 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Photocatalytic CO2 reduction by topologically matched polymer–polymer heterojunction nanosheets†
Catherine M.
Aitchison
 *,
Yu
Zhang
,
Wanpeng
Lu
and
Iain
McCulloch
*,
Yu
Zhang
,
Wanpeng
Lu
and
Iain
McCulloch
Department of Chemistry, University of Oxford, 12 Mansfield Road, Oxford OX1 3TA, UK. E-mail: catherine.aitchison@chem.ox.ac.uk
First published on 15th November 2023
Abstract
Conversion of solar energy into chemical fuel can be achieved through a number of routes but direct conversion, via photocatalysis, is potentially the simplest and cheapest route to the transformation of low-value substances, water and CO2, to useful chemical fuels or feedstocks such as hydrogen, formate, methanol, and syngas. 2D polymers, including carbon nitrides and COFs, have emerged as one of the most promising classes of organic photocatalysts for solar fuels production due to their energetic tunability, charge transport properties and robustness. They are, however, difficult to process and so there have been limited studies into the formation of heterojunction materials incorporating these components. In this work we use our novel templating approach to combine topologically matched imine-based donor polymers with acceptor polymers formed through Knoevenagel condensation. An efficient heterojunction interface was formed by matching the isostructural nodes and linkers that make up the D1 and A1 semiconductors and this was reflected in the increased photocatalytic activity of the heterojunction material T1. Tuning of the templating synthesis route to give heterojunctions with optimised donor![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :acceptor ratios, as well as the photocatalytic conditions, resulted in CO production rates that were between 1.5 and 10 times higher than those of the individual polymers. A further set of polymers A5 and D5 were developed with more optimised structures for CO2 reduction including increased overpotential for the reduction reaction and the presence of co-catalyst chelating groups. These had increased activity compared to the group 1 family and again showed higher activity for CO production by the templated heterojunction, T5, than either individual component or a physical mixture of the donor and acceptor.
:acceptor ratios, as well as the photocatalytic conditions, resulted in CO production rates that were between 1.5 and 10 times higher than those of the individual polymers. A further set of polymers A5 and D5 were developed with more optimised structures for CO2 reduction including increased overpotential for the reduction reaction and the presence of co-catalyst chelating groups. These had increased activity compared to the group 1 family and again showed higher activity for CO production by the templated heterojunction, T5, than either individual component or a physical mixture of the donor and acceptor.
Introduction
Five years after TiO2 was first used as a photoelectrode for water splitting,1 it was shown that when a suspension of the same compound in water was irradiated with UV-light, hydrogen and oxygen were evolved in a 2:1 stoichiometric ratio.2 This was the first example of light-driven overall water splitting though a purely photocatalytic mechanism. In 1979, a further two years later, Fujishima and Honda showed that TiO2 powder suspended in CO2 saturated water could also photocatalyse the reduction of CO2 into carbon products including formate and methanol.3 In the decades since these reports carbon dioxide reduction to liquid and gaseous fuels has, in comparison with water splitting to produce hydrogen, been much less widely studied in a solar fuels context. Electrochemical reduction of CO2, on the other hand, has been a particularly fast-moving field which is on the cusp of industrial relevance. State of the art systems can now produce C1 products such as CO and formate with faradaic efficiency of well over 90%.4 Excitingly, systems utilising Cu catalysts are now also starting to generate C2+ products such as ethylene with reasonable efficiencies,5–7 paving the way for a sustainable production route to a variety of chemical feedstocks.
Using light as a direct driving force for this reaction, i.e. photocatalytic CO2 reduction, offers the potential to form a carbon neutral energy cycle, when used in conjunction with CO2 capture at point of fuel use, or even a mechanism to remove CO2 from the energy cycle when used to make chemical feedstocks for materials such as plastics.
