
Confinement of proteins by thermoresponsive dendronized polymers†
Yi
Yao
 ,
Jintao
Yang
,
Wen
Li
* and
Afang
Zhang
*
,
Jintao
Yang
,
Wen
Li
* and
Afang
Zhang
*
International Joint Laboratory of Biomimetic and Smart Polymers, School of Materials Science and Engineering, Shanghai University, Nanchen Street 333, Shanghai 200444, China. E-mail: wli@shu.edu.cn; azhang@shu.edu.cn
First published on 23rd August 2022
Abstract
The crowding environment created by host polymers plays crucial roles in manipulating interactions with proteins and modulating their bioactivity. Here, we report our investigation on the interactions between polymers and proteins in the confined microenvironments constructed by oligoethylene glycol (OEG)-based dendronized polymers due to the crowded OEG dendrons. Several important characteristics of these dendronized polymers, including their aggregation state, charge state and combination form with biomolecules, were revealed to be the main factors decoding the polymer–protein interactions. To examine the effects of encapsulation and shielding from the dendronized polymers for the biomacromolecules, the guest proteins were combined through either bioconjugation, electrostatic complexation, or just physical mixing. The unprecedented thermoresponsiveness of the dendronized polymers provides tunable crowding and hydrophobicity of the microenvironment conveniently through their thermally induced aggregation, resulting in regulation of the activities for the proteins. This kind of dendronized polymer with structural and topological features is a promising candidate for the construction of intelligent artificial microenvironments to tunably confine biomacromolecules.
Introduction
Biotransformation in cells is often accomplished in confined microenvironments created by crowded biomacromolecules in the vicinity.1,2 The secondary and tertiary structures of biomacromolecules, and even the ways that they associate with each other, will be affected by molecular crowding, resulting in their different behaviours in biological processes.3–5 Moreover, the adverse interactions between the active sites of proteins and crowded molecules could lead to pronounced effects on their folding states, and cause the loss of their activities directly.4,6 Various crowding agents, such as synthetic or natural molecules (polymers,7 polysaccharides,8 liposomes,9etc.), have been used to examine the influence on the folding and stability of proteins caused through their weak hydrophobic interactions.4,8,10,11 The factors affecting the interactions among proteins and crowding agents mainly include: (1) structural features of the macromolecules, such as chain length,12 hydrophobicity,13,14 and even concentration;15 (2) protein properties, such as amino acid sequence/hydrophilicity, and molecular weight;16 and (3) solution conditions, such as temperature and pH.16,17Macromolecules used for biological activity protection should have conformational freedom of the chains in aqueous solutions, like a randomly coiled coil, resulting in the formation of an energetically stable hydration shell wrapped around the proteins and hanging freely in the solution.13,18 For synthetic biomolecule stabilization agents, such as the widely used polyethylene glycols (PEGs),3 their suitable amphiphilicity has been found to be a key parameter that determines their states in aqueous solutions, dictating their activity protection capacity.18,19 PEG analogues are being continuously developed for achieving macromolecules with suitable amphiphilicity.20 Among them, one kind of comb-shaped polymer carrying oligoethylene glycol (OEG) pendants and a methacrylate backbone has attracted wide attention due to its adjustable amphiphilicity, which can be achieved through changing the chain length of the OEGs.7 Compared to their linear OEG counterparts, dendritic OEGs have more advantages in activity protection because of the multi-valency, topological cooperativity, and, most importantly, the densely packed dendritic OEG moieties providing a crowding effect in nanometre dimensions for efficient shielding.4,13,20,21
Recently, we found that OEG-based dendronized polymers exhibit unprecedented thermoresponsiveness, and have shown advantages in forming confined environments at molecular levels for activity protection of biomacromolecules through cooperation interactions and shielding effects.4,10,22 Herein, the interactions between dendronized polymers and proteins in aqueous solutions were investigated in detail. The OEG-based dendronized homopolymers (PG1), copolymers (PG1S), and dendronized chitosans (DCSs) were selected as the models to examine the confinement of proteins through physical mixing, covalent conjugation, or electrostatic complexation (Fig. 1). Moreover, the effects on the hydrophobic interactions between the dendronized polymers and the proteins during thermally induced aggregation of the polymers were revealed by various measurements. On this basis, the protein activity regulation capability of these dendronized polymers was explored under different conditions.
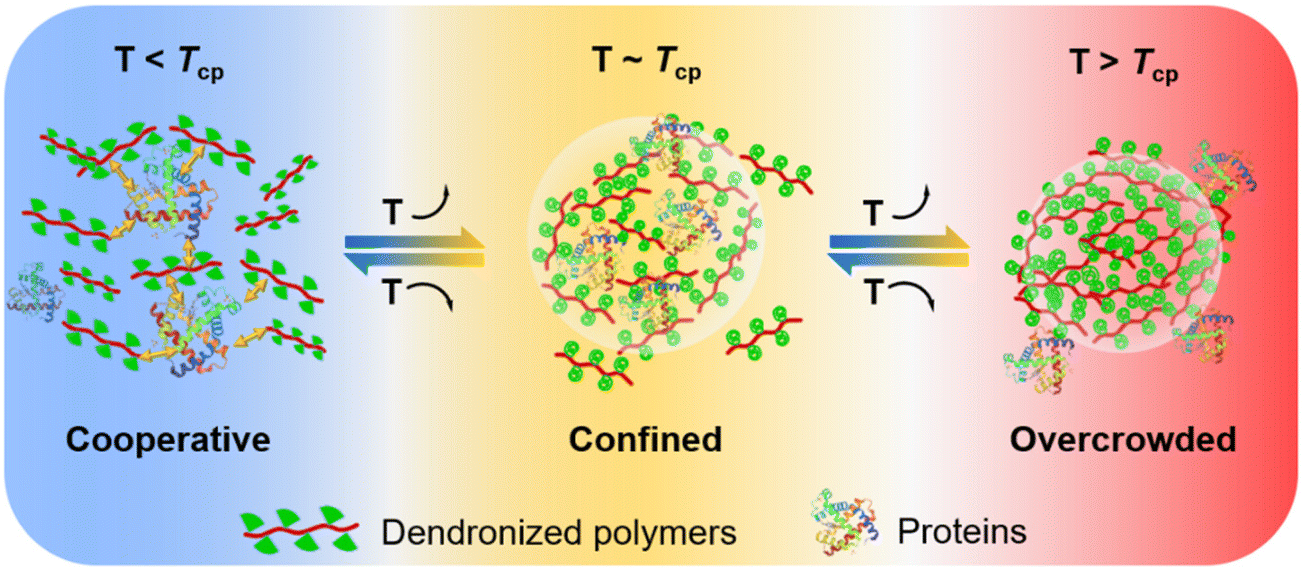 | ||
| Fig. 1 Schematic representation of the possible interaction modes between the dendronized polymers and proteins below, around, and above their cloud points (Tcps). | ||
Results and discussion
Preparation of dendronized polymer–protein conjugates or complexes and their thermoresponsive behaviour
OEG-based dendronized homopolymers (PG1), copolymers (PG1S), and DCSs were selected to examine the confinement of proteins through physical mixing, covalent conjugation, and electrostatic complexation, respectively. Their molecular structures are shown in Fig. S1A.† The synthesis of PG123 and DCSs24 was performed according to previous reports. Dendronized copolymer PG1S pendanted with three-fold dendritic OEG and a 4-nitrophenyl active ester was synthesized through free radical polymerization as shown in Fig. S1B.† Homopolymers and copolymers with similar molecular weights (the Mns of PG1 and PG1S are 2.3 × 105 and 2.2 × 105, respectively, Table S1†) were prepared to reduce the possible effects of molecular size on the interactions.25 Here, the 4-nitrophenyl active ester in MS![[double bond, length as m-dash]](https://www.rsc.org/images/entities/b_char_e001.gif) S was designed for covalent conjugation of proteins via amidation. The copolymerization ratio ([M3]/[MSS]) for PG1S was designed to be 21.5 (Table S1†), which can endow the polymer with a cloud point (Tcp) around physiological temperature. DCSs with a dendron coverage of around 55% were prepared, aiming to keep a suitable amount of positive amino pendants in the CS backbones for complexation with the negatively charged proteins, and simultaneously a proper amount of OEG dendron pendants to provide sufficient shielding effects to form a microenvironment along the polymer chains.
