
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Efficient O-demethylation of lignin monoaromatics using the peroxygenase activity of cytochrome P450 enzymes†
Alix C.
Harlington
 a,
Keith E.
Shearwin
a,
Stephen G.
Bell
*b and
Fiona
Whelan
*a
a,
Keith E.
Shearwin
a,
Stephen G.
Bell
*b and
Fiona
Whelan
*a
aDepartment of Molecular and Biomedical Science, University of Adelaide, Adelaide, SA 5005, Australia. E-mail: fiona.whelan@adelaide.edu.au
bDepartment of Chemistry, University of Adelaide, Adelaide, SA 5005, Australia. E-mail: stephen.bell@adelaide.edu.au
First published on 8th November 2022
Abstract
A crucial reaction in harnessing renewable carbon from lignin is O-demethylation. We demonstrate the selective O-demethylation of syringol and guaiacol using different cytochrome P450 enzymes. These can efficiently use hydrogen peroxide which, when compared to nicotinamide cofactor-dependent monooxygenases and synthetic methods, allows for cheap and clean O-demethylation of lignin-derived aromatics.
The biological valorisation of lignin has the potential to supply useful chemicals and materials traditionally derived from non-renewable feedstocks.1,2 Lignin is comprised of varying proportions of p-coumaryl (H-type), coniferyl (G-type), and sinapyl (S-type) alcohol subunits,3,4 and its depolymerisation generates a heterogenous mixture of aromatic compounds (lignin-derived aromatic compounds; LDACs).5 The majority of LDACs are derived from methoxylated G and S-type subunits which must be O-demethylated to diols before they can be funnelled to high-value ring-opened compounds.6,7 However, O-demethylation is recognised as a rate-limiting reaction limiting the conversion of LDACs to useful compounds in high atom yield.8,9 Thus, efforts have focused on characterisation of demethylase enzymes from bacteria to improve the biological valorisation of lignin.
The cytochrome P450 superfamily of heme-containing enzymes are of interest as O-demethylases for lignin valorisation. These enzymes catalyse C–H bond oxidation reactions on a broad range of substrates.10,11 Recently, a new family of cytochrome P450s – the CYP255A family – have been characterised that catalyse the aromatic O-demethylation of G-type LDACs. The CYP255A2 enzyme (GcoA) catalyses O-demethylation of several LDACs, with highest specificity for guaiacol.12,13 Similarly, CYP255A1 (AgcA) catalyses O-demethylation of guaiacol and 4-alkylguaiacols.13 Moreover, engineered P450 enzymes, non-heme iron oxygenases and tetrahydrofolate dependent enzymes have been reported that can demethylate guaiacol and syringol derivatives.14–16 However, very few S-type LDAC O-demethylases have been characterised, making it difficult to establish efficient microbial valorisation of S-type lignin.17 Accordingly, efforts have been made to expand the substrate range of P450s for S-lignin LDACs.18 The lack of a P450 O-demethylase with native specificity for S-type LDACs limits both our understanding of the enzymatic O-demethylation of S-type LDACs and our capacity to engineer O-demethylases for efficient lignin valorisation.
Here we report the characterisation of a new cytochrome P450 from Amycolatopsis thermoflava N1165 that catalyses the O-demethylation of syringol to 3-methoxycatechol (3-MC). Furthermore, we report for the first time the use of H2O2-driven reactions to efficiently support the catalytic activity of the CYP255 family.
