
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Exploring the mechanisms of drug-delivery by decorated ZnO nanoparticles through predictive ReaxFF molecular dynamics simulations†
Cheherazade
Trouki
 a,
Giovanni
Barcaro
a and
Susanna
Monti
*b
a,
Giovanni
Barcaro
a and
Susanna
Monti
*b
aCNR-IPCF, Institute of Chemical and Physical Processes, Pisa 56124, Italy
bCNR-ICCOM, Institute of Chemistry of Organometallic Compounds, Pisa 56124, Italy. E-mail: sapeptides@gmail.com
First published on 30th August 2022
Abstract
Herein, we study the assembling of a drug delivery nanocarrier through reactive molecular dynamics simulations based on an appropriately tuned force field. First, we focus on the combination of the various components (all selected in agreement with experiments), namely nanoparticle (ZnO), functional chains (oleic acid), drug (carfilzomib), and solvent molecules (ethanol), and then on the ability of the assembled nanotool to release its cargo in a physiological environment (water). The simulation results reveal that reactivity is crucial for characterizing the stability of the functionalized ZnONP, its dynamics, and its interactions with lipid chains and drug molecules. The chains are stably chemisorbed on the ZnONP through monodentate or bidentate binding of the carboxyls to the Zn atoms (the hydrogens are released to the surface oxygens). Chains’ self-interactions reinforce the lipid cover's stability and distribution on the ZnONP interface. The added drug migrates from the solution to the nano assembly and is captured by the lipids. The molecules are entrapped among the oleic acid chains and adsorbed on the uncoated regions of the nanoparticle surface, partially physisorbed or chemisorbed. The analysis of the simulations confirms that the supramolecular assembly is compact and stable in ethanol. However, upon injection into the water, the size of the aggregate gradually increases, and the lipids start to swell with the aqueous medium. The system evolves towards an unpacked structure where the chains are elongated, separated, and prone to release the cargo depending on local water activity and depth of cargo insertion. All the results agree with the literature confirming the reliability of our predictive computational procedure for disclosing the structure and dynamics of complex materials relevant to the medicinal chemistry field.
1. Introduction
Finding effective anticancer therapies, shedding light on how they work, and improving them to avoid drug resistance, which causes an accelerated disease progression, are some of the challenges of the medicinal chemistry sector. The phenomenon of drug resistance, for example, is common in many types of cancer and is particularly evident in multiple myeloma (MM), a hematologic tumor infiltrating the bone marrow by malignant plasma cells. Despite the revolutionary therapeutic procedures available today to fight this condition, patients inevitably suffer from multidrug resistance (MDR).1,2It was found that possible solutions to overcome MDR are: combining different drugs or designing new delivery strategies for in-loco treatment.3 This second option is very appealing and has become one of the targets of modern nanotechnology. Indeed, recent advances in this field have enabled the development of intelligent drug delivery nano vehicles, with different morphologies such as nanorods, nanostars, nanoparticles, etc., made of various materials appropriately combined to undertake their tasks. Functionalized nanoparticles have shown unique pharmacokinetic profiles with longer circulatory half-life and higher tumor uptake, which make them attractive for this therapeutic application.4
The latest innovations include low-toxicity decorated nanoparticle assemblies loaded with one or more drugs5–7 capable of distributing the medication to specific tissues.
Metal oxide nanoparticles are widely used in the biomedical field thanks to their active and passive properties. Indeed, they can act directly as anticancer agents or be functionalized with selected drugs. Iron, titanium, and zinc oxides are the most common; they are approved by global regulatory agencies (FDA and EMA) and are currently under clinical investigation.8
Zinc Oxide (ZnO) nanoparticles (NPs) are very promising for anticancer applications because they can induce the generation of reactive oxygen species (ROS) and thus oxidative stress that leads to antimicrobial activity.9 Recently, an innovative hybrid nanosystem including stimuli-responsive ZnO nanoparticles coated with extracellular vesicles (EVs) equipped with antibodies has been engineered by Dumontel and coworkers10 to reach the target cells more effectively. This nano construct improved the therapeutic effects,11 relying on the combined actions of ZnONPs and the monoclonal antibody carried on the EV shell. Also, Hanley demonstrated the innate ability of ZnONPs to poison cancer cells while sparing the normal ones in 200812 on leukemia cells. Carofiglio et al. confirmed this finding,13 reporting on the enhancement of Fe![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :ZnONPs properties and their use as powerful theranostic agents. Given these premises, it was interesting to expand the research by including predictive computational modeling of the structure and dynamics of the loaded functionalized ZnONP at the atomic scale. We selected carfilzomib (CFZ) for the ZnONPs-drug association because it inhibits the proteasome 20S and has been approved for treating MM.14 CFZ is a tetrapeptide with a labile epoxyketone pharmacophore and a morpholine ring that guarantees solubility.15 The peptide backbone interacts strongly and selectively with the substrate-binding pockets of the proteasome. In contrast, the epoxyketone pharmacophore irreversibly inhibits the activity of another protein subunit through stereospecific interactions with a catalytic residue.16
:ZnONPs properties and their use as powerful theranostic agents. Given these premises, it was interesting to expand the research by including predictive computational modeling of the structure and dynamics of the loaded functionalized ZnONP at the atomic scale. We selected carfilzomib (CFZ) for the ZnONPs-drug association because it inhibits the proteasome 20S and has been approved for treating MM.14 CFZ is a tetrapeptide with a labile epoxyketone pharmacophore and a morpholine ring that guarantees solubility.15 The peptide backbone interacts strongly and selectively with the substrate-binding pockets of the proteasome. In contrast, the epoxyketone pharmacophore irreversibly inhibits the activity of another protein subunit through stereospecific interactions with a catalytic residue.16
From an experimental point of view, dynamic light scattering, transmission electron microscopy, optical spectroscopy, etc. were used to characterize the stability of the nanoparticles and the interactions at the NP surface, as shown in two papers by Carofiglio et al.13 and Barui et al.17 that disclosed the role of oleic acid capped and amine-functionalized ZnONPs. Instead, the computational activity for modeling these nanotools is focused on the interactions between nanoparticles and biomembranes. These are simulated mainly at the mesoscale using coarse-grained and dissipative particle dynamics methods.18 A more detailed characterization at the atomic level of all the interactions at the various interfaces could reveal mechanisms crucial for fine-tuning the vehicle loading and delivery times.
In this work, we have combined quantum chemistry calculations and reactive MD simulations (ReaxFF MD) to disclose: (1) possible supramolecular structures of the nanocarriers; (2) possible binding modes of CFZ to the nanocarriers; (3) possible behavior of the whole system in a stabilizing solution (ethanol – ETH) similar to the experimental environment; (4) possible dynamics of the loaded nanocarrier in a physiological environment.
All these features are crucial for providing a comprehensive picture of an efficient experimental/theoretical design. The validity and potential of our computational procedure have been demonstrated in the last point (bullet 4) by performing MD simulations of the functionalized nanoparticle loaded with CFZ in water. Indeed, we have successfully predicted (in agreement with the experimental observations) the effects of the surrounding environment on the nanocarrier system and possible routes to drug release.
The binding tendencies of CFZ to the functionalized NPs during their preparation and delivery have not been investigated yet. That is why the present work aims at disclosing through reactive simulations, based on a well-tuned force field, the main steps of the adsorption–desorption process of representative CFZ conformations complexed with an oleic acid-functionalized ZnONP, mimicking the experimental setup.
Briefly, a preliminary conformational search to identify preferential structures of CFZ was performed using a molecular mechanics force field coupled with a genetic algorithm. Then, density functional theory (DFT) calculations were employed to refine the sample. All the collected data, and other simplified models, describing fragment adsorption on representative ZnO surfaces and simple reactions helped to support and validate the description of ReaxFF in depicting the behavior of the molecule loaded on the nanoparticle in a reactive environment. Finally, extended ReaxFF MD simulations could confidently explore the dynamics of the created carrier loaded with the cargo in ethanol and then in water solution, predicting a possible delivery scenario, which can assist future experiments.