S was designed for covalent conjugation of proteins via amidation. The copolymerization ratio ([M3]/[MSS]) for PG1S was designed to be 21.5 (Table S1†), which can endow the polymer with a cloud point (Tcp) around physiological temperature. DCSs with a dendron coverage of around 55% were prepared, aiming to keep a suitable amount of positive amino pendants in the CS backbones for complexation with the negatively charged proteins, and simultaneously a proper amount of OEG dendron pendants to provide sufficient shielding effects to form a microenvironment along the polymer chains.
Formation of copolymer–protein conjugates (PG1S-Mb) from PG1S and myoglobin (Mb) was followed by UV/vis measurement. The absorbance at 420 nm changed obviously after the addition of Mb to the solution of PG1S (Fig. S3A†). Moreover, the hydrodynamic radii (Rhs, intensity radii) increased twice after the addition of Mb to PG1S as shown in Fig. 2A. Both results prove that the conjugation of Mb to PG1S was successful. The loading ability of the polymers for proteins was further evaluated. The loading efficiency increased dramatically with the increase of the polymer concentration of PG1S, and exceeded 90% when the mass ratio of MSS to proteins was over 16 (Fig. 2B). Through isothermal titration calorimetry (ITC) measurements, the binding stoichiometry (n) of dendritic OEGs per mole of Mb was found to be 35.5 and 5.2 for PG1 and DCS (Fig. S3B and S3C†), respectively.
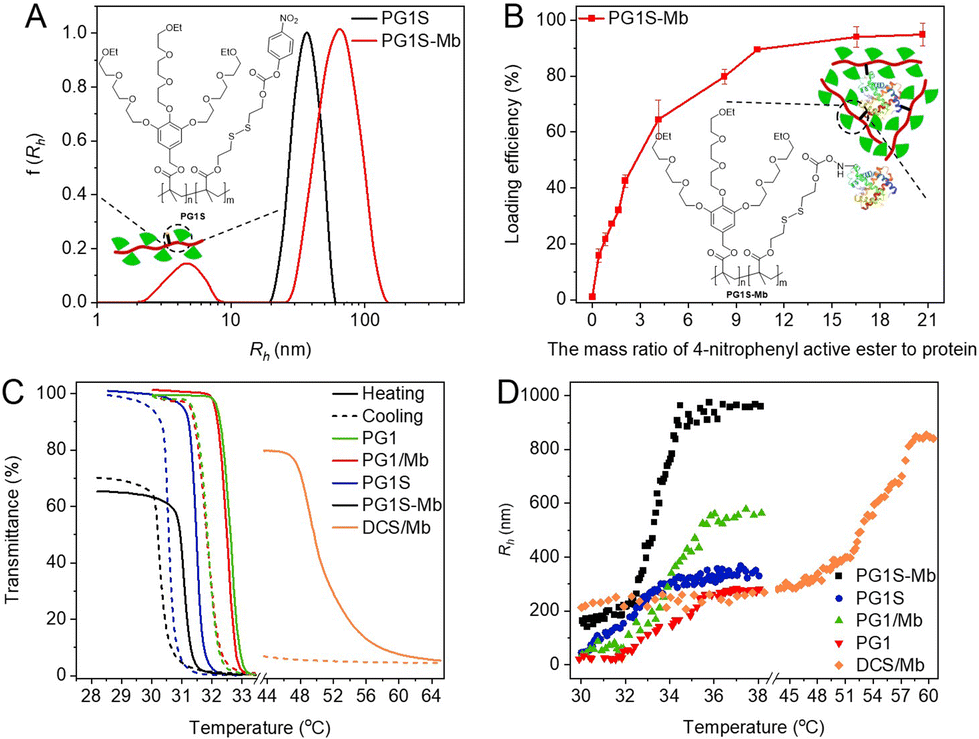 | ||
| Fig. 2 (A) Plots of Rh from PG1S and PG1S-Mb. 25 °C, C = 0.05 mg mL−1. The size polydispersities were 0.40 and 0.59, respectively. (B) Loading efficiency of PG1S for Mb at different concentrations. (C) Plots of transmittance vs. temperature for PG1S, PG1S-Mb, PG1, PG1/Mb, and DCS/Mb. C = 2.5 mg mL−1. (D) Plots of Rh for PG1S, PG1S-Mb, PG1, PG1/Mb, and DCS/Mb between 30 and 60 °C, respectively. C = 0.05 mg mL−1. | ||
The thermoresponsive behaviour of PG1, a physical mixture of PG1 and Mb (PG1/Mb), PG1S, bioconjugate PG1S-Mb, and complex (DCS/Mb) of DCS and Mb was investigated by UV/vis spectroscopy. All of the polymers or mixtures inherited the typical thermoresponsiveness from the OEG-based dendronized homopolymers, irrespective of the presence or absence of proteins, and their transmittance vs. temperature curves are shown in Fig. 2C. The Tcps are 32.6 °C for PG1, 32.5 °C for PG1/Mb, 31.5 °C for PG1S, 31.1 °C for PG1S-Mb, and 49.8 °C for DCS/Mb. PG1/Mb exhibited a similar Tcp to PG1, and only a slight change (less than 0.34 °C) was observed when the concentration of the proteins, including Mb, bovine serum albumin (BSA), and lysozyme (LYZ), increased from 0.05 to 0.25 mg mL−1 (Fig. S4A†). PG1S-Mb also showed nearly the same Tcp as its parent copolymer PG1S. Additionally, the Tcp of DCS/Mb has hardly changed when compared with DCS (49.6 °C, Fig. S4B†). However, DCS/Mb exhibited a relatively broad phase transition when compared to those from the dendronized polymethacrylates due to the stronger hydration. Aqueous solutions of the dendronized polymers became slightly turbid even at room temperature after the introduction of the proteins, as in the cases of PG1S-Mb and DCS/Mb. This suggests that the presence of proteins enhanced the intermolecular associations of the polymers to form aggregates, which forms an interesting feature that proteins can act as a “molecular glue” to induce aggregation of the host polymers through multi-valent interactions. The above suggests that either physical mixing, covalent conjugation, or electrostatic complexation of proteins had hardly any influence on the characteristic dehydration of the polymers carrying dense dendritic OEG pendants.26 This phenomenon is promising, suggesting that these polymers can host the proteins and retain their characteristic thermoresponsiveness. It's necessary to point out that the transmittance of the aqueous solution for both PG1S-Mb and DCS/Mb below its Tcp is lower than 80%, indicating that large and tight aggregates were already formed from the bioconjugate or complexes even at room temperature.
