
Neutral and ionic N-methyl phenylazo-3,5-(di-2-pyridyl)pyrazole photoswitches: probes for reversible pH modulation by light†
Sapna
Singh‡
 ,
Archana
Velloth‡
,
Rajani
,
Manu
Goyal
,
Navneet
Kaur
,
Sanjay
Singh
* and
Sugumar
Venkataramani
*
,
Archana
Velloth‡
,
Rajani
,
Manu
Goyal
,
Navneet
Kaur
,
Sanjay
Singh
* and
Sugumar
Venkataramani
*
Department of Chemical Sciences, Indian Institute of Science Education and Research (IISER) Mohali, Sector 81, SAS Nagar, Knowledge City, Manauli-140306, Punjab, India. E-mail: sanjaysingh@iisermohali.ac.in; sugumarv@iisermohali.ac.in
First published on 23rd August 2024
Abstract
We report the design, synthesis, photoswitching and computational studies of N-methyl arylazo-3,5-(di-2-pyridyl)pyrazole and its N-alkyl pyridinium derivatives with an ionic center proximally located to the azo group. Besides achieving excellent photoswitching characteristics, particularly at longer wavelengths, and tuning Z isomer stability due to the effects of counter ions and pH, the utility of neutral and ionic photoswitches for pH modulation by light was achieved.
Azoheteroarenes in general and azopyrazoles in particular are attractive candidates, well known for their photoswitching characteristics and tunable properties.1 Their light-driven properties have been tuned through heterocycles, substituents, H-bonding, tautomerism, metal coordination, introduction of an ionic character, variations in acid medium or pH.2–5 Along these lines, several intriguing designs of azoheteroarenes have been reported to portray fundamental aspects and equally to realize applications.1c,6 Considering the continuous growth in photoswitches and the increasing demand for them in diverse applications in biology and materials science, newer designs of neutral and ionic photoswitches are inevitable.
Typically, azo photoswitches can be converted into ionic ones by attaching and quaternizing basic units such as triazole or by incorporating acidic groups (COOH, SO3H, etc.) at a remote position.7 Direct alkylation of heterocyclic moieties in azoheteroarene photoswitches is an alternative strategy, which governs tunability in Z isomer stability through which the half-lives can be varied from milliseconds to years. Taking advantage of this possibility, several ionic photoswitches, such as azopyridinium, azopyrazolium, and azoimidazolium, have been synthesized with diverse characteristics and applications (Scheme 1).4 This direct quaternization strategy leads to delocalization of the cationic charge via π-conjugation in E isomers. Despite showing good to excellent reversible E–Z and Z–E photoisomerization, the cationic charge associated with geometrical changes in polar solvents is mainly controlled by intramolecular forces.4a,b To overcome this, we envisioned designing azo photoswitches with an ionic group located proximally to the azo group that can be spatially influenced by photoswitching. In this regard, we report the design, synthesis and studies of N-methyl arylazo-3,5-(di-2-pyridyl)pyrazole and its N-alkyl pyridinium derivatives as neutral and ionic photoswitches. The photoswitching characteristics and Z isomer stability were controlled by varying counter ions and pH. Profiting from the proton accepting ability and cooperativeness of the 2-pyridyl and azo groups, the neutral and ionic photoswitches were also utilized for pH modulation. All these results are presented in this paper.
