRef CAS PubMed.
- G. Dalmasso, L. Charrier-Hisamuddin, H. T. Nguyen, Y. Yan, S. Sitaraman and D. Merlin, PepT1-mediated tripeptide KPV uptake reduces intestinal inflammation, Gastroenterology, 2008, 134, 166–178 CrossRef CAS PubMed.
- K. Kannengiesser, C. Maaser, J. Heidemann, A. Luegering, M. Ross, T. Brzoska, M. Bohm, T. A. Luger, W. Domschke and T. Kucharzik, Melanocortin-derived tripeptide KPV has anti-inflammatory potential in murine models of inflammatory bowel disease, Inflammatory Bowel Dis., 2008, 14, 324–331 CrossRef PubMed.
- N. Kommineni, V. G. S. Sainaga Jyothi, A. Butreddy, S. Raju, T. Shapira, W. Khan, P. Angsantikul and A. J. Domb, SNAC for enhanced oral bioavailability: an updated review, Pharm. Res., 2023, 40, 633–650 CrossRef CAS PubMed.
- C. A. Lipinski, F. Lombardo, B. W. Dominy and P. J. Feeney, Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings, Adv. Drug Delivery Rev., 2001, 46, 3–26 CrossRef CAS PubMed.
- F. J. Caro-Leon, W. Arguelles-Monal, E. Carvajal-Millan, Y. L. Lopez-Franco, F. M. Goycoolea-Valencia, J. San Roman Del Barrio and J. Lizardi-Mendoza, Production and characterization of supercritical CO(2) dried chitosan nanoparticles as novel carrier device, Carbohydr. Polym., 2018, 198, 556–562 CrossRef CAS PubMed.
- A. G. Luque-Alcaraz, J. Lizardi-Mendoza, F. M. Goycoolea, I. Higuera-Ciapara and W. Argüelles-Monal, Preparation of chitosan nanoparticles by nanoprecipitation and their ability as a drug nanocarrier, RSC Adv., 2016, 6, 59250–59256 RSC.
- G. Gao, H. Wang, J. Zhou, P. Rao, L. Ke, J. J. Lin, B. Sun Pan, Y. Zhang and Q. Wang, Isolation and characterization of bioactive proteoglycan-lipid nanoparticles from freshwater clam (Corbicula fluminea Muller) soup, J. Agric. Food Chem., 2021, 69, 1610–1618 CrossRef CAS PubMed.
- A. Zielinska, F. Carreiro, A. M. Oliveira, A. Neves, B. Pires, D. N. Venkatesh, A. Durazzo, M. Lucarini, P. Eder, A. M. Silva, A. Santini and E. B. Souto, Polymeric nanoparticles: production, characterization, toxicology and ecotoxicology, Molecules, 2020, 25, 3731 CrossRef CAS PubMed.
- S. More, V. Bampidis, D. Benford, C. Bragard, T. Halldorsson, A. Hernandez-Jerez, S. H. Bennekou, K. Koutsoumanis, C. Lambre, K. Machera, H. Naegeli, S. Nielsen, J. Schlatter, D. Schrenk, V. Silano, D. Turck, M. Younes, J. Castenmiller, Q. Chaudhry, F. Cubadda, R. Franz, D. Gott, J. Mast, A. Mortensen, A. G. Oomen, S. Weigel, E. Barthelemy, A. Rincon, J. Tarazona and R. Schoonjans, Guidance on technical requirements for regulated food and feed product applications to establish the presence of small particles including nanoparticles, EFSA J., 2021, 19, 6769 Search PubMed.
- EFSA, Outcome of the public consultation on the draft guidance on risk assessment of the application of nanoscience and nanotechnologies in the food and feed chain: Part 1, human and animal health, EFSA Supporting Publ., 2018, 15, 1430E, DOI:10.2903/sp.efsa.2018.EN-1430.
- R. Schoonjans, J. Castenmiller, Q. Chaudhry, F. Cubadda, T. Daskaleros, R. Franz, D. Gott, J. Mast, A. Mortensen, A. G. Oomen, H. Rauscher, S. Weigel, M. C. Astuto, I. Cattaneo, E. Barthelemy, A. Rincon and J. Tarazona, Regulatory safety assessment of nanoparticles for the food chain in Europe, Trends Food Sci. Technol., 2023, 134, 98–111 CrossRef CAS.
- M. Xavier, I. A. Parente, P. M. Rodrigues, M. A. Cerqueira, L. Pastrana and C. Gonçalves, Safety and fate of nanomaterials in food: The role of in vitro tests, Trends Food Sci. Technol., 2021, 109, 593–607 CrossRef CAS.
