
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Advanced electrocatalytic redox processes for environmental remediation of halogenated organic water pollutants
Madeleine K.
Wilsey
a,
Teona
Taseska
b,
Ziyi
Meng
a,
Wanqing
Yu
b and
Astrid M.
Müller
 *abc
*abc
aMaterials Science Program, University of Rochester, Rochester, New York 14627, USA. E-mail: astrid.mueller@rochester.edu
bDepartment of Chemical Engineering, University of Rochester, Rochester, New York 14627, USA
cDepartment of Chemistry, University of Rochester, Rochester, New York 14627, USA
First published on 22nd September 2023
Abstract
Halogenated organic compounds are widespread, and decades of heavy use have resulted in global bioaccumulation and contamination of the environment, including water sources. Here, we introduce the most common halogenated organic water pollutants, their classification by type of halogen (fluorine, chlorine, or bromine), important policies and regulations, main applications, and environmental and human health risks. Remediation techniques are outlined with particular emphasis on carbon–halogen bond strengths. Aqueous advanced redox processes are discussed, highlighting mechanistic details, including electrochemical oxidations and reductions of the water–oxygen system, and thermodynamic potentials, protonation states, and lifetimes of radicals and reactive oxygen species in aqueous electrolytes at different pH conditions. The state of the art of aqueous advanced redox processes for brominated, chlorinated, and fluorinated organic compounds is presented, along with reported mechanisms for aqueous destruction of select PFAS (per- and polyfluoroalkyl substances). Future research directions for aqueous electrocatalytic destruction of organohalogens are identified, emphasizing the crucial need for developing a quantitative mechanistic understanding of degradation pathways, the improvement of analytical detection methods for organohalogens and transient species during advanced redox processes, and the development of new catalysts and processes that are globally scalable.
 Madeleine K. Wilsey | Madeleine Wilsey completed her BS degree in chemistry and physics at Western Illinois University in 2019, where she conducted research under the supervision of Prof. Saisudha Mallur and Prof. J. Scott McConnell. After the completion of her undergraduate studies, Madeleine worked as an R&D intern at the company AP Lazer under the supervision of CEO Tong Li. Madeleine joined Prof. Astrid M. Müller's group at the University of Rochester in summer 2020, and she is currently a PhD candidate investigating sustainable electrocatalysis for selective conversions of hydrocarbons into alcohols, using bimetallic nanocatalysts made by pulsed laser in liquids synthesis. |
 Teona Taseska | Teona Taseska, native of Ohrid, North Macedonia, came to the U.S. for her undergraduate education at the University of Rochester, where she is a rising senior in chemical engineering, clustering in Japanese. She has won the 2021 MLC Book Award for Japanese Language. She has been conducting undergraduate research in the lab of Prof. Astrid M. Müller since her sophomore year. Her research projects focus on opportunities for climate-friendly catalysis technologies to meet human needs on a global scale, hydrogen peroxide electrosynthesis, and the development of processes for the destruction of PFAS chemicals in aqueous systems, electrocatalyzed by laser-made nanomaterials. |
 Ziyi Meng | Ziyi Meng obtained his BSc in Polymer Materials and Engineering (2019) and MSc in Materials Science (2022) at Hubei University of Technology (China), where he conducted research in microbial modified mineral materials, advanced functional super elastic, and inorganic–organic composited proton exchange membrane fuel cells for extreme conditions under the supervision of Prof. Qingting Liu. In 2022, he enrolled at the University of Rochester as a PhD student in the Materials Science Program and joined the group of Prof. Astrid M. Müller to work on the degradation of per- and polyfluoroalkyl substances (PFAS), employing aqueous electrocatalysis and laser-made bimetallic nanomaterials. |
 Wanqing Yu | Wanqing Yu hails from Guangzhou, China, and came to the U.S. for her undergraduate education at the University of Rochester, where she obtained dual-major BS and BA degrees in chemical engineering and dance in 2023. At the University of Rochester, she conducted undergraduate research in the group of Prof. Astrid M. Müller, working on aqueous electrocatalytic defluorination of the PFAS chemical perfluorooctanoic acid, catalyzed by nanomaterials made by pulsed laser in liquids synthesis. In fall 2022, Wanqing received an Undergraduate Teaching Assistant Award in chemical engineering. Currently, Wanqing is a PhD student in chemical engineering at Georgia Institute of Technology. |
 Astrid M. Müller | Astrid M. Müller is an Assistant Professor of Chemical Engineering at the University of Rochester since 2018. Prof. Müller earned a PhD in Physical Chemistry for work on ultrafast reaction dynamics at the Max Planck Institute of Quantum Optics. Her postdoctoral work centred on developing a fundamental understanding of laser–matter interactions. Her independent research focuses on pulsed laser in liquids synthesis of mixed-metal nanomaterials with controlled structural and electronic properties. This uniquely positions Prof. Müller's group to quantitatively understand how nanocatalysts and electrocatalytic mechanisms impact the performance of nanomaterials in sustainable energy, green chemistry, and aqueous PFAS destruction applications. |
Introduction
The industrial revolution led to unprecedented technological progress, human health, prosperity, wellbeing, and population growth, but it also caused climate change1 and environmental pollution on a global scale. Technological innovations in consumer goods, and pharmaceutical, agricultural, and industrial applications released large quantities of halogenated organic pollutants into the environment, including Earth's waterways. Electrocatalytic technologies that remediate halogenated organic water pollutants can learn from other electrocatalysis fields and adapt analogue concepts, for example from water oxidation2–5 and oxygen reduction electrocatalysis,2,6–8 aqueous photoelectrochemical mechanisms,9–12in situ and operando spectroscopies of electrocatalysts,13–15 and computational electrocatalyst design.15–17Halogenated organic water pollutants are hazardous for humans and animals.18–27 They are organic compounds that contain the halogen atoms fluorine, chlorine, or bromine, possess solubility in natural water, and are harmful to the environment. Halogenated organic compounds, also called organohalogens, are typically classified into different categories depending on their structure or chemical properties. Halogenated organic water pollutants include aliphatic or aromatic halogenated hydrocarbons.28 Common organohalogens are shown in Fig. 1. Organohalogens serve as solvents, coatings, degreasing agents, biocides, medical propellants, plasticizers, hydraulic and heat transfer fluids, chemical synthesis intermediates, refrigerants, coolants, and flame retardants.22,23,25,27 Organohalogens possess exceptionally strong carbon–halogen bonds, leading to high heat resistance, low surface tension, high lipophilicity, and chemical inertness. Most organohalogens possess amphiphilic (ionic and neutral) properties and are xenobiotic, although natural organohalogens are known.29 Because of their thermodynamically strong covalent C–X (X = F, Cl, or Br) bonds, these compounds were initially considered nonmetabolizable and nontoxic,30 which turned out to be false, creating global human health risks and an urgent need for environmental remediation, particularly from water sources.
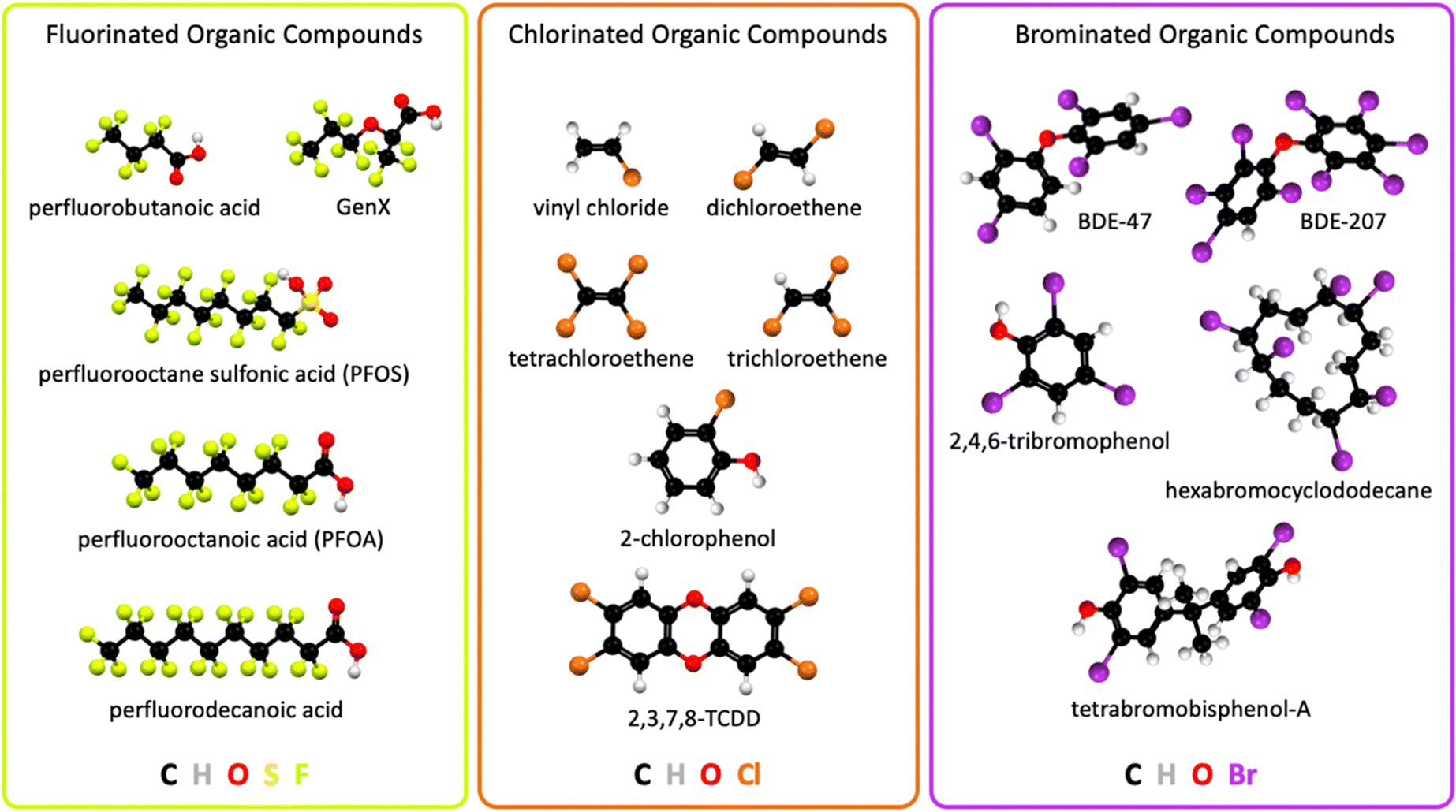 | ||
| Fig. 1 Chemical structures of widespread halogenated organic water pollutants; 2,3,7,8-TCDD, 2,3,7,8-tetrachlorodibenzodioxin; BDE, bromodiphenyl ether. | ||
The discovery and manufacturing of organohalogens has led to revolutionary materials with high utility, such as non-stick Teflon cookware coatings, Rain-X water repellants, fire-retardant, water-proofing, and grease-resisting additives to upholstery and clothing, firefighting foams, and cleaning agents for electronics and microelectronics manufacturing. The widespread use of these chemicals has resulted in inadvertent or purposeful discharge into the environment.31 The high strength of carbon–halogen bonds inhibits biodegradation processes in nature, and leads to extended lifetimes and accumulation of chlorinated, brominated, and fluorinated organic compounds in the environment, animals and humans.20
Policy regulations and restrictions regarding the manufacturing and use of halogenated organics have been instated on the national level and worldwide. For example, restrictions on the production of polychlorinated biphenyls (PCBs) began in 1970s, followed by international implementations through the Stockholm Convention in 2004, which banned the production of PCBs, aiming to phase out PCBs in use by 2025, and ensuring environmentally sound management by 2028.32 Limitations on brominated compounds started in 2009 when the Stockholm Convention listed polybrominated diphenyl ethers as new persistent organic pollutants and banned their production and use.33 Currently, five specific groups of brominated flame retardants are listed in the Stockholm Convention: hexabromobiphenyl, hexabromocyclododecane (HBCD), and the commercial polybrominated diphenyl ethers octabromodiphenyl ether (octaBDE), pentabromodiphenyl ether (pentaBDE), and decabromodiphenyl ether (decaBDE).33
Additional policy efforts have been made to discontinue or limit the production and use of poly-fluorinated compounds. Significant New Use Rules were established in the U.S. to restrict the production and use of per- and polyfluoroalkyl substances (PFAS). The U.S. Environmental Protection Agency worked with leading chemical companies on a global phaseout of perfluorooctanoic acid (PFOA) through the 2010/2015 PFOA Stewardship Program to reduce emissions and residual content of PFAS.34 By 2009, perfluorooctane sulfonic acid (PFOS) and related compounds were listed under Annex B of the Stockholm Convention on Persistent Organic Pollutants.35 The U.S. Environmental Protection Agency's notification levels for PFOA and PFOS are 5.1 and 6.5 ppt, respectively, and the response levels for PFOA and PFOS are 10 and 40 ppt, respectively.36,37
Chlorofluorocarbons (CFCs) and hydrochlorofluorocarbons (HCFCs) are fully or partially halogenated hydrocarbons that are produced from methane, ethane, and propane. CFCs and HCFCs are nontoxic, non-flammable, and long-lived synthetic compounds that contain carbon, hydrogen, chlorine, and fluorine.38 Any production of CFCs was banned in 2010 through the Montreal Protocol to reduce CFC emissions into the atmosphere, where CFCs deplete stratospheric ozone.39
Applications and health risks of halogenated organic water pollutants
Exposure to organohalogens threatens human health.40–42 Resistance to biodegradation causes long lifetimes and accumulation of organohalogens in the environment, together with global distribution via waterways.43 Bioaccumulation through food chains and direct uptake introduce organohalogens into the human body, where they have been linked to Parkinson's disease,44 harm in cognitive function and development, reproductive, hormonal, and metabolic processes, and increased risk for cancer.30,45,46 Specific applications and associated risks of chlorinated, brominated, and fluorinated organic compounds are detailed below.The high stability and environmental persistence of chlorinated organic compounds is attributable to the lipophilicity and high thermodynamic strength of C–Cl bonds.50,51 Common chlorinated organic water pollutants are polychlorinated dibenzodioxins (PCDDs), polychlorinated dibenzofurans (PCDFs), PCBs, and chlorinated paraffins, whose global annual production exceeded 1 million metric tons in 2009,22 the year chlorinated compounds started to get restricted. Chlorinated alkanes are toxic: human exposure can harm the liver, kidneys, lungs, neurological systems, cardiovascular system, and immune system, and they have been linked to increased risk of cancer (Fig. 2).52 Although the production of PCBs has been phased out decades ago, 10 million tons of PCB-containing materials remain across the globe.32 PCBs accumulate in fat, liver, skin, and nerve tissues in humans (Fig. 2).20 Compounds produced by PCB detoxification in the liver have been linked to hormonal interference, reproductive,53,54 metabolic,55 and neurological dysfunction,56 and cancer (Fig. 2).57 The extent of toxicity depends on the chemical structure of chlorinated organic compounds and particularly the position of chlorine atoms on phenyl rings, with the highest toxicity observed for chlorine in meta and para positions.58 Dioxins have been linked to similar reproductive, neurological, and carcinogenic issues as PCBs.20 Chlorinated organic compounds often originate in small organisms and transfer through food chains to accumulate in higher-order species in large doses.20
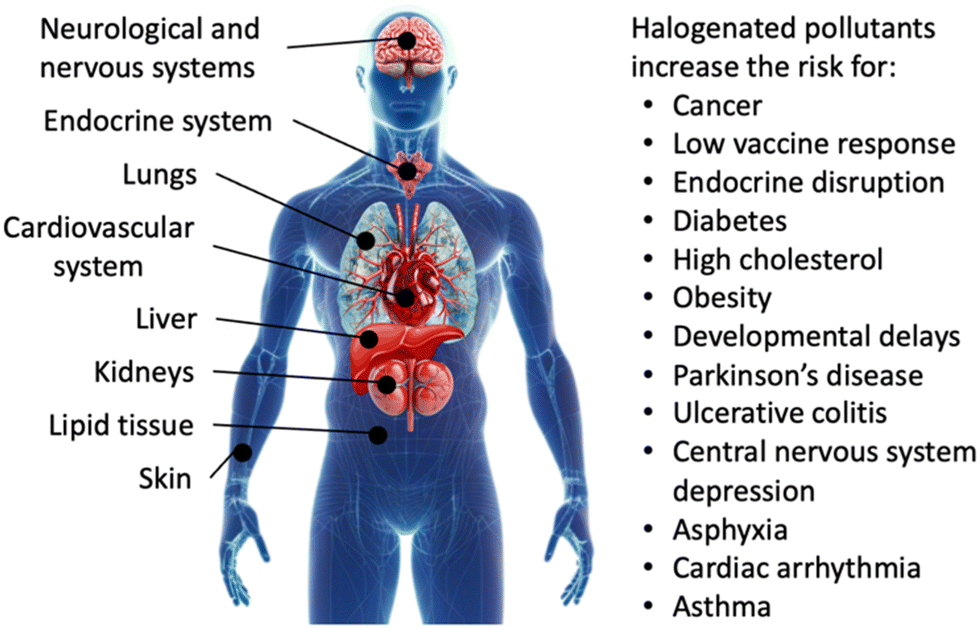 | ||
| Fig. 2 Human health risks upon exposure to halogenated organic water pollutants. | ||
Most brominated organic water pollutants are commonly known as brominated flame retardants (BFRs), which are used across many industries to inhibit or suppress combustion processes. BFRs are often found in plastics, textiles, electronic circuitry, and building materials.49,59 The utility of BFRs has led to a growing market with a global market size of 2.14 billion USD in 2022 and a compound annual growth rate of 5.7%.60 BFRs are lipophilic and bioaccumulate if leached from products, during manufacturing, or during incineration.61 Brominated, like chlorinated, organic pollutants accumulate in the environment and threaten human health by build-up in lipid tissues.20 Specific health effects caused by BFRs are less understood than those of PCBs. BFRs have been linked to interfering with nerve development in fetuses and newborns, and harming infant cognitive function (Fig. 2).20,62
PFAS have been identified in ground water, surface water, drinking water, soil, indoor air, dietary sources, manufacturing locations, and wastewater treatment plants, as well as in human blood, serum, milk, urine, and placenta tissue.82 For example, the PFAS chemical PFOS bioaccumulates in fish that humans consume, making such fish a source of human PFAS exposure.83 Human PFAS exposure can additionally occur via dermal contact because PFAS can be taken up from oil- and water-resistant non-stick coatings on cookware and paper food packaging materials.83 Inhalation of indoor air and household dust is another route for human PFAS uptake.83
Human exposure to PFAS causes reproductive and immune system harm, increased risk of non-Hodgkin's lymphoma and kidney, testicular, prostate, breast, liver, and ovarian cancers, decreased vaccine response, increased risk of asthma in adolescents, increased risk of diabetes and hypertension in women, developmental delays in children, changes in liver enzymes, endocrine disruption, and increased cholesterol levels and/or risk of obesity;80,81,84–89 in unborn children, delayed mammary gland development, reduced response to vaccines, and lower birth weight have been observed (Fig. 2).90,91 PFAS also pose ecological risks through bioaccumulation, food chains, and toxicity in terrestrial and aquatic wildlife.92
Remediation techniques for halogenated organic water pollutants
Maximum contaminant limits in water in the U.S. are set by the U.S. Environmental Protection Agency and published in the National Primary Drinking Water Regulations.93 For most chlorinated, brominated, and fluorinated organic pollutants, the allowable contamination threshold is on the order of parts per billion (ppb) or parts per trillion (ppt), depending on the compound and associated risks.52,94 To achieve safe levels, water remediation techniques are needed for effective removal and destruction of pollutants. Halogenated organic pollutants have a particularly high resistance against degradation because of their strong carbon–halogen bonds. As a result, only a few destruction techniques for complete mineralization of halogenated organic water pollutants have been commercialized, and the field is a very active research area.27,95,96 Several separation techniques have found commercial use to remove halogenated pollutants from water sources; separation only concentrates pollutants,27 after which disposal by destruction is necessary.Concentration vs. destruction techniques
Organohalogen concentrations differ vastly across water sources and mainly depend on the distance from the location of pollutant discharge. For example, PFAS concentrations in water range from 26 to 5200 ng L−1;97,98 for illustration, the lower limit corresponds to approximately one drop in an Olympic swimming pool full of water. Freshwater sources are typically contaminated with PFAS at levels on the order of hundreds of nanograms per liter, whereas concentrations in marine water are at tens of nanograms per liter.98Adsorption techniques. Adsorption utilizes physical or chemical interactions between the surface of a solid (adsorbent) and a solute (adsorbate) to remove pollutants from water sources. In contrast, absorbents are porous materials that take up matter into spaces within and throughout the material. Adsorbable organic halogens are removed from water by adsorption methods,101,102 such as Pd/Fe bimetallic particles to treat activated sludge of chemical dyestuff wastewater.103 Granular activated carbon and powdered activated carbon are the most frequently used adsorbents, but activated alumina and zeolites have also been employed.104 Two common chlorinated groundwater pollutants, tetrachloroethylene and trichloroethylene, have shown significant adsorption on granular activated carbon.105 Likewise, absorbent materials that are capable of removing brominated water pollutants include metal organic frameworks,106 biochar,107 and microplastics.108
Reverse osmosis. Reverse osmosis is a separation technique that passes water through a semi-permeable membrane that discharges a treated stream (permeate) and a rejection stream (concentrate). Reverse osmosis decreased the concentration of 2,3,7,8-substituted polychlorinated dibenzo-p-dioxins (PCDD), dibenzofurans (PCDFs), and dioxin-like PCBs in water.109
| X | R–X | Bond dissociation energy (kJ mol−1) |
|---|---|---|
| F | GenX–F | 536.0 |
| HF2C–F | 534.7 | |
| H5C6–F | 526.0 | |
| H2FC–F | 499.2 | |
| HO2C8F14–F (PFOA) | 451.9–494.1 | |
| H3C–F | 452.3 | |
| O3SC8F16–F (PFOS) | 415.6–461.9 | |
| Cl | H5C6–Cl | 400.0 |
| (H3C)3C–Cl | 359.4 | |
| H3CH2CH2C–Cl | 356.9 | |
| H3CH2C–Cl | 354.4 | |
| H3C–Cl | 334.7 | |
| H2ClC–Cl | 328.4 | |
| HCl2C–Cl | 316.3 | |
| H5C6H2C–Cl | 302.1 | |
| Br | H5C6–Br | 337.0 |
| (H3C)3C–Br | 304.2 | |
| H3C(H2C)2–Br | 303.3 | |
| H3CH2C–Br | 302.9 | |
| H3C–Br | 291.1 | |
| H2BrC–Br | 280.3 | |
| HBr2C–Br | 266.9 | |
| H5C6H2C–Br | 248.5 | |
The bond dissociation energy values, which are a measure for thermodynamic bond strength, show that halogen atoms with higher electronegativity form stronger bonds to carbon atoms (Table 1). Fluorine is the most electronegative element in the periodic table and ergo C–F bonds are the strongest covalent bonds known in organic chemistry. In general, electronegativity of halogens follows F > Cl > Br, and likewise the trend in bond strengths follows C–F > C–Cl > C–Br. The C–F bond dissociation energies in PFAS depend on the extent of fluorination of carbon atoms, the position of C–F bonds, and the nature of the head group in the molecule (Fig. 3).