2. Methods
Typical conformations of CFZ were obtained through the conformational search procedure available in the Balloon software.19 The method is based on distance geometry, a multi-objective genetic algorithm that iteratively changes torsion angles, the stereochemistry of double bonds, tetrahedral chiral centers, ring conformations, and short energy minimizations (reimplementation of the MMFF94 force field20) that frequently remove unrealistic structures. Starting from a conformation of CFZ downloaded from the Protein Data Bank (PDB code: 4R67), we used Balloon (version 1.6.4) to run 200 generations with an initial population of 50 conformers. The rings were frozen, the peptide bonds were constrained to the trans configuration, and the maximum number of conjugate gradient optimization iterations was set to 500 with a convergence criterion of 0.01 kcal mol−1 Å−1. The EEM charge was employed. Nineteen degrees of freedom, namely ten torsional angles of the backbone and nine dihedrals of the side chains, were explored. The program produced 356 conformers that were reduced to 126 structures by excluding similar backbone and side chain arrangements. These structures were favored by intramolecular interactions between neighboring residues (especially hydrogen bonds of the backbones and stacking/T-shaped orientations of the rings). These were reoptimized at the quantum chemistry (QC) M06-2X/6-31G(d) level in the gas phase using Gaussian09 revision A0221 to prepare the sample tuned for the subsequent MD simulations. All the conformations were analyzed and classified in terms of torsional angles, head–tail and ring–ring distances, number/type of intramolecular hydrogen bonds, and secondary structure characteristics (Ramachandran plot22). All the data is included in the ESI.†Instead of using implicit solvent models to characterize CFZ conformational preferences in ethanol and water solutions, we opted for an explicit representation of the solvent and carried out classical MD simulations employing the AMBER16 package.23 Water was depicted through the TIP3P model and ethanol through a pre-parametrized/randomized solvent box.24 Solute and solvent were described through the ff14SB and general amber force field (GAFF) with RESP charges (calculated at the DFT/M06-2X/6-31G(d) level of theory). We chose as starting CFZ conformations the minimum and maximum energy structures identified through the conformational search procedure after optimization at the QC level.
After an initial equilibration, all the simulations were carried out, first, in the NPT ensemble for about 550 ps at constant temperature (300 K) and pressure (1 bar), using Berendsen's barostat25 with isotropic molecule-based scaling and a time constant of 1 ps. The temperature was maintained by coupling the system to a thermal bath with the Andersen algorithm and a time constant of 1 ps. All Lennard-Jones interactions were cut off at 12 Å, and a particle mesh Ewald correction to the long-range electrostatic contribution was applied. The integration step was set to 1 fs. Production simulations were then performed in the NVE ensemble saving the configurations every 0.1 ps for about ten nanoseconds. The analysis was focused on dihedral angles and solute–solvent interactions through atom–atom radial distribution functions (RDFs), spatial distribution functions (SDFs), various types of atom–atom distances, radii of gyration, and intramolecular hydrogen-bond distributions.
Before investigating the complex system made of ZnO nanoparticles covered with oleic acid (OLA) chains and loaded with the drug, we tested the behavior of the reactive force field (combination of the protein26 and ZnO FFs27) by comparing our data with the literature related to the adsorption of OLA on inorganic interfaces27,28 and then focusing on the interactions of CFZ with an OLA matrix mimicking the ZnONP outer shell. This is because, according to the experimental procedure, the nanoparticles were functionalized with OLA before adding the drug. Thus, CFZ could be entirely or partially adsorbed inside available pockets among the chains or on top of them. Considering that simulating the natural insertion of CFZ into the OLA network is a highly time-consuming process, we tested the opposite situation: once inside an OLA pocket, is CFZ prone to remain there?
A simplistic solution was to build a small OLA matrix model consisting of 25 straight parallel chains 5 Å far apart, perpendicularly to a planar (XY) 5 × 5 grid, mimicking the connection to a generic ZnO surface through the COOH group. Periodic boundary conditions were applied in X and Y directions, whereas in Z, a vacuum region of about 40 Å was considered. The five central chains were removed to create a possible pocket, and the minimum energy conformer of CFZ was inserted inside this cleft. MD simulations were carried out in the NVT ensemble at 300 K freezing the positions of all the COOH moieties, first in the gas phase and then filling the empty portion of the simulation box with water or ethanol molecules to test solvent effects.
The ZnONP model used in the ReaxFF-MD simulations (displayed in Fig. S1 in the ESI†) was inspired by both the experimental characterizations of Cauda and coworkers,12,13,29 who suggested spherical shapes and X-ray diffraction patterns similar to the single-phase wurtzite crystalline structure, and previous works of ours focused on partially crystalline nanoparticles (NPs)30 and zinc oxide clusters.31 The starting structure of the ZnONP was a truncated octahedron (spherical shape) made of 1419 Zn and 1419 O atoms (2838 atoms in all, randomly organized) with a maximum diameter of approximately 4 nm and different surface motifs. This was a sound prototype system that could confidently represent the whole dynamic behavior of the smaller experimental nanoparticles at reasonable computational times compatible with our HPC resources.
After geometry optimization, which determined a better relocation of the atoms, the ZnONP was functionalized with OLA by placing 150 extended chains radially with the carboxyl groups close to the ZnONP surface. The approach used to distribute the molecules was similar to that developed by Kamerlin and coworkers,32 which ensures optimal packing and orientation. In our conditions, a four Å separation was the most appropriate choice. Then, twenty CFZ molecules, in their minimum energy conformation, were randomly distributed around the functionalized nanoparticle at a close distance. ReaxFF MD simulations were run in the NVT ensemble to relax the system and equilibrate it at ambient temperature for about 100 ps. Then, the final structure was inserted in an ETH simulation box, where the size was about 90 × 90 × 90 Å3. MDs were carried out firstly in the NVT ensemble at T = 300 K for about 200 ps, then in the NPT ensemble for about 250 ps to reach the correct packing of all the system components and the appropriate density. Finally, the production dynamics was performed in the NVE ensemble for about 500 ps, and system structures were collected every 0.05 ps.
Temperature and pressure were controlled through Berendsen's thermostat and barostat with relaxation constants of 0.1 ps. The time step was set to 0.2 fs. The final portion of the trajectory was analyzed to disclose system stability, molecular clustering, and CFZ capture.