Possible protein-driven aggregation, as well as the thermally induced aggregation, was therefore investigated by dynamic light scattering (DLS) at different temperatures, and the results are plotted in Fig. 2D and S4C.† At room temperature, PG1S-Mb and DCS/Mb showed obvious aggregation below their Tcps with Rhs of 150 and 210 nm, respectively. The above indicates that the presence of proteins induces the aggregation of the dendronized polymers, and this aggregation was more pronounced in the cases of PG1S-Mb and DCS. Possible hydrophobic interactions between the dendritic OEGs and the proteins should have contributed to this aggregation, and covalent linkages in the case of PG1S-Mb or electrostatic interactions in the case of DCS should be helpful to enhance the wrapping of the polymer chains around the proteins.27 On the other hand, the multivalency of the proteins may have provided multiple sites to interact with and physically crosslink the dendronized polymers, resulting in very large aggregates. When the temperature increased above their Tcps, the Rh values of PG1 (20 to 280 nm), PG1S (50 to 330 nm), PG1/Mb (40 to 550 nm), DCS/Mb (210 to 850 nm), and PG1S-Mb (150 to 960 nm) all increased over 4 times. The sizes of the aggregates reached micrometre dimensions, and the latter two were more pronounced. In addition, the size distributions of the aggregates from PG1/protein (Fig. S5A†), PG1S-Mb, and DCS/Mb (Fig. S5B†) were small with narrower peaks at 25 °C, but their peak widths increased significantly above their Tcps. These results indicate that thermally induced dehydration of the dendronized polymers formed more hydrophobic domains, which enhanced the interconnection between the aggregates to form much larger mesoglobules. These hydrophobic domains could be helpful to protect proteins from high-temperature stress and the binding between the protein and substrate.28
Accordingly, we propose that the microconfinement formed through densely grafted dendritic OEGs provides a molecular envelope to impart the protein with a more hydrophobic microenvironment and cooperatively enhances hydrophobic interactions of the proteins with the polymers. To verify this conjecture, the hydrophobicity differences in the microenvironments for these polymers were examined by fluorescence measurements with ANS as a probe. The intensity at the maximum emission (530 nm) for ANS in the cases of PG1 and PG1S was over 10 times higher when compared to that of free proteins at temperatures both below or above their Tcps (Fig. S5C†), indicating that the dendronized polymers have provided the proteins with a more hydrophobic microenvironment.29
Interactions between the dendronized polymers and proteins
The possible interaction sites between the dendronized polymers and proteins were explored by infrared spectroscopy (FT-IR). For free proteins, the protein amide I band was found near 1670 cm−1 (1670 cm−1 for Mb and BSA, 1691 cm−1 for LYZ, Fig. 3A) in the FT-IR spectra.14 Differently, the amide I band from PG1/protein (1647 cm−1 for PG1/Mb, 1657 cm−1 for PG1/BSA, and 1680 cm−1 for PG1/LYZ) observed red-shifting over 11 cm−1, indicating that its stretch was affected. The methylene (CH2) stretching vibrations in the spectra of PG1/protein were found at 2915, 2900, and 2898 cm−1 for PG1/Mb, PG1/BSA, and PG1/LYZ, respectively, which had over 5 cm−1 blue-shifting compared to PG1 (located at 2885 cm−1). In addition, there were several new peaks at 1300–1050 cm−1 (corresponding to the stretching vibration of the ether bond –C–O–) for PG1S-Mb when compared with the other samples, shown as Fig. S6A.