 | ||
| Scheme 1 Representative examples for azoheteroarene-based ionic photoswitches and approaches for pH modulation by light. | ||
Accordingly, arylazopyrazoles with two 2-pyridyl substitutions at the 3,5-positions of the pyrazole moiety were targeted. The purpose of those 2-pyridyl units is to introduce an ionic center proximally (through space), yet separated from the azo group (through bonds) and for protonation. To access those targets, aniline A and 2,6-difluoroaniline B were diazotized, and further reacted with dione derivative C in the presence of sodium acetate (Scheme 2 and see Section S2 in the ESI†). Neutral azo photoswitches 2a and 2b were synthesized from 1a and 1b, respectively, by cyclization using N-methylhydrazine sulfate in the presence of sodium carbonate. Then, alkylation procedures were adopted using dimethyl sulfate or alkyl iodide, to access the targets 2a-Me-MeSO4, 2a-Me-I, 2a-Et-I, and 2b-Et-I. Further, the selected counter ions were exchanged using either Amberlite 440® ion exchange resin or treatment with NaBF4 to obtain 2a-Et-Cl and 2a-Et-BF4. All the targets were fully characterized by various spectroscopic techniques, and the solid-state structures of two of them (2a-Me-I and 2a-Et-I) were elucidated using SC-XRD (see Section S2 in the ESI†).§
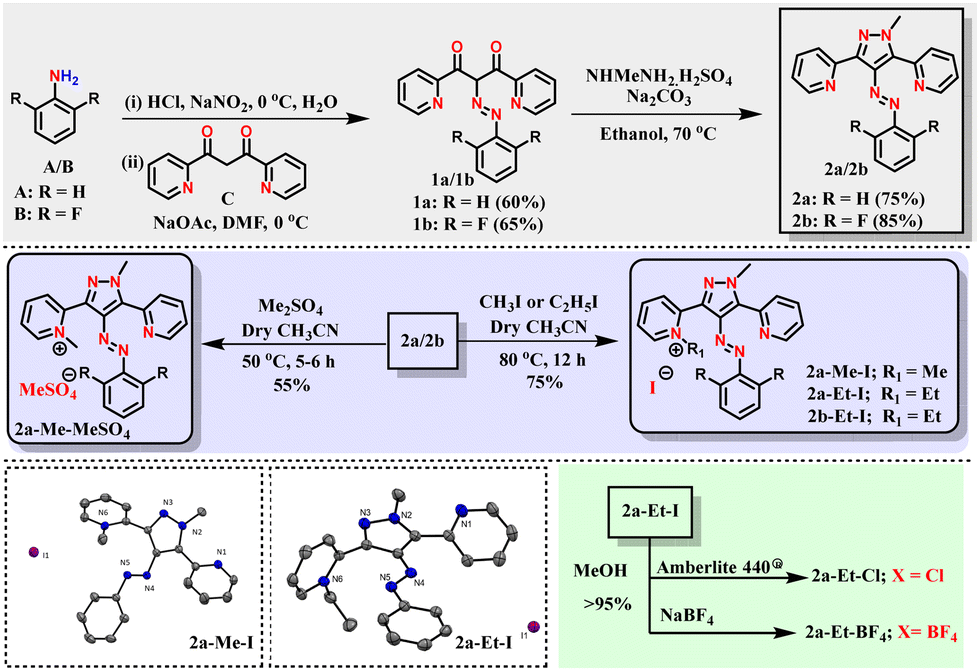 | ||
| Scheme 2 Synthesis of the target photoswitches (neutral and ionic) and SC-XRD data of 2a-Me-I and 2a-Et-I. | ||
All the synthesized targets were studied for reversible photoswitching and thermal relaxation in DMSO and water (except for the neutral compounds) and analyzed by UV-vis and 1H-NMR spectroscopy (see Section S3 in the ESI†). As observed previously, DMSO showed red-shifted π–π* absorption in the neutral and ionic derivatives.4a,b The neutral targets 2a and 2b showed moderate to good forward E–Z photoisomerization by visible light (λ = 525 nm). This can be attributed to the repulsive interaction between the π-cloud of the 2-pyridyl group and/or its nitrogen lone pair and that of azo nitrogen. This was supported by AIM analysis, which revealed a bond critical point (BCP) between the 2-pyridyl nitrogen and the azo nitrogen (see Section S5 in the ESI†). However, the photoswitching is near quantitative (>95%) if the irradiation wavelength corresponds to the π–π* absorption of the azo group. However, upon quaternization, such repulsive interaction was diminished, which enabled only UV (365 nm)-induced photoisomerization despite showing near quantitative photoswitching (>95%). Even the purposeful introduction of 2,6-difluoro at the aryl group was ineffective at inducing visible-light photoswitching in the ionic analogue. Conversely, good to excellent (81–90%) reverse Z–E photoisomerization was observed by employing 490 or 525 nm light. Overall, all the molecules showed excellent photoswitching with moderate quantum yields (Fig. 1, Table 1, see Section S3 in the ESI†).