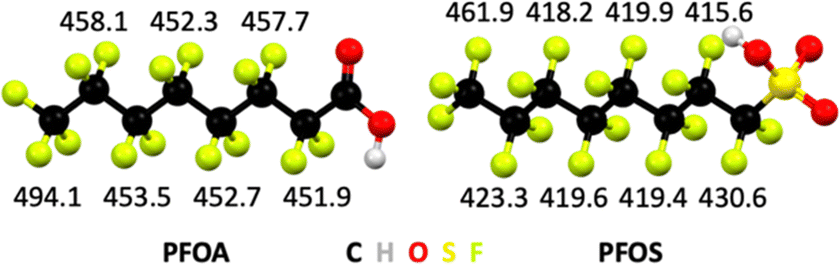 | ||
| Fig. 3 Bond dissociation energies in kJ mol−1 of C–F bonds by position in perfluorooctanoic acid (PFOA) and perfluorooctane sulfonic acid (PFOS). From ref. 122 and 123. | ||
In contrast, in chlorinated and brominated organic compounds, dissociation energies of C–X (X = Cl or Br) bonds depend on the degree of substitution at the carbon of the C–X bond. Thus, the strongest C–X bonds of chlorinated and brominated compounds are found in halogenated phenyls.124
Threshold limit values and response levels of common halogenated organic water pollutants
Threshold limit values (TLVs) serve as important benchmarks for monitoring workplace exposure. These values denote permissible airborne concentrations of chemical substances that the majority of workers can encounter in a repetitive manner throughout their career span, without detrimental health consequences. The American Conference of Governmental Industrial Hygienists (ACGIH) formulates TLV-time-weighted averages (TLV-TWAs) to safeguard against prolonged exposures, as well as short-term exposure limits to shield against sudden spikes in exposure. It is important to note that TLVs are not designed to establish a rigid demarcation between safe and hazardous exposures. Instead, their primary objective is to safeguard workers from potential health impacts.125 The notification level is the concentration of a hazardous substance in the environment, above which specific actions or notifications are required. Notification levels are often used to assess water pollutants because they represent human health-based advisory levels for chemicals in drinking water, which lack maximum contaminant levels.126 Human health-based advisory levels establish the contaminant concentration in drinking water, below which no adverse health effects and/or aesthetic impacts are expected during specific periods of exposure.127 Actions might include reporting the presence of the substance to regulatory authorities or taking steps to mitigate the exposure or contamination. Notification levels are typically set by regulatory agencies and are designed to alert relevant parties when a certain level of contamination has been reached. In contrast, the response level represents a concentration of a hazardous substance in the environment that triggers specific actions or interventions to mitigate risks and protect human health or the ecosystem. Response levels are set to guide the appropriate measures to be taken when contamination reaches a certain level. These measures might include evacuation, containment, or clean-up, to prevent further exposure or harm.126 TLVs and notification levels of common halogenated organic water pollutants that are discussed in this article are in Table 2.| Class | Compound | Notification level | Response level | Ref. | Threshold limit value | Ref. |
|---|---|---|---|---|---|---|
| a Massachusetts drinking water maximum contaminant levels, PFDA combined with other PFAS.156 b California drinking water standards.131 | ||||||
| Fluorinated | PFBA | 100 ppt | 1000 ppt | 128 | 5 ppt | 129 |
| GenX | 30 ppt | 300 ppt | 128 | 5 ppt | ||
| PFDA | 20 ppta | — | 130 | 3 ppt | ||
| PFOS | 6.5 ppt | 40 ppt | 4 ppt | |||
| PFOA | 5.1 ppt | 10 ppt | 4 ppt | |||
| PFHxS | 3 ppt | 20 ppt | 3 ppt | |||
| Fluorinated–chlorinated | Freon-11 | 5 ppbb | 150 ppbb | 131 | 1000 ppm (airborne) | 132 |
| Freon-12 | 1 ppm | 10 ppm | 133 | 1000 ppm (airborne) | 134 | |
| Freon-13 | — | 3000 ppm (airborne) | 135 | Not established | 136 | |
| HCFC-22 | — | — | 1000 ppm (airborne) | 137 | ||
| HCFC-142b | — | — | 1000 ppm (airborne) | 138 | ||
| HCFC-141b | — | — | Not established | 139 | ||
| Chlorinated | VC | 2 ppb | 2 ppb | 140,141 | 1 ppm (airborne) | 142 |
| DCE | 500 pptb | 500 pptb | 131 | 2 ppm (airborne) | 143 | |
| PERC | 500 pptb | 5 ppbb | 131 | 25 ppm (airborne) | 144 | |
| TCE | 5 ppbb | 500 pptb | 131 | 10 ppm (airborne) | 145 | |
| 2-Chlorophenol | 100 ppt (based on water aesthetics) | — | 146 | Not established | 146 and 147 | |
| 2,3,7,8-TCDD | 5 ppqb | 30 ppqb | 30 ppq | 148 | ||
| PCB-47 | 500 pptb | 500 pptb | 131 | 1 ppb | 149 | |
| PCB-77 | 500 pptb | 500 pptb | 131 | 500 ppt | 149 | |
| Brominated | BDE-47 | — | 24 ppt | 150 | 50 mg kg−1 body weight per day | 151 |
| BDE-207 | — | — | 50 mg kg−1 body weight per day | 151 | ||
| TBP | — | — | Not established | 152 | ||
| HBCD | — | — | 100 mg kg−1 body weight per day | 153 | ||
| TBBPA | — | 15 ppb | 154 | 10 ppb | 155 | |
Destructive remediation of chlorinated and brominated organic water pollutants
Destructive methods are necessary to treat waste streams or sludges after concentration of target pollutants by separation processes. Chlorinated and brominated organic pollutants can be destroyed by incineration,157 non-thermal plasma discharge,158 γ-irradiation,159 biological, catalytic, photolytic, photocatalytic, or photochemical processes.Recent progress in the destruction of chlorinated organic water pollutants
Recent progress in the destruction of brominated organic water pollutants
Destructive remediation of fluorinated organic water pollutants including PFAS
The destructive remediation of fluorinated organic compounds, particularly PFAS, is an area of intense research. Methods for defluorination of the common PFAS chemical perfluorooctanoic acid (PFOA) have been reported by electrochemical reduction at a Rh/Ni cathode in dimethyl formamide via hydrodefluorination,224 hazardous γ-irradiation with a 60Co source in an alkaline solution under N2-saturated conditions,159 and mineralization of perfluorocarboxylic acids, including PFOA, via the formation of rapidly decomposing carbanions in polar, aprotic dimethyl sulfoxide electrolyte; the carbanion-based mechanisms can only operate in water-free, polar, aprotic solvent and fail to degrade sulfonic acid PFAS, such as PFOS.225 Further, 19F-NMR was used to quantify PFOA defluorination, which appeared to have a detection threshold of ≥5 mM (approx. 2000 ppm PFOA).225 Globally scalable, viable technologies must work in aqueous media and enable destruction of PFAS with much lower concentrations. Perfluorocarboxylic acids, such as perfluoro-butanoic, pentanoic, hexanoic, heptanoic, and octanoic acid (PFOA), were mineralized using a Ce-doped nanocrystalline PbO2 film electrode;226 the toxicity of lead poses challenges. While these reports are mechanistically intriguing, only cost- and energy-saving aqueous methods that utilize nontoxic, nonprecious materials and renewable electricity will be viable and sustainable on a global scale. Electrooxidation of PFOA and PFOS at Magnéli-phase Ti4O7 ceramic anodes outperformed mineralization at Ce-doped PbO2 and Ti-modified boron-doped diamond electrodes, due to faster oxidation rate.227 PFOA in water was degraded by ultraviolet-visible light assisted ZnxCu1−xFe2O4-oxalic acid system, using a ferrite-based catalyst that allowed for magnetic catalyst recovery.228Technoeconomic analyses of destruction techniques are indispensable to assess viability on a global scale. Significant research has been done to determine the most efficient destruction techniques.27 Energy efficiency is important to lower operational costs and improve carbon footprints, whereas capital expenditures matter for assessing the affordability of units. Estimated energy and capital cost requirements of existing PFAS destruction techniques, based on literature data,167,229–237 are shown in Fig. 4. Capital expenditure values refer to cost of equipment in the United States, are given in U.S. dollars, and are needed to treat at least one cubic meter of polluted water.
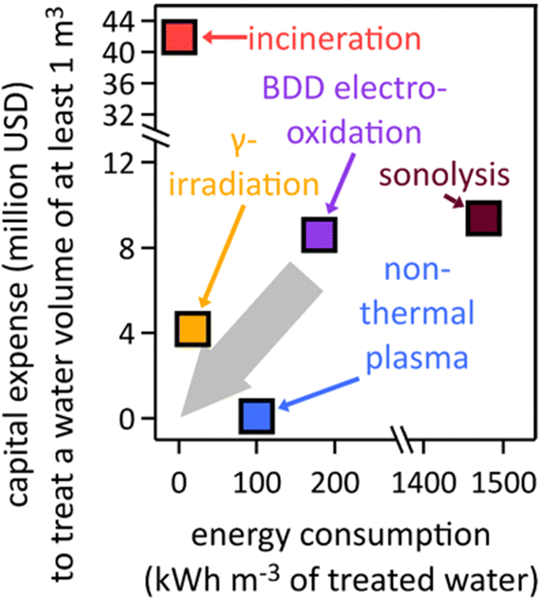 | ||
| Fig. 4 Estimated electrical energy and capital expenditure requirements of existing PFAS destruction techniques, based on literature data;167,229–237 BDD, boron-doped diamond. The grey arrow depicts the direction into which technologies must move in terms of capital expense and energy consumption to become viable on a global scale. | ||
Incineration requires an initial investment of $41![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 938050 for a one-line “turn-key” incineration plant.83 The average amount of energy required for incineration is 0.45 kW h m−3 in the U.S., which has been measured and varies depending on the country and the type of incineration plant used.233 The reported initial investment cost of γ-irradiation is $4176150, which includes the sum of the cost of the electron beam accelerator with lifespan of 15 years and personnel costs needed to run a γ-irradiation facility.238 The equivalent electrical energy requirement to achieve 90% sulfadiazine degradation by γ-irradiation from a 60Co source, taken here as a proxy for PFAS degradation, at a constant dose rate of 6.69 kGy h−1 has been reported to be 18 kW h m−3.239 The capital investment for non-thermal plasma231,237 of $92329 was obtained from a capital cost approximation for industrial wastewater plants.163,240 The energy required to achieve a high removal rate of PFOA from water using non-thermal plasma is 100 kW h m−3, which was deduced from the energy efficiency of non-thermal plasma setups.237 Sonolysis by ultrasonication has a capital investment cost of $9390000, which has been calculated to include the part replacement cost, labor cost, analytical costs, chemical costs, and electrical costs.230 The energy requirement for ultrasonication (1475 kW h m−3) was calculated using the energy efficiency of ultrasonication in g (kW h)−1.235,241
938050 for a one-line “turn-key” incineration plant.83 The average amount of energy required for incineration is 0.45 kW h m−3 in the U.S., which has been measured and varies depending on the country and the type of incineration plant used.233 The reported initial investment cost of γ-irradiation is $4176150, which includes the sum of the cost of the electron beam accelerator with lifespan of 15 years and personnel costs needed to run a γ-irradiation facility.238 The equivalent electrical energy requirement to achieve 90% sulfadiazine degradation by γ-irradiation from a 60Co source, taken here as a proxy for PFAS degradation, at a constant dose rate of 6.69 kGy h−1 has been reported to be 18 kW h m−3.239 The capital investment for non-thermal plasma231,237 of $92329 was obtained from a capital cost approximation for industrial wastewater plants.163,240 The energy required to achieve a high removal rate of PFOA from water using non-thermal plasma is 100 kW h m−3, which was deduced from the energy efficiency of non-thermal plasma setups.237 Sonolysis by ultrasonication has a capital investment cost of $9390000, which has been calculated to include the part replacement cost, labor cost, analytical costs, chemical costs, and electrical costs.230 The energy requirement for ultrasonication (1475 kW h m−3) was calculated using the energy efficiency of ultrasonication in g (kW h)−1.235,241
Chemical oxidation of PFOA by stoichiometric amounts of permanganate,242 hydrogen peroxide, or persulfate26,243 is cost-prohibitive and creates large amounts of chemical waste. Methods based on electrochemical processes have gained popularity because of lower energy demands than physical destruction methods, i.e. incineration, γ-irradiation, non-thermal plasma, and sonolysis. PFAS destruction by supercritical water oxidation244 requires high initial investment;245 we were unable to find numbers for investment costs. The thermal energy requirement for an efficient supercritical water generation facility is 5 MW for 250 metric tons of water per day.246
Electrooxidations of PFAS at expensive, often toxic, specialized anodes are nonviable.226,235,247–257 The inter-electrode distance matters in electrolyzers because smaller distances are concomitant with less ohmic losses.258 A generally accepted upper limit for inter-electrode distance in aqueous systems is 10 cm, which necessitates 10 m2 geometric electrode area to treat 1 m3 of polluted water,258 rendering boron-doped diamond (BDD) electrodes cost-prohibitive.259 At smaller inter-electrode distances, which generally result in higher electrocatalytic performance, the geometric electrode area requirement increases for treatment of a 1 m3 batch, making the economics of BDD electrodes even more unfavorable. The energy required to halve an initial PFOA concentration of 15 mg L−1 with a BDD electrode of 38 cm2 geometric area and an inter-electrode distance of 4 mm was reported as 180 W h L−1 at 50 mA cm−2, for the treatment of 250 mL solution.234 Conversion of these numbers to the treatment of 1 m3 of polluted water requires BDD electrodes of 15.2 m2 at a cost of $8.58 million229 and 180 kW h m−3 electrical energy (Fig. 4). Electrochemical degradation of PFOA on ultra-nanocrystalline BDD coated on niobium electrodes additionally produced toxic perchlorate.234 Ultraviolet-light-assisted electrochemical PFAS defluorination required cost-prohibitive platinum electrodes and N2-saturated electrolyte.260 Ergo, potentially viable PFAS destruction technologies must be more cost-effective and energy-saving to achieve economic feasibility and reduce carbon emissions.
Recent progress in the destruction of PFAS
Perfluorooctanoic acid (PFOA) has been degraded anaerobically to shorter chain perfluoroalkyl carboxylic acids and produced fluoride in biosolids using Acidimicrobium sp. strain A6 and ferrihydrite.261 PFOA has also been degraded efficiently by 80% over ball-milled boron nitride in deionized water or by 60% in simulated drinking water using photocatalysis for 2 hours.262 The photocatalytic degradation of PFOA under UV-A and UV-C illumination with boron nitride was improved by using a composite of boron nitride and titanium oxide instead of neat boron nitride or titanium oxide. However, complete mineralization was not achieved and only shorter chain perfluorinated carboxylic acids were produced. The composite was capable of degrading PFOA by 68% in 7 hours using natural sunlight.263 PFOA was degraded via a photocatalytic process that used a carbon-modified bismuth phosphate composite, absorbing PFOA in 2 hours and achieving nearly complete decomposition in situ in 4 hours under UV irradiation.264 Another composite that has been developed for the photocatalytic degradation of PFOA in water is iron (hydr)oxides/carbon sphere (FeO/CS) composite. This material almost completely adsorbed PFOA in 1 hour, which subsequently underwent photodegradation and defluorination. During this process, PFOA was photodegraded by 95.2% and defluorinated by 57.2% in 4 hours.265 Another photocatalytic process that used UV light and Bi3O(OH)(PO4)2 in acidic conditions has been reported to degrade PFOA. However, this process was unable to degrade shorter chain PFAS unless some changes were made to the reactor system. With this system, challenges for implementation in real waters exist because the reaction was quenched by chloride and sulfate.266 Complete defluorination of PFOA was achieved after 6 hours in water using a dual-frequency ultrasonic activated persulfate. This method can be used for other PFAS compounds, but not as effectively.267 An electrochemical degradation process for PFOA using sodium sulfate has been developed, which enabled 99.5% degradation and 50% fluoride generation after 4 hours.268 A sorptive photocatalyst Fe/TNTs@AC, which was based on activated carbon and titanium oxide, was synthesized and used for PFOA degradation. This catalyst completely absorbed PFOA in 1 hour, degraded it by 90% in 4 hours using UV radiation, and then mineralized 62% of the degraded PFOA to fluoride.269 Another photocatalytic process that has been developed to degrade PFOA in aqueous solutions used In2O3 nanoparticles. This process worked most efficiently in acidic (pH = 2) conditions and successfully decomposed most of PFOA to fluoride and carbon dioxide within 90 minutes under UV irradiation, while also achieving 95.99% of PFOA defluorination.270 Chemical mineralization of perfluorocarboxylic acids is mechanistically intriguing,225 but works only in polar, non-protic electrolytes, such as dimethyl sulfoxide (DMSO), hampering global scalability, and fails to degrade sulfonic acid PFAS, such as PFOS.Perfluorooctane sulfonic acid (PFOS) degradation has been reported, using sonolysis by 96.9%, 93.8%, and 91.2% at 400 kHz, 500 kHz, and 1000 kHz, respectively, in 4 hours.271 Another method to degrade PFOS in an aqueous solution used reverse vortex flow gliding arc plasma; PFOS was degraded by 93.1% in 1 hour. This method can also be used to degrade other PFAS compounds, such as PFOA or PFDA and others.272 Both PFOA and PFOS were defluorinated by 88% in 1 hour and 92% in 24 hours, respectively, using a UV light in a system that contained sulfite and iodide. Adding iodide to the UV light illuminated sulfite system greatly accelerated the defluorination of many PFAS. This system has achieved a complete removal of both PFOA and PFOS from concentrated mixtures in NaCl brine.273 Electrooxidation using titanium suboxide anodes were used to degrade multiple PFAS compounds, such as PFOA and PFOS, whose concentrations decreased very quickly and approached zero in 1 hour.274 Another method to degrade PFOA and PFOS as well as other PFAS compounds is γ-irradiation, using a 60Co source. PFOS degradation was more efficient with branched PFOS isomers compared to linear molecules. Branched PFOS isomers were degraded almost completely, while PFOA was degraded by 87%.275 A non-thermal plasma generator was custom-built to remove PFOA, perfluorohexanoic acid (PFHxA), and PFOS from water in both ultrapure and groundwater. In 30 minutes, PFOS was degraded completely in ultrapure water and by 85% in groundwater. PFHxA was degraded by 35% in ultrapure water and by 40% in groundwater, while PFOA degradation reached about 50% in both water systems.276 PFOS and PFOA were significantly degraded in soil and groundwater by high dose electron beam technology. This process enabled the decrease of PFOS and PFOA concentrations in groundwater by 87.9% and 53.7%, respectively.277 A duo-functional tri-metallic-oxide hybrid photocatalyst has been developed for the degradation of many PFAS compounds.278 It possessed a high adsorption capacity and achieved 99.8% and 99.4% adsorption efficiency for PFOS and PFOA respectively, and it exhibited a high defluorination rate up to 67.6% for PFOS and 74.8% for PFOA. With this catalyst, PFOS was degraded by 95.5% in 5 hours, while PFOA was degraded by 98.9% in 30 minutes.278 Boron-doped graphene sponge anodes have been reported for the degradation of PFAS compounds by electrochemical oxidation. This method had a higher removal efficiency for longer-chain than for short-chain PFAS, but its defluorination efficiency was lower than that of other methods.279 GenX, which is a short-chain PFAS, was mineralized by an electro-Fenton process that paired a graphene-coated nickel foam electrode with a boron doped diamond electrode. This process achieved 92.2% mineralization after 6 hours of treatment.280
Aqueous advanced redox processes
Advanced oxidation processes, advanced reduction processes, and combined hybrid processes have emerged as promising strategies in water remediation due to potentially high organohalogen destruction efficiencies.113,281 These processes can have rapid reaction rates, destroy pollutants without generating solid waste, do not require chemical oxidants, high pressures, or high temperatures, and can be powered by sustainable renewable electricity.282–285 The advantages of advanced redox processes have led to intense research, such as photolytic, photochemical, photocatalytic, cavitation, electrochemical, and ionizing radiation approaches for water remediation, as well as combinations of these methods.113Advanced oxidation processes (AOPs) generate and utilize highly reactive species with high oxidation potentials that are capable of cleaving carbon–halogen bonds of pollutants in wastewaters.286 Oxidants are often radicals under aerobic conditions, such as the hydroxyl radical (˙OH, aqueous lifetime 0.02 μs), superoxide radical anion (OO˙−, aqueous lifetime 1.3 μs), sulfate radical anion (SO4˙−, aqueous lifetime 30–40 μs), and sulfite radical anion (SO3˙−), in addition to potent non-radical species, such as hydrogen peroxide (H2O2) and singlet oxygen (1O2, aqueous lifetime 3.5 μs).285,287–301
Advanced reduction processes (ARPs) produce highly reactive species with high reduction potentials to cleave carbon–halogen bonds of aqueous pollutants.302 Reductants generated by ARPs require completely anaerobic conditions and are comprised of radicals, such as the sulfur dioxide radical anion (SO2˙−), sulfite radical anion (SO3˙−),303 and hydrogen radical (H˙), as well as non-radical hydrated electrons (e−aq, aqueous lifetime 0.43 or 8.6 μs at pH 7.0 or 9.5, respectively).113,287,304 Oxygen-free conditions are often impractical, especially since the water oxidation half reaction can produce O2,305,306 membranes that separate oxidation and reduction half reactions cannot completely exclude oxygen crossover,307 and airtight seals are inherently challenging on a large scale.