- R. K. Thapa and J. O. Kim, Nanomedicine-based commercial formulations: current developments and future prospects, J. Pharm. Invest., 2023, 53, 19–33 CrossRef CAS PubMed.
- P. Uhl, G. Bajraktari-Sylejmani, D. Witzigmann, C. Bay, S. Zimmermann, J. Burhenne, J. Weiss, W. E. Haefeli and M. Sauter, A nanocarrier approach for oral peptide delivery: evaluation of cell–penetrating–peptide–modified liposomal formulations in dogs, Adv. Ther., 2023, 2300021, 1–12 Search PubMed.
- H. Ichikawa, in Nanoparticle Technology Handbook, ed. M. Hosokawa, K. Nogi, M. Naito and T. Yokoyama, Elsevier Science, 2008, pp. 442–450 Search PubMed.
- S. Pistone, F. M. Goycoolea, A. Young, G. Smistad and M. Hiorth, Formulation of polysaccharide-based nanoparticles for local administration into the oral cavity, Eur. J. Pharm. Sci., 2017, 96, 381–389 CrossRef CAS PubMed.
- F. M. Goycoolea-Valencia, G. Lollo, C. Remunan-Lopez, F. Quaglia and M. J. Alonso, Chitosan-Alginate Blended Nanoparticles as Carriers for the Transmucosal Delivery of Macromolecules, Biomacromolecules, 2009, 10, 1736–1743 CrossRef PubMed.
- M. Iqbal, N. Zafar, H. Fessi and A. Elaissari, Double emulsion solvent evaporation techniques used for drug encapsulation, Int. J. Pharm., 2015, 496, 173–190 CrossRef CAS PubMed.
- O. Torres, B. S. Murray and A. Sarkar, Overcoming in vitro gastric destabilisation of emulsion droplets using emulsion microgel particles for targeted intestinal release of fatty acids, Food Hydrocolloids, 2019, 89, 523–533 CrossRef CAS.
- H. Park, D. H. Ha, E. S. Ha, J. S. Kim, M. S. Kim and S. J. Hwang, Effect of stabilizers on encapsulation efficiency and release behavior of exenatide-loaded PLGA microsphere prepared by the W/O/W solvent evaporation method, Pharmaceutics, 2019, 11, 627 CrossRef CAS PubMed.
- T. D. Brown, K. A. Whitehead and S. Mitragotri, Materials for oral delivery of proteins and peptides, Nat. Rev. Mater., 2019, 5, 127–148 CrossRef.
- J. P. Fuenzalida, M. E. Flores, I. Moniz, M. Feijoo, F. Goycoolea, H. Nishide and I. Moreno-Villoslada, Immobilization of hydrophilic low molecular-weight molecules in nanoparticles of chitosan/poly(sodium 4-styrenesulfonate) assisted by aromatic-aromatic interactions, J. Phys. Chem. B, 2014, 118, 9782–9791 CrossRef CAS PubMed.
- G. J. O'Neill, T. Egan, J. C. Jacquier, M. O'Sullivan and E. D. O'Riordan, Whey microbeads as a matrix for the encapsulation and immobilisation of riboflavin and peptides, Food Chem., 2015, 160, 45–52 Search PubMed.
- K. Kettler, K. Veltman, D. van de Meent, A. van Wezel and A. J. Hendriks, Cellular uptake of nanoparticles as determined by particle properties, experimental conditions, and cell type, Environ. Toxicol. Chem., 2014, 33, 481–492 CrossRef CAS PubMed.
- C. Yang, J. Uertz, D. Yohan and B. D. Chithrani, Peptide modified gold nanoparticles for improved cellular uptake, nuclear transport, and intracellular retention, Nanoscale, 2014, 6, 12026–12033 RSC.
- P. Foroozandeh and A. A. Aziz, Insight into cellular uptake and intracellular trafficking of nanoparticles, Nanoscale Res. Lett., 2018, 13, 339 CrossRef PubMed.
- Y. Yun, Y. W. Cho and K. Park, Nanoparticles for oral delivery: targeted nanoparticles with peptidic ligands for oral protein delivery, Adv. Drug Delivery Rev., 2013, 65, 822–832 CrossRef CAS PubMed.
- J. P. Pearson, P. I. Chater and M. D. Wilcox, The properties of the mucus barrier, a unique gel–how can nanoparticles cross it?, Ther. Delivery, 2016, 7, 229–244 CrossRef CAS PubMed.