As with water splitting, research into this reaction has typically focused on inorganic semiconductors based on metals oxides8 or metal chalcogenides.9,10 However, hybrid inorganic–organic materials, such as MOFs,11 are increasingly popular candidates for photocatalytic CO2 reduction due to their easily adjustable chemistry and ability to integrate light absorbing and electrocatalytic components into a single material. For example, polyoxometalate-based materials have been shown to be capable of high methane production rates and selectivity12 whilst very recently mixed zinc-porphyrin and cobalt-porphyrin nanomiscelles have shown sustained methane production over 30 days and turnover number greater than 6000.13 Whilst some linear conjugated polymers have been investigated for photocatalytic CO2 reduction,14,15 2D polymers have been at the forefront of research into organic semiconductors for this application due to their highly tuneable structures, stability and high porosity. Carbon nitride is the most widely studied organic semiconductor for solar fuels applications and CO2 reduction rates have increased by orders of magnitude over the past decade via various doping and nano-structuring strategies.16 In particular, optimisation of cocatalyst integration has led to a step change in efficiencies as well as expansion of the reduction products accessible through photocatalysis. For instance, phosphine-containing carbon nitrides with Cu catalytic sites have even been shown to be active for the production of both ethane17 and ethylene.18 These results are impressive but the inherent synthetic limitations associated with carbon nitride mean that optimisation of the polymer backbone is difficult. Covalent triazine frameworks19 offer more monomer flexibility and they, along with cross-linked polymers formed via metal-catalysed cross coupling14,20 have been shown to be active for CO2 reduction. However covalent organic frameworks (COFs) are increasingly becoming the organic semiconductor of choice for this reaction; COFs with a variety of different semi-reversible linkages have been studied including azine, hydrazone, imine and C–C based materials.21 These are generally combined with a metal cocatalyst, such as Re,22,23 although high activity has been observed with more sustainable metals such a Ni24 and Co.25,26
These materials microporous and crystalline structures are ideally suited to high CO2 accessible surface areas and good energy transfer between the point of excitation and the active site. However, the insoluble nature of these 2D materials means that no post-synthesis solvent processing is possible. In contrast we have shown that processibility is highly beneficial to some linear polymer based photocatalysts as they can be blended into donor–acceptor heterojunction materials. These are capable of separating photogenerated excitons into the “free” polaron species required for more challenging electrochemical reactions.27 Instead of solution processing we have recently shown that effective type II donor–acceptor heterojunctions can be generated from 2D polymers using a templation strategy.28 This involved formation of thin “seed” sheets of one phase followed by epitaxial growth of the second polymer phase onto the first. Effective donor–acceptor interface formation was enabled by choosing polymers with topologically similar structures. Polymers A1 and D1 (Fig. 1) were shown to both be moderately active photocatalysts for light driven hydrogen evolution from water but when combined into a heterojunction via templated synthesis the photocatalytic activity was vastly improved in comparison to the single component semiconductors. Transient absorption spectroscopy revealed that the improvement in hydrogen evolution rates was due to enhanced charge separation in the templated material with the formation of long-lived polarons that could be effectively extracted to drive proton reduction and scavenger oxidation respectively. The improved charge separation accessible through this templated heterojunction formation should not be limited to proton reduction applications. In this work, we study the templated heterojunctions for CO2 reduction to CO, firstly through a study of our previously published materials A1, D1 and their templated heterojunction T1, and then by development of a new templated system involving donor and acceptor polymers, D5 and A5, with structures and energy levels better suited to carbon dioxide reduction.