† These results indicate that there are indeed hydrophobic interactions between the polymer backbone or dendritic OEGs and proteins.30 The FT-IR spectra of PG1S-Mb were further recorded at different temperatures. As shown in Fig. 3B, the peak shape in the range of 1260–1030 cm−1 gradually became smoother with increasing temperature. The peak shape of PG1S-Mb almost overlapped with those of PG1S at temperatures above 40 °C, which is much higher (>10 °C) than their Tcps (Fig. S6B†). This indicates that the enhanced hydrophobic interactions between the polymer chains due to the extensive dehydration should have reduced their interactions with the proteins, which could eventually cause exposure of the active center of the proteins through collapse and aggregation of the polymer chains.28 This also suggests that the hydrophobic interactions between the dendritic OEGs and proteins changed dynamically during thermally induced aggregation of the polymers. This is a valuable finding, implying that hydrophobic interactions between dendronized polymers and proteins can be regulated through their characteristic thermoresponsiveness, resulting in the adjustment of the confinement of the proteins.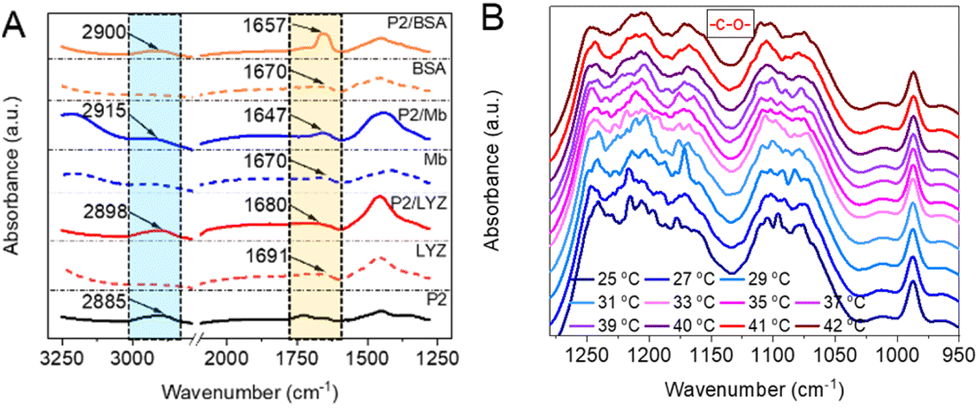 | ||
| Fig. 3 FT-IR spectra of (A) PG1/protein, free proteins, and PG1, at 25 °C (the different colour areas are a guide for the eye), and (B) PG1S-Mb at different temperatures. All samples were prepared in D2O. | ||
The quartz crystal microbalance with dissipation technique (QCM-D) was used to provide information on the adsorbed mass and the viscoelastic properties during the interactions between the dendronized polymers and the proteins at different temperatures.31 The gold chip was modified by the proteins before testing according to the method reported previously (Fig. S7A†).32 The frequency shift (Δf) for the adsorption process of PG1 took 5.3 h to reach equilibrium at 25 °C (Fig. 4A, red curve), but changed continuously at 38 °C (Fig. 4B, red curve). The adsorbed mass Δm (calculated according to Sauerbrey model and Δf) of PG1 on Mb at a given time (5.3 h) was 30.98 and 1971.71 ng cm−2 at 25 and 38 °C, respectively. The energy dissipation (ΔD) of PG1/Mb at 5.3 h was 0.461 × 10–6 at 25 °C (Fig. 4A, blue curve), which increased dramatically to 157.67 × 10−6 at 38 °C (Fig. 4B, blue curve). In addition, the Δf of DCS/Mb (at 42 min) was found to be −16.8 at 25 °C, which decreased to −31.2 Hz at 60 °C. The above results suggest that both PG1 and DCS show different affinities to the protein at temperatures below and above their Tcps, which remarkably influenced their interactions.33 The dendronized polymers adsorbed on the protein to form a relatively stiff layer with low dissipation capacity at temperatures below the Tcp (Fig. 4C). This is comparable to properties reported for OEG-alkanethiol self-assembled monolayers (SAMs) on gold or silver surfaces.34–36 Differently, as the temperature increases above the Tcp, the adsorption layer of dendronized polymers on the protein showed viscoelastic and strongly dissipating properties, which are comparable to those of the widely studied PEG-based brush assemblies.31,37,38
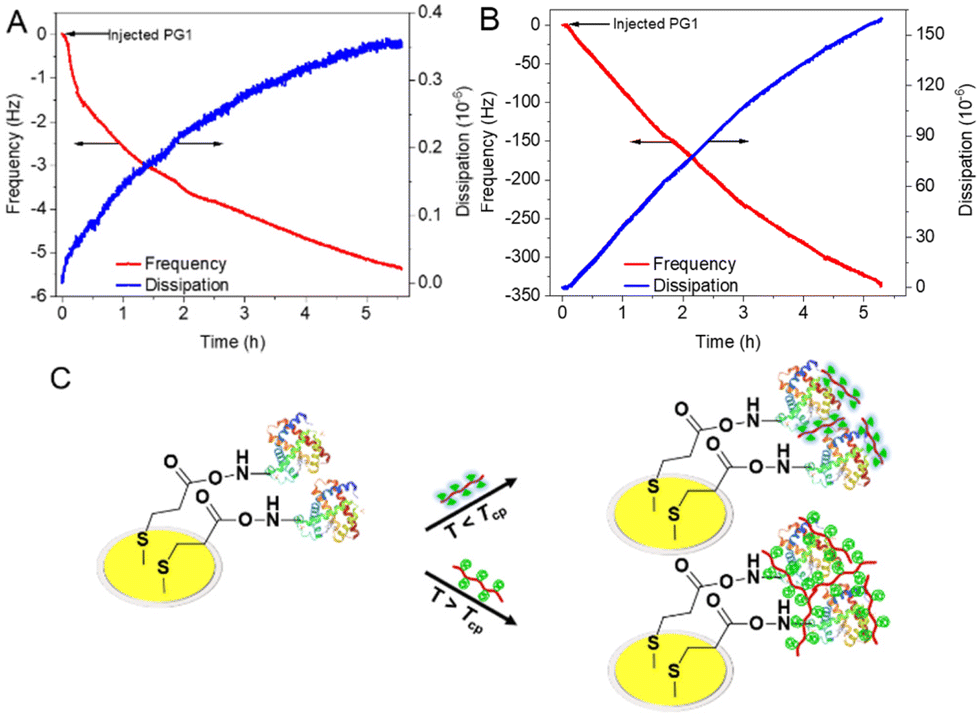 | ||
| Fig. 4 Changes of frequency Δf (red curves) and dissipation ΔD (blue curves) in a QCM cell (modified by Mb) over time after injection of PG1 at (A) 25 °C and (B) 38 °C. (C) Possible interactions between the proteins and the polymers. | ||
Influence on protein secondary structure in the presence of dendronized polymers
Circular dichroism (CD) was employed to monitor the effect of dendronized polymers on the secondary structures of the proteins. There is a weaker CD signal at 210–220 nm (mainly from α-helical structures) in the spectrum of free Mb (Fig. 5A, curve in light pink).39 For PG1/protein (including Mb, BSA, and LYZ, Fig. S8A and B†), the negative CD signal near 210 nm was enhanced with the increase of the polymer concentration. This should be mainly due to the enhanced interactions of the proteins with PG1. The hydrophobicity of the densely crowded dendritic chains from PG1 created a hydrophobic microenvironment,27,39 resulting in weakening solvation of the proteins, and accordingly enhancing their chiral conformation.40,41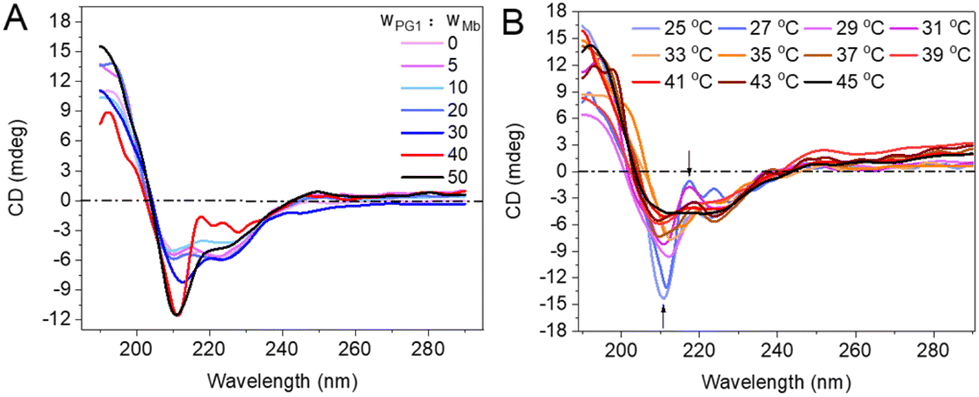 | ||