Several species of bacteria have the capacity to degrade and assimilate S-type LDACs.1,8,19,20 However, the catabolic pathways for the degradation of S-type LDACs remain largely unelucidated. We hypothesised that certain bacterial species may encode various cytochrome P450 O-demethylases with specificity for different LDACs to support degradation of different lignin monoaromatics. We noticed that A. thermoflava N1165 contained two genes that encode CYP255 enzymes. One of these is essentially identical to the GcoA from Amycolatopsis sp. strain ATCC 39116, except for a single point mutation (K206R), and therefore could be assigned as a guaiacol O-demethylase GcoA. The second gene (WP_037322545) encodes a CYP255 enzyme which shares 47% sequence identity with GcoA and we hypothesised that this enzyme could be a potential S-type LDAC O-demethylase (herein referred to as SyoA). Phylogenetic analysis revealed that SyoA shares a common ancestor with the CYP255A and CYP1232 families of O-demethylases (Fig. 1).12,21
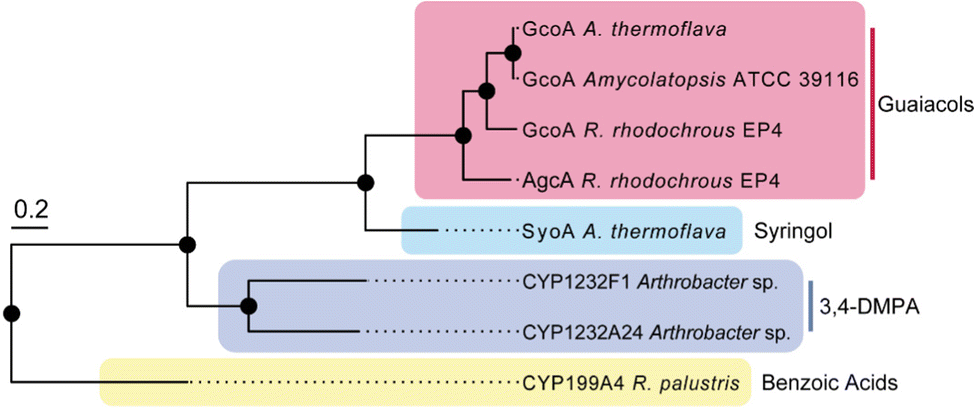 | ||
| Fig. 1 Phylogenetic relationships of the CYP255A, CYP199A and CYP1232 family of cytochromes P450s. The tree includes characterised CYP255A enzymes GcoA and AgcA (red box) that catalyse the O-demethylation of guaiacols; CYP199A4 that catalyses the demethylation of 4-methoxybenzoic acids; the CYP1232 family of O-demethylases involved in the demethylation of 3,4-dimethoxyphenylacetic acid (3,4-DMPA); and the uncharacterised cytochrome P450 SyoA from A. thermoflava. The tree scale represents the number of substitutions per site. | ||
The SyoA and GcoA enzymes were produced in E. coli and purified using affinity and anion exchange chromatography (see ESI,† for details). UV-vis spectroscopic characterisation of SyoA and GcoA was conducted to compare their substrate specificity. The UV-vis spectra of the ferric, ferrous and ferrous-CO forms of SyoA and GcoA were typical for cytochrome P450 enzymes (Fig. S1 and S2, ESI†). The ferrous CO spectra were used to estimate the extinction coefficients (GcoA ε417nm = 119 mM−1 cm−1; SyoA ε417nm = 123 mM−1 cm−1).
The binding of guaiacol and syringol – the base units of G and S-type LDACs – to the enzymes was analysed spectroscopically. As expected, addition of guaiacol to GcoA induces a large type I shift in the Soret peak to ∼390 nm of ∼90%. Addition of syringol to GcoA induces a much smaller ∼15% shift (Fig. 2a). In contrast, addition of guaiacol and syringol to ferric SyoA induce a ∼20% and ∼70% type I shift, respectively (Fig. 2b). The substrate binding affinities of guaiacol and syringol to both enzymes were determined by UV-vis analysis with titration of substrates. GcoA binds guaiacol with a higher affinity (KD = 0.4 ± 0.1 μM) than syringol (KD = 3.3 ± 0.3 μM). Furthermore, SyoA binds syringol with a higher affinity (KD = 16 ± 1 μM) than guaiacol (KD = 37 ± 2 μM) (Fig. 2c, d and Fig. S3, ESI†).