Results and discussion
Analysis of group 1 materials
Fig. 1 shows imine-based polymer D1 and Knoevenagel polymer A1. These polymers and their heterojunction material T1 were formed as previously described,28 the latter by condensation of the A1 monomers (M3 and M4) onto pre-formed D1 sheets such that the donor acceptor ratio was 50:50 wt%.
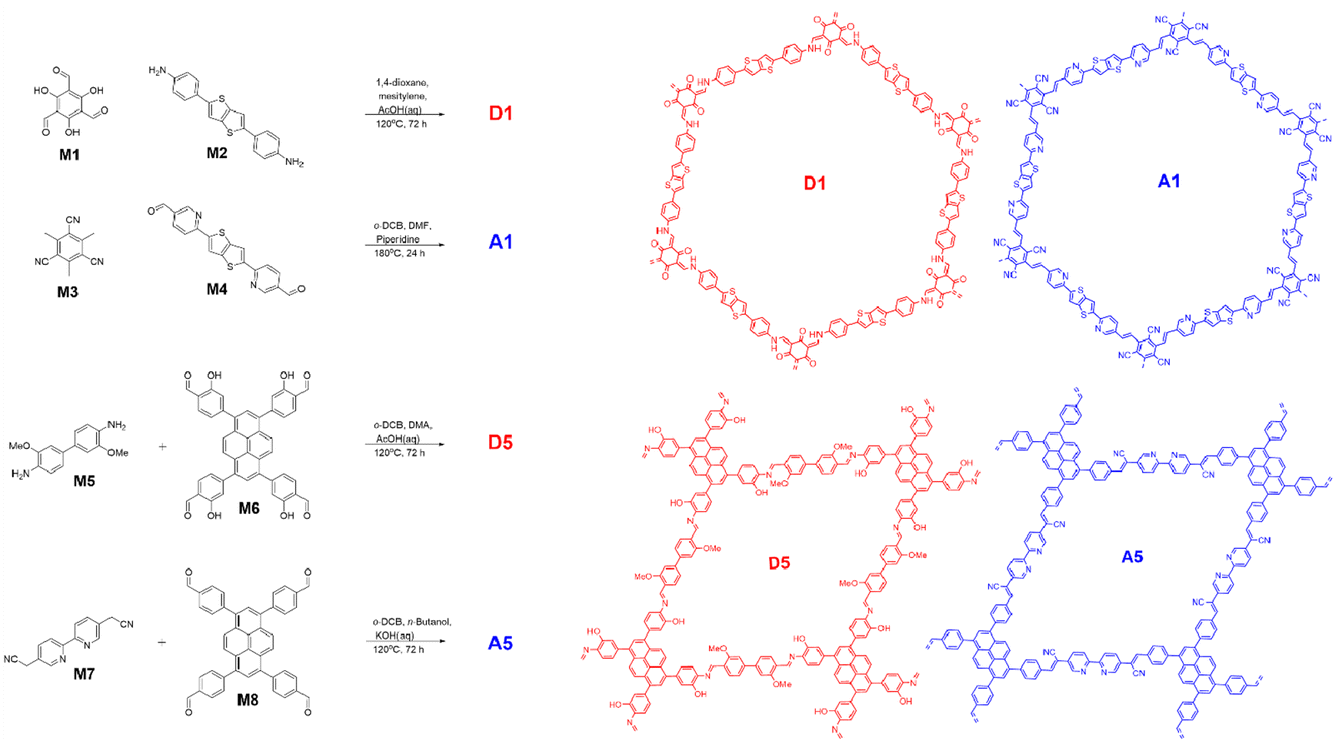 | ||
| Fig. 1 Synthesis and chemical structures of the polymers. | ||
One of the advantages of studying these condensation polymers for photocatalytic activity is that they generally contain very low levels of residual palladium (as opposed to polymers formed via Pd-catalysed cross-coupling reactions). This is beneficial as Pd, like Pt, is a highly active catalyst for the competing proton reduction reaction.29 To favour CO2 reduction a cobalt based co-catalyst was used instead. Cobalt species have been shown to be active for carbon dioxide (photo)electroreduction in various forms including as molecular catalysts, single atoms and clusters.30 Unlike Pt or Pd (for hydrogen production) or other highly active CO2 reduction catalysts based on Ir, Ru or Re, cobalt is a cheap, earth abundant element. Thanks to its range of easily accessible oxidations states (Co0, CoI, CoII, CoIII and CoIV), and intermediate CO2 adsorption it can reduce carbon dioxide to carbon monoxide with high activity and selectivity. Organic semiconductor photocatalysts have previously employed Co/bipyridine via surface contact15 and by direct coordination anchoring.25 The chemical structures of A1 and D1 do not have the double nitrogen chelating groups required for the latter strategies and so cobalt was deposited on the materials by sonicating the polymers with CoCl2·7H2O. Analysis of these materials by EDX indicated that cobalt loading amounts were very similar at 0.46, 0.38 and 0.34 wt% for D1, A1 and T1 respectively. Typically, nitrogen coordination of cobalt species is required to give high reduction rates and so during photocatalytic testing a free bipyridine ligand was added as a coordination agent.
Photocatalytic activity was measured by suspending the polymers in a mixture of acetonitrile and water (6:1) with an ascorbic acid (0.1 M) hole scavenger. The suspensions were illuminated under an atmosphere of CO2 using 1 sun irradiation (300 W Xe lamp fitted with an AM 1.5 filter). All three materials produced CO continuously (Fig. 2). A1 produced the least, just 0.02 μmol over the 15 hour illumination period, with a maximum rate of 0.002 μmol h−1. D1 was significantly more active, producing 0.15 μmol, with an order of magnitude higher maximum rate of 0.017 μmol h−1. After a significantly longer induction period T1 eventually produced 0.28 μmol of CO, and has a maximum rate of 0.040 μmol h−1, over twice as high as either single component material.
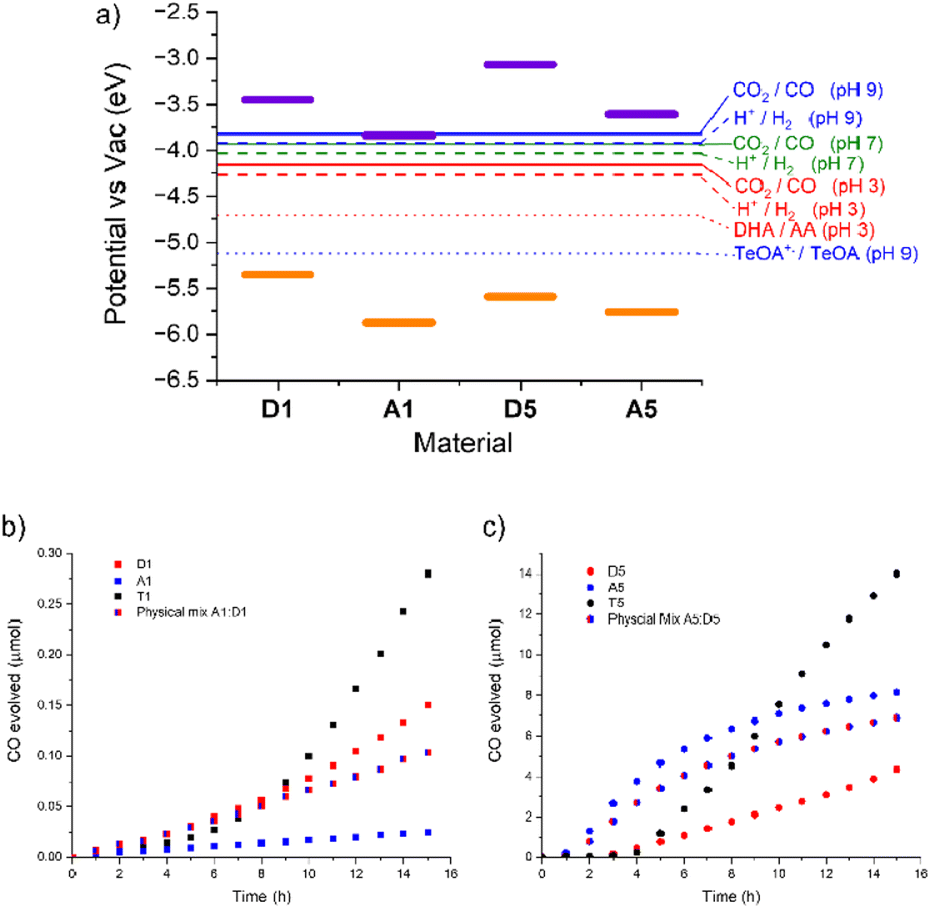 | ||
| Fig. 2 (a) IPs (orange) and EAs (purple) of the materials along with relevant electrochemical potentials. (b) Photocatalysis of the Co-loaded group 1 polymers (2.5 mg) and 2,2′-bipyridine (2 mg) in MeCN:water (6:1, 35 mL) with Ascorbic acid (616 mg) under an atmosphere of CO2. (c) Photocatalysis of the Co-loaded group 5 polymers (2.5 mg) and 2,2′-bipyridine (2 mg) in MeCN (32 mL) with TeOA (3 mL) under an atmosphere of CO2. | ||
We attribute the higher activity of the templated heterojunction material to the improved charge separation and extraction previously shown in T1 compared to its constituent component materials. We have previously shown via transient absorption spectroscopy measurements that the yield of charge separated polaron states is higher in T1 and that they persist for longer.28 Whilst long-lived excited states were observed in the A1 component material, measurements with variable fluence indicated that these were not free, non-geminate charges but instead were trapped states that were inaccessible for photocatalysis. In contrast the