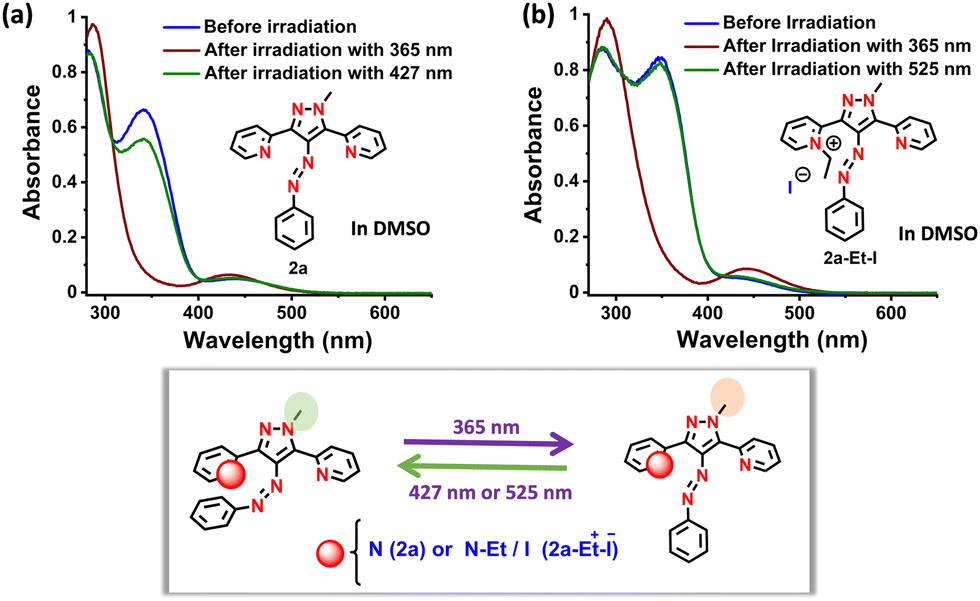 | ||
| Fig. 1 Photoswitching of (a) 2a and (b) 2a-Et-I followed by UV-vis spectroscopy (55 and 71 μM, respectively). A scheme representing reversible photoisomerization is also illustrated. | ||
| S. no. | Targets | Electronic absorption data (λmax, nm/ε, M−1 cm−1) | PSS datab (%E![[thin space (1/6-em)]](https://www.rsc.org/images/entities/i_char_2009.gif) :%Z) :%Z) |
Quantum yield | k (s−1) | t 1/2 (d) | |||
|---|---|---|---|---|---|---|---|---|---|
| π–π* | n–π* | E → Z | Z → E | E → Z | Z → E | ||||
| a Performed in DMSO due to insolubility in water. (Absorption data: Normal - E isomer; Bold - Z isomer; quantum yields are estimated in DMSO). b Estimated using 1H-NMR data in D2O (for 2a, 2b and 2a-Et-BF4 in DMSO-d6; concentration: 7–20 mM). c Thermal reverse (Z–E) isomerization kinetics data obtained by UV-vis spectroscopy; concentration: 40–81 μM (for photoisomerization studies, E–Z: 365 nm; Z–E: 427 or 490 or 525 nm). d For 2b, the π−π* band overlaps with the solvent. | |||||||||
| 1 | 2a | 341/15![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 539 ± 421 (287) 539 ± 421 (287) |
440 (432) | 4:96 |
70:30 |
0.40 ± 0.03 | 0.58 ± 0.03 | 3.8 × 10−8 | 211 |
| 2 | 2a-Et-I | 336/9692 ± 355 (281) | 430 (445) | 5:95 |
86:14 |
0.42 ± 0.02 | 0.53 ± 0.02 | 3.1 × 10−7 | 26 |
| 3 | 2a-Me-I | 336/9847 ± 370 (282) | 430 (444) | 5:95 |
86:14 |
0.48 ± 0.03 | 0.50 ± 0.03 | 2.3 × 10−7 | 28 |
| 4 | 2a-Et-BF4 | 336/16431 ± 505a (289) |
429 (444) | 6:94 |
86:14 |
0.38 ± 0.03 | 0.57 ± 0.04 | 2.1 × 10−7 | 38 |
| 5 | 2a-Et-Cl | 338/6314 ± 226 (281) | 430 (436) | 5:95 |
86:14 |
0.38 ± 0.01 | 0.53 ± 0.02 | 3.8 × 10−7 | 21 |
| 6 | 2a-Me-MeSO4 | 336/8947 ± 285 (284) | 429 (434) | 6:94 |
85:15 |
0.39 ± 0.04 | 0.62 ± 0.05 | 1.4 × 10−7 | 59 |
| 7 | 2b , | 353/11341 ± 300 |
435 (432) | 5:95 |
65:35 |
0.45 ± 0.05 | 0.51 ± 0.04 | 4.5 × 10−8 | 179 |
| 8 | 2b-Et-I | 350/9800 ± 290 (285) | 436 (443) | 5:95 |
81:19 |
0.48 ± 0.03 | 0.50 ± 0.03 | 5.4 × 10−7 | 15 |
Next, all the targets were studied for their thermal Z–E relaxation by subjecting them to kinetics studies in DMSO and water (except 2a and 2b). All the ionic photoswitches were also studied to understand the effect of counter ions. Both the Z isomers of the neutral derivatives 2a and 2b showed excellent thermal stability with estimated half-lives of 65 and 48 min, respectively, at 90 °C in DMSO, whereas the corresponding ionic derivatives