- S. Salatin, S. M. Dizaj and A. Y. Khosroushahi, Effect of the surface modification, size, and shape on cellular uptake of nanoparticles, Cell Biol. Int., 2015, 39, 881–890 CrossRef CAS PubMed.
- S. Sreekumar, F. M. Goycoolea, B. M. Moerschbacher and G. R. Rivera-Rodriguez, Parameters influencing the size of chitosan-TPP nano- and microparticles, Sci. Rep., 2018, 8, 4695 CrossRef PubMed.
- L. Pravinata, M. Akhtar, P. J. Bentley, T. Mahatnirunkul and B. S. Murray, Preparation of alginate microgels in a simple one step process via the Leeds Jet Homogenizer, Food Hydrocolloids, 2016, 61, 77–84 CrossRef CAS.
- E. Seyrek, J. Hierrezuelo, A. Sadeghpour, I. Szilagyi and M. Borkovec, Molecular mass dependence of adsorbed amount and hydrodynamic thickness of polyelectrolyte layers, Phys. Chem. Chem. Phys., 2011, 13, 12716–12719 RSC.
- A. O'Connell, F. M. Goycoolea, A. Gulotta, P. Holmqvist, P. Schuetz and J. Mattsson, , The structure and dynamics of locust bean gum in aqueous solution, Food Hydrocolloids, 2023, 138, 108446 CrossRef.
- N. I. Anaraki, A. Sadeghpour, K. Iranshahi, C. Toncelli, U. Cendrowska, F. Stellacci, A. Dommann, P. Wick and A. Neels, New approach for time-resolved and dynamic investigations on nanoparticles agglomeration, Nano Res., 2020, 13, 2847–2856 CrossRef CAS.
- I. M. Adjei, B. Sharma and V. Labhasetwar, Nanoparticles: cellular uptake and cytotoxicity, Adv. Exp. Med. Biol., 2014, 811, 73–91 CrossRef PubMed.
- M. C. Okeudo-Cogan, B. S. Murray, R. Ettelaie, S. D. Connell, S. J. Radford, S. Micklethwaite and A. Sarkar, Understanding the microstructure of a functional meat analogue: Demystifying interactions between fungal hyphae and egg white protein, Food Hydrocolloids, 2023, 140, 108606 CrossRef CAS.
- A. R. Mackie, F. M. Goycoolea, B. Menchicchi, C. M. Caramella, F. Saporito, S. Lee, K. Stephansen, I. S. Chronakis, M. Hiorth, M. Adamczak, M. Waldner, H. M. Nielsen and L. Marcelloni, Innovative methods and applications in mucoadhesion research, Macromol. Biosci., 2017, 17, 1600534 CrossRef PubMed.
- J. Chu, P. Metcalfe, H. V. Linford, S. Zhao, F. M. Goycoolea, S. Chen, X. Ye, M. Holmes and C. Orfila, Short-time acoustic and hydrodynamic cavitation improves dispersibility and functionality of pectin-rich biopolymers from citrus waste, J. Cleaner Prod., 2022, 330, 129789 CrossRef CAS PubMed.
- D. Sanver, A. Sadeghpour, M. Rappolt, F. Di Meo and P. Trouillas, Structure and dynamics of dioleoyl-phosphatidylcholine bilayers under the influence of quercetin and rutin, Langmuir, 2020, 36, 11776–11786 CrossRef CAS PubMed.
- T. Zhao, L. Zheng, Q. Zhang, S. Wang, Q. Zhao, G. Su and M. Zhao, Stability towards the gastrointestinal simulated digestion and bioactivity of PAYCS and its digestive product PAY with cognitive improving properties, Food Funct., 2019, 10, 2439–2449 RSC.
- W. Liao, H. Fan, P. Liu and J. Wu, Identification of angiotensin converting enzyme 2 (ACE2) up-regulating peptides from pea protein hydrolysate, J. Funct. Foods, 2019, 60, 103395 CrossRef CAS.
- M. Mirzaei, S. Mirdamadi, M. Safavi and N. Soleymanzadeh, The stability of antioxidant and ACE-inhibitory peptides as influenced by peptide sequences, LWT – Food, 2020, 130, 109710 CrossRef CAS.
- J. Shaji and V. Patole, Protein and peptide drug delivery: oral approaches, Indian J. Pharm. Sci., 2008, 70, 269–277 CrossRef PubMed.