Mechanistic aspects of electrocatalytic aqueous advanced redox processes
A quantitative mechanistic understanding of aqueous advanced redox processes is lacking to date, albeit urgently needed to accelerate the development of viable aqueous electrocatalytic organohalogen destruction techniques. Radicals and reactive oxygen species play a major role in aqueous advanced redox processes, and the mechanisms of their formation and reactions with organohalogens must be understood in detail. Electrocatalysts significantly lower energy requirements for electrochemical transformations,305 especially in aqueous systems, where potential-leveling by proton-coupled electron transfer (PCET) decreases needed energy inputs.308Reactive oxygen species of the water–oxygen system
Aerobic aqueous processes that produce H2O2, ˙OH, HOO˙, or OO˙− are based on the electrochemical transformations of the water–oxygen system (Fig. 5). Most of the transformations are electrochemical oxidations or reductions that proceed through energy-saving, potential-leveling PCET steps, which require that equal numbers of protons and electrons are transferred.308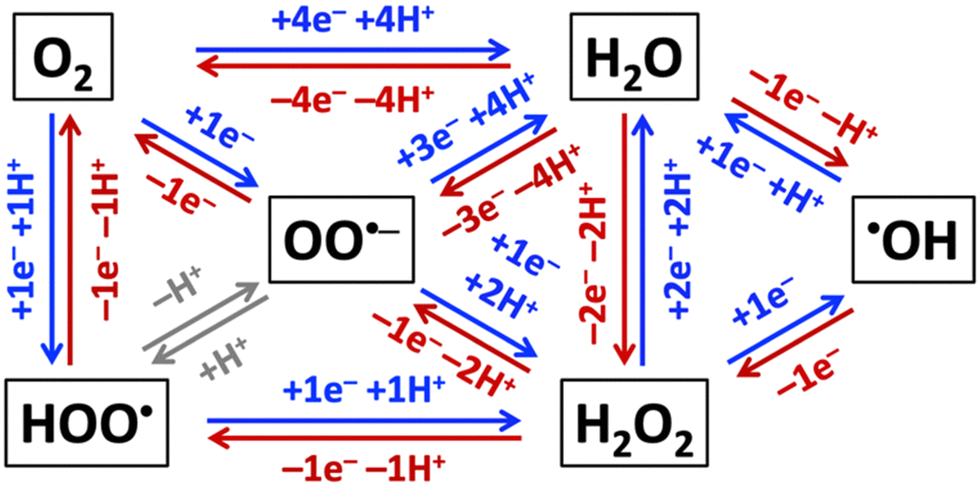 | ||
| Fig. 5 Electrochemical reductions (blue) and oxidations (red) of non-surface-bound species in the H2O–O2-system. | ||
Reactive oxygen species that are relevant for organohalogen destruction, i.e. H2O2, ˙OH, HOO˙, and OO˙−, can be produced directly or via H2O2 decomposition. Water oxidation can only produce O2, H2O2, or ˙OH through direct electrocatalysis. The one-electron-one-proton or two-electron-two-proton water oxidation reactions, given here together with standard potentials E0 in V vs. the standard reference electrode (SHE), i.e. H2O → ˙OH(aq) + (H+ + e−), E0 = 2.38 VSHE, or 2H2O → H2O2 + 2(H+ + e−), E0 = 1.76 VSHE, are kinetically easier than the four-electron-four-proton reaction, i.e. 2H2O → O2 + 4(H+ + e−), E0 = 1.23 VSHE, but thermodynamically more uphill.3 Molecular oxygen can be reduced to H2O2, ˙OH, HOO˙, or OO˙− (Fig. 5).309 Electrochemical reductions and oxidations that involve the transfer of less electrons and protons are kinetically faster than those that require the movement of more electrons and protons in the H2O–O2-system (Fig. 5). The electronic and chemical structures of intermediates of one-electron-one-proton transformations are known from computational density functional theory work, which cannot accurately capture concerted two-electron steps.2,3,6 Together with the thermodynamic potentials below, these kinetic considerations allow as a function of electrolyte pH estimates which reactant species are likely formed in the highest concentrations for efficient destruction of halogenated organic water pollutants.
Hydrogen peroxide is a precursor for ˙OH, HOO˙, or OO˙− radical generation (Fig. 5) and, therefore, often called a pre- or pro-radical species. Hydrogen peroxide is a strong oxidant but reacts slowly with organics, so that it is often activated by alkali, activated carbon, heat, ultraviolet light, or transition metals,296 such as iron-containing catalysts, which can in the presence of H2O2 serve as initiators for propagation reactions that generate ˙OH, HOO˙, OO˙−, and HO2− species via Fenton, Haber–Weiss, or photo-Fenton reactions.310–313 Addition of H2O2 to the electrolyte was found to quench the production of toxic perchlorate at BDD electrodes.314
Thermodynamic potentials of reactive oxygen species
The thermodynamic potentials of reactive oxygen species determine their oxidation or reduction strengths in AOPs and ARPs of halogenated organic water pollutants. The protonation state and thermodynamic potential of aqueous reactive oxygen species depend critically on the electrolyte pH (Table 3). For example, at pH 0, ˙OH and HOO˙ are strong aqueous oxidants with standard potentials (i.e. thermodynamic potentials at pH 0) of 2.73 and 1.66, respectively, and both radicals possess sufficient thermodynamic driving force for PFAS oxidation. In contrast, at pH 14, the deprotonated hydroxyl radical anion (O˙−) possesses a thermodynamic potential of 1.77 V, whereas the thermodynamic potential of the superoxide radical anion (OO˙−) is only 0.65 V, which is insufficient to oxidize PFAS.| Reduction reaction at pH 0 | E (V) at pH 0 | Reduction reaction at pH 7 | E (V) at pH 7 | Reduction reaction at pH 14 | E (V) at pH 14 |
|---|---|---|---|---|---|
| O2 → HOO˙ | −0.05 | O2 → OO˙− | −0.33 | O2 → OO˙− | −0.33 |
| O2 → H2O2 | +0.695 | O2 → H2O2 | +0.281 | O2 → HOO− | −0.065 |
| O2 → (H2O +) ˙OH | +0.73 | O2 → (H2O +) ˙OH | −0.31 | O2 → (OH− +) O˙− | −0.03 |
| O2 → H2O | +1.229 | O2 → H2O | +0.815 | O2 → OH− | +0.401 |
| HOO˙ → H2O2 | +1.44 | OO˙− → H2O2 | +0.89 | OO˙− → HOO− | +0.20 |
| HOO˙ → (H2O +) ˙OH | +1.13 | OO˙− → (H2O +) ˙OH | +0.64 | OO˙− → (OH− +) O˙− | +0.09 |
| HOO˙ → H2O | +1.66 | OO˙− → H2O | +1.20 | OO˙− → OH− | +0.65 |
| H2O2 → (H2O +) ˙OH | +0.80 | H2O2 → (H2O +) ˙OH | +0.38 | HOO− → (OH− +) O˙− | −0.03 |
| H2O2 → H2O | +1.763 | H2O2 → H2O | +1.349 | HOO− → OH− | +0.867 |
| ˙OH → H2O | +2.72 | ˙OH → H2O | +2.31 | O˙− → OH− | +1.77 |
The protonation state of chemical species depends on their pKa value and the pH of the aqueous solution; if the pH value is larger than the pKa value of a species, the molecule is deprotonated. Aqueous pKa values of reactants and chemicals that are relevant for PFAS destruction are shown in Table 4.
Mechanistic role of oxygen radicals for PFAS destruction
Reactive oxygen radicals, such as ˙OH, O˙−, HO2˙, OO˙−, are key species in AOPs and ARPs for C–C and C–F bond cleavage.113,322 Superoxide radical anions (OO˙−) can oxidize PFAS molecules; OO˙− can also act as a reductant and is the predominant species at pH > 5, as HOO˙ has an aqueous pKa of 4.8.309 Hydroxyl radicals in aqueous solution are widely considered insufficient for PFOA or PFOS degradation310,323,324 because ˙OH radicals have an approximately 10× shorter aqueous diffusion distance than OO˙− radicals.325,326 Quenching experiments with saturated alcohols that selectively scavenged solution ˙OH radicals had no effect on the degradation of PFAS.327,328 The exact role of ˙O(H) radicals in aqueous electrocatalytic PFAS destruction is still debated. Recently, decreased degradation efficiency was observed in the presence of unsaturated allyl alcohol that is capable of quenching ˙OH radicals in the vicinity of the anode, suggesting that adsorbed ˙OH radicals played a significant role in PFAS degradation.328,329 Adsorbed ˙OH radicals are physisorbed; analog surface-bound species are non-radical hydroxide anions (OH−) that lack the thermodynamic driving force for PFAS destruction.309Auxiliary radicals and non-radical species
Addition of suitable solutes to aqueous media enables the formation of auxiliary radicals and non-radical redox agents in aqueous AOPs and ARPs. Reactive sulfur radicals are generated through dissociative reactions of persulfate, peroxomonosulfate, sulfate, or bisulfate to produce sulfate radicals, which are capable of assisting PFAS breakdown,26,243,301,304 whereas the dissociation of bisulfite or sulfite anions produces sulfite radicals and hydrated electrons in strictly oxygen-free conditions.113,301,330–332 Addition of iodide, dithionite, or ferrocyanide to anaerobic electrolytes can aid the detachment mechanism to produce hydrated electrons.260,333 Dissociation of dithionite will produce reductive sulfur dioxide radicals.302 Sulfite assists PFAS degradation through formation of persulfite.323,334 Stoichiometric PFOA oxidation by permanganate has been observed.242 Borate is known to react with H2O2, to produce peroxoborates that are stable at pH 8 to 12 and highly reactive towards nucleophiles.335 Standard potentials of PFAS-relevant auxiliary species in aqueous solution are in Table 5.285,288–299| Reaction | E 0 (VSHE) |
|---|---|
| SO4˙− → SO42− | 2.44 |
| SO3˙− → SO32− | 0.73 |
| S2O82− → SO4˙− + SO42 | 1.44 |
| SO5˙− → SO52− | 0.81 |
| H2BO3− + 5H2O → BH4− + 8OH− (pH 14) | −1.24 |
| MnO4− → MnO2 | 1.70 |
| e− → e−aq | −2.88 |
| H+ → H˙ | −2.31 |
Mechanistically, the initial attack on the pollutant occurs by radical addition to a carbon–carbon double bond or by the abstraction of a carbon-bound hydrogen after the generation of auxiliary radicals and species.336–338 In the case of perfluoro-compounds, such as PFOA and PFOS, that do not contain carbon-bound hydrogens and C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C double bonds, a different initial oxidation step is required that starts with the elimination of the head group in photo- or electrochemical processes.282,339,340 An electron transfer from the carboxylic acid to the anode creates a carboxyl radical that undergoes decarboxylation to produce the carbon-centered perfluoroalkyl radical (˙CnF2n+1) and CO2.176,183,274 Likewise, perfluorinated sulfonic acids are desulfonated via an analogue initial electron transfer.256,282,341,342 The generated carbon-centered radicals quickly react with surrounding dissolved oxygen, water, or other radicals to form smaller carbonyl species. Once initiated, subsequent propagation reactions between the parent and daughter species of the decomposing pollutant with highly reactive, strong oxidants will ultimately lead to complete mineralization of perfluorinated compounds.286
C double bonds, a different initial oxidation step is required that starts with the elimination of the head group in photo- or electrochemical processes.282,339,340 An electron transfer from the carboxylic acid to the anode creates a carboxyl radical that undergoes decarboxylation to produce the carbon-centered perfluoroalkyl radical (˙CnF2n+1) and CO2.176,183,274 Likewise, perfluorinated sulfonic acids are desulfonated via an analogue initial electron transfer.256,282,341,342 The generated carbon-centered radicals quickly react with surrounding dissolved oxygen, water, or other radicals to form smaller carbonyl species. Once initiated, subsequent propagation reactions between the parent and daughter species of the decomposing pollutant with highly reactive, strong oxidants will ultimately lead to complete mineralization of perfluorinated compounds.286
Reductive dehalogenation via hydrated electrons that are generated by ultraviolet photolysis from sulfite requires oxygen-free conditions, which are impractical on a large scale. Added sulfite can effectively deoxygenate aqueous solutions, forming sulfate, which in turn can form SO4˙− for AOPs. Sulfite promotes the formation of SO3˙−, which is a mild oxidant343 that reacts rapidly with oxygen to produce SO5˙− and subsequently SO4˙− and ˙OH radicals, which are stronger oxidants and can more effectively degrade organohalogen pollutants.301
Electrocatalytic aqueous generation mechanisms of reactive species
Electrocatalytic AOP and ARP mechanisms proceed through direct electrocatalysis at materials surfaces,96,115,283,344 indirect solution reactions,176,345 or assisted reactions at materials and in solution.332,346,347 In aqueous electrolyte without auxiliary solutes, aerobic electrocatalysis encompasses anodic water oxidation to H2O2 (precursor for AOPs), ˙OH radicals (for AOPs), or O2 (loss process),3 and cathodic oxygen reduction at suitable catalysts348,349 to H2O2, ˙OH or OO˙− radicals, or water; H2O2 reduction to water can also occur (see Fig. 5 and 6A). Produced reactive species can diffuse into the bulk solution if they are sufficiently long-lived (see aqueous advanced redox processes) and react in indirect reactions to produce additional reactive species. The aqueous lifetime of ˙OH is 0.02 μs, whereas that of OO˙− is 1.3 μs, resulting in a diffusion distance of OO˙− radicals (30 nm) that is approximately 10× longer than that of ˙OH radicals (4.5 nm).325,326 The diffusion distances of both ˙OH and OO˙− radicals are too short for diffusion from electrodes into the bulk solution, but solution ˙OH and OO˙− radicals can be created by decomposition of H2O2 in strongly acidic or alkaline water.350,351 In the absence of dioxygen, i.e. in anaerobic media, anodic water oxidation produces ˙OH radicals, H2O2, or O2, which requires deoxygenation of the aqueous electrolyte, e.g. by sulfite,343 to ensure completely oxygen-free conditions. Direct, unassisted cathodic electrocatalysis in anaerobic electrolyte consists of H2O2 reduction to water (Fig. 6B). Deep-ultraviolet light,352 radiolysis,297 or sonolysis297 enable assisted electrocatalytic production of reactive oxygen radicals and, in O2-free aqueous environment, hydrated electrons, e−aq, and hydrogen radicals, H˙. Assisted aerobic electrocatalysis creates ˙OH radicals, H2O2, or O2 anodically by water oxidation, and H2O2, ˙OH, or OO˙− by cathodic oxygen reduction; produced H2O2 immediately decomposes to ˙OH radicals by deep-ultraviolet light,352 or ˙OH or OO˙− radicals by radiolysis297 or sonolysis297 processes that assist electrocatalysis (Fig. 6C). Assisted anaerobic electrocatalysis produces ˙OH radicals, H2O2, or O2 by anodic water oxidation (Fig. 6D). Elimination of O2 to ensure anaerobic conditions and immediate decomposition of H2O2 to ˙OH radicals by deep-ultraviolet light,352 or ˙OH or OO˙− radicals by radiolysis297 or sonolysis297 leave only ˙OH radicals (or OO˙− radicals if radiolysis or sonolysis were used) as reducible species for the cathode half reaction (Fig. 6D); the highly energetic assisting processes continuously generate ˙OH (or OO˙−) radicals in the entire electrochemical cell, including in the vicinity of the cathode. Assisted anaerobic electrocatalysis (Fig. 6D) additionally enables the production of hydrated electrons, e−aq, and hydrogen radicals, H˙, which are needed for ARPs, and which are only accessible in completely O2-free aqueous electrolyte.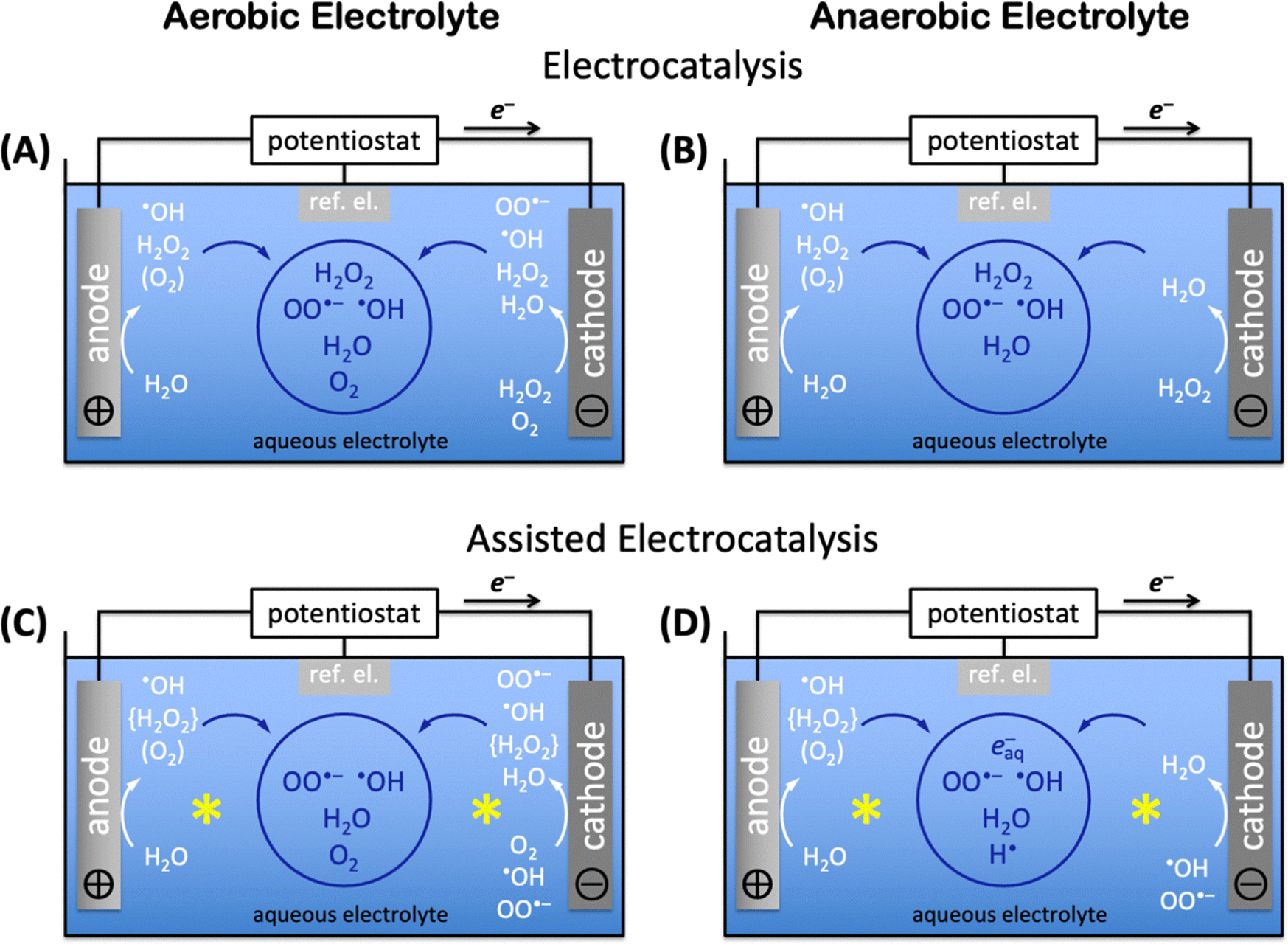 | ||
| Fig. 6 Generation of radicals and reactive species in aqueous electrocatalysis for use in advanced redox processes; ref. el., reference electrode. Species produced at electrodes are in white, species produced in bulk solution are in blue in the blue circles; the yellow stars indicate assisting processes, such as deep ultraviolet light irradiation, radiolysis, or sonolysis. Anodic dioxygen (O2) may be formed, depending on the water oxidation catalyst. Species in braces are short-lived intermediates. (A) Electrocatalysis in aerobic electrolyte, (B) electrocatalysis in anaerobic electrolyte, (C) assisted electrocatalysis in aerobic electrolyte, (D) assisted electrocatalysis in anaerobic electrolyte; e−aq, hydrated electron. | ||
The aqueous generation mechanisms of reactive oxygen species and radicals are highly entangled, creating complex reaction networks. Much research has been dedicated to the elucidation of mechanisms. Pollutant degradation via AOPs and ARPs involves three principal steps: (i) reactive species generation, (ii) initial attack on the pollutant, and (iii) subsequential attacks on the pollutant until mineralization is complete.113 Direct reactions occur at the electrode surface through electron transfers between the electrode and the chemical substrate.283 In electrochemical oxidation reactions, the anodically generated holes must have sufficient electrochemical potential to create oxidizing agents, without turning on the four-electron-four-proton water oxidation electrocatalysis to dioxygen. Suppression of O2 evolution requires applied potentials at or below the oxygen evolution potential, which is comprised of the thermodynamic potential and kinetic overpotential at the chosen pH conditions and catalyst materials.96,305,306 Direct oxidation reactions are often slow because substrate adsorption at the anode controls the reaction rate. Due to these slow kinetics and limited useful applied anodic potential, direct electrocatalytic reactions typically do not result in complete mineralization of pollutants.96
Quasi-direct redox reactions occur at the electrode–electrolyte interface via physisorbed or chemisorbed redox species,115,344 which are typically generated through reactions between the electrode and the aqueous electrolyte. Thus, the oxidizing strength in these processes is governed by the thermodynamic potential of the produced reactive species. Quasi-direct reactions have inherent mass transport limitations to and from the bulk solution because these processes must take place in the vicinity of the electrode–electrolyte interface.
Indirect processes occur in the bulk electrolyte, to where mediators migrate after electrochemical generation at the electrode–electrolyte interface, to react with pollutant species.176,345 Reactive species for indirect AOPs and ARPs are typically long-lived to enable long diffusion distances into the bulk solution. Therefore, reactive oxygen species generated at the electrode–aqueous electrolyte interface are of limited use in these reactions due to their proclivity for recombination and short lifetimes despite being among the strongest oxidizing agents.353 Some halogenated redox agents have longer lifetimes in comparison to those of reactive oxygen species, however, their selective reactivity and lower oxidation strength limits their reaction efficiency in the bulk electrolyte. Other advanced redox species, like sulfate radicals, possess longer lifetimes and simultaneously similar or greater oxidation strengths compared to reactive oxygen species, enabling indirect redox reations.165,354,355
Indirect processes have been coupled with other advanced redox activation processes to further increase the degradation efficiency of electrochemical systems.332,346,347,356 Ultraviolet or visible light irradiation of electrochemical systems has most often been used. These coupled processes maintain similar reaction networks as dark electrochemical systems, with photo-activation of electrochemically generated mediators, so that increased concentrations of redox agents are produced at the electrode surface and within the bulk electrolyte.332,346,347
Photochemistry is frequently used to destroy pollutants and can be classified as three processes: photolysis, photochemical, and photocatalysis. Photo-assisted AOPs and ARPs have similar advantages as electrochemical processes, such as operation at ambient temperature and pressure, low operating costs, and no generation of waste streams.