long-lived signals in T1 were more fluence dependant and ascribed to a free electron polaron that was extractable and quenched upon addition of a Pt cocatalyst ‘electron sink’. Whilst this again shows the benefit of heterojunction formation on photocatalysis, it was noted that the rates of CO evolved in these experiments were very low at 0.040 μmol h−1 (16 μmol h−1 g−1). By comparison, conjugated linear polymers loaded with a similar cobalt cocatalyst had CO evolution rates of between 100–1000 μmol h−1 g−1.15 Those experiments and most CO2 reduction reactions – photochemical and electrochemical – are performed at alkaline pH.31 Whilst there are some reports using ascorbic acid as a hole scavenger for photocatalytic CO2 reduction,32 it is much more typical to use basic triethanolamine (TeOA).
Photocatalytic testing of A1, D1 and T1 with TeOA (ESI, Fig. S5†), however, all resulted in lower CO evolution than with ascorbic acid; A1 produced no measurable CO at all whilst T1 produced 0.08 μmol h−1 with a maximum rate of 0.005 μmol h−1, nearly an order of magnitude lower than with ascorbic acid. Interestingly D1 was slightly more active than T1 under these conditions and has a similar rate to that with acid conditions. D1 produced 0.12 μmol of CO in 15 hours with a TeOA hole scavenger, at a maximum rate of 0.010 μmol h−1. This is consistent with our previous results that found that at pH 7 the hydrogen evolution rate of A1, and subsequently the T1 system, was limited by a reduced driving force for proton reduction. Examining Fig. 2 we can see that the electrochemical potential for proton reduction at pH 7 is less than 0.1 eV below the LUMO of A1. The CO2 reduction potential to CO at pH 9 (approximately the pH of the 10% TeOA solution) lies at an even more positive value meaning the driving force for this half reaction by A1 is even smaller. We note that there is some debate about the correct conversion factor between electrochemical potentials pinned to SHE and FMO energies vs. vacuum and that values can vary significantly depending on testing conditions.33 Photocatalytic measurements are also run in a primarily MeCN electrolyte which, along with the variation in proton concentration, will affect the electrochemical potentials. However, it has been estimated the CO2/CO potential is similar in MeCN and aqueous conditions.34,35
The electrochemical potentials indicate that the lower activity of T1 under these conditions is due to the lack of contribution from the acceptor phase in this material. The activity is lower than the single D1 component due to parasitic absorption by the unproductive acceptor phase as well as the more limited exposure of the “core” donor phase to the electrolyte.28
Given this inherent limitation to the energetics of this initial T1 system alternative donor and acceptor phases were developed.
Development and analysis of group 5 materials
To operate CO2 reduction at high pH an acceptor phase with a shallowed LUMO is required. At the same time a type II offset in donor and acceptor energy levels must be maintained, as well as a donor HOMO that is deep enough to drive oxidation of our TeOA hole scavenger. A5 (Fig. 1) has previously been shown to be active for CO2 reduction, has, according to DFT modelling, the requisite energy levels and contains free 2,2′bipyridine linker sites that are ideal for anchoring cocatalyst groups. To generate a topologically matched donor polymer to template A5 with we replaced the Knoevenagel-type linkages with chemically orthogonal imine bonds. The carbanion forming 2,2’-([2,2′-bipyridine]-5,5′-diyl)diacetonitrile was swapped for a more electron rich dianisidine nucleophile, whilst electron donating alcohol groups were added to the pyrene-based electrophile to give monomer M8. Whilst in Fig. 1, D5 is drawn as the imine form, these alcohol groups also likely facilitate tautomerisation of the imine bonds into the highly stable β-keto–enol form.36A5 and D5 were first synthesised as single component materials; A5via a modified literature procedure using KOH(aq) catalysed Knoevenagel condensation and D5via acetic acid catalysed imine condensation. Both polymers formed powders that were insoluble in all common solvents.