Instead, MD simulations in water solutions were used to investigate possible drug delivery actions in a biomimetic environment. The starting structure was the final configuration of the MD in the ETH solution, and the simulation protocol was similar to the one used for ETH. All the ReaxFF MD simulations were carried out with the Amsterdam Density Functional (ADF) modeling suite.33
3. Results and discussion
3.1. CFZ conformations
It is evident from the examination of structural descriptors and the energy differences of the molecules in the gas phase, reported in Fig. S2 and S3 in the ESI,† and in solution (Fig. S4 in the ESI† and Fig. 1) that CFZ has remarkable flexibility not only in the side chains but also in the backbone that can adopt, as shown in the Ramachandran plots19 of Fig. 1, parallel and antiparallel β-sheet, polyproline-II, collagen-like, ribbon configurations (corresponding to the top areas of the plots in the range: −180° < ϕ < 0°, 60° < ψ < 180°), but also α-helix (a), 310-helix, and π-helix configurations (corresponding to the lower areas of the plots in the range: −180° < ϕ < 0°, −60° < ψ < 30°). In the case of ETH, left-handed α-helix configurations (corresponding to the top area of the plot in the range: 30° < ϕ < 90°, −30° < ψ < 60°) are probable as well (Fig. 1).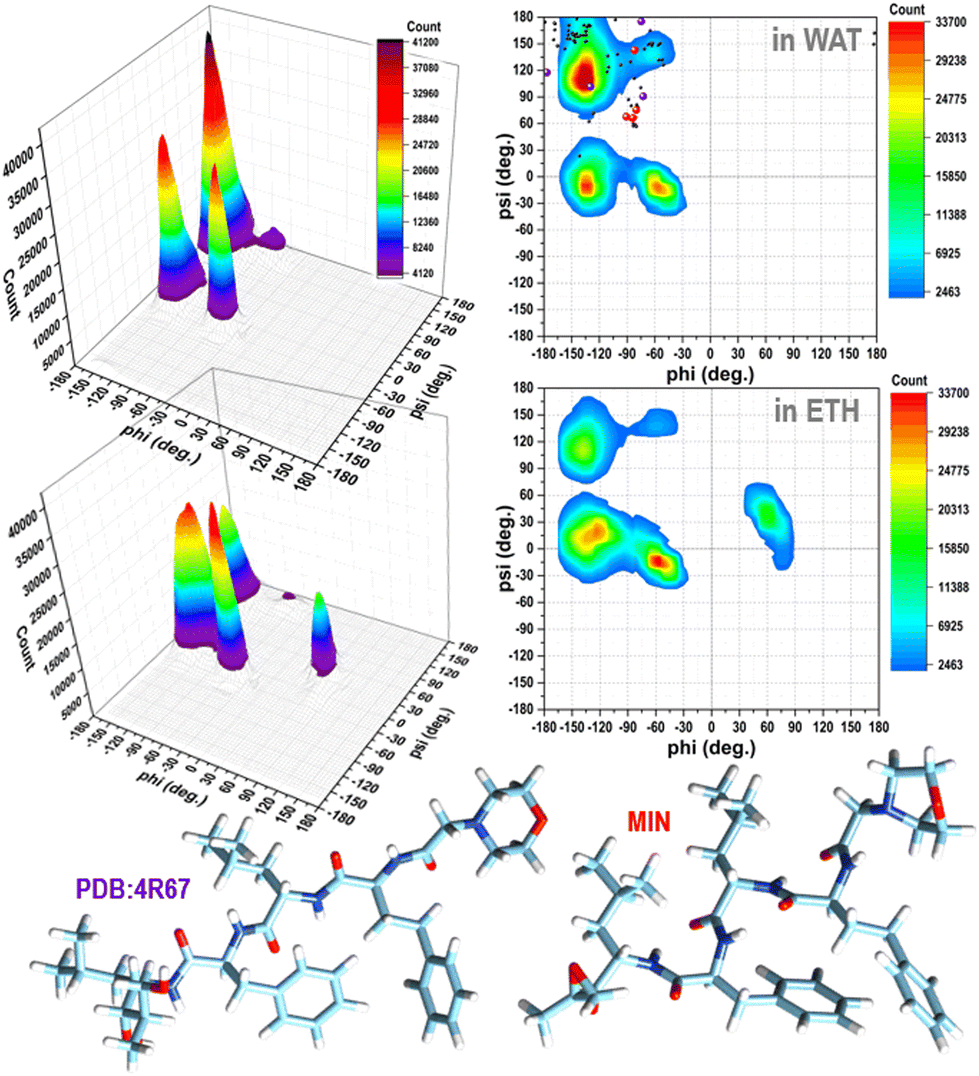 | ||
| Fig. 1 Ramachandran plots (contours with the respective 3D representations on the left) of CFZ in water and ethanol solutions (from classical MD simulations considering 200000 conformations, in each case – 800000 torsion angles pairs). In the plot in water, the torsion angle pairs of the twenty-one conformers within 10 kcal mol−1 of the minimum, the lowest energy minimum, and the structure bound to the 20S proteasome (4R67) are shown as black, red, and violet circles, respectively. | ||
The minimum energy structures within 6 kcal mol−1 of the lowest energy minimum are stabilized by intramolecular hydrogen bonds (three at most) between the N–H and C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O groups of the backbone. Favorable interactions between the sidechains due to the rings of the hPHE and PHE residues, which adopt a T-shaped orientation (improper hydrogen bond between a hydrogen atom and the π density of the other ring), improve the stabilization. These structures have an average gyration radius (Rgyr) of about 5.3 Å (range: 5–7 Å), indicating a compact folded shape. On the contrary, no intramolecular hydrogen bonds are present in the higher energy conformations. The backbone is usually extended, the side chains are located far from each other, and Rgyr is about 6.5 Å, suggesting a partial unfold. The torsional angles of the backbone (ϕ, ψ) of CFZ are mainly in the β-strand and right-handed α-helix regions (dark red areas in Fig. 1). The head–tail groups can be close (about 6 Å) or far from each other (about 17 Å), independently of the energy of the structure (Fig. S3 in the ESI†).
O groups of the backbone. Favorable interactions between the sidechains due to the rings of the hPHE and PHE residues, which adopt a T-shaped orientation (improper hydrogen bond between a hydrogen atom and the π density of the other ring), improve the stabilization. These structures have an average gyration radius (Rgyr) of about 5.3 Å (range: 5–7 Å), indicating a compact folded shape. On the contrary, no intramolecular hydrogen bonds are present in the higher energy conformations. The backbone is usually extended, the side chains are located far from each other, and Rgyr is about 6.5 Å, suggesting a partial unfold. The torsional angles of the backbone (ϕ, ψ) of CFZ are mainly in the β-strand and right-handed α-helix regions (dark red areas in Fig. 1). The head–tail groups can be close (about 6 Å) or far from each other (about 17 Å), independently of the energy of the structure (Fig. S3 in the ESI†).
This picture is confirmed by the dynamics of the molecule in solution, where the distributions of Rgyr are broader (Fig. S4 in the ESI†).
Their peaks, however, suggest a preference for folded structures, especially in water where both the rings and terminus groups are inclined to stay closer (Fig. S4 in the ESI†). Mobility and flexibility are less pronounced in ethanol. The molecule is surrounded by approximately 35 molecules (against the 45 in water) that form a cage constraining the rings and head–tail groups at longer distances (Fig. S4 and S5 in the ESI†). The tendency to opt for a more compact structure in water is also confirmed by the Ramachandran plots (Fig. 1).
3.2. CFZ inside an OLA layer
Moving to the behavior of CFZ inside a pre-formed pocket in a representative OLA matrix (Fig. S6 in the ESI† and Fig. 2), it was noticed that the solvent influenced the orientation of the OLA chains, binding affinity, location, and conformation of CFZ (Fig. S7 and S8 in the ESI†). Indeed, as Fig. S7 and S8 in the ESI† show, the initial pocket volume was reduced by about 65% in the gas phase and 47% in ethanol. Instead, in water, an increase of about 16% was obtained. This is because solvent molecules entered the cleft and surrounded CFZ.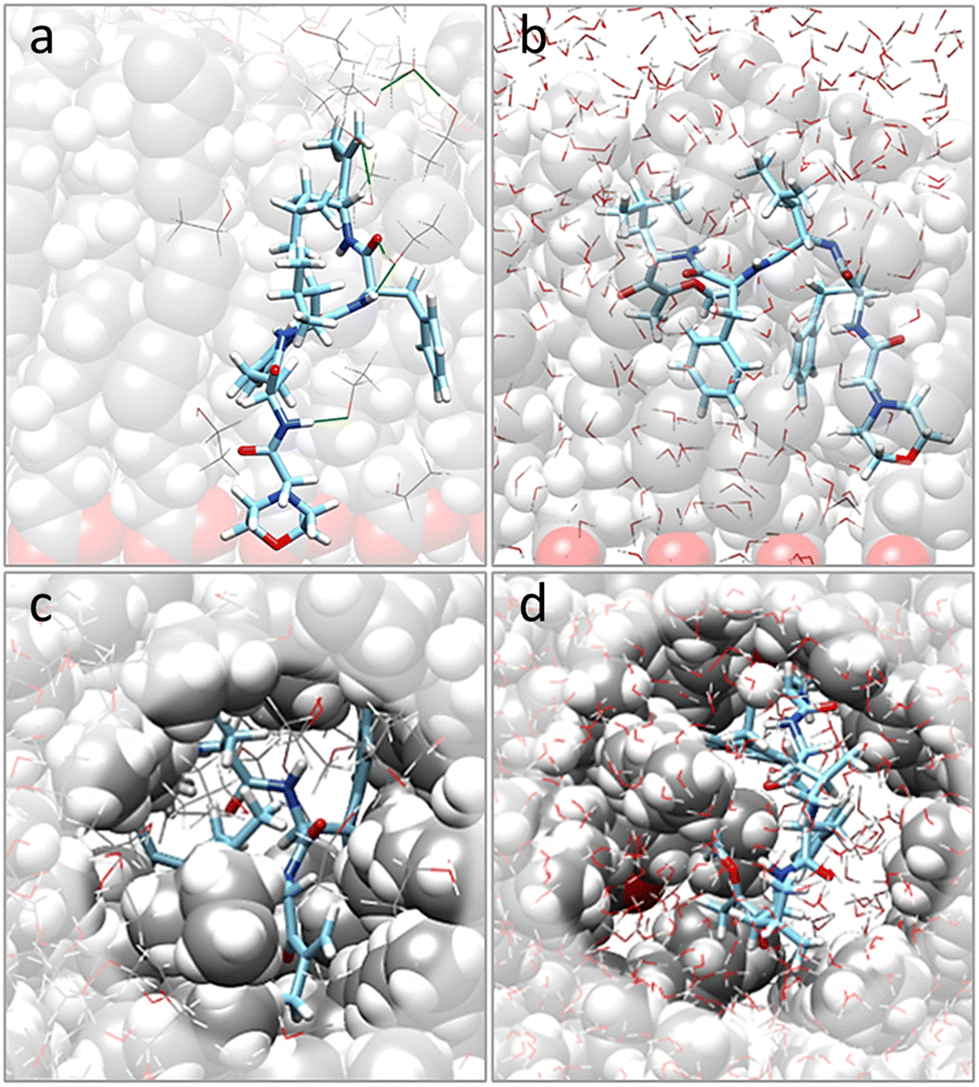 | ||
| Fig. 2 Final arrangements of CFZ inside a pocket of an OLA matrix model in ethanol [(a) side and (c) top views] and water [(b) side and (d) top views] solutions. OLA chains are depicted by gray-white-red (C, H, O) slightly transparent vdW spheres, CFZ by sticks (C = light blue, N = blue, H = white, O = red), and solvent molecules by lines. | ||
In the case of ethanol, the surrounding molecules were just a few, whereas, in water, a complete first hydration shell and a partial second layer around CFZ (Fig. 2) were present. In ethanol, the drug remained buried inside the matrix, maintaining its intramolecular hydrogen bond. In contrast, in water, CFZ was hydrogen-bonded to the solvent molecule and tended to migrate towards the opening of the pocket driven by the action of the solvent. The slight reduction of the CFZ radius of gyration could be ascribed to the accompanying packing of the rings and the shortening of the head–tail distance.