Photolysis is the direct absorption of light by chemical substrates for direct degradation via homolytic bond scission or direct light absorption by water to produce highly reactive redox agents for indirect degradation.113 The mechanism of photolysis consists of three steps: (i) light absorption that excites electrons in the molecule, (ii) primary photochemical processes that transform photoexcited molecules or result in relaxation back to the ground state, and (iii) secondary thermal reactions that transform the intermediates that were produced in step (ii).357 Photolysis is limited to pollutants that exhibit large molar absorption cross sections and quantum yields, which restricts the overall applicability of photolysis.358 Previous research has demonstrated applications of photolysis for pollutant degradation,359–361 but photolysis is mainly used for the inactivation of pathogenic microorganisms.362,363
Direct ultraviolet photolysis of anaerobic water to form ˙OH radicals, H˙ atoms, and hydrated electrons, e−aq, (see Tables 3 and 5 for thermodynamic potentials) has been demonstrated with vacuum ultraviolet (VUV) wavelengths <200 nm, albeit with low quantum yields and very slow breakdown of PFAS.364,365 Use of VUV irradiation is impractical because of the high absorption cross section of most materials at wavelengths below 200 nm. Deep ultraviolet irradiation at 254 nm is inefficient for direct photolysis of PFOA.366 But deep ultraviolet light is capable of enhancing PFAS decomposition by AOPs of auxiliary chemicals (H2O2, SO42−, SO32−) via radical generation and indirect pollutant oxidation.323,324,367
Addition of transient chemical oxidants to water can overcome photolysis challenges and enhance the overall degradation efficiency of persistent pollutants95,368 in a process known as photochemical degradation. Mechanistically, photochemical processes proceed through similar pathways as photolysis, i.e., photoactivation of transient chemicals to create highly reactive species, which then interact with pollutants and other molecules in the surrounding bulk electrolyte solution, creating complex reaction networks.369 Typical oxidants in aqueous photochemical organohalogen destruction processes are H2O2, ozone, peroxosulfate, peroxomonosulfate, and sulfite.113
Photocatalysis employs a semiconductor catalyst to lower the activation energy required to photoactivate water and initiate AOPs and ARPs for pollutant degradation. Semiconducting photocatalysts for oxidation reactions must be n-type to take advantage of the band bending at the semiconductor–electrolyte interface to extract photogenerated holes that can perform oxidation reactions; conversely, photocatalysts for reduction reactions must be p-type to enable enhanced injection of photogenerated electrons into reducible species at the semiconductor–electrolyte interface. Band bending occurs under equilibrium conditions at the junction between a conductor and a semiconductor,370–373 here the conducting electrolyte and the solid-state semiconductor photocatalyst. When a conductor and semiconductor are in contact, free electrons will transfer between the conductor and semiconductor because of the work function difference, to align the Fermi levels of both materials. The Fermi level, i.e., the total electrochemical potential of electrons, can be considered as the hypothetical energy level of an electron. Under equilibrium conditions, a Helmholtz double layer forms at the conductor–semiconductor interface, where the conductor and semiconductor carry opposite charges near their surfaces due to electrostatic induction. A charge imbalance arises because semiconductors have a low concentration of free charge carriers; therefore, the electric field at conductor–semiconductor junctions cannot effectively be screened in the semiconductor, which causes the free charge carrier concentration near the semiconductor surface to be depleted relative to the bulk. This interfacial region is called the space charge region.374 In n-type semiconductors, the Fermi level is closer to the conduction band than the valence band, and the electron concentration is larger than the hole concentration. Electrons are the majority charge carriers. Therefore, electrons are depleted in the space charge region, leading to excess positive charges, i.e. photo-holes that can perform oxidations at the interface. In the space charge region, the energy band edges in the semiconductor are continuously bent upwards if the semiconductor work function is smaller than that of the adjoining conducting medium, i.e. the electrolyte. This happens due to the charge transfer induced by the electric field at the junction, and the effect is called band bending. Besides different Fermi levels at a semiconductor junction, an external electric field, adsorbed species, or surface states (due to termination of lattice periodicity of a material at the surface) can also induce band bending near the semiconductor interface.374 Band bending can significantly decrease detrimental electron–hole pair recombination rates and enhance carrier transport to the semiconductor surface,374 where redox reactions occur.
The general mechanism of photocatalysis starts with adsorption of light with a photon energy equal to or greater than the bandgap of the semiconductor to photoexcite an electron in the photocatalyst material from the valence band to the conduction band, leaving a hole in the valence band.375,376 This photogenerated hole initiates oxidation reactions with the surrounding electrolyte to release reactive redox agents into the solution. Continually regenerated photo-holes and reactive species result in the oxidative destruction of pollutants.375,377 Homogeneous and heterogenous photocatalytic processes have been reported for aqueous AOPs and ARPs. The most often used homogenous process is the photo-Fenton process, which employs iron complexes that undergo photochemical reduction to ferrous iron.378 Ferrous iron catalyses H2O2 conversion to ˙OH radicals.378–380 This process is effective for persistent organic pollutant destruction,378 but homogeneous processes suffer from inherent separation costs.381–383 Heterogeneous photocatalysis employs solid catalyst materials that are readily recoverable and reusable, reducing separation expenses.383 Halogenated organic pollutants have completely been mineralized to environmentally benign products, using photocatalysis.376,384,385
Advanced redox processes for chlorinated and brominated organic compounds
Brominated and chlorinated pollutants are often degraded cathodically via ARPs because oxidation of liberated bromide or chloride produces toxic liquid bromine or chlorine gas, respectively.110,180–182 Nevertheless, Fenton-based AOPs have extensively been studied for the degradation of brominated and chlorinated pollutants. Fenton-based technology utilizes the reaction of ferrous ions with hydrogen peroxide, at an optimum pH, to generate hydroxyl radicals that react with pollutants in solution.386 While Fenton-based methods are popular due to their wide application range, cost effectiveness of iron, strong anti-interference ability, simple operation, and rapid degradation,387,388 significant disadvantages exist, such as the narrow working pH range and the generation of significant amounts of iron sludge.389 Brominated flame retardants were completely mineralized,390–392 whereas chlorinated trichloroethylene was not completely degraded.393 Fenton methods in pyrite suspension have been developed to enhance reaction kinetics of H2O2 decomposition to ˙OH radicals for more efficient pollutant degradation.393 Iron ions activated persulfate anions to produce sulfate radicals in a Fenton-like reaction for the chemical degradation of trichloroethylene.394 Photoelectrochemical systems have demonstrated enhanced AOP performance, synergistically degrading halogenated pollutants. Specifically, a photoelectrochemical system has been developed to degrade a series of chlorinated organic molecules to carbon dioxide, carbon monoxide, and chloride ions.395 Pulsed potential electrolysis has been shown to significantly increase mineralization efficiencies compared to constant potential electrolysis for chlorinated organic pollutants.395 In a gas diffusion device, 4-chlorophenol and 4-bromophenol were electrochemically dehalogenated to phenol.175Pairing Fenton processes with ultraviolet irradiation capitalizes on parallel reactive oxygen generation pathways. An ultraviolet-Fenton system achieved complete degradation of tetrabromobisphenol-A.396 Hybrid Fenton-like processes have been paired with other AOPs and ARPs, such as sonolysis by ultrasound.397,398 Hybrid systems have garnered much attention recently because of enhanced overall degradation rates. Sonophotocatalysis utilizes ultrasonic radiation and photocatalysis to synergistically generate more ˙OH radicals than either individual method by itself does, evidenced by the degradation of 2-chlorophenol.399 Hybrid systems that utilized ozone and H2O2 in a tube reactor nearly completely mineralized trichloroethylene and perchloroethylene in aqueous solution.400
Advanced redox processes for fluorinated organic compounds including PFAS
Conventional wastewater treatment processes are inefficient for PFAS destruction because of the exceptional stability of C–F bonds (see Table 1 and Fig. 3).90 PFAS can be destroyed by AOPs and ARPs, particularly when aided by photo-assisted processes.90 Direct photolysis is ineffective for treating many PFAS chemicals because the optical absorption of most PFAS molecules is limited to the UV-C region (<220 nm); UV-C irradiation requires specialized equipment and skin-cancer safety precautions; system lifetimes were inherently lower than those of systems irradiated with longer wavelengths, adding to overall capital investments.90 Photocatalysis enhanced PFAS degradation, as was reported for 254 nm (deep ultraviolet) illumination of the benchmark photocatalyst TiO2 for PFOA decomposition; irradiation with 315–400 nm light significantly decreased degradation performance, suggesting that the ability of the photocatalyst to generate sufficiently strong oxidants, such as ˙OH or OO˙−, controlled defluorination efficiency.401 Addition of persulfate to aqueous systems increased PFAS defluorination from negligible to nearly complete mineralization, via direct photolysis of persulfate to sulfate radicals.402–404 Photo-assisted electrochemical processes demonstrated enhanced degradation efficiencies of fluorinated compounds compared to dark electrochemical processes.260,341,405,406 The photo-Fenton process was utilized to increase transient radical concentrations compared to those generated by the dark Fenton process407 and concomitantly enhanced PFAS degradation.408 Addition of auxiliary chemicals to the electrochemical system benefitted the overall degradation efficiency of PFAS chemicals.280,330 Direct electrochemical and indirect hybrid electrochemical systems have been investigated for fluorinated pollutant degradation.234,260,280,330,339,341,405,406,409,410 Reported direct electrochemical PFAS degradation processes were most efficient when they occurred on boron-doped diamond (BDD) electrodes,234,339,409,410 which are cost-prohibitive on a large scale (see Fig. 4).Mechanisms of electrochemical aqueous PFAS destruction
All PFAS electrooxidation pathways start with a direct electron transfer to the anode, which is rate-limiting according to density functional theory calculations,282 followed by decarboxylation (perfluorinated carboxylic acids, such as PFOA)176,183,274 or desulfonation (perfluorinated sulfonic acids, such as PFOS)256,282,341,342 to form a ˙CnF2n+1 radical, from which CF2 moieties are unzipped by ˙OH or OO˙− radicals to form shorter-chain perfluoroalkyl radicals, ultimately producing CO2 and HF, which can safely be mineralized as calcium-containing solids.411 Several reaction pathways have been proposed, depending on the chemical nature of the oxidant (Fig. 7).304,310,412 All reported mechanisms of aqueous electrochemical degradation of PFOA start with electron transfer to the anode and decarboxylation, liberating CO2, to form the ˙C7F15 radical, which then further reacts with ˙OH to form C7F15OH in the hydroxyl radical mechanism or with oxygen and protons in the hydrogen peroxide mechanism.304,310,412 Other auxiliary anions in aqueous solution, such as sulfate or borate, enhance the generation of superoxide radial anions or hydroxyl radicals, opening pathways that proceed through the hydrogen peroxide mechanism or hydroxyl radical mechanism, respectively (Fig. 7A). The intermediate C7F15OH (structurally equal to C6F13CF2OH) is unstable and undergoes intramolecular rearrangement and hydrolysis, by which C6F13COO− is produced, effectively unzipping one CF2 moiety as one CO2 and two HF molecules from the original C7F15COO− reactant.304 Alternatively, the ˙C7F15 radical can react with dioxygen in the oxygen mechanism (Fig. 7A) to form the C7F15OO˙ radical, followed by decomposition of C7F15O˙ to perfluoorohexyl radical and carbonyl fluoride.412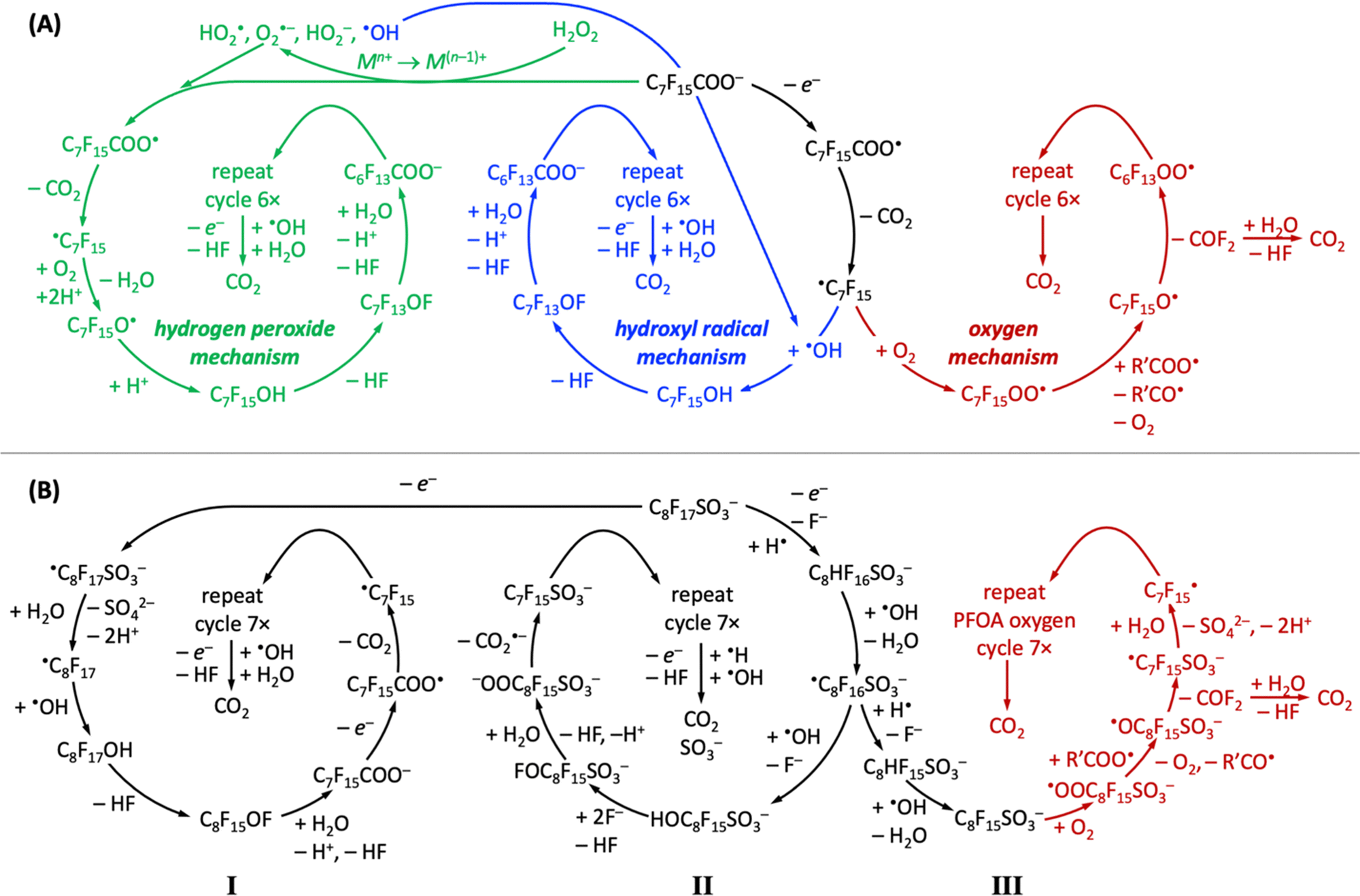 | ||
| Fig. 7 Overview of reported proposed reaction pathways of electrochemical aqueous oxidation of PFOA (A) and PFOS (B), adapted from ref. 304, 310 and 412 (PFOA) and ref. 412–414 (PFOS). | ||
Reported aqueous electrochemical degradation of PFOS starts with a direct electron transfer to the anode. Because the bond dissociation energy of the C–S bond (272 kJ mol−1) in PFOS is lower than that of the C–C bonds (346 kJ mol−1)415 or that of the C–F bonds (464.5–481.5 kJ mol−1),416 desulfonation occurs after the initial electron transfer (mechanisms I and III).412–414 Nevertheless, mechanism II has been proposed, which unzips CF2 moieties before desulfonation.412 Desulfonation can follow different mechanistic pathways (Fig. 7B). In mechanism I, the initial electron transfer to the anode is followed by the formation of a very unstable ˙C8F17SO3− radical that reacts with water to form ˙C8F17, which undergoes hydroxylation and hydrolysis reactions to form deprotonated PFOA and HF. This cycle repeats seven times to produce CO2 and HF.413,414 In the presence of hydrogen radicals (H˙), aqueous electrocatalytic PFOS degradation follows mechanisms II or III, where PFOS (C8F17SO3−) reacts upon electron transfer with H˙ under fluoride abstraction, to form C8HF16SO3−, which reacts further with ˙OH radicals to form ˙C8F16SO3− and water. We note that H˙ radicals only exist under anaerobic conditions (Fig. 6). In mechanism II, the intermediate ˙C8F16SO3− reacts with ˙OH radicals, undergoes hydrolysis and decarboxylation reactions, to form C7F15SO3− upon liberation of CO2˙−, whose spectroscopic signature is known,417 and the cycle repeats seven times until only CO2, HF, and SO3− remain of PFOS. In contrast, in mechanism III, the intermediate ˙C8F16SO3− undergoes an H/F exchange and a decarboxylation reaction to form ˙C7F15, which is an intermediate of aqueous electrocatalytic PFOA degradation (Fig. 7A). In mechanism III, PFOS-derived ˙C7F15 has been reported to decompose along the oxygen mechanism pathway of PFOA until CO2 and HF are obtained as final products (Fig. 7B).412 Likewise, ˙C7F15 decomposition can also proceed through the PFOA hydroxyl radical mechanism to arrive at the desired products CO2 and HF.
Carbon-based radicals are longer-lived than oxygen-based radicals;418 in aqueous electrolyte, carbon-based radicals are at PFAS molecules, and oxygen-based radicals are the reactive oxygen species that stem from electrolytes. The protonation state, which depends on electrolyte pH and pKa values of reactants and intermediates (see Table 4), critically affects mechanistic pathways. For example, the C7F15OH intermediate of the hydrogen peroxide and hydroxyl radical mechanisms in Fig. 7A can only be formed at pH values below 2 (see Table 4).
Future research directions
More research is needed to develop viable, globally scalable technologies for the destruction of halogenated organic water pollutants. Systems must work in aqueous media, consist of nonprecious materials, minimize capital expenditures and energy requirements, and be capable of being powered by renewable electricity for sustainability. A quantitative mechanistic understanding of organohalogen destruction via aqueous advanced redox processes is urgently needed, for which analytical detection methods for organohalogens and transient species must be improved, and the development of new catalysts and processes must be advanced.Analytical PFAS detection methods
Detection of low concentrations of organohalogens in the ppb to ppt range is challenging, particularly in natural water samples, in which ubiquitous abundant ions, such as chloride, can interfere.419,420 Defluorination performance is typically assessed by fluoride ion quantification by ion selective electrode measurements.421 Nuclear magnetic resonance (NMR) spectroscopy has been used since the early 1960s to detect PFOA and PFOS, with a limit of detection of 1.5 μM at that time.422 Gas chromatography (GC) and liquid chromatography (LC) have gradually gained more importance as separation techniques prior to compound identification and quantification. GC can only separate neutral and volatile analytes with low molecular weight, and is limited by low sensitivity and long separation times.423,424 Compared to GC, LC is more widely utilized in quantitative organohalogen analysis, owing to its ability to separate semi- or non-volatile, polar, higher molecular weight, and thermally unstable compounds.47 A widely used analysis technique is LC coupled with mass spectrometry (LC-MS) to detect halogenated contaminants,425 which is often used together with reference standards that are currently available for one hundred of the hundreds of potentially relevant PFAS that are harmful to human health.426 High-resolution mass spectrometry (HRMS) has emerged as a key tool for identifying legacy and novel PFAS. HRMS can detect more than 750 PFAS.427 LC and GC, coupled with various HRMS-based techniques, have been widely used for the identification and quantification of organohalogens.428 LC/GC-HRMS has extremely high selectivity, high resolving power, and is capable of detecting unknown PFAS. The lowest limit of PFAS detection for LC/GC-HRMS that has been reported was 100 nM.429 The high cost of HRMS instrumentation and challenges with respect to differentiation of PFAS isomers are the main disadvantages of LC/GC-HRMS.26 PFAS concentrations down to 10 nM are detectable by mobility spectrometry combined with LC and MS, which provides high selectivity and sensitivity as well as fast detection, but is expensive and unable to distinguish PFAS isomers, which limits broad usability.430Catalyst development
The development of new, nonprecious, efficient, selective, and robust catalysts is critical for the advancement of halogenated pollutant destruction. Catalyst materials are the foundation of degradation technologies because catalysts lower kinetic barriers in the conversion of supplied energy into chemical energy that initiates the degradation process. New synthetic and computational methods are needed to advance the design of robust, active, selective, and globally scalable catalysts, predicated on a quantitative mechanistic understanding.6,16,17,305,306,431–435 Advanced operando spectroscopies are essential to reveal the electronic and physical structures of catalysts under turnover, which together with catalyst property–performance relationships will accelerate catalyst discovery through rational design.435–441 Understanding how surface species bind to catalysts through first principles and machine learning computations is key to inform and direct experimental investigations in the vast materials space.442,443The electronic and geometric structure of catalysts, including changes during turnover and bonding motifs of reaction intermediates can be determined by advanced in situ or operando spectroscopies, combined with first principles and machine learning. In situ techniques monitor changes of the catalyst material under turnover, whereas operando spectroscopies additionally identify and quantify generated products during catalysis, thus enabling direct catalyst property–performance relationships during catalytic cycling. In situ and operando spectroscopies are often employed to gain a mechanistic understanding of electron, ion, and mass transport at catalyst interfaces,444 providing insights into the surface and bulk structure of catalysts, their composition, oxidation states, and adsorbed intermediates under reaction conditions.436,441,445In situ and operando spectroscopies additionally enable critical insights into reaction pathways,446 especially when coupled with computational studies.
Some of the most frequently used in situ and operando characterization techniques of catalyst materials are in situ Raman and surface enhanced Raman spectroscopy (SERS),445,447 Fourier transform infrared spectroscopy (FTIR),446 and the synchrotron-based techniques X-ray diffraction,446 X-ray absorption spectroscopy,448 near ambient pressure X-ray photoelectron spectroscopy,446 high-energy-resolution fluorescence-detection X-ray absorption spectroscopy,449 and X-ray absorption fine structure spectroscopy,449 sometimes used in grazing incidence angle configuration to enhance catalyst surface specific information.450 To gain a more accurate and quantitative understanding of catalysts and to reveal critical insights into the thermodynamics and kinetics of species during electrocatalysis, theoretical approaches, such as first principles density functional theory (DFT) calculations and microkinetic modelling simulations are often combined with in situ and operando characterization techniques.436,444,451,452 First-principles calculations, rooted in quantum mechanical electronic structure theory, have been utilized to analyse the intimate and dynamic relation between the microscopic processes and the meso- to macroscopic environment.453 DFT is the most commonly used first-principle method and utilizes functionals of spatially dependent electron density calculations to investigate the electronic structure of atoms, molecules, and solids.454 First-principle calculations are especially important in the context of catalyst discovery because they enable screening for new catalysts based on user-defined criteria and hypotheses, such as optimal activity, stability of elements under specific conditions, formation energy, selectivity, and material phases.2,455 These computational screening tools have been employed to analyze ensemble effects and electronic effects, to determine catalytic activity and selectivity.455 Electronic effects control the binding of the reaction intermediates.455 DFT calculations can be used to predictively design advanced catalysts, utilizing functional mechanisms of existing catalysts, comparing them, and making predictions which of these mechanisms is most suitable for each future reaction.442,455 Theoretical predictions from first-principle calculations are based on properties calculated from basic physical quantities and do not consider experimental results. Thus, it is challenging to quantitatively understand catalysts by advanced in situ or operando spectroscopies or theoretical models alone. Therefore, experimental results and theoretical models are nowadays often combined with machine learning algorithms that enable a better understanding of the nature of chemical bonding and its variation in strength across physically tuneable factors.442 Data-driven artificial intelligence models are capable of being integrated into active and iterative learning schemes that incorporate experimental results to improve the extrapolative, i.e., predictive, capabilities of models.456,457 Machine learning can also be combined with multiscale simulations and quantum mechanics to predict the performance of surface sites of catalysts.458
Advanced in situ or operando spectroscopies, together with computational first principles and machine learning approaches enable much-needed quantitative understanding of catalytically active sites, reactions centres, and reaction mechanisms.446,451,458,459 Right now, detailed DFT calculations and machine learning studies have challenges, such as high computational cost and potential loss of physical intuition since most models consist of complicated mathematical formulations that are difficult to interpret.442 Furthermore, reporting of experimental and computational information in much detail must be ensured to overcome challenges regarding choosing suitable functionals in DFT calculations.460,461 Multi-disciplinary approaches that combine experimental electrochemical performance assessments, operando spectroscopies, and computational approaches have to date the greatest potential to advance catalyst development.451
Process development
The thermodynamics, kinetics, mass transport, and chemistries of a system (i.e. the four pillars of chemical engineering) govern the overall electrocatalysis process and are tunable by the electrocatalytic process parameters outlined in Fig. 8. The electrolyte pH critically affects the thermodynamic potentials and chemistries, including generation mechanisms and protonation state, of reactive species produced during catalytic turnover (see Table 3). Electrolytes composed of small ions enhance mass transport, and electrolyte composition affects chemistries, particularly with respect to auxiliary solutes that can form transient radicals with high oxidation or reduction strengths. The electrolyte temperature impacts kinetics and mass transport of the system. The reaction time influences reaction kinetics, and electrolyte agitation increases mass transport. The applied potential controls the thermodynamics, kinetics, and chemistries by making different mechanistic pathways accessible. | ||
| Fig. 8 Electrocatalytic process parameters, color-coded to visualize which chemical engineering descriptor they affect. | ||
The scaleup of halogenated pollutant destruction technologies from the laboratory scale to industrially relevant scales demands that investigated systems are operated in water at pollutant concentrations that occur in the field, or at least at concentrations that are achievable by separation techniques,462,463 necessitating optimization of substrate mass transport.