The absorption spectra of the polymers were recorded in acetonitrile suspension (ESI, Fig. S8†). The imine polymer D5 was found to have an onset at 491 nm whilst A5 was red shifted in comparison with an onset 578 nm. Photoelectron Emission Spectra in Air (PESA) was conducted on the single component materials (ESI Fig. S6 and S7†) to determine their ionization potentials (IPs). The IP of D5 was determined as −5.59 eV, whilst the electron poor A5 has an IP of −5.76 eV. Electron affinities (EAs) were calculated by the addition of the optical band gap to the IP, giving −3.07 and −3.61 eV for D5 and A5, respectively. The frontier molecular orbital (FMO) energies are summarized in Table 1. The 0.54 eV and 0.17 eV offset between the materials LUMOs and HOMOs respectively suggest they are suitable candidates to form a type II heterojunction. Crucially the FMO energies of both materials straddle the CO2 reduction potential and the TeOA oxidation potential (Fig. 2.), indicating they should be able to facilitate CO production. To confirm this, photocatalysis was first conducted on single component materials.
| Material | IPa (eV) | EAb (eV) | CO evolved using AAc (μmol) | Max CO evolution rate using AA (μmol h−1) | CO evolved using TeOAc (μmol) | Max CO evolution rate using TeOA (μmol h−1) |
|---|---|---|---|---|---|---|
| a Derived from PESA. b Derived from adding the optical gap to the IP. c Over 15 hours of illumination. | ||||||
| D1 | −5.36 | −3.45 | 0.15 | 0.017 | 0.12 | 0.010 |
| A1 | −5.88 | −3.84 | 0.02 | 0.002 | 0 | 0 |
| T1 | — | — | 0.28 | 0.040 | 0.08 | 0.005 |
| D5 | −5.59 | −3.07 | — | — | 4.4 | 0.45 |
| A5 | −5.76 | −3.61 | — | — | 8.2 | 1.2 |
| T5 | — | — | — | — | 14 | 1.5 |
Cobalt cocatalyst was loaded onto the polymers via the same sonication method as described previously. In these material Co loading was generally more efficient. A5 contained 1.03 wt% cobalt as determined by EDX but D5 contained slightly more at 1.60 wt%. EDX mapping of the two materials showed an even distribution of Co throughout the materials (ESI, Fig. S9 and S10†). The cobalt loaded polymers were suspended in acetonitrile with TeOA (10 vol%) hole scavenger and illuminated under an atmosphere of CO2 using 1 sun irradiation (300 W Xe lamp fitted with an AM 1.5 filter). These materials were significantly more active than the group 1 polymers, even the heterojunction material T1. D5 produced 4.4 μmol of CO in 15 hours with, after a short induction period, a maximum rate of 0.45 μmol h−1. A5 produced 8.2 μmol with a high maximum rate of 1.2 μmol h−1.
As both single components materials were active for CO2 reduction, we proceeded to combine them into a donor–acceptor heterojunction via the same templation method as T1. We have previously shown that a heterojunction with higher photocatalytic activity can be formed via templating the acceptor onto the donor or by templating the donor onto the acceptor, although the former was shown to have higher hydrogen evolution rates.28 We therefore initially attempted to form the group 5 template in the same manner, by adding D5 seed sheets to the mixture of M7 and M8 required to form A5. However, the harsh reaction conditions required to form A5 – including KOH(aq) (4 M) – appeared to be incompatible with the semi-reversible linkages in D5 and resulted in breakdown of this component and low yields of the template material. Instead T5 was formed by templating D5 onto A5 seed sheets. Our previous study indicated that addition of more than 50 wt% of the second phase resulted in less active materials, possibly due to blocking of the internal phase’s interaction with the electrolyte. As such, T5 was formed with 70 wt% A5 internal phase and 30 wt% D5 external phase (in terms of M5 and M6 starting monomer quantities).
T5 was loaded with cobalt in the same manner as A5 and D5 and analysed by EDX. This showed a cobalt content of 2.25 wt%. Photocatalytic testing was again conducted with TeOA in MeCN solvent and, as with the group 1 templated material, produced significantly more CO than both A5 and D5. T5 produced 14 μmol of CO in 15 hours, more than 3 times the amount produced by D5 and 1.7 times the amount produced by A5. This improvement of the templated materials indicates some synergistic positive effect between the donor and acceptor materials. Interestingly the maximum rate of 1.5 μmol h−1 is only slightly higher than A5 due the latter materials gradual slowing of activity. This small improvement suggests the heterojunction in this system is less efficient at charge separation than that shown in the group 1 materials. To assess the importance of the templating synthesis route to the improvement in photocatalytic activity a physical mixture of the two materials A5 and D5 was sonicated together in the same proportions as present in T5 and was also tested for CO2 reduction. This physical mixture did not appear to have any heterojunction benefit and displayed activity similar to the major A5 component producing 6.9 μmol CO over the same time period with a maximum rate of 0.95 μmol h−1. The higher activity of A5 over D5 is likely due to its ability to chelate the Co cocatalyst with the embedded 2,2′-bipyridine linker. However, the fact that D5, the polymer with the most limited light absorption in this study, still has relatively high activity (more than an order of magnitude higher than all group 1 materials) indicates the importance of a large overpotential for CO2 reduction.