| Fig. 5 Far-UV CD spectra of Mb from PG1/Mb (A) with different concentrations of polymer, and (B) at different temperatures (polymer/protein = 40, w/w). | ||
Possible effects of thermally induced aggregation of PG1 on the helical conformation of the proteins were further monitored. As shown in Fig. 5B, the intensities of the negative (near 210 nm) and positive (near 220 nm) Cotton effects from the CD spectra of PG1/Mb decreased with increase of the solution temperature. The spectra of PG1/protein (Fig. S8C and D†) were almost overlapping with those of the free protein when the temperature well exceeds (>40 °C) its Tcp. This is further evidence that thermally induced aggregation of the polymer changed the hydrophobic interactions between PG1 and Mb.39,42
Activity regulation of the proteins by the dendronized polymers
In myoglobin, the interactions between the protein side chain and heme result in a strong absorption near 410 nm and will determine its activity.39 Hence, the absorption spectra of PG1/Mb solutions were recorded at different temperatures to explore the effect of dendronized polymers on the bioactivity. As shown in Fig. S9A,† the absorbance near 410 nm in spectra of Mb at 25 °C only showed a slight decrease (from 0.45 to 0.41) when the concentration of PG1 was 50 times higher than that of the protein. The spectra of PG1/Mb were overall upward shifted when the temperature was higher than 35 °C (>Tcp) without changing their shape (Fig. S9B†), and can be restored to their original state after temperature recovery to 25 °C. This process can be repeated multiple times (Fig. S9C†), indicating that the thermally induced aggregation process of the dendronized polymers hardly caused any destruction of the protein.39,43The catalytic activity of Mb within the mixture was further investigated at different temperatures, and the results are shown in Fig. 6A. The activity of Mb from all samples increased linearly with the temperature between 25–45 °C from 30% to 61% for Mb, 40% to 73% for PG1/Mb, 42% to 79% for DCS/Mb, and 41% to 90% for PG1S-Mb, respectively. Compared to other groups, the activity of PG1S-Mb increased more obviously between 30–37 °C. For DCS/Mb, it increased obviously from 79% to nearly 100% between 45–60 °C. These temperatures correspond to their Tcps, indicating that around the phase transitions, the proteins exhibited the highest activities. However, the activity of the free proteins, PG1/Mb and PG1S-Mb decreased obviously to lower than 34% with further increase to 60 °C. We ascribe these abnormal increases of activity to the thermally induced dehydration of the dendritic OEGs from the polymers, which collapsed and aggregated to form hydrophobic microenvironments to enhance the hydrophobic interactions between the polymer and proteins. This leads to enhancement of the enveloping and shielding capability for the polymer chains towards Mb, simultaneously preventing the solvation of Mb, which is helpful to the binding of the proteins to the substrate. Moreover, the activity of the samples decreased at temperatures much higher than their Tcps. This should be due to the collapsing between the polymer chains, which caused the exposure of the active center of the proteins, eventually resulting in the loss of their activity.28 Differently, in the case of DCS/Mb, the effective electrostatic interactions between the CS mainchains and Mb was supportive of confinement on the proteins and regulation of their bioactivity. This complexation of the protein by DCS would make it benefit from the shielding effect from dendritic OEGs even at high temperature.