Detection of transient ARP and AOP species
Quantitative analytical techniques are needed for the detection of short-lived, highly reactive redox species352,464–466 that are produced during assisted or unassisted electrocatalytic ARPs and AOPs, to develop a quantitative mechanistic understanding of organohalogen destruction processes. Electron paramagnetic resonance (EPR) spectroscopy is the premier method for radical detection.467–469 Freeze or spin trapping preserves radical species long enough to detect them in an EPR spectrometer.335 Spin traps are chemicals that react with radicals to create longer-lived, paramagnetic, EPR-active species, which are used to identify radicals with short lifetimes.335 Qualitative radical identification is important for understanding which species are involved in ARP and AOP mechanisms. Nevertheless, quantification is necessary to deeply understand mechanisms, and optimize reaction conditions and overall pollutant degradation. While EPR is typically used for identification of radical species, an external standard, such as 2,2,6,6-tetramethylpiperidine 1-oxyl (TEMPO), can be utilized to obtain quantitative data.470 Other quantitative detection methods are fluorescence and optical spectroscopy that utilize turn-on or turn-off dyes to determine radical concentrations.352,471,472 These dyes typically react directly with radical intermediates, resulting in a change in the fluorescence or absorption spectrum of that dye, enabling the determination of concentrations of radical intermediates. Faster, more cost-effective, and more sensitive detection methods must be developed to accelerate the innovation of viable aqueous electrocatalytic organohalogen destruction techniques.Conclusions
Halogenated organic compounds are widespread due to their exceptional utility and because they were initially considered nonmetabolizable and nontoxic, owing to the extreme stability of carbon–halogen bonds. However, prolonged use has led to their accumulation in the environment and organisms globally. We have introduced various classes of these pollutants categorized by halogen type (fluorine, chlorine, bromine), discussed important policies and regulations, outlined their applications, and highlighted associated environmental and human health risks. We discussed remediation techniques, focusing on carbon–halogen bond strengths, capital expense and energy needs for destruction, and electrocatalytic aqueous advanced redox processes. We highlighted mechanistic details of electrocatalysis, including oxidations and reductions of the water–oxygen system, as well as thermodynamic potentials, protonation states, and lifetimes of radicals and reactive oxygen species in aqueous electrolytes, importantly, at different pH conditions. We pointed out that advanced reduction processes necessitate anaerobic conditions, which are impractical beyond the laboratory scale because electrocatalytic water oxidation can produce dioxygen, membranes partitioning oxidation and reduction half-reactions cannot completely prevent oxygen crossover, and airtight seals are inherently challenging in large-scale applications; ergo, advanced oxidation processes appear to be more promising. We reviewed aqueous advanced redox processes for different halogenated compounds and PFAS (per- and polyfluoroalkyl substances), detailing potential mechanisms. Future research directions require quantitative understanding of destruction mechanisms, improved detection methods, advanced catalyst development, and energy-efficient processes. Scalable systems using nonprecious materials, powered by renewable electricity, are crucial. We outlined the interconnectedness of electrocatalytic process parameters and their effect on the chemical engineering descriptors thermodynamics, kinetics, mass transport, and chemistries, all vital in steering complex reaction networks. Finally, we suggested strategies to accelerate the development of effective aqueous electrocatalytic techniques for organohalogen destruction.Abbreviations
| ACGIH | American Conference of Governmental Industrial Hygienists |
| AOP | Advanced oxidation process |
| ARP | Advanced reduction process |
| BDD | Boron-doped diamond |
| BDE-15 | 4,4′-Dibromodiphenyl ether |
| BDE-207 | 2,2′,3,3′,4,4′,5,6,6′-Nonabromodiphenyl ether |
| BDE-22 | 2,3,4′-Tribromodiphenyl ether |
| BDE-28 | 2,4,4′-Tribromodiphenyl ether |
| BDE-3 | 4-Monobromodiphenyl ether |
| BDE-47 | 2,2′,4,4′-Tetrabromodiphenyl ether |
| BFR | Brominated flame retardant |
| CFC | Chlorofluorocarbon |
| CFC-11 | Trichloro(fluoro)methane |
| CFC-12 | Dichlorodi(fluoro)methane |
| CFC-13 | Chloro(trifluoro)methane |
| CIPAH | Chlorinated polycyclic aromatic hydrocarbon |
| DCE | 1,2-Dichloroethane |
| decaBDE | Decabromodiphenyl ether |
| DFT | Density functional theory |
| DMSO | Dimethyl sulfoxide |
| e-waste | electronic waste |
| EPR | Electron paramagnetic resonance |
| Freon-11 | Trichloro(fluoro)methane |
| Freon-12 | Dichlorodi(fluoro)methane |
| Freon-13 | Chloro(trifluoro)methane |
| Fs | Furans |
| FTIR | Fourier transform infrared spectroscopy |
| GC | Gas chromatography |
| GenX | Hexafluoropropylene oxide dimer acid |
| HBCD | Hexabromocyclododecane |
| HCFC | Hydrochlorofluorocarbon |
| HCFC-141b | 1,1-Dichloro-1-fluoroethane |
| HCFC-142b | Chloro-1,1-difluoroethane |
| HCFC-22 | Chloro(difluoro)methane |
| HFC | Hydrofluorocarbon |
| HRMS | High-resolution mass spectrometry |
| LC | Liquid chromatography |
| MS | Mass spectrometry |
| NMR | Nuclear magnetic resonance |
| octaBDE | Octabromodiphenyl ether |
| PBB | Polybrominated biphenyl |
| PBDD | Polybrominated dibenzo-p-dioxins |
| PBDE | Polybrominated diphenyl ethers |
| PCB | Polychlorinated biphenyls |
| PCB-47 | 2,2′,4,4′-Tetrachlorobiphenyl |
| PCB-77 | 1,2-Dichloro-4-(3,4-dichlorophenyl)benzene |
| PCDD | Polychlorinated dibenzo-p-dioxins |
| PCDF | Polychlorinated dibenzofuran |
| PCE | Perchloroethylene = tetrachloroethene |
| PCET | Proton-coupled electron transfer |
| pentaBDE | Pentabromodiphenyl ether |
| PERC | Tetrachloroethene |
| PFAS | Per- and polyfluoroalkyl substances |
| PFBA | Perfluorobutanoic acid |
| PFDA | Perfluorodecanoic acid |
| PFHxA | Perfluorohexanoic acid |
| PFOA | Perfluorooctanoic acid |
| PFOS | Perfluorooctane sulfonic acid |
| PHB | Poly-β-hydroxybutyrate |
| ppb | Parts per billion (μg L−1) |
| ppm | Parts per million (mg L−1) |
| ppq | Parts per quadrillion (pg L−1) |
| ppt | Parts per trillion (ng L−1) |
| R-11 | Trichloro(fluoro)methane |
| R-12 | Dichlorodi(fluoro)methane |
| R-13 | Chloro(trifluoro)methane |
| SERS | Surface-enhanced Raman spectroscopy |
| TBBPA | Tetrabromobisphenol-A |
| TBP | 2,4,6-Tribromophenol |
| 2,3,7,8-TCDD | 2,3,7,8-Tetrachlorodibenzodioxin |
| TCE | Trichloroethene |
| TEMPO | 2,2,6,6-Tetramethylpiperidine 1-oxyl |
| TLV | Threshold limit value |
| TWA | Time-weighted average |
| UV | Ultraviolet |
| VC | Vinyl chloride |
| VUV | Vacuum ultraviolet |
Author contributions
Madeleine K. Wilsey, writing – original draft preparation, writing – review and editing, visualization; Teona Taseska, writing – original draft preparation, visualization; Ziyi Meng, writing – original draft preparation; Wanqing Yu, writing – original draft preparation; Astrid M. Müller, conceptualization, writing – original draft preparation, writing – review and editing, visualization, supervision, project administration, funding acquisition.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Acknowledgment is made to the Donors of the American Chemical Society Petroleum Research Fund for support of this research (grant number 62578-DNI5). A. M. M. is grateful for start-up funds from the University of Rochester.Notes and references
- T. Taseska, W. Yu, M. K. Wilsey, C. P. Cox, Z. Meng, S. S. Ngarnim and A. M. Müller, Top. Catal., 2023, 66, 338–374 CrossRef CAS PubMed.
- M. Busch, N. B. Halck, U. I. Kramm, S. Siahrostami, P. Krtil and J. Rossmeisl, Nano Energy, 2016, 29, 126–135 CrossRef CAS.
- S. Siahrostami, G.-L. Li, V. Viswanathan and J. K. Nørskov, J. Phys. Chem. Lett., 2017, 8, 1157–1160 CrossRef CAS PubMed.
- H. Lv, J. Song, Y. V. Geletii, J. W. Vickers, J. M. Sumliner, D. G. Musaev, P. Kögerler, P. F. Zhuk, J. Bacsa, G. Zhu and C. L. Hill, J. Am. Chem. Soc., 2014, 136, 9268–9271 CrossRef CAS PubMed.
- Y. V. Geletii, B. Botar, P. Kogerler, D. A. Hillesheim, D. G. Musaev and C. L. Hill, Angew. Chem., 2008, 120, 3960 CrossRef.
- A. Kulkarni, S. Siahrostami, A. Patel and J. K. Nørskov, Chem. Rev., 2018, 118, 2302–2312 CrossRef CAS PubMed.
- X. Yang, Y. Zeng, W. Alnoush, Y. Hou, D. Higgins and G. Wu, Adv. Mater., 2022, 34, 2107954 CrossRef CAS PubMed.
- D. Higgins, P. Zamani, A. Yu and Z. Chen, Energy Environ. Sci., 2016, 9, 357–390 RSC.
- B. Kumar, M. Llorente, J. Froehlich, T. Dang, A. Sathrum and C. P. Kubiak, Annu. Rev. Phys. Chem., 2012, 63, 541–569 CrossRef CAS PubMed.
- C. W. Machan, M. D. Sampson, S. A. Chabolla, T. Dang and C. P. Kubiak, Organometallics, 2014, 33, 4550–4559 CrossRef CAS.
- D. K. Dogutan and D. G. Nocera, Acc. Chem. Res., 2019, 52, 3143–3148 CrossRef CAS PubMed.
- T. Faunce, S. Styring, M. R. Wasielewski, G. W. Brudvig, A. W. Rutherford, J. Messinger, A. F. Lee, C. L. Hill, H. Degroot and M. Fontecave, Energy Environ. Sci., 2013, 6, 1074–1076 RSC.
- W. Li, J. Wu, D. C. Higgins, J.-Y. Choi and Z. Chen, ACS Catal., 2012, 2, 2761–2768 CrossRef CAS.
- E. Nikolla, J. Schwank and S. Linic, J. Am. Chem. Soc., 2009, 131, 2747–2754 CrossRef CAS PubMed.
- X.-K. Gu, J. S. Carneiro, S. Samira, A. Das, N. M. Ariyasingha and E. Nikolla, J. Am. Chem. Soc., 2018, 140, 8128–8137 CrossRef CAS PubMed.
- M. Karamad, R. Magar, Y. Shi, S. Siahrostami, I. D. Gates and A. B. Farimani, Phys. Rev. Mater., 2020, 4, 093801 CrossRef CAS.
- S. Siahrostami, C. Tsai, M. Karamad, R. Koitz, M. García-Melchor, M. Bajdich, A. Vojvodic, F. Abild-Pedersen, J. K. Nørskov and F. Studt, Catal. Lett., 2016, 146, 1917–1921 CrossRef CAS.
- S. Rondinini, C. Locatelli, A. Minguzzi and A. Vertova, Electrochemical water and wastewater treatment, Elsevier, 2018, pp. 3–28 Search PubMed.
- E. T. Martin, C. M. McGuire, M. S. Mubarak and D. G. Peters, Chem. Rev., 2016, 116, 15198–15234 CrossRef CAS PubMed.
- S. Manzetti, E. R. van der Spoel and D. van der Spoel, Chem. Res. Toxicol., 2014, 27, 713–737 Search PubMed.
- W. Guo, B. Pan, S. Sakkiah, G. Yavas, W. Ge, W. Zou, W. Tong and H. Hong, Int. J. Environ. Res. Public Health, 2019, 16, 4361 Search PubMed.
- B. Yuan, K. Vorkamp, A. M. Roos, S. Faxneld, C. Sonne, S. E. Garbus, Y. Lind, I. Eulaers, P. Hellström and R. Dietz, Environ. Sci. Technol., 2019, 53, 3526–3537 CrossRef CAS PubMed.
- A. Covaci, S. Harrad, M. A.-E. Abdallah, N. Ali, R. J. Law, D. Herzke and C. A. de Wit, Environ. Int., 2011, 37, 532–556 CrossRef CAS PubMed.
- D. C. Muir and P. H. Howard, Environ. Sci. Technol., 2006, 40, 7157–7166 CrossRef CAS PubMed.
- Z. Chen, Y. Liu, W. Wei and B.-J. Ni, Environ. Sci. Nano, 2019, 6, 2332–2366 RSC.
- C. K. da Silva-Rackov, W. A. Lawal, P. A. Nfodzo, M. M. Vianna, C. A. do Nascimento and H. Choi, Appl. Catal. B: Environ., 2016, 192, 253–259 CrossRef CAS.
- J. N. Meegoda, B. Bezerra de Souza, M. M. Casarini and J. A. Kewalramani, Int. J. Environ. Res. Public Health, 2022, 19, 16397 CrossRef PubMed.
- P. Adriaens, C. Gruden and M. McCormick, Treatise Geochem., 2003, 9, 612 Search PubMed.
- G. W. Gribble, J. Chem. Educ., 2004, 81, 1441 CrossRef CAS.
- P. R. S. Kodavanti and B. G. Loganathan, The international encyclopedia of public health, ed. S. R. Quah, W. C. Cockerham, 2017, vol. 5, pp. 359–366 Search PubMed.
- M. Dorner, S. Lokesh, Y. Yang and S. Behrens, Sci. Total Environ., 2022, 158381 CrossRef CAS PubMed.
- L. Melymuk, J. Blumenthal, O. E. Sáňka, A. Shu-Yin, V. Singla, K. I. Šebková, K. Pullen Fedinick and M. L. Diamond, Environ. Sci. Technol., 2022, 56, 9029–9040 CrossRef CAS PubMed.
- M. Sharkey, S. Harrad, M. A.-E. Abdallah, D. S. Drage and H. Berresheim, Environ. Int., 2020, 144, 106041 CrossRef CAS PubMed.
- U. EPA, United States Environmental Protection Agency, Washington, DC, 2010 Search PubMed.
- U. Unep, United Nations environment programme: Stockholm convention on persistent organic pollutants, Geneva, 2009, p. 112 Search PubMed.
- M. G. Alalm and D. C. Boffito, Chem. Eng. J., 2022, 138352 CrossRef.
- F. Liu, X. Guan and F. Xiao, J. Hazard. Mater., 2022, 129580 CrossRef CAS PubMed.
- P. Höhener, D. Werner, C. Balsiger and G. Pasteris, Worldwide occurrence and fate of chlorofluorocarbons in groundwater, 2003 Search PubMed.
- L. M. Western, M. K. Vollmer, P. B. Krummel, K. E. Adcock, P. J. Fraser, C. M. Harth, R. L. Langenfelds, S. A. Montzka, J. Mühle and S. O’Doherty, Nat. Geosci., 2023, 1–5 Search PubMed.
- L. Yin, Z. Shen, J. Niu, J. Chen and Y. Duan, Environ. Sci. Technol., 2010, 44, 9117–9122 CrossRef CAS PubMed.
- E. O. Igbinosa, E. E. Odjadjare, V. N. Chigor, I. H. Igbinosa, A. O. Emoghene, F. O. Ekhaise, N. O. Igiehon and O. G. Idemudia, Sci. World J., 2013, 2013, 60215 Search PubMed.
- P. H. Howard, Handbook of environmental degradation rates, CRC Press, 1991 Search PubMed.
- A. J. Jamieson, T. Malkocs, S. B. Piertney, T. Fujii and Z. Zhang, Nat. Ecol. Evol., 2017, 1, 1–4 CrossRef PubMed.
- W. M. Caudle, T. S. Guillot, C. R. Lazo and G. W. Miller, Neurotoxicology, 2012, 33, 178–188 CrossRef CAS PubMed.
- S. Ullah, M. W. Javed, M. Shafique and S. F. Khan, J. Himal. Earth Sci., 2014, 47, 163–174 Search PubMed.
- M. V. Russo, I. Notardonato, A. Rosada, G. Ianiri and P. Avino, Int. J. Environ. Res. Public Health, 2021, 18, 1628 CrossRef CAS PubMed.
- S.-L. Badea, E.-I. Geana, V.-C. Niculescu and R.-E. Ionete, Sci. Total Environ., 2020, 722, 137914 CrossRef CAS PubMed.
- G. R. Chaudhry and S. Chapalamadugu, Microbiol. Rev., 1991, 55, 59–79 CrossRef CAS PubMed.
- H. G. Ni, H. Zeng, S. Tao and E. Y. Zeng, Environ. Toxicol. Chem., 2010, 29, 1237–1247 CAS.
- S. S. Zumdahl and S. A. Zumdahl, Chemistry, Cengage Learning, 2013 Search PubMed.
- Y. Guo, Y. Li and Z. Wang, Water Res., 2023, 119810 CrossRef CAS PubMed.
- C. Choi, X. Wang, S. Kwon, J. L. Hart, C. L. Rooney, N. J. Harmon, Q. P. Sam, J. J. Cha, W. A. Goddard III and M. Elimelech, Nat. Nanotechnol., 2023, 18, 160–167 CrossRef CAS PubMed.
- C. Wolf, C. Lambright, P. Mann, M. Price, R. L. Cooper, J. Ostby and L. E. Gray Jr, Toxicol. Ind. Health, 1999, 15, 94–118 CrossRef PubMed.
- G. Buck Louis, J. Weiner, B. Whitcomb, R. Sperrazza, E. Schisterman, D. Lobdell, K. Crickard, H. Greizerstein and P. Kostyniak, Hum. Reprod., 2005, 20, 279–285 CrossRef CAS PubMed.
- H. I. Zeliger, Rev. Environ. Health, 2013, 28, 9–20 CrossRef CAS PubMed.
- S. L. Schantz, J. J. Widholm and D. C. Rice, Environ. Health Perspect., 2003, 111, 357–576 CrossRef CAS PubMed.
- V. J. Cogliano, Environ. Health Perspect., 1998, 106, 317–323 CrossRef CAS PubMed.
- J. A. Goldstein, P. Hickman, H. Bergman, J. D. Mckinney and M. P. Walker, Chem.-Biol. Interact., 1977, 17, 69–87 CrossRef CAS PubMed.
- B. McHugh, R. Poole, J. Corcoran, P. Anninou, B. Boyle, E. Joyce, M. B. Foley and E. McGovern, Chemosphere, 2010, 79, 305–313 CrossRef CAS PubMed.
- Material and Chemicals – Brominated Flame Retardant (BFR) Market, https://www.reportsanddata.com/report-detail/brominated-flame-retardant-market, accessed June 20, 2023.
- C. A. De Wit, Chemosphere, 2002, 46, 583–624 CrossRef CAS PubMed.
- J. Chevrier, K. G. Harley, A. Bradman, M. Gharbi, A. Sjödin and B. Eskenazi, Environ. Health Perspect., 2010, 118, 1444–1449 CrossRef CAS PubMed.
- T. Okazoe, Proc. Jpn. Acad., Ser. B, 2009, 85, 276–289 CrossRef CAS PubMed.
- S. M. S. Hussain, A. A. Adewunmi, A. Mahboob, M. Murtaza, X. Zhou and M. S. Kamal, Adv. Colloid Interface Sci., 2022, 102634 CrossRef CAS PubMed.
- Z. Yue, H. Dunya, S. Aryal, C. U. Segre and B. Mandal, J. Power Sources, 2018, 401, 271–277 CrossRef CAS.
- D. Schrenk, M. Bignami, L. Bodin, J. K. Chipman, J. del Mazo, B. Grasl-Kraupp, C. Hogstrand, L. Hoogenboom and J. C. Leblanc, EFSA J., 2020, 18, e06223 CAS.
- R. Lohmann, I. T. Cousins, J. C. DeWitt, J. Gluge, G. Goldenman, D. Herzke, A. B. Lindstrom, M. F. Miller, C. A. Ng and S. Patton, Environ. Sci. Technol., 2020, 54, 12820–12828 CrossRef CAS PubMed.
- E. Gagliano, M. Sgroi, P. P. Falciglia, F. G. Vagliasindi and P. Roccaro, Water Res., 2020, 171, 115381 CrossRef CAS PubMed.
- L. N. Plummer and E. Busenberg, Environmental tracers in subsurface hydrology, 2000, pp. 441–478 Search PubMed.
- M. K. Vollmer, J. Mühle, S. Henne, D. Young, M. Rigby, B. Mitrevski, S. Park, C. R. Lunder, T. S. Rhee and C. M. Harth, Proc. Natl. Acad. Sci. U. S. A., 2021, 118, e2010914118 CrossRef CAS PubMed.
- M. Wernke, Encyclopedia of Toxicology, 3rd edn, 2014, pp. 664–666 Search PubMed.
- M. McFarland, Proc. Natl. Acad. Sci. U. S. A., 1992, 89, 807–811 CrossRef CAS PubMed.
- W.-T. Tsai, Chemosphere, 2005, 61, 1539–1547 CrossRef CAS PubMed.
- E. A. Ural, Process Saf. Prog., 2003, 22, 65–73 CrossRef CAS.
- Forever Chemicals and the Advancement of Filtration Standards, https://www.nsf.org/knowledge-library/forever-chemicals-advancement-filtration-standards, accessed June 14, 2023.
- J. Glüge, M. Scheringer, I. T. Cousins, J. C. DeWitt, G. Goldenman, D. Herzke, R. Lohmann, C. A. Ng, X. Trier and Z. Wang, Environ. Sci.: Process Impacts, 2020, 22, 2345–2373 Search PubMed.
- J. W. Washington and T. M. Jenkins, Environ. Sci. Technol., 2015, 49, 14129–14135 CrossRef CAS PubMed.
- J. Garnett, C. Halsall, H. Winton, H. Joerss, R. Mulvaney, R. Ebinghaus, M. Frey, A. Jones, A. Leeson and P. Wynn, Environ. Sci. Technol., 2022, 56, 11246–11255 CrossRef CAS PubMed.
- M. E. Andersen, B. Hagenbuch, U. Apte, J. C. Corton, T. Fletcher, C. Lau, W. L. Roth, B. Staels, G. L. Vega and H. J. Clewell, Toxicology, 2021, 459, 152845 CrossRef CAS PubMed.