Conclusions
The formation of 2D donor–acceptor polymer heterojunctions via templation was found to give materials with higher photocatalytic activity than the individual semiconductor components. In our previously studied group 1 materials the improved charge separation found in the templated material T1 enables CO evolution rates over twice as high as the D1 and A1 polymers as individual materials. However, these materials have low rates compared to established literature systems and are not compatible with the basic photocatalytic conditions optimal for favouring CO2, rather than proton, reduction. Group 5 polymers were developed and showed higher activity for CO2 reduction than the group 1 materials. This was most likely due to a combination of their increased overpotential and the ability of the acceptor component to chelate the Co co-catalyst and enable efficient energy transfer between the semiconductor and the active site. A templated heterojunction of A5 and D5, showed higher photocatalytic activity than either single component material. This is likely due to the formation of a donor–acceptor interface that enables more efficient charge separation. However, further study via spectroscopic methods would be required to support this. The CO production rate of the most active heterojunction material, T5 at 1.5 μmol h−1 (600 μmol h−1 g−1), is similar to the most active single organic semiconductor systems in the literature. However, this is without the use of any separate photosensitiser. Photosensitisers are frequently noble metal complexes and are often crucial to charge separation. In systems without separate photosensitisers, the organic semiconductor component often shows very limited activity.25,37 This study indicates that the use of a templated heterojunction may be a viable strategy to facilitate photogenerated charge formation without these unsustainable components. Equally, these tests were conducted with a transition metal based cocatalyst, indicating that high CO evolution rates need not rely on expensive Re or Ru based active sites.Experimental
Synthesis
A1, D1 and T1 were synthesised as described previously.28 2,2′-([2,2′-bipyridine]-5,5′-diyl)diacetonitrile (M7) and o-dianisidine (M5) were purchased from Ambeed and used without further purification.4,4′,4′′,4′′′-(Pyrene-1,3,6,8-tetrayl)tetrakis(2-hydroxybenzaldehyde), M6
1,3,6,8-Tetrabromopyrene (420 mg, 0.81 mmol), 2-hydroxy-4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)benzaldehyde (1.01 g, 4.06 mmol), 1,4-dioxane (20 mL), water (2 mL) and K2CO3 (420 mg, 3.8 mmol) were added to a flask and degassed for 45 minutes by N2 bubbling. Pd(PPh3)4 (28 mg, 0.02 mmol, 2.5 mol%) was added and the mixture degassed for a further 10 minutes by N2 bubbling before heating at 110 °C for 48 hours under an atmosphere of N2. The mixture was allowed to cool before pouring into water (300 mL). After stirring for 1 hour, the yellow precipitate was filtered off and washed with water (300 mL). The crude solid was recrystallised in NMP/MeOH to give the pure product as a yellow powder (845 mg, 72%). 1H NMR (400 MHz, DMSO-d6): δ(ppm) = 10.99 (bs, 4H), 10.39 (s, 4H), 8.24 (s, 4H), 8.04 (s, 2H), 7.88 (d, J = 7 Hz, 4H), 7.31 (s, 4H), 7.29 (d, J = 7 Hz, 4H). Solubility of M6 was too low for 13C NMR analysis.4,4′,4′′,4′′′-(Pyrene-1,3,6,8-tetrayl)tetrabenzaldehyde, M8
1,3,6,8-Tetrabromopyrene (1.00 g, 1.93 mmol), 4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)benzaldehyde (2.32 g, 10 mmol), DMF (50 mL) and K2CO3 (2 M, 10 mL) were added to a flask and degassed for 45 minutes by N2 bubbling. Pd(PPh3)4 (50 mg, 0.04 mmol, 2 mol%) was added and the mixture degassed for a further 10 minutes by N2 bubbling before heating at 130 °C for 24 hours under an atmosphere of N2. The mixture was allowed to cool before pouring into water (300 mL). After stirring for 1 hour, the yellow precipitate was filtered off and washed with water (300 mL) and THF (300 mL). The crude solid was recrystallised in hot chloroform/hexane to give the pure product as a yellow powder (1.05 g, 88%). 1H NMR (400 MHz, CDCl3): δ(ppm) = 10.16 (s, 4H), 8.18 (s, 4H), 8.10 (d, J = 7 Hz, 8H), 8.04 (s, 2H), 7.86 (d, J = 7 Hz, 8H). Solubility of M8 was too low for 13C NMR analysis.D5