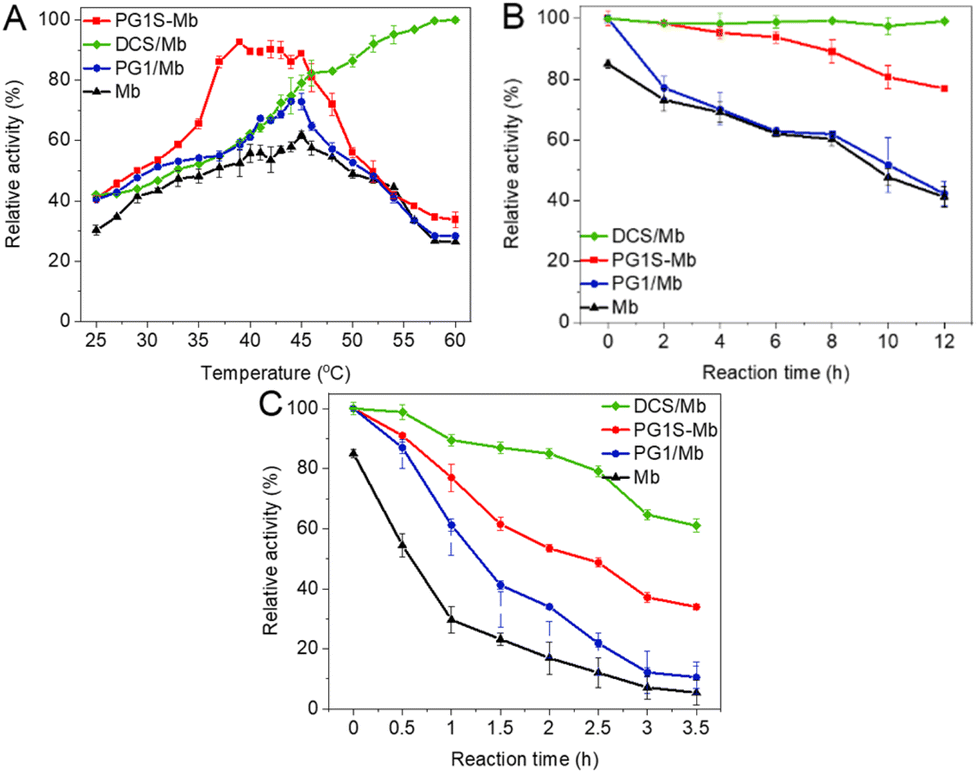 | ||
| Fig. 6 Quantitative relative activity of Mb from different samples at different temperatures (A), as well as after being kept at (B) 25 °C and (C) 60 °C for different time intervals. | ||
The bioactivity protection capability of the polymer was further explored at different temperatures for different time intervals. As shown in Fig. 6B, the activity for the free protein, PG1/Mb, PG1S-Mb, and DCS/Mb after being kept at 25 °C for 12 h was 41%, 42%, 76%, and 97%, respectively. Notably, the bioactivity of the protein in the cases of PG1S-Mb and DCS/Mb was almost two times higher than the other cases, indicating better protection capability of the protein. Furthermore, after being kept at 60 °C for 3.5 h, the bioactivity of the protein in the cases of free Mb and PG1/Mb only remained 6% and 11% (Fig. 6C), respectively, indicating significant loss of activity at elevated temperatures. In contrast, the bioactivity in the case of DCS/Mb was over 2 times that for PG1S-Mb at a high temperature, and was found to be 34% and 61% for PG1S-Mb and DCS/Mb, respectively. This indicates that the DCSs showed much better protection capability towards the protein at high temperature. This is understandable since the aggregated dendronized polymers provided a more hydrophobic and crowded microenvironment, which protected the proteins from strong solvation and was more suitable for proteins to maintain their high activities, especially at elevated temperatures.44,45 On the other hand, the effective electrostatic interactions between the CS mainchains and Mb form stable complexes, which should be favourable for Mb to compensate against solvation at high temperature.
Conclusions
The interactions between thermoresponsive polymers and proteins within the confined microenvironments created by OEG-based dendronized polymers were investigated. The proteins were combined with the polymers through physical mixing, bioconjugation or ionic complexation, aiming at examining possible effects of the forms of linkage of the proteins on the confinement. The thermally mediated aggregation of the dendronized polymers and their combination forms with biomolecules were found to play an important role in modulating the polymer–protein interactions. The confinement of proteins by the dendronized polymers is better achieved through their covalent conjugation and electrostatic complexation. This favours the enhancement of cooperative, encapsulation, and shielding effects from the dendritic OEGs. The crowding level and hydrophobicity of the microenvironment can be adjusted conveniently during the thermally induced aggregation of the polymers, which has exhibited advantages for activity regulation of the biomacromolecules. Due to the suitable number of amino pendants in the CS backbone to provide binding sites for complexation with proteins, and a proper amount of OEG dendron pendants to provide sufficient shielding effects to form a microenvironment along the polymers, DCS exhibited the best activity protection capability towards the proteins. We therefore believe that these stimuli-responsive polymers are promising for the construction of a bionic system to ensure biomacromolecule activity.Experimental
Materials and reagents
The OEG-based dendronized methacrylate monomer (M3),23 dendronized homopolymer (PG1),23 dendronized chitosans (DCSs),24 and comonomer carrying a 4-nitrophenyl active ester (MSS)29 were synthesized according to previous reports. Myoglobin (Mb), bovine serum albumin (BSA), and lysozyme (LYZ) were purchased from Sigma-Aldrich. Dithiobis (succinimidylpropionate) (DTSP) and 2,2′-azino-bis (3-ethylbenzothiazoline-6-sulfonic acid) (ABTS) were purchased from Tokyo Chemical Industry, Japan. Other reagents were purchased at reagent grade and used without further purification. Doubly deionized water was used in all experiments.
Synthesis of PG1S
M3 (200 mg, 0.284 mmol), MSS (5.5 mg, 0.014 mmol), and AIBN (1.0 mg) were dissolved in dry DMF (0.3 mL) in a Schlenk tube. The solution was deoxygenated and stirred at 65 °C for copolymerization. After 4 h, the mixture was cooled to room temperature. Then, the copolymer was purified by column chromatography (silica gel, DCM as eluent) to yield the copolymer as a colourless oil (125 mg, 61%). 1H NMR (500 MHz, DMSO-d6, Fig. S2†): δ = 1.04 (br, CH3), 1.25–1.31 (m, CH2), 3.49–3.67 (m, CH2), 3.99 (br, CH2), 4.78 (d, CH2), 6.56 (br, Ar–H), 7.56 (br, Ar–H), 8.25 (br, Ar–H).
Preparation of dendronized copolymer–protein conjugates and measurement of the loading efficiency for the proteins
The preparation of copolymer–protein conjugates was achieved according to a method reported previously.46,47 PB solution (pH = 8.0) containing Mb (0.5 mg mL−1) was mixed with PG1S at different mass ratios (at 25 °C) and stirred for 6 h. Then, the mixed solution was separated by using a filter membrane (0.1 μm). Meanwhile, the content of free protein remaining in the filtrate was determined by the Bradford method, to determine the loading efficiency of PG1S. The loading efficiency was calculated according to the following formula (1): | (1) |
Characterization
1H NMR spectra were recorded on a Bruker ADVANCE NEO 500 M (1H: 500 MHz) spectrometer at 60 °C, and chemical shifts were reported as values (ppm) relative to TMS.Gel permeation chromatography (GPC) measurements were carried out on a Waters GPC e2695 instrument with a 3 column set (Styragel HR3 + HR4 + HR5) equipped with a refractive index detector (Waters 2414), and DMF (containing 1 g L−1 LiBr) as eluent at 45 °C. The calibration was performed with poly(methyl methacrylate) standards in the range of Mp = 2580–981![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 (Polymer Standards Service-USA Inc.).
000 (Polymer Standards Service-USA Inc.).
UV/vis absorption spectra (between 350 and 450 nm) and turbidity measurements were performed on a JASCO V750 UV/vis spectrophotometer with a thermo-controlled bath. Polymer solutions were placed in the spectrophotometer (path length 1 cm) under the heating or cooling rate of 0.2 °C min−1. Tcp is determined at which the transmittance had reached 50% of its initial value at λ = 700 nm. The concentration of the polymer is 2.5 mg mL−1. The samples were all prepared in buffer solution (PBS, pH 7.0, 50 mM).