- S. Kurwadkar, J. Dane, S. R. Kanel, M. N. Nadagouda, R. W. Cawdrey, B. Ambade, G. C. Struckhoff and R. Wilkin, Sci. Total Environ., 2022, 809, 151003 CrossRef CAS PubMed.
- H. N. Phong Vo, H. H. Ngo, W. Guo, T. M. Hong Nguyen, J. Li, H. Liang, L. Deng, Z. Chen and T. A. Hang Nguyen, J. Water Process. Eng., 2020, 36, 101393 CrossRef.
- S. Sharma, N. P. Shetti, S. Basu, M. N. Nadagouda and T. M. Aminabhavi, Chem. Eng. J., 2022, 430, 132895 CrossRef CAS.
- D. Benford, J. De Boer, A. Carere, A. Di Domenico, N. Johansson, D. Schrenk, G. Schoeters, P. De Voogt and E. Dellatte, EFSA J., 2008, 653, 1–131 Search PubMed.
- K. Steenland and A. Winquist, Environ. Res., 2021, 194, 110690 CrossRef CAS PubMed.
- E. M. Sunderland, X. C. Hu, C. Dassuncao, A. K. Tokranov, C. C. Wagner and J. G. Allen, J. Expo. Sci. Environ. Epidemiol., 2019, 29, 131–147 CrossRef CAS PubMed.
- M. Averina, J. Brox, S. Huber, A.-S. Furberg and M. Sørensen, Environ. Res., 2019, 169, 114–121 CrossRef CAS PubMed.
- N. Ding, C. A. Karvonen-Gutierrez, B. Mukherjee, A. M. Calafat, S. D. Harlow and S. K. Park, Hypertension, 2022, 79, 1876–1886 CrossRef CAS PubMed.
- P. Grandjean, E. W. Andersen, E. Budtz-Jørgensen, F. Nielsen, K. Mølbak, P. Weihe and C. Heilmann, JAMA, 2012, 307, 391–397 CrossRef CAS PubMed.
- B. M. Sharma, G. K. Bharat, S. Tayal, T. Larssen, J. Bečanová, P. Karásková, P. G. Whitehead, M. N. Futter, D. Butterfield and L. Nizzetto, Environ. Pollut., 2016, 208, 704–713 CrossRef CAS PubMed.
- D. Leonello, M. A. Fendrich, F. Parrino, N. Patel, M. Orlandi and A. Miotello, Appl. Sci., 2021, 11, 8458 CrossRef CAS.
- Z. Zeng, B. Song, R. Xiao, G. Zeng, J. Gong, M. Chen, P. Xu, P. Zhang, M. Shen and H. Yi, Environ. Int., 2019, 126, 598–610 CrossRef CAS PubMed.
- J. Zodrow, U. Vedagiri, T. Sorell, L. McIntosh, E. Larson, L. Hall, M. Dourson, L. Dell, D. Cox and K. Barfoot, Remediation, 2022, 32, 29–44 CrossRef.
- National Primary Drinking Water Regulations, https://www.epa.gov/ground-water-and-drinking-water/national-primary-drinking-water-regulations#Organic, accessed June 20, 2023.
- J. A. Roberson, Forever Chemicals, CRC Press, 2021, pp. 87–97 Search PubMed.
- M. Gmurek, M. Olak-Kucharczyk and S. Ledakowicz, Chem. Eng. J., 2017, 310, 437–456 CrossRef CAS.
- S. Garcia-Segura, J. D. Ocon and M. N. Chong, Process Saf. Environ. Prot., 2018, 113, 48–67 CrossRef CAS.
- E. Hepburn, C. Madden, D. Szabo, T. L. Coggan, B. Clarke and M. Currell, Environ. Pollut., 2019, 248, 101–113 CrossRef CAS PubMed.
- G. M. Sinclair, S. M. Long and O. A. Jones, Chemosphere, 2020, 258, 127340 CrossRef CAS PubMed.
- E. Kostyal, M. Borsanyi, L. Rigottier-Gois and M. S. Salkinoja-Salonen, Appl. Microbiol. Biotechnol., 1998, 50, 612–622 CrossRef CAS PubMed.
- A. J. Toth, E. Haáz, T. Nagy, A. J. Tarjani, D. Fozer, A. Andre, N. Valentinyi and P. Mizsey, Desalination Water Treat, 2018, 120, 2–29 Search PubMed.
- R. Xu, Y. Xie, J. Tian and L. Chen, J. Clean. Prod., 2021, 283, 124645 CrossRef CAS.
- M. N. Rashed, Adsorption technique for the removal of organic pollutants from water and wastewater, 2013 Search PubMed.
- C. Xu, R. Liu, W. Zheng, L. Lin and L. Chen, Water, 2021, 13, 1543 CrossRef CAS.
- S. Sharma and A. Bhattacharya, Appl. Water Sci., 2017, 7, 1043–1067 CrossRef CAS.
- K. Urano, E. Yamamoto, M. Tonegawa and K. Fujie, Water Res., 1991, 25, 1459–1464 CrossRef CAS.
- X. Li, H. Liu, X. Jia, G. Li, T. An and Y. Gao, Sci. Total Environ., 2018, 621, 1533–1541 CrossRef CAS PubMed.
- G. Liu, H. Sheng, Y. Fu, Y. Song, M. Redmile-Gordon, Y. Qiao, C. Gu, L. Xiang and F. Wang, Chemosphere, 2019, 214, 176–183 CrossRef CAS PubMed.
- Y. Zhu, X. Li, L. Wang, N. Hui, J. Ma and F. Chen, Water Air Soil Pollut., 2021, 232, 494 CrossRef CAS.
- C. Rodriguez, A. Cook, B. Devine, P. Van Buynder, R. Lugg, K. Linge and P. Weinstein, Int. J. Environ. Res. Public Health, 2008, 5, 356–367 CrossRef CAS PubMed.
- C. A. Martínez-Huitle, M. A. Rodrigo, I. Sirés and O. Scialdone, Chem. Rev., 2015, 115, 13362–13407 CrossRef PubMed.
- A. Urtiaga, Curr. Opin. Electrochem., 2021, 27, 100691 CrossRef CAS.
- S. C. E. Leung, P. Shukla, D. Chen, E. Eftekhari, H. An, F. Zare, N. Ghasemi, D. Zhang, N.-T. Nguyen and Q. Li, Sci. Total Environ., 2022, 153669 CrossRef CAS PubMed.
- S. Khan, M. Sayed, M. Sohail, L. A. Shah and M. A. Raja, Advances in water purification techniques, 2019, pp. 135–164 Search PubMed.
- A. Yazdanbakhsh, A. Eslami, G. Moussavi, M. Rafiee and A. Sheikhmohammadi, Chemosphere, 2018, 191, 156–165 CrossRef CAS PubMed.
- C. Comninellis, A. Kapalka, S. Malato, S. A. Parsons, I. Poulios and D. Mantzavinos, J. Chem. Technol. Biotechnol., 2008, 83, 769–776 CrossRef CAS.
- C. Cao, Sci. China, Ser. B: Chem., 2009, 52, 943–951 CrossRef CAS.
- A. S. Menon, G. P. Wood, D. Moran and L. Radom, J. Phys. Chem. A, 2007, 111, 13638–13644 CrossRef CAS PubMed.
- X.-M. Zhang, J. Chem. Soc., Perkin Trans. 2, 1993, 2275–2279 RSC.
- K. Daasbjerg, J. Chem. Soc., Perkin Trans. 2, 1994, 1275–1277 RSC.
- F. Li, J. Duan, S. Tian, H. Ji, Y. Zhu, Z. Wei and D. Zhao, Chem. Eng. J., 2020, 380, 122506 CrossRef CAS.
- X. Xiong, Y. Shang, L. Bai, S. Luo, T. W. Seviour, Z. Guo, L. D. Ottosen and Z. Wei, Water Res., 2023, 235, 119829 CrossRef CAS PubMed.
- Y. Su, U. Rao, C. M. Khor, M. G. Jensen, L. M. Teesch, B. M. Wong, D. M. Cwiertny and D. Jassby, ACS Appl. Mater. Interfaces, 2019, 11, 33913–33922 CrossRef CAS PubMed.
- J. Liu, D. J. Van Hoomissen, T. Liu, A. Maizel, X. Huo, S. R. Fernández, C. Ren, X. Xiao, Y. Fang and C. E. Schaefer, Environ. Sci. Technol. Lett., 2018, 5, 289–294 CrossRef CAS.
- G. Cavallo, P. Metrangolo, R. Milani, T. Pilati, A. Priimagi, G. Resnati and G. Terraneo, Chem. Rev., 2016, 116, 2478–2601 CrossRef CAS PubMed.
- H. N. Lynch, R. L. Prueitt and J. E. Goodman, Regul. Toxicol. Pharmacol., 2018, 97, 189–196 CrossRef CAS PubMed.
- Drinking Water Notification Levels, https://www.waterboards.ca.gov/drinking_water/certlic/drinkingwater/NotificationLevels.html, accessed August 8, 2023.
- EPA, Drinking Water Health Advisories (HAs), https://www.epa.gov/sdwa/drinking-water-health-advisories-has, accessed August 26, 2023.
- Amendment of Subpart 5-1 of Title 10 NYCRR (Maximum Contaminant Levels (MCLs)), https://regs.health.ny.gov/regulations/proposed-rule-making, accessed August 8, 2023.
- US EPA, Fifth Unregulated Contaminant Monitoring Rule, https://www.epa.gov/dwucmr/fifth-unregulated-contaminant-monitoring-rule, accessed August 8, 2023.
- PFAS: Per- and Polyfluoroalkyl Substances, https://www.waterboards.ca.gov/drinking_water/certlic/drinkingwater/pfas.html, accessed August 8, 2023.
- California Drinking Water Standards, https://www.gswater.com/sites/main/files/file-attachments/california-drinking-water-standards-rev-21218.pdf?1658793570, accessed August 27, 2023.
- New Jersey Department of Health and Senior Services, Hazardous Substance Fact Sheet: Trichlorofluoro-Methane https://nj.gov/health/eoh/rtkweb/documents/fs/1891.pdf, accessed August 8, 2023.
- California Water Boards, Drinking Water Notification Levels and Response Levels: An Overview, https://www.waterboards.ca.gov/drinking_water/certlic/drinkingwater/NotificationLevels.html, accessed August 8, 2023.
- New Jersey Department of Health and Senior Services, Hazardous Substance Fact Sheet: Dichlorodifluoro-Methane, https://nj.gov/health/eoh/rtkweb/documents/fs/0649.pdf, accessed August 8, 2023.
- Chlorotrifluoromethane, https://cameochemicals.noaa.gov/chemical/2934, accessed August 27, 2023.
- New Jersey Department of Health and Senior Services, Hazardous Substance Fact Sheet: Chlorotrifluoro-Methane, https://www.nj.gov/health/eoh/rtkweb/documents/fs/0425.pdf, accessed August 8, 2023.
- New Jersey Department of Health, Hazardous Substance Fact Sheet: Chlorodifluoromethane, https://nj.gov/health/eoh/rtkweb/documents/fs/0386.pdf, accessed August 8, 2023.
- National Refrigerants, Inc., Safety Data Sheet: 1,Chloro-1,1difluoroethane, https://refrigerants.com/wp-content/uploads/2020/01/SDS-R142b.pdf, accessed August 8, 2023.
- US OSHA, Occupational Chemical Database: 1,1-Dichloro-1-fluoroethane, https://www.osha.gov/chemicaldata/814, accessed August 8, 2023.
- EPA, Consumer Factsheet on: Vinyl Chloride, https://archive.epa.gov/water/archive/web/pdf/archived-consumer-fact-sheet-on-vinyl-chloride.pdf, accessed August 27, 2023.
- Vinyl Chloride and Drinking Water, https://www.health.state.mn.us/communities/environment/risk/docs/guidance/gw/vinylchlorideinfo.pdf, accessed August 27, 2023.
- New Jersey Department of Health, Hazardous Substance Fact Sheet: Vinyl Chloride, https://nj.gov/health/eoh/rtkweb/documents/fs/2001.pdf, accessed August 8, 2023.
- US EPA, Acute Exposure Guidelines Levels: 1,2-Dichloroethene, https://www.epa.gov/sites/default/files/2014-11/documents/1_2_dichloroethylene_final_volume_8.pdf, accessed August 8, 2023.
- New Jersey Department of Health, Hazardous Substance Fact Sheet: Tetrachloroethylene, https://nj.gov/health/eoh/rtkweb/documents/fs/1810.pdf, accessed August 8, 2023.
- New Jersey Department of Health, Hazardous Substance Fact Sheet: trichloroethylene, https://nj.gov/health/eoh/rtkweb/documents/fs/1890.pdf, accessed August 8, 2023.
- Ambient Water Quality Criteria for 2-chlorophenol, https://www.epa.gov/sites/default/files/2018-12/documents/ambient-wqc-2chlorophenol.pdf accessed August 27, 2023.
- Agilent, Safety Data Sheet: 2-Chlorophenol, https://www.agilent.com/cs/library/msds/RCP-001_NAEnglish.pdf, accessed August 8, 2023.
- US EPA, Consumer Fact Sheet on: Dioxin (2,3,7,8-TCDD), https://archive.epa.gov/water/archive/web/pdf/archived-consumer-fact-sheet-on-dioxin.pdf, accessed August 8, 2023.
- US OSHA, Permissible Exposure Limits – Annotated Tables, https://www.osha.gov/annotated-pels/table-z-1, accessed August 8, 2023.
- Canadian Environmental Protection Act, 1999, https://www.ec.gc.ca/ese-ees/default.asp?lang=En&n=05DF7A37-1, accessed August 27, 2023.
- Bluesign, Threshold Limit Values for Chemical Substances in Chemical Products, https://www.bluesign.com/downloads/bsbl/2023/bsbl_v50.pdf, accessed August 8, 2023.
- US Department of Health and Human Services, NTP Nomination History and Review: 2,4,6-Tribromophenol, https://ntp.niehs.nih.gov/sites/default/files/ntp/htdocs/chem_background/exsumpdf/tribromophenol_508.pdf, accessed August 8, 2023.
- Amending Regulation (EC) No. 850/2004 of the European Parliament and of the Council on Persistent Organic Pollutants as Regard Annex 1, https://eur-lex.europa.eu/legal-content/EN/TXT/PDF/?uri=CELEX:32016R0293&from=DE, accessed August 8, 2023.
- Tetrabromobisphenol A, https://ntp.niehs.nih.gov/sites/default/files/ntp/htdocs/chem_background/exsumpdf/tetrabromobisphenola_508.pdf, accessed August 27, 2023.
- US Department of Health and Human Services, Review of Toxicological Literature: Tetrabromobisphenol A, https://ntp.niehs.nih.gov/sites/default/files/ntp/htdocs/chem_background/exsumpdf/tetrabromobisphenola_508.pdf, accessed August 8, 2023.
- Agricultural Operations Notifications and Reporting Requirement https://media.defense.gov/2021/Nov/19/2002896203/-1/-1/0/AGRICULTURAL-OPERATIONS-NOTIFICATIONS-AND-REPORTING-REQUIREMENT.PDF, accessed August 27, 2023.
- S.-I. Sakai, K. Hayakawa, H. Takatsuki and I. Kawakami, Environ. Sci. Technol., 2001, 35, 3601–3607 CrossRef CAS PubMed.
- Q. Huang and C. Fang, Sci. Total Environ., 2020, 739, 139926 CrossRef CAS PubMed.
- Z. Zhang, J.-J. Chen, X.-J. Lyu, H. Yin and G.-P. Sheng, Sci. Rep., 2014, 4, 1–6 Search PubMed.
- N. R. Council, Waste incineration and public health, 2000.
- L. Makarichi, W. Jutidamrongphan and K.-A. Techato, Renewable Sustainable Energy Rev., 2018, 91, 812–821 CrossRef CAS.
- A. G. Capodaglio, Appl. Sci., 2020, 10, 4549 CrossRef CAS.
- K.-I. Naicker, P. Kaweesa, M. O. Daramola and S. A. Iwarere, Appl. Sci., 2023, 13, 6243 CrossRef CAS.
- M. A. Malik, Plasma Chem. Plasma Process., 2010, 30, 21–31 CrossRef CAS.
- X. Hou, G. Zhan, X. Huang, N. Wang, Z. Ai and L. Zhang, Chem. Eng. J., 2020, 382, 122355 CrossRef CAS.
- D. C. Schmelling, D. L. Poster, M. Chaychian, P. Neta, J. Silverman and M. Al-Sheikhly, Environ. Sci. Technol., 1998, 32, 270–275 CrossRef CAS.
- R. M. Morrison, An economic analysis of electron accelerators and cobalt-60 for irradiating food, US Department of Agriculture, Economic Research Service, Rockville, MD, USA, 1989 Search PubMed.
- T. A. Saleh, M. Mustaqeem and M. Khaled, Environ. Nanotechnol. Monit. Manag, 2022, 17, 100617 CAS.
- H. Md Anawar and R. Chowdhury, Sustainability, 2020, 12, 7017 CrossRef CAS.
- X. Bai, X. Zhu, H. Jiang, Z. Wang, C. He, L. Sheng and J. Zhuang, Water, 2020, 12, 1054 CrossRef CAS.
- W.-x Zhang, C.-B. Wang and H.-L. Lien, Catal. Today, 1998, 40, 387–395 CrossRef CAS.
- K.-C. Huang, G. E. Hoag, P. Chheda, B. A. Woody and G. M. Dobbs, J. Hazard. Mater., 2001, 87, 155–169 CrossRef CAS PubMed.
- Z. Ronen and A. Abeliovich, Appl. Environ. Microbiol., 2000, 66, 2372–2377 CrossRef CAS PubMed.
- J. Radjenovic and D. L. Sedlak, Environ. Sci. Technol., 2015, 49, 11292–11302 CrossRef CAS PubMed.
- H. Wang and J. Wang, Appl. Catal. B: Environ., 2007, 77, 58–65 CrossRef CAS.
- C. A. Martinez-Huitle and S. Ferro, Chem. Soc. Rev., 2006, 35, 1324–1340 RSC.
- J. Qiao and Y. Xiong, J. Water Process Eng., 2021, 44, 102308 CrossRef.
- B. Yang, H. Li, Z. Zhang, K. Xiao, M. Yang, F. Zhang, M. Wang, X. Guo, Q. Li and W. Fu, Chem. Eng. J., 2022, 427, 131719 CrossRef CAS.
- L. Yang, C. Huang, Z. Yin, J. Meng, M. Guo, L. Feng, Y. Liu, L. Zhang and Z. Du, Chemosphere, 2021, 264, 128515 CrossRef CAS PubMed.
- M. P. Rayaroth, C. T. Aravindakumar, N. S. Shah and G. Boczkaj, Chem. Eng. J., 2022, 430, 133002 CrossRef CAS.
- H. Feng, X. Liao, R. Yang, S. Chen, Z. Zhang, J. Tong, J. Liu and X. Wang, Water Res., 2023, 230, 119531 CrossRef CAS PubMed.
- E. Domingues, E. Fernandes, J. Gomes and R. C. Martins, Sci. Total Environ., 2021, 776, 145958 CrossRef CAS.
- M. Lei, N. Wang, S. Guo, L. Zhu, Y. Ding and H. Tang, Chem. Eng. J., 2018, 345, 586–593 CrossRef CAS.
- Y. Wei, Y. Gong, X. Zhao, Y. Wang, R. Duan, C. Chen, W. Song and J. Zhao, Environ. Sci. Nano, 2019, 6, 1585–1593 RSC.
- R. A. Torres-Palma and E. A. Serna-Galvis, Advanced oxidation processes for waste water treatment, Elsevier, 2018, pp. 177–213 Search PubMed.
- R. Xiao, Z. Wei, D. Chen and L. K. Weavers, Environ. Sci. Technol., 2014, 48, 9675–9683 CrossRef CAS PubMed.
- J. Blotevogel, S. M. Thagard and S. Mahendra, Curr. Opin. Chem. Eng., 2023, 41, 100944 CrossRef.
- A. Asghar, A. A. A. Raman and W. M. A. W. Daud, J. Clean. Prod., 2015, 87, 826–838 CrossRef CAS.
- J. González-García, V. Sáez, I. Tudela, M. I. Díez-Garcia, M. Deseada Esclapez and O. Louisnard, Water, 2010, 2, 28–74 CrossRef.
- M. H. Lim, S. H. Kim, Y. U. Kim and J. Khim, Ultrason. Sonochem., 2007, 14, 93–98 CrossRef CAS PubMed.
- D. Panda, V. Sethu and S. Manickam, Environ. Technol. Innovation, 2020, 17, 100605 CrossRef CAS.
- D. Panda and S. Manickam, Sep. Purif. Technol., 2019, 209, 914–920 CrossRef CAS.
- H. Wang, C. Zhang, X. Zhang, S. Wang, Z. Xia, G. Zeng, J. Ding and N. Ren, Chem. Eng. J., 2022, 429, 132445 CrossRef CAS.
- S. Khammar, N. Bahramifar and H. Younesi, J. Hazard. Mater., 2020, 394, 122422 CrossRef CAS PubMed.
- Q. Zheng, Z. Liu, L. Yang, J. Lu, C. Qiu, X. Guan and K. Yan, Chem. Eng. J., 2020, 387, 124005 CrossRef CAS.
- A. T. Dao, S. J. Loenen, K. Swart, H. T. Dang, A. Brouwer and T. E. de Boer, Chemosphere, 2021, 263, 128280 CrossRef CAS PubMed.
- N. B. Varzaghani, S. Shokrollahzadeh and A. Farazmand, J. Environ. Chem. Eng., 2021, 9, 105098 CrossRef CAS.
- F. Escalona-Durán, M. Muñoz-Morales, F. Souza, C. Sáez, P. Cañizares, C. Martínez-Huitle and M. Rodrigo, Chemosphere, 2021, 279, 130525 CrossRef PubMed.
- N. D. Mohamad, Z. M. Zaki and A. Amir, Chem. Eng. J., 2020, 393, 124760 CrossRef CAS.
- Y. Lyu, S. Lyu, P. Tang, W. Jiang, Y. Sun, M. Li and Q. Sui, Chemosphere, 2020, 257, 127223 CrossRef CAS PubMed.
- Y. Sun, M. Danish, M. Ali, A. Shan, M. Li, Y. Lyu, Z. Qiu, Q. Sui, X. Zang and S. Lyu, Chem. Eng. J., 2020, 394, 124830 CrossRef CAS.
- M. Ali, M. Danish, M. Tariq, A. Ahmad, K. S. Ayub and S. Lyu, Chem. Eng. J., 2020, 399, 125754 CrossRef CAS.
- F. Li, D. Deng, L. Zeng, S. Abrams and M. Li, Sci. Total Environ., 2021, 774, 145118 CrossRef CAS PubMed.
- A. Venkateshaiah, D. Silvestri, S. Wacławek, R. K. Ramakrishnan, K. Krawczyk, P. Saravanan, M. Pawlyta, V. V. Padil, M. Černík and D. D. Dionysiou, Environ. Sci. Water Res. Technol., 2022, 8, 162–172 RSC.
- M. Dutschke, T. Schnabel, F. Schütz and C. Springer, J. Environ. Manage., 2022, 304, 114236 CrossRef CAS PubMed.