M5 (24.4 mg, 0.10 mmol), M6 (34.1 mg, 0.05 mmol), o-dichlorobenzene (0.8 mL), N,N-dimethylacetamide (2.5 mL) and AcOH(aq) (6 M, 0.15 mL) were loaded into a glass vial and subjected to three cycles of freeze–pump–thaw. The vial was put under vacuum and flame sealed before heating to 120 °C for 72 hours. The mixture was poured into THF (50 mL) and stirred for 30 minutes before filtering off the red precipitate. The solid was washed with water (20 mL) and MeOH (20 mL) before Soxhlet extraction in THF for five hours. The solid was dried under vacuum at 90 °C for 16 hours to yield the product as a red powder (49 mg, 89%).A5
M7 (23.4 mg, 0.10 mmol), M8 (30.9 mg, 0.05 mmol), o-dichlorobenzene (1 mL), n-butanol (1 mL) and KOH(aq) (4 M, 0.2 mL) were loaded into a glass vial and subjected to three cycles of freeze–pump–thaw. The vial was put under vacuum and flame sealed before heating to 120 °C for 72 hours. The mixture was poured in THF (50 mL) and stirred for 30 minutes before filtering off the orange precipitate. The solid was washed with HCl (1 M, 10 mL), water (20 mL) and THF (20 mL) before Soxhlet extraction in THF for five hours. The solid was dried under vacuum at 90 °C for 16 hours to yield the product as an orange powder (35 mg, 69%).T5
A5 (20 mg), M5 (4.1 mg, 0.017 mmol), M6 (5.7 mg, 0.008 mmol), o-dichlorobenzene (0.4 mL), DMF (1.3 mL) and AcOH(aq) (6 M, 0.08 mL) were loaded into a glass vial and subjected to three cycles of freeze–pump–thaw. The vial was put under vacuum and flame sealed before heating to 120 °C for 72 hours. The mixture was poured in THF (50 mL) and stirred for 30 minutes before filtering off the red precipitate. The solid was washed with water (20 mL) and MeOH (20 mL) before Soxhlet extraction in THF for five hours. The solid was dried under vacuum at 90 °C for 16 hours to yield the product as a red-orange powder (26 mg, 67% WRT imine).Cocatalyst loading
Polymer (20 mg) and cobalt chloride heptahydrate (10 mg) were added to MeCN (5 mL). The dispersion was sonicated for 5 hours (Branson 550 W, 30% power 45:15 second on:off pulses). The solid was collected by filtration and washed with more MeCN (100 mL) and dried under vacuum to yield the cobalt loaded polymer.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors would like to acknowledge financial support from KAUST Office of Sponsored Research CRG10, by EU Horizon 2020 grant agreement no. 952911, BOOSTER, grant agreement no. 862474, and grant agreement no. 101007084 CITYSOLAR, as well as EPSRC Projects EP/T026219/1 and EP/W017091/1.References
- A. Fujishima and K. Honda, Nature, 1972, 238, 37–38 CrossRef CAS.
- G. N. Schrauzer and T. D. Guth, J. Am. Chem. Soc., 1977, 99, 7189–7193 CrossRef CAS.
- T. Inoue, A. Fujishima, S. Konishi and K. Honda, Nature, 1979, 277, 637–638 CrossRef CAS.
- M. B. Ross, P. De Luna, Y. Li, C. T. Dinh, D. Kim, P. Yang and E. H. Sargent, Nat. Catal., 2019, 2, 648–658 CrossRef CAS.
- C. Choi, S. Kwon, T. Cheng, M. Xu, P. Tieu, C. Lee, J. Cai, H. M. Lee, X. Pan, X. Duan, W. A. Goddard and Y. Huang, Nat. Catal., 2020, 3, 804–812 CrossRef CAS.
- C. Xiao and J. Zhang, ACS Nano, 2021, 15, 7975–8000 CrossRef CAS PubMed.
- A. R. Woldu, Z. Huang, P. Zhao, L. Hu and D. Astruc, Coord. Chem. Rev., 2022, 454, 214340 CrossRef CAS.
- K. K. Ghuman, L. B. Hoch, P. Szymanski, J. Y. Y. Loh, N. P. Kherani, M. A. El-Sayed, G. A. Ozin and C. V. Singh, J. Am. Chem. Soc., 2016, 138, 1206–1214 CrossRef CAS PubMed.
- X. Li, Y. Sun, J. Xu, Y. Shao, J. Wu, X. Xu, Y. Pan, H. Ju, J. Zhu and Y. Xie, Nat. Energy, 2019, 4, 690–699 CrossRef CAS.
- S. Wang, B. Y. Guan, Y. Lu and X. W. Lou, J. Am. Chem. Soc., 2017, 139, 17305–17308 CrossRef CAS PubMed.
- J. Li, H. Huang, W. Xue, K. Sun, X. Song, C. Wu, L. Nie, Y. Li, C. Liu, Y. Pan, H. L. Jiang, D. Mei and C. Zhong, Nat. Catal., 2021, 4, 719–729 CrossRef CAS.
- Q. Huang, Q. Niu, X. F. Li, J. Liu, S. N. Sun, L. Z. Dong, S. L. Li, Y. P. Cai and Y. Q. Lan, Sci. Adv., 2022, 8, eadd559 Search PubMed.
- J. Yu, L. Huang, Q. Tang, S. B. Yu, Q. Y. Qi, J. Zhang, D. Ma, Y. Lei, J. Su, Y. Song, J. C. Eloi, R. L. Harniman, U. Borucu, L. Zhang, M. Zhu, F. Tian, L. Du, D. L. Phillips, I. Manners, R. Ye and J. Tian, Nat. Catal., 2023, 6, 464–475 CrossRef CAS.
- S. Wang, X. Hai, X. Ding, S. Jin, Y. Xiang, P. Wang, B. Jiang, F. Ichihara, M. Oshikiri, X. Meng, Y. Li, W. Matsuda, J. Ma, S. Seki, X. Wang, H. Huang, Y. Wada, H. Chen and J. Ye, Nat. Commun., 2020, 11, 1149 CrossRef.
- Z. Fu, A. Vogel, M. A. Zwijnenburg, A. I. Cooper and R. S. Sprick, J. Mater. Chem. A, 2021, 9, 4291–4296 RSC.
- A. Kumar, P. Raizada, V. Kumar Thakur, V. Saini, A. Aslam Parwaz Khan, N. Singh and P. Singh, Chem. Eng. Sci., 2021, 230, 116219 CrossRef CAS.