3.3. ZnONP functionalized with OLA
All these data were crucial for tuning the subsequent simulations of the complex system consisting of the functionalized ZnONP loaded with several molecules of CFZ in an ETH solution.As a first step, the equilibrated model of the ZnONP was analyzed by checking its morphology (surface atom relocation and entire shape) by visual inspection, and its crystallinity by comparing the calculated diffractograms of the initial and final geometries (Fig. S1 in the ESI†) with characteristic experimental spectra.29,34 The random combinations of the atoms of the different facets in the starting model, which were responsible for tensioned surface configurations, were replaced by reorganized stable arrangements alternating oxygen and zinc atoms (Fig. S1 in the ESI†). The whole structure maintained a spherical shape.
Comparing the two calculated diffractograms reveals no significant difference in crystallinity between the two structures. Both geometries present the characteristic reflections (slightly shifted) of single-phase wurtzite ZnO nanoparticles at 2θ = 32, 34, 36, 47, and 57. However, other signals among the canonical reflections suggest the presence of amorphous regions and confirm the model's semicrystalline traits.
After randomly positioning all around the ZnONP straight OLA chains perpendicular to the ZnONP surface (radial orientation) (Fig. 3), we equilibrated the whole complex at ambient temperature through ReaxFF MD.
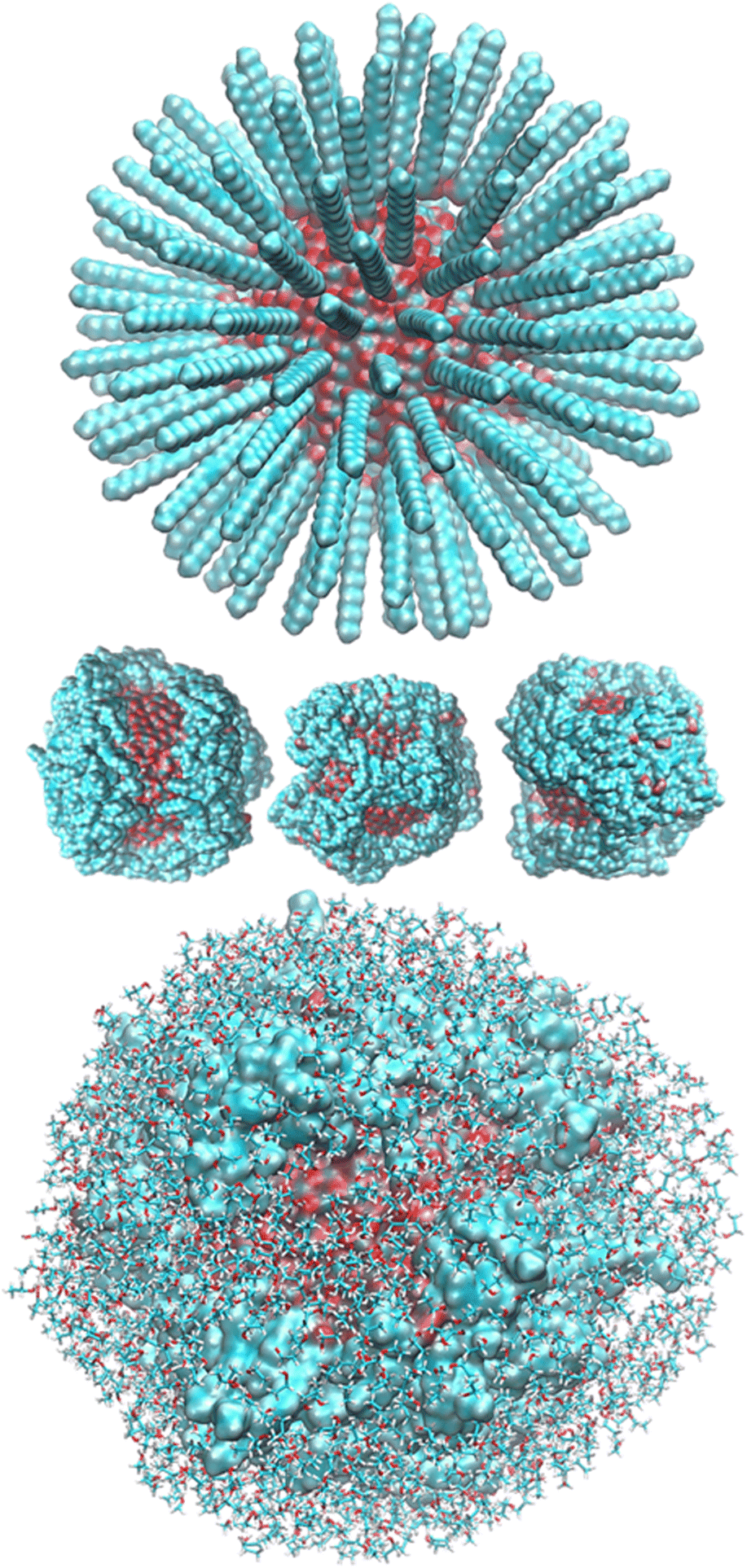 | ||
| Fig. 3 Initial and final arrangements of ZnONP functionalized with OLA. In the top and middle images, ETH molecules have been undisplayed for clarity, and in contrast, the bottom image shows them through the stick model. The ZnONP surface has been rendered through the solvent-accessible surface and OLA chains with the vdW representation. Color codes: C cyan, O red, H white, Zn bluish. | ||
We observed a significant reorganization of the molecules that, besides self-interacting, moved to more favorable positions where they could adsorb the carboxyl groups through monodentate and even bidentate coordination to the Zn atoms (Fig. 4).