- Y.-C. Chang, M. V. Reddy and D. Choi, Green Chem., 2021, 23, 2729–2737 RSC.
- Z. Chen, L. Wang, H. Xu and Q. Wen, Chem. Eng. J., 2020, 389, 124345 CrossRef CAS.
- Q. Wang, T. Wang, G. Qu, Y. Zhang, Q. Sun, X. Guo and H. Jia, Sep. Purif. Technol., 2020, 240, 116615 CrossRef CAS.
- C. Hou, W. Chen, L. Fu, S. Zhang, C. Liang and Y. Wang, Carbohydr. Polym., 2020, 247, 116731 CrossRef CAS PubMed.
- C. Fang, X. Lou, Y. Tang, L. Tian, Y. Cai, D. Xiao, Y. Guo and J. Liu, J. Environ. Chem. Eng., 2020, 8, 103998 CrossRef CAS.
- L. Fu, F. Lide, Y. Ding, C. Wang, J. Jiang and J. Huang, Sep. Purif. Technol., 2022, 285, 120282 CrossRef CAS.
- T.-H. Chang, R. Wang, Y.-H. Peng, T.-H. Chou, Y.-J. Li and Y.-H. Shih, Chemosphere, 2020, 246, 125621 CrossRef CAS PubMed.
- T. Li, Y. Lu, L. Liu, Y. He, J. Huang and X. Peng, J. Hazard. Mater., 2023, 131739 CrossRef CAS PubMed.
- Y. Jiang, S. Yang, J. Liu, T. Ren, Y. Zhang and X. Sun, Chemosphere, 2020, 244, 125536 CrossRef CAS PubMed.
- Y. Jiang, S. Yang, M. Wang, Y. Xue, J. Liu, Y. Li and D. Zhao, Chemosphere, 2021, 279, 130520 CrossRef CAS PubMed.
- L. Wei, B. Zhou, K. Xiao, B. Yang, G. Yu, J. Li, C. Zhu, J. Zhang and H. Duan, J. Hazard. Mater., 2020, 385, 121551 CrossRef CAS PubMed.
- J. Liang, H. Liu, M. Zou, X. Tao, J. Zhou, Z. Dang and G. Lu, Chemosphere, 2023, 325, 138396 CrossRef CAS PubMed.
- K. Huang, H. Liu, J. He, Y. He, X. Tao, H. Yin, Z. Dang and G. Lu, J. Environ. Chem. Eng., 2021, 9, 105077 CrossRef CAS.
- X. Xu, M. Liu, Y. Zhang and Y. Zhang, S. Afr. J. Bot., 2023, 153, 94–101 CrossRef CAS.
- X. Zhang, M. Feng, F. Liu, L. Qin, R. Qu, D. Li and Z. Wang, Environ. Sci. Pollut. Res., 2014, 21, 1924–1935 CrossRef CAS PubMed.
- Z. Wei, J. Xi, S. Gao, X. You, N. Li, Y. Cao, L. Wang, Y. Luan and X. Dong, Sci. Rep., 2018, 8, 5423 CrossRef PubMed.
- T. Pan, Y. Wang, X. Yang, X.-F. Huang and R.-L. Qiu, Chem. Eng. J., 2020, 384, 123248 CrossRef CAS.
- Z. Guo, H. Yin, X. Wei, M. Zhu, G. Lu and Z. Dang, J. Hazard. Mater., 2021, 415, 125698 CrossRef CAS PubMed.
- J. Zhu, Y. Chen, Y. Gu, H. Ma, M. Hu, X. Gao and T. Liu, J. Hazard. Mater., 2022, 422, 126953 CrossRef CAS PubMed.
- B. Trang, Y. Li, X.-S. Xue, M. Ateia, K. Houk and W. R. Dichtel, Science, 2022, 377, 839–845 CrossRef CAS PubMed.
- J. Niu, H. Lin, J. Xu, H. Wu and Y. Li, Environ. Sci. Technol., 2012, 46, 10191–10198 CrossRef CAS PubMed.
- H. Lin, J. Niu, S. Liang, C. Wang, Y. Wang, F. Jin, Q. Luo and Q. Huang, Chem. Eng. J., 2018, 354, 1058–1067 CrossRef CAS.
- S. Verma, B. Mezgebe, E. Sahle-Demessie and M. N. Nadagouda, Chemosphere, 2021, 280, 130660 CrossRef CAS PubMed.
- I. K. A. Electrochemistry Kit: Boron Doped Diamond, https://www.ika.com/en/Products-Lab-Eq/Electrochemistry-Kit-csp-516/Boron-doped-diamond,-Set-of-2-cpdt-40002856/, accessed July 7, 2022.
- N. N. Mahamuni and Y. G. Adewuyi, Ultrason. Sonochem., 2010, 17, 990–1003 CrossRef CAS PubMed.
- A. Blumberga, D. Blumberga, J. Pubule and F. Romagnoli, Energy Procedia, 2015, 72, 170–174 CrossRef.
- D. Panepinto, S. Fiore, G. Genon and M. Acri, J. Clean. Prod., 2016, 137, 1323–1329 CrossRef.
- A. Guerrini, G. Romano and A. Indipendenza, Sustainability, 2017, 9, 1126 CrossRef.
- C. E. Schaefer, C. Andaya, A. Burant, C. W. Condee, A. Urtiaga, T. J. Strathmann and C. P. Higgins, Chem. Eng. J., 2017, 317, 424–432 CrossRef CAS.
- T. X. H. Le, H. Haflich, A. D. Shah and B. P. Chaplin, Environ. Sci. Technol. Lett., 2019, 6, 504–510 CrossRef CAS.
- X. Pang, J. T. Davis, A. D. Harvey and D. V. Esposito, Energy Environ. Sci., 2020, 13, 3663–3678 RSC.
- D. Palma, C. Richard and M. Minella, Chem. Eng. J. Adv., 2022, 100253 CrossRef CAS.
- A. Kimura, M. Taguchi, Y. Ohtani, Y. Shimada, H. Hiratsuka and T. Kojima, Radiat. Phys. Chem., 2007, 76, 699–706 CrossRef CAS.
- I. B. Rivas-Ortiz, G. Cruz-González, A. M. Lastre-Acosta, M. Manduca-Artiles, M. Rapado-Paneque, A. Chávez-Ardanza, A. C. S. C. Teixeira and U. J. Jáuregui-Haza, J. Radioanal. Nucl. Chem., 2017, 314, 2597–2607 CrossRef CAS.
- L. Barillas, presented in part at the Journal of Physics: Conference Series, 2015.
- J.-C. Lin, C.-Y. Hu and S.-L. Lo, Ultrason. Sonochem., 2016, 28, 130–135 CrossRef CAS PubMed.
- C. Liu, K. Shih and F. Wang, Sep. Purif. Technol., 2012, 87, 95–100 CrossRef CAS.
- Y.-C. Lee, S.-L. Lo, J. Kuo and Y.-L. Lin, Chem. Eng. J., 2012, 198, 27–32 CrossRef.
- M. J. Krause, E. Thoma, E. Sahle-Damesessie, B. Crone, A. Whitehill, E. Shields and B. Gullett, J. Environ. Eng., 2022, 148, 05021006 CrossRef CAS PubMed.
- J. Yang, S. Wang, Y. Li, Y. Zhang and D. Xu, J. Environ. Manage., 2019, 233, 131–140 CrossRef CAS PubMed.
- V. Vadillo, J. Sánchez-Oneto, J. R. Portela and E. J. M. de la Ossa, Advanced Oxidation Processes for Waste Water Treatment, Elsevier, 2018, pp. 333–358 Search PubMed.
- Q. Zhuo, S. Deng, B. Yang, J. Huang and G. Yu, Environ. Sci. Technol., 2011, 45, 2973–2979 CrossRef CAS PubMed.
- H. Lin, J. Niu, S. Ding and L. Zhang, Water Res., 2012, 46, 2281–2289 CrossRef CAS PubMed.
- H. Zhao, J. Gao, G. Zhao, J. Fan, Y. Wang and Y. Wang, Appl. Catal., 2013, 136–137, 278–286 CrossRef CAS.
- B. Yang, C. Jiang, G. Yu, Q. Zhuo, S. Deng, J. Wu and H. Zhang, J. Hazard. Mater., 2015, 299, 417–424 CrossRef CAS PubMed.
- Q. Zhuo, M. Luo, Q. Guo, G. Yu, S. Deng, Z. Xu, B. Yang and X. Liang, Electrochim. Acta, 2016, 213, 358–367 CrossRef CAS.
- Z. Xu, Y. Yu, H. Liu and J. Niu, Sci. Total Environ., 2017, 579, 1600–1607 CrossRef CAS PubMed.
- Q. Zhuo, Q. Xiang, H. Yi, Z. Zhang, B. Yang, K. Cui, X. Bing, Z. Xu, X. Liang, Q. Guo and R. Yang, J. Electroanal. Chem., 2017, 801, 235–243 CrossRef CAS.
- H. Lin, J. Niu, S. Liang, C. Wang, Y. Wang, F. Jin, Q. Luo and Q. Huang, Chem. Eng. J., 2018, 354, 1058–1067 CrossRef CAS.
- S. Sharma, N. P. Shetti, S. Basu, M. N. Nadagouda and T. M. Aminabhavi, Chem. Eng. J., 2022, 430, 132895 CrossRef CAS.
- K. E. Carter and J. Farrell, Environ. Sci. Technol., 2008, 42, 6111–6115 CrossRef CAS PubMed.
- J. N. Uwayezu, I. Carabante, T. Lejon, P. van Hees, P. Karlsson, P. Hollman and J. Kumpiene, J. Environ. Manage., 2021, 290, 112573 CrossRef CAS PubMed.
- E. Amores, J. Rodriguez and C. Carreras, Int. J. Hydrogen Energy, 2014, 39, 13063–13078 CrossRef CAS.
- F. Gaied, B. Louhichi, M. Bali and M. R. Jeday, Songklanakarin J. Sci. Technol., 2019, 41, 1084–1092 CAS.
- U. Rao, Y. Su, C. M. Khor, B. Jung, S. Ma, D. M. Cwiertny, B. M. Wong and D. Jassby, Environ. Sci. Technol., 2020, 54, 10668–10677 CrossRef CAS PubMed.
- S. Huang, M. Sima, Y. Long, C. Messenger and P. R. Jaffé, J. Hazard. Mater., 2022, 424, 127699 CrossRef CAS PubMed.
- L. Duan, B. Wang, K. Heck, S. Guo, C. A. Clark, J. Arredondo, M. Wang, T. P. Senftle, P. Westerhoff and X. Wen, Environ. Sci. Technol. Lett., 2020, 7, 613–619 CrossRef CAS.
- L. Duan, B. Wang, K. N. Heck, C. A. Clark, J. Wei, M. Wang, J. Metz, G. Wu, A.-L. Tsai and S. Guo, Chem. Eng. J., 2022, 448, 137735 CrossRef CAS.
- T. Xu, Y. Zhu, J. Duan, Y. Xia, T. Tong, L. Zhang and D. Zhao, Chem. Eng. J., 2020, 395, 124991 CrossRef CAS.
- T. Xu, H. Ji, Y. Gu, T. Tong, Y. Xia, L. Zhang and D. Zhao, Chem. Eng. J., 2020, 388, 124230 CrossRef CAS.
- M. Qanbarzadeh, D. Wang, M. Ateia, S. P. Sahu and E. L. Cates, ACS ES&T Engineering, 2020, 1, 239–248 Search PubMed.
- Y.-J. Lei, Y. Tian, Z. Sobhani, R. Naidu and C. Fang, Chem. Eng. J., 2020, 388, 124215 CrossRef CAS.
- J. N. Uwayezu, I. Carabante, T. Lejon, P. van Hees, P. Karlsson, P. Hollman and J. Kumpiene, J. Environ. Manage., 2021, 290, 112573 CrossRef CAS PubMed.
- F. Li, Z. Wei, K. He, L. Blaney, X. Cheng, T. Xu, W. Liu and D. Zhao, Water Res., 2020, 185, 116219 CrossRef CAS PubMed.
- W. Zhang, H. Efstathiadis, L. Li and Y. Liang, J. Environ. Sci., 2020, 93, 48–56 CrossRef CAS PubMed.
- R. J. Wood, T. Sidnell, I. Ross, J. McDonough, J. Lee and M. J. Bussemaker, Ultrason. Sonochem., 2020, 68, 105196 CrossRef PubMed.
- A. J. Lewis, T. Joyce, M. Hadaya, F. Ebrahimi, I. Dragiev, N. Giardetti, J. Yang, G. Fridman, A. Rabinovich and A. A. Fridman, Environ. Sci. Water Res. Technol., 2020, 6, 1044–1057 RSC.
- Z. Liu, Z. Chen, J. Gao, Y. Yu, Y. Men, C. Gu and J. Liu, Environ. Sci. Technol., 2022, 56, 3699–3709 CrossRef CAS PubMed.
- Y. Wang, H. Shi, C. Li and Q. Huang, Environ. Sci. Water Res. Technol., 2020, 6, 144–152 RSC.
- D. Patch, N. O'Connor, I. Koch, T. Cresswell, C. Hughes, J. B. Davies, J. Scott, D. O'Carroll and K. Weber, Sci. Total Environ., 2022, 832, 154941 CrossRef CAS PubMed.
- D. Palma, D. Papagiannaki, M. Lai, R. Binetti, M. Sleiman, M. Minella and C. Richard, Molecules, 2021, 26, 924 CrossRef CAS PubMed.
- J. Lassalle, R. Gao, R. Rodi, C. Kowald, M. Feng, V. K. Sharma, T. Hoelen, P. Bireta, E. F. Houtz and D. Staack, Radiat. Phys. Chem., 2021, 189, 109705 CrossRef CAS.
- M. S. Samuel, M. Shang and J. Niu, Chemosphere, 2022, 293, 133568 CrossRef CAS PubMed.
- N. Duinslaeger and J. Radjenovic, Water Res., 2022, 213, 118148 CrossRef CAS PubMed.
- H. Olvera-Vargas, Z. Wang, J. Xu and O. Lefebvre, Chem. Eng. J., 2022, 430, 132686 CrossRef CAS.
- C. A. Martínez-Huitle and E. Brillas, Appl. Catal. B: Environ., 2009, 87, 105–145 CrossRef.
- J. Niu, H. Lin, C. Gong and X. Sun, Environ. Sci. Technol., 2013, 47, 14341–14349 CrossRef CAS PubMed.
- M. Panizza, Electrochemistry for the Environment, 2010, pp. 25–54 Search PubMed.
- J. Yang, J. Wang and J. Jia, Environ. Sci. Technol., 2009, 43, 3796–3802 CrossRef CAS PubMed.
- Y. Deng and R. Zhao, Curr. Pollut. Rep., 2015, 1, 167–176 CrossRef CAS.
- K. G. Linden and M. Mohseni, Advanced Oxidation Processes: Applications in Drinking Water Treatment, Comprehensive Water Quality and Purification, 2014, 2, 148–172, DOI:10.1016/B978-0-12-382182-9.00031-1.
- B. P. Vellanki, B. Batchelor and A. Abdel-Wahab, Environmental engineering science, 2013, vol. 30, pp. 264–271 Search PubMed.
- D. Zhou, H. Zhang and L. Chen, J. Chem. Technol. Biotechnol., 2015, 90, 775–779 CrossRef CAS.
- Q. Yi, J. Ji, B. Shen, C. Dong, J. Liu, J. Zhang and M. Xing, Environ. Sci. Technol., 2019, 53, 9725–9733 CrossRef CAS PubMed.
- P. Attri, Y. H. Kim, D. H. Park, J. H. Park, Y. J. Hong, H. S. Uhm, K.-N. Kim, A. Fridman and E. H. Choi, Sci. Rep., 2015, 5, 1–8 Search PubMed.
- P. Neta, R. E. Huie and A. B. Ross, J. Phys. Chem. Ref. Data, 1988, 17, 1027–1284 CrossRef CAS.
- S. Guerra-Rodríguez, E. Rodríguez, D. N. Singh and J. Rodríguez-Chueca, Water, 2018, 10, 1828 CrossRef.
- T. N. Das, R. E. Huie and P. Neta, J. Phys. Chem. A, 1999, 103, 3581–3588 CrossRef CAS.
- S. G. Mayhew, Eur. J. Biochem., 1978, 85, 535–547 CrossRef CAS PubMed.
- H. A. Schwarz, Free radicals generated by radiolysis of aqueous solutions, ACS Publications, 1981 Search PubMed.
- P. Devi, U. Das and A. K. Dalai, Sci. Total Environ., 2016, 571, 643–657 CrossRef CAS PubMed.
- M. Hayyan, M. A. Hashim and I. M. AlNashef, Chem. Rev., 2016, 116, 3029–3085 CrossRef CAS PubMed.
- D. A. Armstrong, R. E. Huie, W. H. Koppenol, S. V. Lymar, G. Merényi, P. Neta, B. Ruscic, D. M. Stanbury, S. Steenken and P. Wardman, Pure Appl. Chem., 2015, 87, 1139–1150 CrossRef CAS.
- X. Wang and L. Zhang, RSC Adv., 2018, 8, 40632–40638 RSC.
- R. Ossola, O. M. Jönsson, K. Moor and K. McNeill, Chem. Rev., 2021, 121, 4100–4146 CrossRef CAS PubMed.
- T. Luo, Z. Wang, Y. Wang, Z. Liu and I. P. Pozdnyakov, Molecules, 2019, 24, 2307 CrossRef CAS PubMed.
- Q. Xiao, S. Yu, L. Li, T. Wang, X. Liao and Y. Ye, J. Hazard. Mater., 2017, 324, 230–240 CrossRef CAS PubMed.
- K. Hofman, N. W. Liu and G. Manolikakes, Chem. – Eur. J., 2018, 24, 11852–11863 CrossRef CAS PubMed.
- Y. Liu, X. Fan, X. Quan, Y. Fan, S. Chen and X. Zhao, Environ. Sci. Technol., 2019, 53, 5195–5201 CrossRef CAS PubMed.
- B. M. Hunter, H. B. Gray and A. M. Muller, Chem. Rev., 2016, 116, 14120–14136 CrossRef CAS PubMed.
- R. C. Forsythe and A. M. Müller, Catal. Today, 2022, 388, 329–332 CrossRef.
- S. Chabi, K. M. Papadantonakis, N. S. Lewis and M. S. Freund, Energy Environ. Sci., 2017, 10, 1320–1338 RSC.
- S. Hammes-Schiffer, Chem. Rev., 2010, 110, 6937–6938 Search PubMed.
- D. T. Sawyer, Oxygen radicals in biology and medicine, 1988, pp. 11–20 Search PubMed.
- S. M. Mitchell, M. Ahmad, A. L. Teel and R. J. Watts, Environ. Sci. Technol. Lett., 2014, 1, 117–121 CrossRef CAS.
- W. H. Koppenol, The Haber-Weiss cycle–70 years later, 2001 Search PubMed.
- G. Ruppert, R. Bauer and G. Heisler, J. Photochem. Photobiol. A: Chem., 1993, 73, 75–78 CrossRef CAS.
- S. R. Pouran, A. A. Aziz and W. M. A. W. Daud, J. Ind. Eng. Chem., 2015, 21, 53–69 CrossRef.
- S. Yang, S. Fernando, T. M. Holsen and Y. Yang, Environ. Sci. Technol. Lett., 2019, 6, 775–780 CrossRef CAS.
- D. R. Lide, CRC handbook of chemistry and physics, CRC Press, 2004 Search PubMed.
- A. J. Bard, L. R. Faulkner and H. S. White, Electrochemical methods: fundamentals and applications, John Wiley & Sons, 2022 Search PubMed.
- J. Robertson, 1.14 Free Radicals and Reactive Oxygen Species, 2010 Search PubMed.
- J. L. Weeks and J. Rabani, J. Phys. Chem., 1966, 70, 2100–2106 CrossRef CAS.
- S. Yang, A. Verdaguer-Casadevall, L. Arnarson, L. Silvioli, V. Colic, R. Frydendal, J. Rossmeisl, I. Chorkendorff and I. E. Stephens, ACS Catal., 2018, 8, 4064–4081 CrossRef CAS.
- Q. Yu, R. Zhang, S. Deng, J. Huang and G. Yu, Water Res., 2009, 43, 1150–1158 CrossRef CAS PubMed.
- Y. Zhang, A. Moores, J. Liu and S. Ghoshal, Environ. Sci. Technol., 2019, 53, 8672–8681 CrossRef CAS PubMed.
- M. Trojanowicz, A. Bojanowska-Czajka, I. Bartosiewicz and K. Kulisa, Chem. Eng. J., 2018, 336, 170–199 CrossRef CAS.
- M. Umar, Water, 2021, 13, 3185 CrossRef CAS.
- C. Fang, M. Megharaj and R. Naidu, J. Adv. Oxid. Technol., 2017, 20, 20170014 CAS.
- Y. G. Song, B. Liu, L. F. Wang, M. H. Li and Y. Liu, Photosynth. Res., 2006, 90, 67–78 CrossRef CAS PubMed.
- J. Dumanović, E. Nepovimova, M. Natić, K. Kuča and V. Jaćević, Front. Plant Sci., 2021, 11, 552969 CrossRef PubMed.
- T. Li, C. Wang, T. Wang and L. Zhu, Appl. Catal. B: Environ., 2020, 268, 118442 CrossRef CAS.
- H. Shi, Y. Wang, C. Li, R. Pierce, S. Gao and Q. Huang, Environ. Sci. Technol., 2019, 53, 14528–14537 CrossRef CAS PubMed.
- L. Wang, J. Lu, L. Li, Y. Wang and Q. Huang, Water Res., 2020, 170, 115254 CrossRef CAS PubMed.
- F. Zhang, Z. Sun and J. Cui, RSC Adv., 2020, 10, 33928–33936 RSC.
- X. Liu, S. Yoon, B. Batchelor and A. Abdel-Wahab, Chem. Eng. J., 2013, 215, 868–875 CrossRef.
- J. D. García-Espinoza, I. Robles, V. Gil, E. Becerril-Bravo, J. A. Barrios and L. A. Godínez, J. Environ. Chem. Eng., 2019, 7, 103228 CrossRef PubMed.
- M. C. Sauer, R. A. Crowell and I. A. Shkrob, J. Phys. Chem. A, 2004, 108, 5490–5502 CrossRef CAS.
- C. Fang, M. Megharaj and R. Naidu, J. Adv. Oxid. Technol., 2017, 20, 20170014 CAS.
- M. J. Davies, Methods, 2016, 109, 21–30 CrossRef CAS PubMed.
- S. Khan, X. He, J. A. Khan, H. M. Khan, D. L. Boccelli and D. D. Dionysiou, Chem. Eng. J., 2017, 318, 135–142 CrossRef CAS.
- P. Thanekar, M. Panda and P. R. Gogate, Ultrason. Sonochem., 2018, 40, 567–576 CrossRef CAS PubMed.
- K. E. O’Shea and D. D. Dionysiou, J. Phys. Chem. Lett., 2012, 3, 2112–2113 CrossRef.
- T. Ochiai, Y. Iizuka, K. Nakata, T. Murakami, D. A. Tryk, A. Fujishima, Y. Koide and Y. Morito, Diamond Relat. Mater., 2011, 20, 64–67 CrossRef CAS.