showed reasonably lower Z–E thermal half-lives from 8 to 12 min at 90 °C in DMSO. However, the thermal stability of the ionic photoswitches increased upon changing the solvent to water, where the half-lives were determined to be 16 to 28 min at 90 °C. Further variable temperature kinetics measurements were performed and the experimental energy barriers for the thermal reverse isomerization steps were calculated using Arrhenius and Eyring plots (see Section S4 in the ESI†). Based on the data, the Gibbs free energy of the barrier for the neutral compounds was estimated to be 27.5 (for 2a) and 27.4 kcal mol−1 (for 2b) in DMSO, whereas the corresponding values dropped to 25.8–25.9 kcal mol−1 for all the ionic derivatives. Notably, the barriers for ionic derivatives changed to 25.7–26.6 kcal mol−1 in water, indicating the impact of both counter ions and solvent on the thermal stability of the Z isomers.
Density functional theory (DFT) calculations were performed to disentangle the effects of counter ions, solvent effects, Z isomer stability, and activation energies for the thermal reverse Z–E isomerization channels. Firstly, barriers in gas phase and PCM model (in DMSO) of the neutral 2a and the corresponding ionic derivatives 2a-Me-X (X = Cl−, I−, BF4−, and MeSO4−) demonstrated a higher inversion barrier for the former than the latter (see Section S5, Fig. S5.2–S5.6 and Table S5.2 in the ESI†). To decipher the origin of the difference in the stability of 2a-Me-X (X = Cl−, I−, BF4−, and MeSO4−), the E and Z isomers were optimized with counter ions. The geometrical features were analyzed, and non-covalent interactions (NCI) were identified through the atoms in molecule (AIM) formalism. Importantly, cation–π, cation–anion (electrostatic interactions), anion–π, lone pair–cation, and lone pair–π interactions were recognized through NCI analysis (see Section S5, Fig. S5.7–S5.22, and Table S5.3 in the ESI†). In terms of contact distance, electron density at the BCP, and significant anion–π interactions, the Z isomer with the MeSO4 counter ion is the most stable, and the Z isomer stability for the corresponding E isomer followed the order: MeSO4− (11.3) > BF4− (11.7) > I− (11.9) > Cl− (12.4) kcal mol−1. This is in good agreement with the experimental observations. Additionally, the dispersive contribution for the MeSO4 counter ion is found to be the highest, with an estimated energy of 6.4 kcal mol−1 (Table S5.4 in the ESI†).