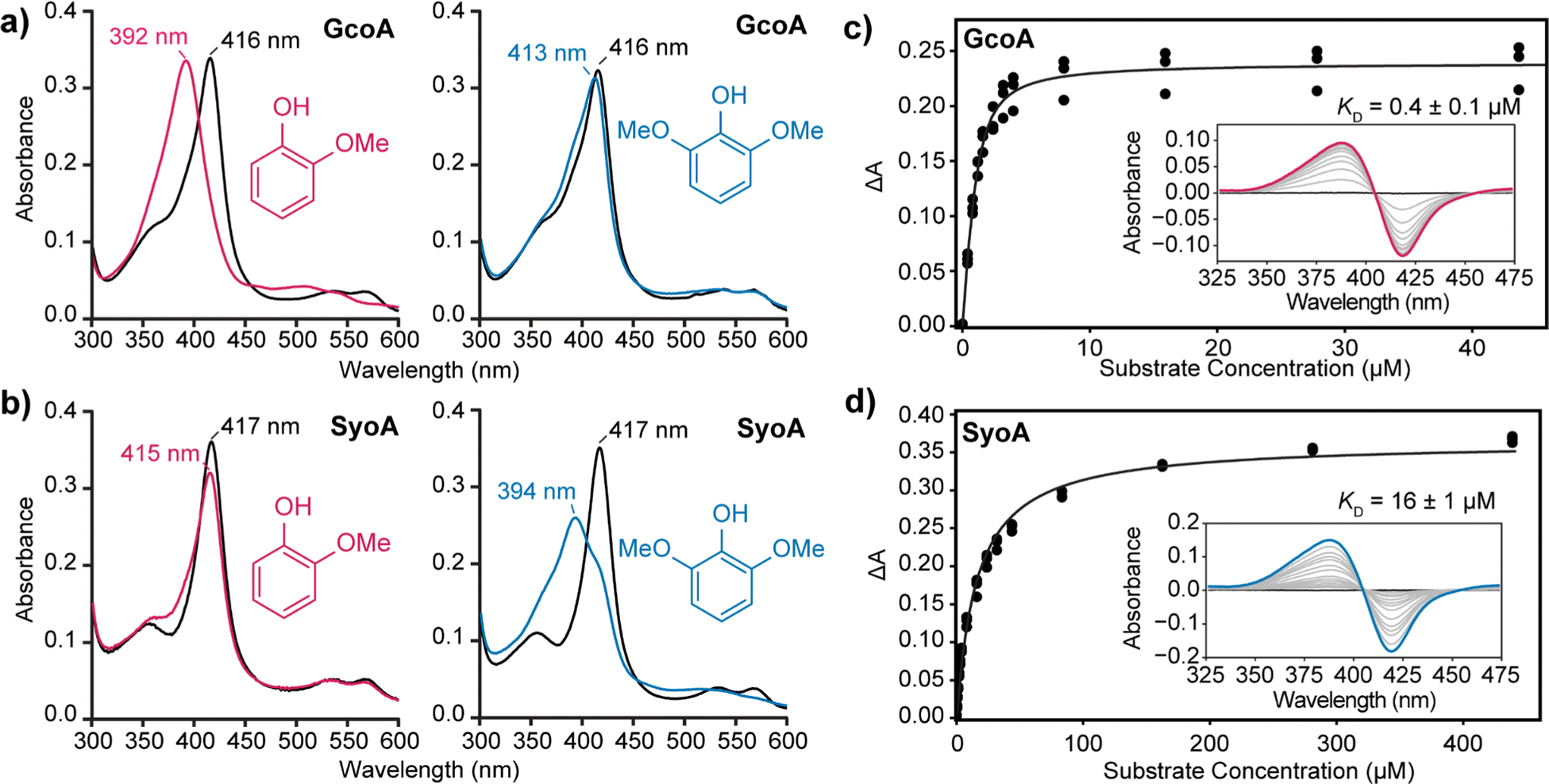 | ||
| Fig. 2 Analysis of substrate binding of guaiacol and syringol to GcoA and SyoA. In vitro spin state shifts of (a) GcoA and (b) SyoA showing preferential binding of GcoA to guaiacol and SyoA to syringol. A micromolar excess of guaiacol or syringol was titrated into a solution containing GcoA or SyoA and the spectra monitored prior to addition of ligand (black), post addition of guaiacol (pink) or syringol (blue). The binding affinity of (c) guaiacol to GcoA and (d) syringol to SyoA was determined by plotting the change in absorbance (ΔA) from the substrate-free to substrate-bound spectra. | ||
These results show that GcoA has greater specificity for guaiacol, while SyoA demonstrates preferential binding of syringol, supporting the idea that A. thermoflava may encode both cytochrome P450s to broaden its catalytic spectrum for lignin. A structure-based sequence alignment using GcoA (PDB accession code 5ncb) revealed that the active site residue F169 is replaced with an isoleucine in SyoA (Fig. S4, ESI†). The inclusion of this smaller hydrophobic residue likely allows for accommodation of the extra methoxy group of syringol into the active site of SyoA.18
Typically, P450 enzymes require electron transfer partners and NAD(P)H to supply electrons to active dioxygen (O2) to oxidise their substrate.22 Both the GcoA and SyoA encoding genes in A. thermoflava N1165 are associated with an electron transfer protein encoding gene. However, neither could be produced efficiently in a fully reconstituted form using E. coli as a host. Usually, P450s have an acid–alcohol pair within the I-helix that are responsible for the delivery of protons required to complete the catalytic cycle (Fig. 3a).23 We observed that neither GcoA nor SyoA have the conserved acid–alcohol pair, which is replaced in these enzymes with a glutamine–glutamate pair of residues (Fig. 3b). Interestingly, other heme-containing peroxygenases that use H2O2 contain a residue capable of acid-base chemistry (histidine, aspartate, or glutamate) on the distal side of the heme.24–26 Recent reports have demonstrated that mutation of the conserved threonine of the acid–alcohol pair to glutamic acid, confers peroxygenase activity in several members of the P450 superfamily.27,28 Given the presence of this glutamic acid residue in the active sites of GcoA and SyoA, we reasoned that these enzymes may be capable of peroxygenase activity using H2O2 to O-demethylate their substrates.