- S. C. Panchangam, A. Y.-C. Lin, K. L. Shaik and C.-F. Lin, Chemosphere, 2009, 77, 242–248 CrossRef CAS PubMed.
- C. Li, Y. Wang, Y. Wang, Z. Wang and Q. Huang, J. Hazard. Mater., 2022, 436, 129091 CrossRef CAS PubMed.
- J. Radjenovic, N. Duinslaeger, S. S. Avval and B. P. Chaplin, Environ. Sci. Technol., 2020, 54, 14815–14829 CrossRef CAS PubMed.
- P. Neta and R. E. Huie, Environ. Health Perspect., 1985, 64, 209–217 CrossRef CAS PubMed.
- K. G. Serrano, Electrochemical water and wastewater treatment, Elsevier, 2018, pp. 133–164 Search PubMed.
- D. Kanakaraju, B. D. Glass and M. Oelgemöller, J. Environ. Manage., 2018, 219, 189–207 CrossRef CAS PubMed.
- M. J. M. de Vidales, C. Sáez, J. F. Pérez, S. Cotillas, J. Llanos, P. Canizares and M. A. Rodrigo, J. Appl. Electrochem., 2015, 45, 799–808 CrossRef.
- S. Cotillas, M. J. M. de Vidales, J. Llanos, C. Sáez, P. Canizares and M. A. Rodrigo, J. Hazard. Mater., 2016, 319, 93–101 CrossRef CAS PubMed.
- X. Shi, S. Back, T. M. Gill, S. Siahrostami and X. Zheng, Chem, 2021, 7, 38–63 CAS.
- S. Siahrostami, Chem. Catal, 2023, 3, 100568 CrossRef CAS.
- K.-T. Lu, C.-C. Yang and P.-C. Lin, J. Hazard. Mater., 2006, 135, 319–327 CrossRef CAS PubMed.
- O. Špalek, J. Balej and I. Paseka, J. Chem. Soc., Faraday Trans. 1, 1982, 78, 2349–2359 RSC.
- Y. Nosaka and A. Y. Nosaka, Chem. Rev., 2017, 117, 11302–11336 CrossRef CAS PubMed.
- G. Li, X. Liu, H. Zhang, T. An, S. Zhang, A. R. Carroll and H. Zhao, J. Catal., 2011, 277, 88–94 CrossRef CAS.
- D. Zhi, Y. Lin, L. Jiang, Y. Zhou, A. Huang, J. Yang and L. Luo, J. Environ. Manage., 2020, 260, 110125 CrossRef CAS PubMed.
- C.-W. Wang and C. Liang, Chem. Eng. J., 2014, 254, 472–478 CrossRef CAS.
- F. L. Souza, C. Sáez, P. Canizares, A. J. Motheo and M. A. Rodrigo, Appl. Catal. B: Environ., 2014, 144, 121–128 CrossRef CAS.
- J. G. Speight, Environmental organic chemistry for engineers, Butterworth-Heinemann, 2016 Search PubMed.
- S. Parsons, Advanced oxidation processes for water and wastewater treatment, IWA Publishing, 2004 Search PubMed.
- H. D. Burrows, J. Santaballa and S. Steenken, J. Photochem. Photobiol. B: Biol., 2002, 67, 71–108 CrossRef CAS PubMed.
- K. Li, M. I. Stefan and J. C. Crittenden, Environ. Sci. Technol., 2004, 38, 6685–6693 CrossRef CAS PubMed.
- P. Chelme-Ayala, M. G. El-Din and D. W. Smith, Water Res., 2010, 44, 2221–2228 CrossRef CAS PubMed.
- W. Hijnen, E. Beerendonk and G. J. Medema, Water Res., 2006, 40, 3–22 CrossRef CAS PubMed.
- A. Kumar, A. Sagdeo and P. R. Sagdeo, Photodiagnosis Photodyn. Ther., 2021, 34, 102234 CrossRef CAS PubMed.
- M. Gonzalez and A. Braun, Res. Chem. Intermed., 1995, 21, 837–859 CrossRef CAS.
- T. Yamamoto, Y. Noma, S.-I. Sakai and Y. Shibata, Environ. Sci. Technol, 2007, 41, 5660–5665 CrossRef CAS PubMed.
- R. R. Giri, H. Ozaki, T. Okada, S. Taniguchi and R. Takanami, Chem. Eng. J., 2012, 180, 197–203 CrossRef CAS.
- M. C. López, M. I. Fernández, C. Martínez and J. A. Santaballa, Pure Appl. Chem., 2013, 85, 1437–1449 CrossRef.
- G. Liu, S. You, Y. Tan and N. Ren, Environ. Sci. Technol., 2017, 51, 2339–2346 CrossRef CAS PubMed.
- J. Prousek, Chem. Listy, 1996, 90, 307–315 CAS.
- N. F. Mott, Mathematical Proceedings of the Cambridge Philosophical Society, Cambridge University Press, 1938, vol. 34, pp. 568–572 Search PubMed.
- W. Schottky, Naturwissenschaften, 1938, 26, 843 CrossRef CAS.
- N. F. Mott, Proc. R. Soc. London, Ser. A, 1939, 171, 281–285 CAS.
- W. Schottky, Z. Phys., 1939, 113, 367–414 CrossRef CAS.
- Z. Zhang and J. T. Yates Jr, Chem. Rev., 2012, 112, 5520–5551 CrossRef CAS PubMed.
- V. Hasija, P. Raizada, A. Sudhaik, K. Sharma, A. Kumar, P. Singh, S. B. Jonnalagadda and V. K. Thakur, Appl. Mater. Today, 2019, 15, 494–524 CrossRef.
- M. N. Chong, B. Jin, C. W. Chow and C. Saint, Water Res., 2010, 44, 2997–3027 CrossRef CAS PubMed.
- L. Jiang, X. Yuan, Y. Pan, J. Liang, G. Zeng, Z. Wu and H. Wang, Appl. Catal. B: Environ., 2017, 217, 388–406 CrossRef CAS.
- L. Clarizia, D. Russo, I. Di Somma, R. Marotta and R. Andreozzi, Appl. Catal. B: Environ., 2017, 209, 358–371 CrossRef CAS.
- J. R. Domínguez, T. González, P. Palo and E. M. Cuerda-Correa, Ind. Eng. Chem. Res., 2012, 51, 2531–2538 CrossRef.
- A. Babuponnusami and K. Muthukumar, J. Environ. Chem. Eng., 2014, 2, 557–572 CrossRef CAS.
- A. Mirzaei, Z. Chen, F. Haghighat and L. Yerushalmi, Chemosphere, 2017, 174, 665–688 CrossRef CAS PubMed.
- J. A. L. Perini, A. L. Tonetti, C. Vidal, C. C. Montagner and R. F. P. Nogueira, Appl. Catal. B: Environ., 2018, 224, 761–771 CrossRef CAS.
- M. Antonopoulou, C. Kosma, T. Albanis and I. Konstantinou, Sci. Total Environ., 2021, 765, 144163 CrossRef CAS PubMed.
- A. Sudhaik, P. Raizada, P. Shandilya, D.-Y. Jeong, J.-H. Lim and P. Singh, J. Ind. Eng. Chem., 2018, 67, 28–51 CrossRef CAS.
- S. Sharma, V. Dutta, P. Singh, P. Raizada, A. Rahmani-Sani, A. Hosseini-Bandegharaei and V. K. Thakur, J. Clean. Prod., 2019, 228, 755–769 CrossRef CAS.
- Y. Deng and J. D. Englehardt, Water Res., 2006, 40, 3683–3694 CrossRef CAS PubMed.
- L. Chen, J. Ma, X. Li, J. Zhang, J. Fang, Y. Guan and P. Xie, Environ. Sci. Technol., 2011, 45, 3925–3930 CrossRef CAS PubMed.
- N. Wang, T. Zheng, G. Zhang and P. Wang, J. Environ. Chem. Eng., 2016, 4, 762–787 CrossRef CAS.
- M.-H. Zhang, H. Dong, L. Zhao, D.-X. Wang and D. Meng, Sci. Total Environ., 2019, 670, 110–121 CrossRef CAS PubMed.
- J. Dong, G. Li, J. Gao, H. Zhang, S. Bi, S. Liu, C. Liao and G. Jiang, Sci. Total Environ., 2022, 157695 CrossRef CAS PubMed.
- M. Xiang, M. Huang, H. Li, W. Wang, Y. Huang, Z. Lu, C. Wang, R. Si and W. Cao, Chem. Eng. J., 2021, 417, 129208 CrossRef CAS.
- L. Zhou, H. Zhang, L. Ji, Y. Shao and Y. Li, RSC Adv., 2014, 4, 24900–24908 RSC.
- H. Che, S. Bae and W. Lee, J. Hazard. Mater., 2011, 185, 1355–1361 CrossRef CAS PubMed.
- C. Liang, C. J. Bruell, M. C. Marley and K. L. Sperry, Chemosphere, 2004, 55, 1213–1223 CrossRef CAS PubMed.
- T. Ohsaka, K. Shinozaki, K. Tsuruta and K. Hirano, Chemosphere, 2008, 73, 1279–1283 CrossRef CAS PubMed.
- Y. Zhong, X. Liang, Z. He, W. Tan, H. He, R. Zhu, Y. Zhong, J. Zhu, P. Yuan and Z. Jiang, J. Nanosci. Nanotech., 2014, 14, 7307–7314 CrossRef CAS PubMed.
- L. Tan, S. Lu, Z. Fang, W. Cheng and E. P. Tsang, Appl. Catal. B: Environ., 2017, 200, 200–210 CrossRef CAS.
- S. Luo, S. Yang, Y. Xue, F. Liang and C. Sun, J. Hazard. Mater., 2011, 192, 1795–1803 CrossRef CAS PubMed.
- M. Mrowetz, C. Pirola and E. Selli, Ultrason. Sonochem., 2003, 10, 247–254 CrossRef CAS PubMed.
- M. Sunder and D.-C. Hempel, Water Res., 1997, 31, 33–40 CrossRef CAS.
- M. Sansotera, F. Persico, C. Pirola, W. Navarrini, A. Di Michele and C. L. Bianchi, Appl. Catal. B: Environ, 2014, 148, 29–35 CrossRef.
- M. J. Bentel, Z. Liu, Y. Yu, J. Gao, Y. Men and J. Liu, Environ. Sci. Technol. Lett., 2020, 7, 351–357 CrossRef CAS.
- R. Tenorio, J. Liu, X. Xiao, A. Maizel, C. P. Higgins, C. E. Schaefer and T. J. Strathmann, Environ. Sci. Technol., 2020, 54, 6957–6967 CrossRef CAS PubMed.
- C. J. Liu, G. McKay, D. Jiang, R. Tenorio, J. T. Cath, C. Amador, C. C. Murray, J. B. Brown, H. B. Wright and C. Schaefer, Water Res., 2021, 205, 117677 CrossRef CAS PubMed.
- D. Zhang, Y. Li, X. Chen, C. Li, L. Dong and Z. Wang, Sep. Purif. Technol., 2022, 287, 120521 CrossRef CAS.
- Y.-P. Peng, H. Chen and C. Huang, Appl. Catal. B: Environ., 2017, 209, 437–446 CrossRef CAS.
- Z. U. Zango, K. S. Khoo, A. Garba, H. A. Kadir, F. Usman, M. U. Zango, W. Da Oh and J. W. Lim, Environ. Res., 2023, 115326 CrossRef CAS PubMed.
- H. Tang, Q. Xiang, M. Lei, J. Yan, L. Zhu and J. Zou, Chem. Eng. J., 2012, 184, 156–162 CrossRef CAS.
- A. B. Nienhauser, M. S. Ersan, Z. Lin, F. Perreault, P. Westerhoff and S. Garcia-Segura, J. Environ. Chem. Eng., 2022, 10, 107192 CrossRef CAS.
- B. Gomez-Ruiz, S. Gómez-Lavín, N. Diban, V. Boiteux, A. Colin, X. Dauchy and A. Urtiaga, Chem. Eng. J., 2017, 322, 196–204 CrossRef CAS.
- F. Wang, K. Shih, X. Lu and C. Liu, Environ. Sci. Technol., 2013, 47, 2621–2627 CrossRef CAS PubMed.
- M. Pierpaoli, M. Szopińska, B. K. Wilk, M. Sobaszek, A. Łuczkiewicz, R. Bogdanowicz and S. Fudala-Książek, J. Hazard. Mater., 2021, 403, 123606 CrossRef CAS PubMed.
- B. Yang, J. Wang, C. Jiang, J. Li, G. Yu, S. Deng, S. Lu, P. Zhang, C. Zhu and Q. Zhuo, Chem. Eng. J., 2017, 316, 296–304 CrossRef CAS.
- X. Duan, W. Wang, Q. Wang, X. Sui, N. Li and L. Chang, Chemosphere, 2020, 260, 127587 CrossRef CAS PubMed.
- A. Kugler, H. Dong, C. Li, C. Gu, C. E. Schaefer, Y. J. Choi, D. Tran, M. Spraul and C. P. Higgins, Water Res., 2021, 200, 117221 CrossRef CAS PubMed.
- A. H. da, S. Filho and G. L. de Souza, Phys. Chem. Chem. Phys., 2020, 22, 17659–17667 RSC.
- I. Janik and G. Tripathi, J. Chem. Phys., 2016, 144, 154307 CrossRef PubMed.
- H. Zipse, Radicals in Synthesis I, 2006, pp. 163–189 Search PubMed.
- C. E. Schaefer, C. Andaya, A. Urtiaga, E. R. McKenzie and C. P. Higgins, J. Hazard. Mater., 2015, 295, 170–175 CrossRef CAS PubMed.
- H. N. P. Vo, H. H. Ngo, W. Guo, T. M. H. Nguyen, J. Li, H. Liang, L. Deng, Z. Chen and T. A. H. Nguyen, J. Water Process Eng., 2020, 36, 101393 CrossRef.
- R. De Marco, G. Clarke and B. Pejcic, Electroanalysis, 2007, 19, 1987–2001 CrossRef CAS.
- D. R. Taves, Nature, 1968, 217, 1050–1051 CrossRef CAS PubMed.
- J. L. Barber, U. Berger, C. Chaemfa, S. Huber, A. Jahnke, C. Temme and K. C. Jones, J. Environ. Monit., 2007, 9, 530–541 RSC.
- K. L. Rodriguez, J.-H. Hwang, A. R. Esfahani, A. A. Sadmani and W. H. Lee, Micromachines, 2020, 11, 667 CrossRef PubMed.
- A. U. Rehman, M. Crimi and S. Andreescu, Trends Environ. Anal. Chem., 2023, e00198 CrossRef CAS.
- Z. Wang, J. C. DeWitt, C. P. Higgins and I. T. Cousins, Environ. Sci. Technol., 2017, 2508–2518 CrossRef CAS PubMed.
- Y. Liu, L. A. D'Agostino, G. Qu, G. Jiang and J. W. Martin, TrAC, Trends Anal. Chem., 2019, 121, 115420 CrossRef CAS.
- I. Kourtchev, S. Hellebust, E. Heffernan, J. Wenger, S. Towers, E. Diapouli and K. Eleftheriadis, Sci. Total Environ., 2022, 835, 155496 CrossRef CAS PubMed.
- D. Camdzic, R. A. Dickman, A. S. Joyce, J. S. Wallace, P. L. Ferguson and D. S. Aga, Anal. Chem., 2023, 95, 5484–5488 CrossRef CAS PubMed.
- J. N. Dodds, Z. R. Hopkins, D. R. Knappe and E. S. Baker, Anal. Chem., 2020, 92, 4427–4435 CrossRef CAS PubMed.
- R. C. Forsythe, C. P. Cox, M. K. Wilsey and A. M. Muller, Chem. Rev., 2021, 121, 7568–7637 CrossRef CAS PubMed.
- M. K. Wilsey, C. P. Cox, R. C. Forsythe, L. R. McCarney and A. M. Müller, Catal. Sci. Technol., 2021, 11, 416–424 RSC.
- P. C. Vesborg and T. F. Jaramillo, RSC Adv., 2012, 2, 7933–7947 RSC.
- Z. W. Seh, J. Kibsgaard, C. F. Dickens, I. Chorkendorff, J. K. Nørskov and T. F. Jaramillo, Science, 2017, 355, eaad4998 CrossRef PubMed.
- C. C. McCrory, S. Jung, J. C. Peters and T. F. Jaramillo, J. Am. Chem. Soc., 2013, 135, 16977–16987 CrossRef CAS PubMed.
- S. Nitopi, E. Bertheussen, S. B. Scott, X. Liu, A. K. Engstfeld, S. Horch, B. Seger, I. E. Stephens, K. Chan and C. Hahn, Chem. Rev., 2019, 119, 7610–7672 CrossRef CAS PubMed.
- R. Schlögl, Angew. Chem., Int. Ed., 2015, 54, 3465–3520 CrossRef PubMed.
- H. Topsøe, J. Catal., 2003, 216, 155–164 CrossRef.
- B. M. Weckhuysen, Phys. Chem. Chem. Phys., 2003, 5, 4351–4360 RSC.
- Y.-W. Choi, H. Mistry and B. R. Cuenya, Curr. Opin. Electrochem., 2017, 1, 95–103 CrossRef CAS.
- B. M. Hunter, N. B. Thompson, A. M. Müller, G. R. Rossman, M. G. Hill, J. R. Winkler and H. B. Gray, Joule, 2018, 2, 747–763 CrossRef CAS.
- N. Omidvar, H. S. Pillai, S.-H. Wang, T. Mou, S. Wang, A. Athawale, L. E. Achenie and H. Xin, J. Phys. Chem. Lett., 2021, 12, 11476–11487 CrossRef CAS PubMed.
- S. Gusarov, S. R. Stoyanov and S. Siahrostami, J. Phys. Chem. C, 2020, 124, 10079–10084 CrossRef CAS.
- Y. Yang, C. R. Peltier, R. Zeng, R. Schimmenti, Q. Li, X. Huang, Z. Yan, G. Potsi, R. Selhorst and X. Lu, Chem. Rev., 2022, 122, 6117–6321 CrossRef CAS PubMed.
- S. Liu, L. D'Amario, S. Jiang and H. Dau, Curr. Opin. Electrochem., 2022, 35, 101042 CrossRef CAS.
- L. Lukashuk, N. Yigit, R. Rameshan, E. Kolar, D. Teschner, M. Hävecker, A. Knop-Gericke, R. Schlögl, K. Föttinger and G. N. Rupprechter, ACS Catal., 2018, 8, 8630–8641 CrossRef CAS PubMed.
- B. S. Yeo and A. T. Bell, J. Phys. Chem. C, 2012, 116, 8394–8400 CrossRef CAS.
- D. Friebel, M. W. Louie, M. Bajdich, K. E. Sanwald, Y. Cai, A. M. Wise, M.-J. Cheng, D. Sokaras, T.-C. Weng and R. Alonso-Mori, J. Am. Chem. Soc., 2015, 137, 1305–1313 CrossRef CAS PubMed.
- H.-J. Lewerenz, M. F. Lichterman, M. H. Richter, E. J. Crumlin, S. Hu, S. Axnanda, M. Favaro, W. Drisdell, Z. Hussain and B. S. Brunschwig, Electrochimica Acta, 2016, 211, 711–719 CrossRef CAS.
- S. J. Blair, M. Doucet, V. A. Niemann, K. H. Stone, M. E. Kreider, J. F. Browning, C. E. Halbert, H. Wang, P. Benedek, E. J. McShane, A. C. Nielander, A. Gallo and T. F. Jaramillo, Energy Environ. Sci., 2023, 16, 3391–3406 RSC.
- F. Dionigi, Z. Zeng, I. Sinev, T. Merzdorf, S. Deshpande, M. B. Lopez, S. Kunze, I. Zegkinoglou, H. Sarodnik and D. Fan, Nat. Commun., 2020, 11, 2522 CrossRef CAS PubMed.
- C. D. Taylor, S. A. Wasileski, J.-S. Filhol and M. Neurock, Phys. Rev. B, 2006, 73, 165402 CrossRef.
- A. Bruix, J. T. Margraf, M. Andersen and K. Reuter, Nat. Catal., 2019, 2, 659–670 CrossRef CAS.
- T. Van Mourik, M. Bühl and M.-P. Gaigeot, Phil. Trans. R. Soc. A, 2014, 372, 20120488 CrossRef PubMed.
- S. Siahrostami, A. Verdaguer-Casadevall, M. Karamad, D. Deiana, P. Malacrida, B. Wickman, M. Escudero-Escribano, E. A. Paoli, R. Frydendal and T. W. Hansen, Nat. Mater., 2013, 12, 1137–1143 CrossRef CAS PubMed.
- A. D. White and G. A. Voth, J. Chem. Theory Comput., 2014, 10, 3023–3030 CrossRef CAS PubMed.
- J. T. Margraf, Angew. Chem., 2023, e202219170 CAS.
- Y. Chen, Y. Huang, T. Cheng and W. A. Goddard III, J. Am. Chem. Soc., 2019, 141, 11651–11657 CrossRef CAS PubMed.
- M. Neurock, M. Janik and A. Wieckowski, Faraday Discuss., 2009, 140, 363–378 RSC.
- E. I. Solomon, R. A. Scott and R. B. King, Computational inorganic and bioinorganic chemistry, John Wiley & Sons, 2013 Search PubMed.
- D. Rappoport, N. R. Crawford, F. Furche and K. Burke, Encyclopedia of Inorganic Chemistry, 2006 Search PubMed.
- K. Kim, P. Baldaguez Medina, J. Elbert, E. Kayiwa, R. D. Cusick, Y. Men and X. Su, Adv. Funct. Mater., 2020, 30, 2004635 CrossRef CAS.
- M. J. Bentel, Y. Yu, L. Xu, H. Kwon, Z. Li, B. M. Wong, Y. Men and J. Liu, Environ. Sci. Technol., 2020, 54, 2489–2499 CrossRef CAS PubMed.
- Y. Zhang, M. Dai and Z. Yuan, Anal. Methods, 2018, 10, 4625–4638 RSC.
- R. Guo, B. Xi, C. Guo, N. Lv and J. Xu, Environ. Funct. Mater., 2022, 67–91 Search PubMed.
- J. Wang and S. Wang, Chem. Eng. J., 2020, 401, 126158 CrossRef CAS.
- S. Stoll and D. Goldfarb, EPR spectroscopy: fundamentals and methods, John Wiley & Sons, 2018 Search PubMed.
- M. M. Roessler and E. Salvadori, Chem. Soc. Rev., 2018, 47, 2534–2553 RSC.
- S. Stoll and R. D. Britt, Phys. Chem. Chem. Phys., 2009, 11, 6614–6625 RSC.
- H. E. Williams, V. C. Loades, M. Claybourn and D. M. Murphy, Anal. Chem., 2006, 78, 604–608 CrossRef CAS PubMed.
- A. Gomes, E. Fernandes and J. L. Lima, J. Biochem. Biophys. Methods, 2005, 65, 45–80 CrossRef CAS PubMed.
- J. Wang, Y. Guo, B. Liu, X. Jin, L. Liu, R. Xu, Y. Kong and B. Wang, Ultrason. Sonochem., 2011, 18, 177–183 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2023 |