Upon introducing the solvents, DMSO, and water using the PCM model, the Z isomers gained stability in water for all counter ions; however, the Cl ion remained stable in the gas phase rather than in the presence of a solvent. The subtle interplay of weak non-bonding interactions is responsible for such changes and was unravelled from NCI analysis in conjugation with the reduced density gradient (RDG) plot (Fig. S5.15–S5.18 in the ESI†). Furthermore, the transition states for the inversion channel along the phenyl and pyrazole units were examined to understand the thermal reverse Z–E isomerization mechanism. In the case of 2a-Me-X, the inversion mechanism along the phenyl ring was found to be the lowest energy pathway for all the compounds analyzed (see Table S5.2 in the ESI†). The computed Gibbs free energy of activation in water followed this order: MeSO4− (26.0) ≈ BF4− (26.0) > I− (25.7) > Cl− (25.2) kcal mol−1; this trend is consistent with the experimentally observed barriers.
The salient feature of the targets is the availability of the 2-pyridyl groups with basic nitrogen capable of binding to protons. Accordingly, their cooperative binding ability with the assistance of azo nitrogen and tuning through photoisomerization between E and Z isomers were considered as reasons enabling them to reversibly modulate the pH of the medium. Considering the importance of these phenomena in biology and materials science,8 we studied this phenomenon selectively in neutral 2a and its ionic derivative 2a-Et-I over a wide range of pH (2.8 to 8.0). In this regard, the targets were dissolved in phosphate buffer solutions and were adjusted to different pH values. Subsequently, the solutions were irradiated reversibly between cis and trans isomers. In the case of 2a, the photoswitching was effective over a wider pH range (2.8, 5.0, 7.4, 8.0, and 10.8) in both directions. Significantly, a 16 nm red shift (after deconvolution) in the π–π* absorbance in the trans isomer was observed upon lowering the pH (7.4 to 2.8), whereas the shift was minimal in the case of the cis isomer (4 nm) (see Section S6 in the ESI†). Computationally, protonation at the 2-pyridyl group exhibited a similar shift. Moreover, the thermal stability of the Z isomers decreased with increasing acidity (low pH) (see Section S6 in the ESI†). Remarkably, different pH levels in the buffer solution showed a consistent modulation between a low (in the Z isomeric state) and a higher pH value (in the E isomeric state) during reversible photoisomerization steps. As the destabilizing effects of Z isomers in acidic conditions are well documented,5b we hypothesized that protonation in the E isomer cooperatively occurs between azo N and 2-pyridyl N, which is further supported by the higher proton affinity (−257.9 kcal mol−1) predicted by computation. Conversely, the Z isomer showed a lower proton affinity (−250.7 kcal mol−1), indicating the dominance of Z isomer geometry without protonation in equilibrium (see Section S6 in the ESI†). These results corroborated the experimental and computational estimates of pKa (Fig. 2a and b, see Section S6 in the ESI†).5a,d Furthermore, the decreasing buffer concentration (Fig. 2c) and increasing concentration of 2a at identical pH (see Section S6 in the ESI†) improves the pH range for modulation during photoisomerization. This can be attributed to the differential proton affinity of the trans and cis isomers of 2a (Fig. 2d). Extension of similar studies using 2a-Et-I also showed a similar trend, but the pH could only be modulated to a limited extent, indicating the less favourable equilibrium conditions arising due to protonation of an already charged cationic system.