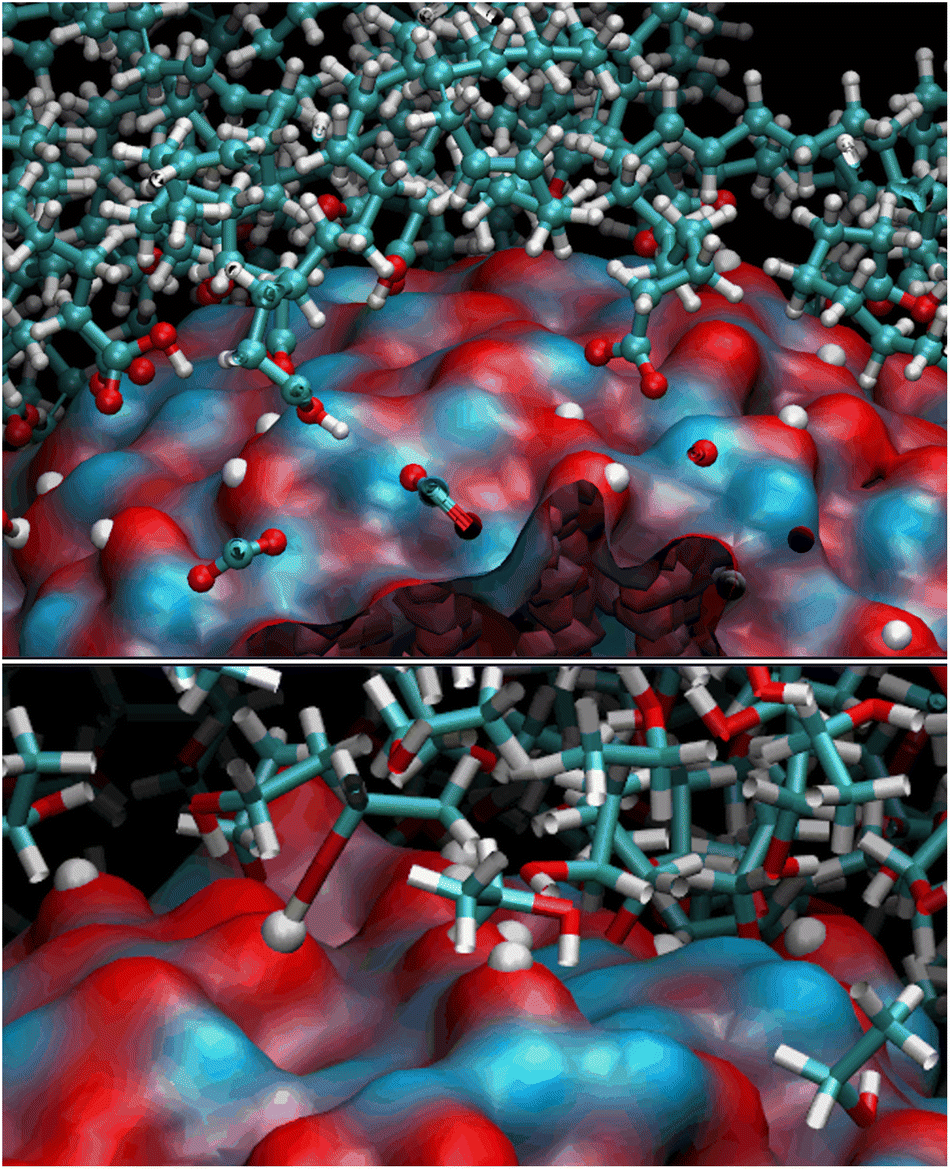 | ||
| Fig. 4 Binding modes of OLA and ETH molecules to the ZnONP surface (rendered through its solvent-accessible surface where the bluish and red patches are Zn and O atoms, respectively). Hydrogen atoms transferred to the oxygens of the ZnONP surface are white spheres. | ||
The final coverage was about 75%, and the bidentate coordination of the carboxyl groups to the Zn atoms was favored (72%). In this adsorption type, the COOH moieties transferred their hydrogens to the surrounding surface oxygens. In the monodentate coordination (28%), only one carboxyl oxygen was bonded to a Zn of the surface, and the hydrogens remained connected to the other oxygen. The aliphatic chains bent towards the surface, leaving a few surface patches uncovered (see Fig. 3 – red and bluish regions of the solvent-accessible surface). These small areas were promptly covered with ETH molecules, contributing to the surface passivation (10% of ETH within 12 Å of the ZnONP released their hydroxylic hydrogens to the nanoparticle surface).
At the end of the equilibration/production dynamics, the OLA layer was stably adsorbed on the ZnONP favored by the ethanol solution. The chains remained in their locations due to extended self-interconnections.
3.4. OLA functionalized ZnONP loaded with CFZ in ETH
OLA enveloped the whole ZnONP, and the OLA cover captured most of the surrounding CFZ molecules, randomly distributed around the functionalized ZnONP (Fig. 5).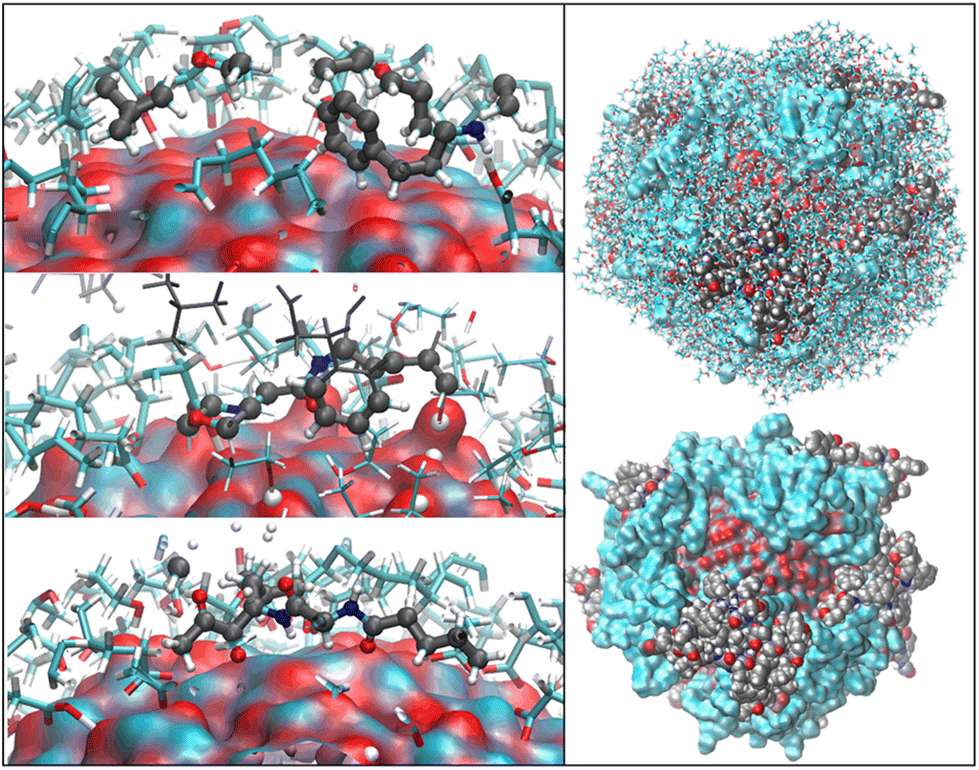 | ||
| Fig. 5 OLA-functionalized ZnONP with adsorbed CFZ molecules surrounded by ETH molecules (only the solvent molecules within four Å of the complex are displayed). Portions of CFZ directly connected to the ZnONP surface through their oxygens and hydrogens are shown on the left (CFZ carbons are dark gray). | ||
Only a few CFZ (26%) reached those vacant portions of the ZnONP surface occupied by the solvent, displaced the solvent molecules, and established binding interactions between the backbone atoms (O(CO) and H(NH)) and the hydrogens of the rings with the Zn and O sites at the interface, remaining at the same time in contact with the OLA layer (Fig. 5).
This behavior is illustrated in Fig. 6, where the spatial density distributions of the various species around the ZnONP are displayed. When CFZ molecules were adsorbed on the functionalized ZnONP, their conformations had reduced flexibility due to the interactions with OLA chains, solvent molecules, and their tendency to self-assembly (CFZ could form aggregates as it is apparent from the extension of the CFZ density regions – Fig. 6). Indeed, their structures had to readapt to the local environment, as is evidenced in the Ramachandran plot (Fig. S9 or ESI†). The backbone angles of the CFZ residues explore not only the regions visited in ethanol and water solution, namely the β-strand and the αR-helix sectors, but also other areas rarely explored in the case of proteins. Examination of the distributions of the radius of gyration (Fig. S9 in the ESI†) suggests that both extended and bent CFZ conformations are loaded on the particle, and the trend of head–tail distances confirms this view (Fig. 7).