DLS measurements were performed on DynaPro Nanostar (Wyatt Technology Corporation). The solutions of the polymers and their mixtures with proteins were placed under the heating or cooling rate of 0.2 °C min−1. All Rhs were calculated according to the intensity.
Fluorescence experiments were carried out on a Horiba Jobin Yvon FluorologR-3 with FluorEssence. The spectra of all samples were obtained with the addition of ANS (30 μM) at the excitation conditions of 350 nm and emission was scanned between 410 and 650 nm with a 4 nm band-pass of the excitation and emission slits.
FT-IR spectra were recorded using a Nicolet Is50 spectrophotometer with a diamond/ZnSe universal ATR sampling accessory. The spectra of all samples in deuteroxide were obtained in transmission mode from 400–4000 cm−1 at a resolution of 4 cm−1 averaging 64 scans. All samples were prepared in D2O.
CD spectra were recorded in the far-UV region using a JASCO-815 spectrometer (Jasco, Japan). The CD spectra were obtained in the range between 190 and 290 nm. Quartz 0.5 cm path length cells were used for all CD experiments. The recording parameters were: scan speed 200 nm min−1, response time 4 s, slit width 1 mmol L−1, bandwidth 4.0 nm, and step resolution 0.2 nm. From 3 to 6 scans were run for each sample.
QCM-D measurements were carried out on a Q-Sense E4 (Q-Sense, Sweden) equipped with a multichannel pump (IPC Ismatec SA, Switzerland). The grafting of protein to the gold cells (5 MHz) was performed according to the method reported previously.31 Firstly, the bare gold surface was exposed to a DMSO solution of 2 mg mL−1 DTSP. DTSP forms SAMs spontaneously through covalent interactions between the sulfur group and the metal surface. Then, the surface was rinsed with pure DMSO several times and dried in a nitrogen stream. In the next step, the gold cells with SAMs was immersed in a PBS solution of Mb (10 mL, 0.1 mg L−1) in an ice bath for 6 h. The gold cells were further purified by deionized water several times. Subsequently, the cells were installed into their corresponding channels. The PBS (for the acquisition of the base line) and dendronized polymers were injected successively, and the changes of Δf and ΔD were recorded over time. The changes of the resonance frequency (Δf) of the crystal reflect the adsorbed mass, while the energy dissipation (ΔD) provides information on the viscoelastic properties of the adsorbed layer.31 Δm = C × Δf, where Δm = adsorbed mass [ng cm−2], C = −17.7 ng (cm2 Hz)−1.
ITC measurements were performed on an isothermal titration calorimeter (TA instruments, TAM IV), to evaluate the thermodynamics of the binding process between the samples and Mb aqueous solutions. The instrument was calibrated electrically with a precision better than (±0.1%). The PG1 or DCS in the syringes was titrated into the solutions containing myoglobin. The titrating solution was automatically added in aliquots of 4.974 μL with a 30 min interval between each injection, and the system was stirred at 60 rpm with a gold propeller. Each experiment was repeated at least 3 times, and the heat evolved per injection was integrated using the software especially designed for TAM IV. The data was analysed using the ITC software with the “independent” model and Boltzmann formula y = A2 + (A1 − A2)/(1 + exp((x − x0)/dx)) was used for curve fitting.
Mb activity
The activity data of Mb was obtained according to its reaction rate with 2,2′-azino-bis (3-ethylbenzothiazoline-6-sulfonic acid) (ABTS, 1 mg mL−1) and H2O2 (0.3%) as the substrate within 1 min. The Mb solution (0.05 mg mL−1) was mixed with different concentrations of PG1 or DCS and kept (mass ratio of polymers to protein from 10 to 50) at 25 °C and 60 °C for different time intervals. Subsequently, 1 mL of solution of PG1/Mb or DCS/Mb was mixed with ABTS (1 mL) and H2O2 (1 mL), and the absorbance changes of the solutions at 405 nm were tracked with the stirring speed at 200 rpm for 1 min immediately. Then, their activity was calculated through formula (2). | (2) |
Author contributions
Y. Y. and J. Y. conducted all experiments. W. L. and A. Z. designed and directed the project. Y. Y. wrote the draft, and W. L. and A. Z. cowrote the paper. All authors discussed the results, commented on the manuscript, and have given approval to the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support from the National Natural Science Foundation of China (No. 21971161), Shanghai Pujiang Program (No. 19PJ1403700), and Program for Professor of Special Appointment (Eastern Scholar) at Shanghai Institutions of Higher Learning (TP2019039) is acknowledged.References
- F. C. Simmel, Nat. Nanotechnol., 2013, 8, 545–546 CrossRef CAS PubMed.
- C. Tan, S. Saurabh, M. P. Bruchez, R. Schwartz and P. Leduc, Nat. Nanotechnol., 2013, 8, 602–608 CrossRef CAS.
- S. Nakano, D. Miyoshi and N. Sugimoto, Chem. Rev., 2014, 114, 2733–2758 CrossRef CAS PubMed.
- G. Xu, K. Liu, B. Xu, Y. Yao, W. Li, J. Yan and A. Zhang, Macromol. Rapid Commun., 2020, 41, 2000325 CrossRef CAS PubMed.
- W. Colón, J. Church, J. Sen, J. Thibeault, H. Trasatti and K. Xia, Biochemistry, 2017, 56, 6179–6186 CrossRef PubMed.
- Q. Fu and X. Bao, Nat. Catal., 2019, 2, 834–836 CrossRef CAS.
- J. Lutz, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 3459–3470 CrossRef CAS.
- I. M. Le-Deygen, O. E. Musatova, V. N. Orlov, N. S. Melik-Nubarov and I. D. Grozdova, Biomacromolecules, 2021, 22, 681–689 CrossRef CAS PubMed.
- M. Mohamed, A. S. A. Lila, T. Shimizu, E. Alaaeldin, A. Hussein, H. A. Sarhan, J. Szebeni and T. Ishida, Sci. Technol. Adv. Mater., 2019, 20, 710–724 CrossRef CAS PubMed.
- Y. Yao, J. Wu, S. Cao, B. Xu, J. Yan, D. Wu, W. Li and A. Zhang, Chin. J. Polym. Sci., 2020, 38, 1164–1170 CrossRef CAS.