 | ||
| Fig. 2 (a) and (b) Effect of pH in the absorption spectra in the trans and cis isomers of 2a, respectively (phosphate buffer, 10 mM). (Inset: Sigmoidal fit of the plot, absorption changes vs. pH to determine the pKa.) (c) Reversible pH modulation by light at different buffer concentrations (2 vs. 10 mM) of 2a (1 mM). (d) Overall summary of pH modulation by light in 2a. | ||
In summary, we synthesized two neutral and six ionic 2-pyridyl appended azoheteroarene-based photoswitches with excellent photoswitching characteristics. The Z isomer stability of ionic systems was strongly influenced by solvent, pH, and counter anions. Computations predicted the dominant anion–π and other non-covalent interactions in stabilizing the Z isomers and increasing the barriers for thermal relaxation. Furthermore, the presence of 2-pyridyl substituents and their protonation capabilities of E and Z isomers were exploited to reversibly modulate the pH using neutral derivative 2a. Through control experiments and extensive computations, the significant difference in the proton affinity and pKa of the E and Z isomers was attributed to light-driven pH modulation. This phenomenon could be useful in a variety of applications such as catalysis, sensors, controlling biochemical processes, and photocaging.
S. V. thanks Science and Engineering Research Board (SERB), New Delhi (CRG/2023/003861) for funding. S. S. and M. G. thank MoE for the fellowships. A. V. and R. thank SERB (NPDF/2022/000605) and UGC for fellowships. The departmental and central research facilities including FIST NMR are greatly acknowledged.
Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Notes and references
- (a) T. Wendler, C. Schütt, C. Näther and R. Herges, J. Org. Chem., 2012, 77, 3284–3287 CrossRef PubMed; (b) C. E. Weston, R. D. Richardson, P. R. Haycock, A. J. P. White and M. J. Fuchter, J. Am. Chem. Soc., 2014, 136, 11878–11881 CrossRef PubMed; (c) S. Crespi, N. A. Simeth and B. König, Nat. Rev. Chem., 2019, 3, 133–146 CrossRef.
- (a) J. Calbo, C. E. Weston, A. J. White, H. S. Rzepa, J. Contreras-Garcia and M. J. Fuchter, J. Am. Chem. Soc., 2017, 139, 1261–1274 CrossRef PubMed; (b) S. Devi, M. Saraswat, S. Grewal and S. Venkataramani, J. Org. Chem., 2018, 83(8), 4307–4322 CrossRef PubMed; (c) A. H. Heindl and H. A. Wegner, Chem. – Eur. J., 2020, 26, 13730–13737 CrossRef PubMed; (d) N. A. Simeth, S. Crespi, M. Fagnoni and B. König, J. Am. Chem. Soc., 2018, 140, 2940–2946 CrossRef PubMed; (e) R. Lin, P. K. Hashim, S. Sahu, A. S. Amrutha, N. M. Cheruthu, S. Thazhathethil, K. Takahashi, T. Nakamura, T. Kikukawa and N. Tamaoki, J. Am. Chem. Soc., 2023, 145(16), 9072–9080 CrossRef PubMed.
- D. Gupta, A. K. Gaur, H. Kumar, S. Singh and S. Venkataramani, ChemPhotoChem, 2023, 7(9), e202300068 CrossRef.