- L. Egger, O. Ménard, C. Delgado-Andrade, P. Alvito, R. Assunção, S. Balance, R. Barberá, A. Brodkorb, T. Cattenoz, A. Clemente, I. Comi, D. Dupont, G. Garcia-Llatas, M. J. Lagarda, S. Le Feunteun, L. JanssenDuijghuijsen, S. Karakaya, U. Lesmes, A. R. Mackie, C. Martins, A. Meynier, B. Miralles, B. S. Murray, A. Pihlanto, G. Picariello, C. N. Santos, S. Simsek, I. Recio, N. Rigby, L.-E. Rioux, H. Stoffers, A. Tavares, L. Tavares, S. Turgeon, E. K. Ulleberg, G. E. Vegarud, G. Vergères and R. Portmann, The harmonized INFOGEST in vitro digestion method: From knowledge to action, Food Res. Int., 2016, 88, 217–225 CrossRef CAS.
- M. Minekus, M. Alminger, P. Alvito, S. Ballance, T. Bohn, C. Bourlieu, F. Carriere, R. Boutrou, M. Corredig, D. Dupont, C. Dufour, L. Egger, M. Golding, S. Karakaya, B. Kirkhus, S. Le Feunteun, U. Lesmes, A. Macierzanka, A. Mackie, S. Marze, D. J. McClements, O. Menard, I. Recio, C. N. Santos, R. P. Singh, G. E. Vegarud, M. S. Wickham, W. Weitschies and A. Brodkorb, A standardised static in vitro digestion method suitable for food - an international consensus, Food Funct., 2014, 5, 1113–1124 RSC.
- A. E. Ross, M. Y. Tang and R. A. Gemeinhart, Effects of molecular weight and loading on matrix metalloproteinase-2 mediated release from poly(ethylene glycol) diacrylate hydrogels, AAPS J., 2012, 14, 482–490 CrossRef CAS PubMed.
- D. W. Cushman and H. S. Cheung, Spectrophotometric assay and properties of the angiotensin-converting enzyme of rabbit lung, Biochem. Pharmacol., 1971, 20, 1637–1648 CrossRef CAS PubMed.
- E. Schnaith, Determination of the pepsin activity in human gastric juice, using defined oligopeptides as substrates, Clin. Biochem., 1989, 22, 91–98 CrossRef CAS PubMed.
- R. J. Simpson, Fragmentation of Protein Using Trypsin, Cold Spring Harb. Protoc., 2006, 2006(5) DOI:10.1101/pdb.prot4550.
- L. Graff, L. Szilagyi and I. Venekei, in Handbook of Proteolytic Enzymes, ed. Barrett A., N. Rawlings and J. Woessner, Elsevier Ltd, Oxford, UK, 3rd edn, 2013 Search PubMed.
- D. S. Auld, in Handbook of Proteolytic Enzymes, ed. A. Barrett, J. Woessner and N. Rawlings, Elsevier, Academic Press, Oxford, UK, 2004 Search PubMed.
- B. S. Murray, R. Ettelaie, A. Sarkar, A. R. Mackie and E. Dickinson, The perfect hydrocolloid stabilizer: Imagination versus reality, Food Hydrocolloids, 2021, 117, 106696 CrossRef CAS.
- V. S. Goncalves, P. Gurikov, J. Poejo, A. A. Matias, S. Heinrich, C. M. Duarte and I. Smirnova, Alginate-based hybrid aerogel microparticles for mucosal drug delivery, Eur. J. Pharm. Biopharm., 2016, 107, 160–170 CrossRef CAS PubMed.
- J. Chen, V. Gaikwad, M. Holmes, B. Murray, M. Povey, Y. Wang and Y. Zhang, Development of a simple model device for in vitro gastric digestion investigation, Food Funct., 2011, 2, 174–182 RSC.
- H. Kozu, Y. Nakata, M. Nakajima, M. A. Neves, K. Uemura, S. Sato, I. Kobayashi and S. Ichikawa, Development of a human gastric digestion simulator equipped with peristalsis function for the direct observation and analysis of the food digestion process, Food Sci. Technol. Res., 2014, 20, 225–233 CrossRef CAS.
- M. Minekus, P. Marteau, R. Havenaar and J. H. J. Huis in't Veld, A multicompartmental dynamic computer-controlled model simulating the stomach and small intestine, ATLA, Altern. Lab. Anim., 1995, 23, 197–209 CrossRef.
- S. Zhang, B. S. Murray, M. Holmes, R. Ettelaie and A. Sarkar, Gastrointestinal fate and fatty acid release of pickering emulsions stabilized by mixtures of plant protein microgels + cellulose particles: an in vitro static digestion study, Food Biophys., 2022, 18, 120–132 CrossRef.
| This journal is © The Royal Society of Chemistry 2024 |