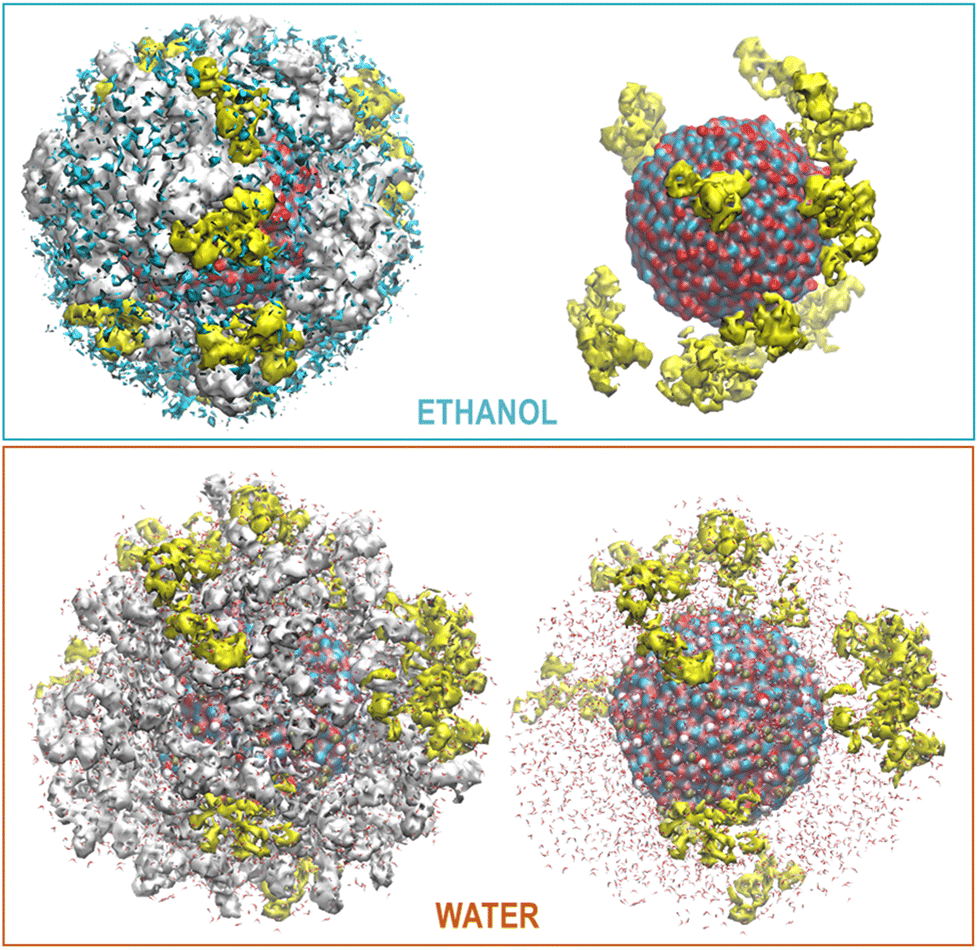 | ||
| Fig. 6 Maximum Density distribution of CFZ (yellow), OLA (white), and ETH (cyan) around the ZnONP (Zn dark cyan, O red). The water density is not displayed but only the molecules. | ||
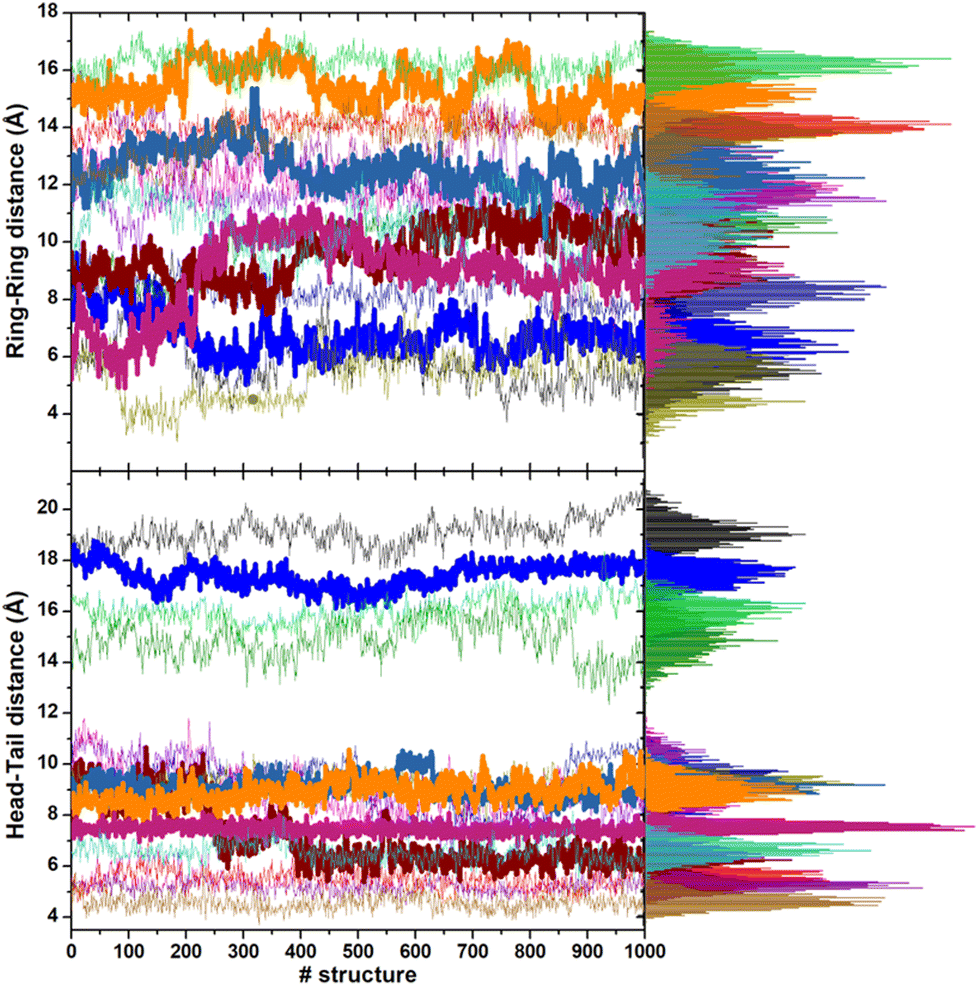 | ||
| Fig. 7 Evolution of the ring–ring and head–tail distances of the adsorbed CFZ molecules during the last 200 ps production dynamics in ETH solution. Thicker lines highlight the trends of these descriptors for those molecules directly connected to the ZnO surface. On the right, the distributions are also shown. | ||
In most structures, this descriptor had minor oscillations around average values at long and short distances, indicating that the terminus regions are actively involved in the adsorption process. On the contrary, the rings are more mobile (the distance distributions are broader) and prone to readjust their orientation frequently, as evidenced by the evolution of the ring–ring distance (Fig. 7). As indicated by the plots describing the evolution of the distance of the center of mass of each CFZ molecule from the center of mass of the ZnONP (Fig. 8), ethanol stabilizes the system.