- (a) A. K. Gaur, D. Gupta, A. Mahadevan, P. Kumar, H. Kumar, D. N. Nampoothiry, N. Kaur, S. K. Thakur, S. Singh, T. Slanina and S. Venkataramani, J. Am. Chem. Soc., 2023, 145, 10584–10594 CrossRef CAS PubMed; (b) A. K. Gaur, D. Gupta, D. N. Nampoothiry, A. Velloth, R. Kaur, N. Kaur and S. Venkataramani, Chem. – Eur. J., 2024, 30, e202401239 CrossRef PubMed; (c) M. Nakagawa, M. Rikukawa, M. Watanabe, K. Sanui and N. Ogata, Bull. Chem. Soc. Jpn., 1997, 70, 737–744 CrossRef CAS; (d) J. G. Amorós, W. A. Massad, S. Nonell and D. Velasco, Org. Lett., 2010, 12(15), 3514–3517 CrossRef PubMed; (e) J. G. Amorós, S. Nonell and D. Velasco, Chem. Commun., 2012, 48, 3421–3423 RSC; (f) T. Yoshida, T. Monji, D. Kawamori, N. Akai, K. Shibuya and A. Kawai, Chem. Lett., 2013, 42, 1490–1492 CrossRef; (g) D. Prischich, A. M. J. Gomila, S. Milla-Navarro, G. Sangüesa, R. Diez-Alarcia, B. Preda, C. Matera, M. Batlle, L. Ramírez, E. Giralt, J. Hernando, E. Guasch, J. J. Meana, P. de la Villa and P. Gorostiza, Angew. Chem., Int. Ed., 2021, 60, 3625–3631 CrossRef PubMed.
- (a) C. E. Weston, R. D. Richardson and M. J. Fuchter, Chem. Commun., 2016, 52, 4521–4524 RSC; (b) L. Kortekaas, J. Simke, N. B. Arndt, M. Bockmann, N. L. Doltsinis and B. J. Ravoo, Chem. Sci., 2021, 12, 11338–11346 RSC; (c) S. Ludwanowski, M. Ari, K. Parison, S. Kalthoum, P. Straub, N. Pompe, S. Weber, M. Walter and A. Walther, Chem. – Eur. J., 2020, 26, 13203–13212 CrossRef CAS PubMed; (d) S. Samanta, A. Babalhavaeji, M.-X. Dong and G. A. Woolley, Angew. Chem., Int. Ed., 2013, 52, 14127–14130 CrossRef CAS PubMed.
- S. Grewal, D. Gupta, A. K. Gaur, M. Saraswat and S. Venkataramani, in Photoisomerization: Causes, Behavior and Effects, ed. D. Sampedro, Nova Publishers, New York, 2019, vol. 4, pp. 111–188 Search PubMed.
- For example, see (a) C. Wang, K. Hashimoto, R. Tamate, H. Kokubo and M. Watanabe, Angew. Chem., Int. Ed., 2018, 57, 227–230 CrossRef CAS PubMed; (b) I. Orellana-Maldonado, A. Aspee, P. Barrias, P. A. Santana and C. Tirapegui, New J. Chem., 2022, 46, 12825–12835 RSC.
- (a) P. K. Patel, J. E. Arias, R. S. Gongora, F. E. Hernandez, A. Moncomble, S. Aloısec and K. Y. Chumbimuni-Torres, Phys. Chem. Chem. Phys., 2018, 20, 26804–26808 RSC; (b) O. Rifaie-Graham, J. Yeow, A. Najer, R. Wang, R. Sun, K. Zhou, T. N. Dell, C. Adrianus, C. Thanapongpibul, M. Chami, S. Mann, J. R. de Alaniz and M. M. Stevens, Nat. Chem., 2023, 15, 110–118 CrossRef CAS PubMed; (c) H. Ren, X.-P. Qiu, Y. Shi, P. Yang and F. M. Winnik, Macromolecules, 2019, 52, 2939–2948 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental and computational methods and all related data are included here. CCDC 2365862 and 2365863. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4cc03455d |
| ‡ Equally contributed. |
| § Crystallographic data (excluding structure factors) of 2a-Et-I and 2a-Me-I have been deposited to the Cambridge Crystallographic Data Centre: CCDC 2365862 (2a-Et-I) and CCDC 2365863 (2a-Me-I) (see Section S2 in the ESI† for further details) |
| This journal is © The Royal Society of Chemistry 2024 |