- J. Wu, C. Zhao, W. Lin, R. Hu, Q. Wang, H. Chen, L. Li, S. Chen and J. Zheng, J. Mater. Chem. B, 2014, 2, 2983–2992 RSC.
- M. A. Carignano and I. Szleifer, Colloids Surf., B, 2000, 18, 169–182 CrossRef CAS.
- S. R. Benhabbour, L. Liu, H. Sheardown and A. Adronov, Macromolecules, 2008, 41, 2567–2576 CrossRef CAS.
- L. Bekale, D. Agudelo and H. A. Tajmir-Riahi, Colloids Surf., B, 2015, 130, 141–148 CrossRef CAS.
- P. M. Mendes, Chem. Soc. Rev., 2008, 37, 2512–2529 RSC.
- Y. Nagasaki, Polym. J., 2011, 43, 949–958 CrossRef CAS.
- M. Rabe, D. Verdes and S. Seeger, Adv. Colloid Interface Sci., 2011, 162, 87–106 CrossRef CAS PubMed.
- F. M. Veronese, Biomaterials, 2001, 22, 405–417 CrossRef CAS.
- Z. Yang, J. A. Galloway and H. Yu, Langmuir, 1999, 15, 8405–8411 CrossRef CAS.
- I. Ozer, A. Tomak, H. M. Zareie, Y. Baran and V. Bulmus, Biomacromolecules, 2017, 18, 2699–2710 CrossRef CAS PubMed.
- N. Badi, Prog. Polym. Sci., 2017, 66, 54–79 CrossRef CAS.
- M. Tully, M. Dimde, C. Weise, P. Pouyan, K. Licha, M. Schirner and R. Haag, Biomacromolecules, 2021, 22, 1406–1416 CrossRef CAS PubMed.
- G. Xu, J. Zhang, R. Jia, W. Li and A. Zhang, Macromolecules, 2022, 55, 630–642 CrossRef CAS.
- Y. Yao, K. Zhang, J. Chen, W. Li and A. Zhang, ACS Sustainable Chem. Eng., 2022, 10, 8265–8274 CrossRef CAS.
- N. V. Efremova, S. R. Sheth and D. E. Leckband, Langmuir, 2001, 17, 7628–7636 CrossRef CAS.
- A. Vergara, L. Paduano and R. Sartorio, Macromolecules, 2002, 35, 1389–1398 CrossRef CAS.
- D. Moatsou, J. Li, A. Ranji, A. Pitto-Barry, I. Ntai, M. C. Jewett and R. K. O'Reilly, Bioconjugate Chem., 2015, 26, 1890–1899 CrossRef CAS PubMed.
- W. Yang, L. Zhu, Y. Cui, H. Wang, Y. Wang, L. Yuan and H. Chen, ACS Appl. Mater. Interfaces, 2016, 8, 15967–15974 CrossRef CAS PubMed.
- Y. Yao, S. Cao, X. Zhang, J. Yan, W. Li, A. Whittaker and A. Zhang, ACS Appl. Nano Mater., 2022, 5, 4350–4359 CrossRef CAS.
- E. Froehlich, J. S. Mandeville, C. J. Jennings, R. Sedaghat-Herati and H. A. Tajmir-Riahi, J. Phys. Chem. B, 2009, 113, 6986–6993 CrossRef CAS.
- T. Gillich, E. M. Benetti, E. Rakhmatullina, R. Konradi, W. Li, A. Zhang, A. D. Schlüter and M. Textor, J. Am. Chem. Soc., 2011, 133, 10940–10950 CrossRef CAS PubMed.
- K. Ataka, F. Giess, W. Knoll, R. Naumann, S. Haber-Pohlmeier, B. Richter and J. Heberle, J. Am. Chem. Soc., 2004, 126, 16199–16206 CrossRef CAS.
- D. Schwendel, R. Dahint, S. Herrwerth, M. Schloerholz, W. Eck and M. Grunze, Langmuir, 2001, 17, 5717–5720 CrossRef CAS.
- T. Hayashi, Y. Tanaka, Y. Koide, M. Tanaka and M. Hara, Phys. Chem. Chem. Phys., 2012, 14, 10196–10206 RSC.
- S. J. Sofia, V. Premnath and E. W. Merrill, Macromolecules, 1998, 31, 5059–5070 CrossRef CAS.
- M. W. A. Skoda, F. Schreiber, R. M. J. Jacobs, J. R. P. Webster, M. Wolff, R. Dahint, D. Schwendel and M. Grunze, Langmuir, 2009, 25, 4056–4064 CrossRef CAS.
- K. Sato, S. Kobayashi, M. Kusakari, S. Watahiki, M. Oikawa, T. Hoshiba and M. Tanaka, Macromol. Biosci., 2015, 15, 1296–1303 CrossRef CAS PubMed.
- N. S. Murthy, W. Wang, S. D. Sommerfeld, D. Vaknin and J. Kohn, Langmuir, 2019, 35, 9769–9776 CrossRef CAS PubMed.
- K. Nasreen, S. Ahamad, F. Ahmad, M. I. Hassan and A. Islam, Int. J. Biol. Macromol., 2018, 106, 130–139 CrossRef CAS PubMed.
- Y. Wang and O. Annunziata, J. Phys. Chem. B, 2007, 111, 1222–1230 CrossRef CAS.
- B. Farruggia, B. Nerli and G. Picó, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2003, 798, 25–33 CrossRef CAS.
- M. Ionov, A. Ihnatsyeukachan, S. Michlewska, N. Shcharbina, D. Shcharbin, J. Majoral and M. Bryszewska, Int. J. Pharm., 2016, 499, 247–254 CrossRef CAS PubMed.
- D. Tomioka, H. Nakatsuji, S. Miyagawa, Y. Sawa and M. Matsusaki, Chem. Commun., 2021, 57, 5131–5134 RSC.
- C. Hu, Y. Yu and J. Wang, Chem. Commun., 2017, 53, 4173–4186 RSC.
- P. Batys, M. Nattich-Rak and Z. Adamczyk, Phys. Chem. Chem. Phys., 2020, 22, 26764–26775 RSC.
- K. Dutta, D. Hu, B. Zhao, A. E. Ribbe, J. Zhuang and S. Thayumanavan, J. Am. Chem. Soc., 2017, 139, 5676–5679 CrossRef CAS PubMed.
- Y. Guo, Y. Zhang, Z. Niu and Y. Yang, Colloids Surf., B, 2019, 184, 110526 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2py00957a |
| This journal is © The Royal Society of Chemistry 2022 |