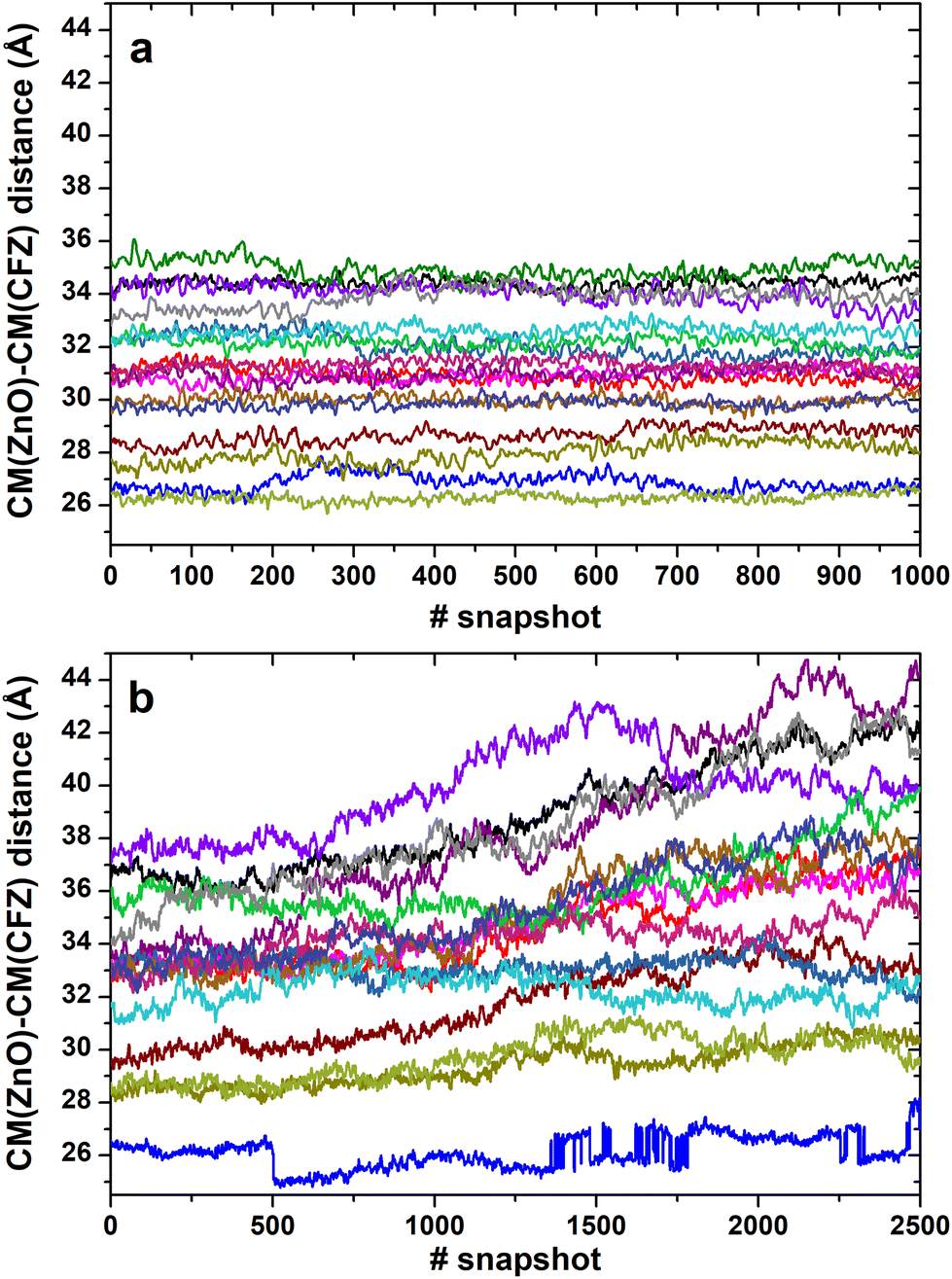 | ||
| Fig. 8 Evolution of the distance between the center of mass of the ZnONP and the center of mass of each adsorbed CFZ molecule (represented by the various color lines) in ethanol (last 200 ps) (a) and water (last 500 ps) (b) solutions. | ||
3.5. OLA functionalized ZnONP loaded with CFZ in WAT
Thus, it was interesting to test the behavior of the loaded carrier in a more biocompatible environment and possibly disclose the initial stages of the CFZ release. Therefore, we removed ethanol and inserted the model into a water simulation box. Reactive molecular dynamics simulations were run in the NPT ensemble at T = 310 K for hundreds of picoseconds. The evolution of the structures was monitored by checking the radius of gyration of the functionalized ZnONP (Fig. 9) to evaluate the OLA packing and the distance of the center of mass of each CFZ molecule from the center of mass of the ZnONP to estimate CFZ release (Fig. 8 bottom). From the parallel of these descriptors with those in ethanol solution (Fig. 8 top), it is clear that a slow degradation occurred in water. More specifically, a swollen state replaced the compact OLA arrangement in ethanol due to the higher chain mobility of OLA in the aqueous environment (Fig. 9).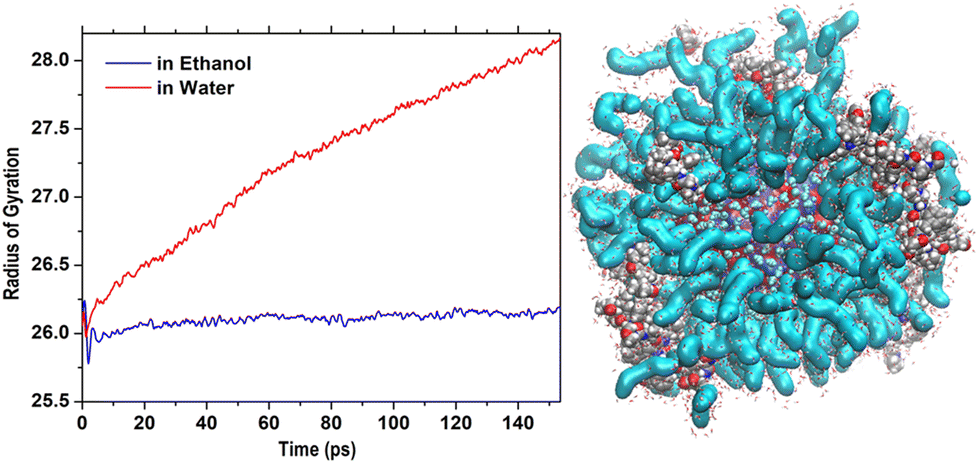 | ||
| Fig. 9 Initial steps of the evolution of Rgyr of the whole OLA-functionalized ZnONP in ethanol and water solution. On the right, structure in water after 500 ps (Rgyr = 30.3 Å). | ||
Consequently, the delivery of CFZ from the particle took place in more stages connected to the degree of penetration of the molecule inside the ZnONP cover. First, the loosely merged molecules started their way toward the solvent. These CFZ molecules were essentially adsorbed on top of the functionalizing chains close to the tails. At the same time, water molecules penetrated the OLA packing that became swollen. Then, the molecules connected to the ZnO surface, reached by water and also perturbed by the OLA motion, were displaced from their locations. They slowly took a few steps from the particle core and started their journey to the solvent.
4. Conclusions
We have designed an efficient computational strategy that can describe and predict the behavior of ZnO-based nanocarriers during the different stages of their preparation, in line with the experimental results. We have tested and demonstrated the stability of the supramolecular assemblies in ethanol solution and evaluated possible configurations of the nanocarriers after the addition of the drug. The structures are stable and compact, and the CFZ molecules can bind the functionalized nanoparticle by entrapment among the OLA chains or by direct contact with the reachable portions of the ZnONP surface. There, they can be physisorbed or chemisorbed, depending on their distance from the surface and orientation. We can speculate that due to this variety of adsorption modes and penetration depths, the release of CFZ should be gradual. Thus, we tested the behavior of the nanocarrier in water, where it is supposed to release its cargo, by removing ethanol and inserting the system in a simulation box of water. The simulation results confirm our view. We have identified various stages of CFZ release together with the initial disassembly steps of the composite system. All these findings suggest that many aspects can be considered to upgrade the nanocarriers. For example, acting on the composition of the vehicle by adsorbing other functional groups on the ZnO surface could modulate the drug's binding modes, or adding layer-by-layer pre-mixed OLA–CFZ assemblies with various CFZ concentrations could tune the particle for a more controlled release.Conflicts of interest
There are no conflicts to declare.Acknowledgements
All the authors acknowledge the MITHoS project (2020MHL8S9) funded by MUR (Ministero Italiano dell'Università e della Ricerca) within the PRIN 2020 program.References
- S. Das, N. Juliana, N. A. A. Yazit, S. Azmani and I. F. Abu, Multiple Myeloma: Challenges Encountered and Future Options for Better Treatment, Int. J. Mol. Sci., 2022, 23, 1649–1667 CrossRef CAS PubMed
.