- G. Wang, Z. Chen, T. Wang, D. Wang and J. Mao, Angew. Chem., Int. Ed., 2022, 61, e202210789 CrossRef CAS PubMed.
- W. Xie, K. Li, X. H. Liu, X. Zhang and H. Huang, Adv. Mater., 2023, 35, 2208132 CrossRef CAS.
- C. Yang, W. Huang, L. C. da Silva, K. A. I. Zhang and X. Wang, Chem. - Eur. J., 2018, 24, 17454–17458 CrossRef CAS PubMed.
- H. P. Liang, A. Acharjya, D. A. Anito, S. Vogl, T. X. Wang, A. Thomas and B. H. Han, ACS Catal., 2019, 9, 3959–3968 CrossRef CAS.
- H. Wang, H. Wang, Z. Wang, L. Tang, G. Zeng, P. Xu, M. Chen, T. Xiong, C. Zhou, X. Li, D. Huang, Y. Zhu, Z. Wang and J. Tang, Chem. Soc. Rev., 2020, 49, 4135 RSC.
- S. Yang, W. Hu, X. Zhang, P. He, B. Pattengale, C. Liu, M. Cendejas, I. Hermans, X. Zhang, J. Zhang and J. Huang, J. Am. Chem. Soc., 2018, 140, 14614–14618 CrossRef CAS.
- Z. Fu, X. Wang, A. M. Gardner, X. Wang, S. Y. Chong, G. Neri, A. J. Cowan, L. Liu, X. Li, A. Vogel, R. Clowes, M. Bilton, L. Chen, R. S. Sprick and A. I. Cooper, Chem. Sci., 2020, 11, 543–550 RSC.
- M. Zhou, Z. Wang, A. Mei, Z. Yang, W. Chen, S. Ou, S. Wang, K. Chen, P. Reiss, K. Qi, J. Ma and Y. Liu, Nat. Commun., 2023, 14, 2473 CrossRef CAS.
- X. Wang, Z. Fu, L. Zheng, C. Zhao, X. Wang, S. Y. Chong, F. McBride, R. Raval, M. Bilton, L. Liu, X. Wu, L. Chen, R. S. Sprick and A. I. Cooper, Chem. Mater., 2020, 32, 9107–9114 CrossRef CAS.
- Q. Zhang, S. Gao, Y. Guo, H. Wang, J. Wei, X. Su, H. Zhang, Z. Liu and J. Wang, Nat. Commun., 2023, 14, 1147 CrossRef CAS PubMed.
- J. Kosco, S. Gonzalez-Carrero, C. T. Howells, T. Fei, Y. Dong, R. Sougrat, G. T. Harrison, Y. Firdaus, R. Sheelamanthula, B. Purushothaman, F. Moruzzi, W. Xu, L. Zhao, A. Basu, S. De Wolf, T. D. Anthopoulos, J. R. Durrant and I. McCulloch, Nat. Energy, 2022, 7, 340–351 CrossRef CAS.
- C. M. Aitchison, S. Gonzalez-Carrero, S. Yao, M. Benkert, Z. Ding, N. P. Young, B. Willner, F. Moruzzi, Y. Lin, J. Tian, P. D. Nellist, J. R. Durrant and I. McCulloch, Adv. Mater., 2023, 2300037 CrossRef.
- J. Kosco, M. Sachs, R. Godin, M. Kirkus, L. Francas, M. Bidwell, M. Qureshi, D. Anjum, J. R. Durrant and I. McCulloch, Adv. Energy Mater., 2018, 8, 1802181 CrossRef.
- C. Li, X. Tong, P. Yu, W. Du, J. Wu, H. Rao and Z. M. Wang, J. Mater. Chem. A, 2019, 7, 16622–16642 RSC.
- J. Li and N. Kornienko, Chem Catal., 2022, 2, 29–38 CrossRef CAS.
- J. Shipp, S. Parker, S. Spall, S. L. Peralta-Arriaga, C. C. Robertson, D. Chekulaev, P. Portius, S. Turega, A. Buckley, R. Rothman and J. A. Weinstein, Inorg. Chem., 2022, 61, 13281–13292 CrossRef CAS.
- J. Bertrandie, J. Han, C. S. P. De Castro, E. Yengel, J. Gorenflot, T. Anthopoulos, F. Laquai, A. Sharma and D. Baran, Adv. Mater., 2022, 34, 2202575 CrossRef CAS PubMed.
- M. L. Pegis, J. A. S. Roberts, D. J. Wasylenko, E. A. Mader, A. M. Appel and J. M. Mayer, Inorg. Chem., 2015, 54, 11883–11888 CrossRef CAS.
- Y. Matsubara, ACS Energy Lett., 2019, 4, 1999–2004 CrossRef CAS.
- S. Kandambeth, A. Mallick, B. Lukose, M. V. Mane, T. Heine and R. Banerjee, J. Am. Chem. Soc., 2012, 134, 19524–19527 CrossRef CAS.
- W. Zhong, R. Sa, L. Li, Y. He, L. Li, J. Bi, Z. Zhuang, Y. Yu and Z. Zou, J. Am. Chem. Soc., 2019, 141, 7615–7621 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3fd00143a |
| This journal is © The Royal Society of Chemistry 2024 |