- L. N. Davis and D. W. Sherbenou, Emerging therapeutic strategies to overcome drug resistance in multiple myeloma, Cancers, 2021, 13, 1686–1707 CrossRef CAS
- N. Chatterjee and T. G. Bivona, Polytherapy and targeted cancer drug resistance, Trends Cancer, 2019, 5, 170–182 CrossRef CAS
- C. M. J. Hu and L. Zhang, Nanoparticle-based combination therapy toward overcoming drug resistance in cancer, Biochem. Pharmacol., 2012, 83, 1104–1111 CrossRef CAS
- M. I. Khan, M. I. Hossain, M. K. Hossain, M. H. K. Rubel, K. M. Hossain, A. M. U. B. Mahfuz and M. I. Anik, Recent Progress in Nanostructured Smart Drug Delivery Systems for Cancer Therapy: A Review, ACS Appl. Bio Mater., 2022, 5, 971–1012 CrossRef CAS PubMed
- R. Liu, C. Luo, Z. Pang, J. Zhang, S. Ruan, M. Wu, L. Wang, T. Sun, N. Li and L. Han,
et al., Advances of nanoparticles as drug delivery systems for disease diagnosis and treatment, Chin. Chem. Lett., 2022 DOI:10.1016/j.cclet.2022.05.032
- V. Harish, D. Tewari, M. Gaur, A. B. Yadav, S. Swaroop, M. Bechelany and A. Barhoum, Review on nanoparticles and nanostructured materials: bioimaging, biosensing, drug delivery, tissue engineering, antimicrobial, and agro-food applications, Nanomaterials, 2022, 12, 457–500 CrossRef CAS
- V. Chandrakala, V. Aruna and G. Angajala, Review on metal nanoparticles as nanocarriers: current challenges and perspectives in drug delivery systems, Emergent Mater., 2022, 1–23 CAS
-
N. Ghosh, S. Chatterjee, M. Kundu and P. C. Sil, Oxidative Stress-Dependent Anticancer Potentiality of Nanotherapeutic Zinc Oxide, in Handbook of Oxidative Stress in Cancer: Therapeutic Aspects, Springer Singapore, Singapore, 2022, pp. 1–22 Search PubMed
- B. Dumontel, F. Susa, T. Limongi, V. Vighetto, D. Debellis, M. Canta and V. Cauda, Nanotechnological engineering of extracellular vesicles for the development of actively targeted hybrid nanodevices, Cell Biosci., 2022, 12, 1–18 CrossRef PubMed
- N. Wiesmann, W. Tremel and J. Brieger, Zinc oxide nanoparticles for therapeutic purposes in cancer medicine, J. Mater. Chem. B, 2020, 8, 4973–4989 RSC
- C. Hanley, J. Layne, A. Punnoose, K. M. Reddy, I. Coombs, A. Coombs, K. Feris and D. Wingett, Preferential killing of cancer cells and activated human T cells using ZnO nanoparticles, Nanotechnology, 2008, 19, 295103 CrossRef PubMed
- M. Carofiglio, M. Laurenti, V. Vighetto, L. Racca, S. Barui, N. Garino, R. Gerbaldo, F. Laviano and V. Cauda, Iron-Doped ZnO Nanoparticles as Multifunctional Nanoplatforms for Theranostics, Nanomaterials, 2021, 11, 2628 CrossRef CAS PubMed
- K. Groen, N. W. C. J. van De Donk, C. A. M. Stege, S. Zweegman and I. S. Nijhof, Carfilzomib for relapsed and refractory multiple myeloma, Cancer Manage. Res., 2019, 11, 2663–2675 CrossRef CAS PubMed
- K. B. Kim and C. M. Crews, From epoxomicin to carfilzomib: chemistry, biology, and medical outcomes, Nat. Prod. Rep., 2013, 30, 600–604 RSC
- S. P. E. Jayaweera, S. P. W. Kanakanamge, D. Rajalingam and G. N. Silva, Carfilzomib: A Promising Proteasome Inhibitor for the Treatment of Relapsed and Refractory Multiple Myeloma, Front. Oncol., 2021, 11, 740796 CrossRef PubMed
- S. Barui, R. Gerbaldo, N. Garino, R. Brescia, F. Laviano and V. Cauda, Facile Chemical Synthesis of Doped ZnO Nanocrystals Exploiting Oleic Acid, Nanomaterials, 2020, 10, 1150 CrossRef CAS
- M. Ramezanpour, S. S. W. Leung, K. H. Delgado-Magnero, B. Y. M. Bashe, J. Thewalt and D. P. Tieleman, Computational and experimental approaches for investigating nanoparticle-based drug delivery systems, Biochim. Biophys. Acta, Biomembr., 2016, 1858, 1688–1709 CrossRef CAS PubMed
- M. J. Vainio and M. S. Johnson, Generating Conformer Ensembles Using a Multi-objective Genetic Algorithm, J. Chem. Inf. Model., 2007, 47, 2462–2474 CrossRef CAS PubMed
- T. A. Halgren, Merck Molecular Force Field. 1. Basis, Form, Scope, Parameterization, and Performance of MMFF94, J. Comput. Chem., 1996, 17, 490–519 CrossRef CAS
-
M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, et al., Gaussian 09, Revision A.02, Gaussian Inc., Wallingford, CT, 2009 Search PubMed
- S. A. Hollingsworth and P. A. Karplus, A fresh look at the Ramachandran plot and the occurrence of standard structures in proteins, Biomol. Concepts, 2010, 1, 271–283 CAS
-
D. A. Case, R. M. Betz, D. S. Cerutti, T. E. Cheatham III, T. A. Darden, R. E. Duke, T. J. Giese, H. Gohlke, A. W. Goetz, N. Homeyer, et al., AMBER 2016, University of California, San Francisco, CA, 2016 Search PubMed
- P. Liu, J. Liu and M. Wang, Adsorption of ethanol molecules on the Al(1 1 1) surface: a molecular dynamics study, R. Soc. Open Sci., 2019, 6, 181189 CrossRef CAS PubMed
- H. J. C. Berendsen, J. P. M. Postma, W. F. van Gunsteren, A. DiNola and J. R. Haak, Molecular Dynamics with Coupling to an External Bath, J. Chem. Phys., 1984, 81, 3684–3690 CrossRef CAS
- S. Monti, A. Corozzi, P. Fristrup, K. L. Joshi, Y. K. Shin, P. Oelschlaeger, A. C. T. van Duin and V. Barone, Exploring the conformational and reactive dynamics of biomolecules in solution using an extended version of the glycine reactive force field, Phys. Chem. Chem. Phys., 2013, 15, 15062–15077 RSC
- M. Y. Sengul, C. A. Randall and A. C. T. van Duin, ReaxFF Molecular Dynamics Study on the Influence of Temperature on Adsorption, Desorption, and Decomposition at the Acetic Acid/Water/ZnO(10
![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) 0) Interface Enabling Cold Sintering, ACS Appl. Mater. Interfaces, 2018, 10, 37717–37724 CrossRef CAS PubMed
0) Interface Enabling Cold Sintering, ACS Appl. Mater. Interfaces, 2018, 10, 37717–37724 CrossRef CAS PubMed - B. Chen, J. Z. Liu, S. Shan and W. J. Yang, Adsorption mechanism of oleic acid on the surface of aluminum nanoparticle: ReaxFF molecular dynamics simulation and experimental study, Colloids Surf., A, 2021, 618, 126500 CrossRef CAS
- A. Ancona, B. Dumontel, N. Garino, B. Demarco, D. Chatzitheodoridou, W. Fazzini, H. Engelke and V. Cauda, Lipid-Coated Zinc Oxide Nanoparticles as Innovative ROS-Generators for Photodynamic Therapy in Cancer Cells, Nanomaterials, 2018, 8, 143–158 CrossRef PubMed
- S. Monti, G. Barcaro, L. Sementa, V. Carravetta and H. Ågren, Dynamics and self-assembly of bio-functionalized gold nanoparticles in solution: Reactive molecular dynamics simulations, Nano Res., 2018, 11, 1757–1767 CrossRef CAS
- G. Barcaro, S. Monti, L. Sementa and V. Carravetta, Modeling Nucleation and Growth of ZnO Nanoparticles in a Low Temperature Plasma by Reactive Dynamic, J. Chem. Theory Comput., 2019, 15, 2010–2021 CrossRef CAS PubMed
- D. M. Krüger and S. C. L. Kamerlin, Micelle Maker: An Online Tool for Generating Equilibrated Micelles as Direct Input for Molecular Dynamics Simulations, ACS Omega, 2017, 8, 4524–4530 CrossRef
-
ADF Modeling Suite, SCM Theoretical
Chemistry, Vrije Universiteit, Amsterdam, The Netherlands, 2017 Search PubMed
- P. Basnet, D. Samanta, T. I. Chanu, J. Mukherjee and S. Chatterjee, Assessment of synthesis approaches for tuning the photocatalytic property of ZnO nanoparticles, SN Appl. Sci., 2019, 1, 633 CrossRef
Footnote |
| † Electronic supplementary information (ESI) available: Further details of the ZnONP structure and CFZ optimized conformations in the gas phase. Further results from all the MD simulations, such as Ramachandran plots and distance distributions, etc. See DOI: https://doi.org/10.1039/d2nr03941a |
| This journal is © The Royal Society of Chemistry 2022 |