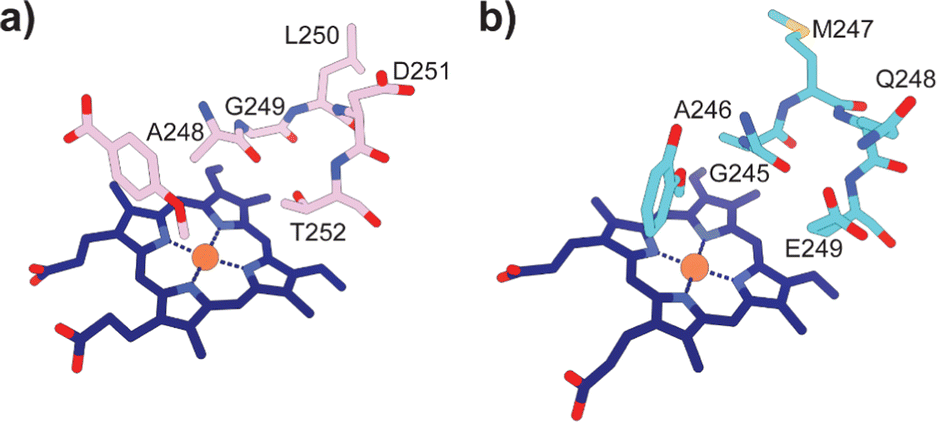 | ||
| Fig. 3 Active site structures of (a) CYP199A4 bound to 4-methoxybenzoic acid (PDB: 4do1) and (b) GcoA bound to guaiacol (PDB: 5ncb) highlighting the oxygen binding groove of the I-helix. The conserved acid–alcohol pair (D251 and T252) responsible for the monooxygenase activity of most P450s is replaced with an amide and acid pair (Q248 and E249) in GcoA. | ||
Addition of H2O2 to P450 enzymes can result in destruction of the heme cofactor, leaving the enzyme inactive.29 Heme bleaching assays were used to assess the stability of GcoA and SyoA to the presence of H2O2, and by extension, their potential as peroxygenases. The destruction of heme was determined by monitoring change in absorbance of the Soret heme peak by UV-vis spectroscopy. Addition of 40 mM H2O2 to ∼1 μM substrate-free GcoA and SyoA resulted in destruction of the heme over ∼15–20 min (Fig. S5, ESI†). Addition of the preferred substrates improved stability of both enzymes, with the Soret heme peak present after 30 min (Fig. S6, ESI†). These experiments demonstrated that compared to many heme enzymes, GcoA and SyoA have some resistance to H2O2.29,30 Interestingly, after addition of 40 mM H2O2 to guaiacol-bound GcoA and syringol-bound SyoA, the solutions changed colour to red and the UV-vis absorbance spectrum changed over the course of 30 min, indicating a potential catalytic reaction (Fig. S6, ESI†).
High-performance liquid chromatography (HPLC) analysis of 10–20 mM H2O2-driven enzyme (1 μM) reactions was performed to identify the predicted O-demethylated products of guaiacol and syringol. HPLC analysis of the reactions with GcoA and guaiacol showed the formation of a product with a retention time of 7.2 min. This product exhibited a retention time identical to a commercial sample of catechol and it was determined that ∼59% (295 ± 12 μM, total turnover number, TTN 295) of guaiacol was O-demethylated to catechol by 120 min (Fig. 4a–c). The reaction with GcoA and syringol yielded no products that matched the retention time of 3-MC, indicating syringol could not be efficiently O-demethylated by GcoA (Fig. S7, ESI†).
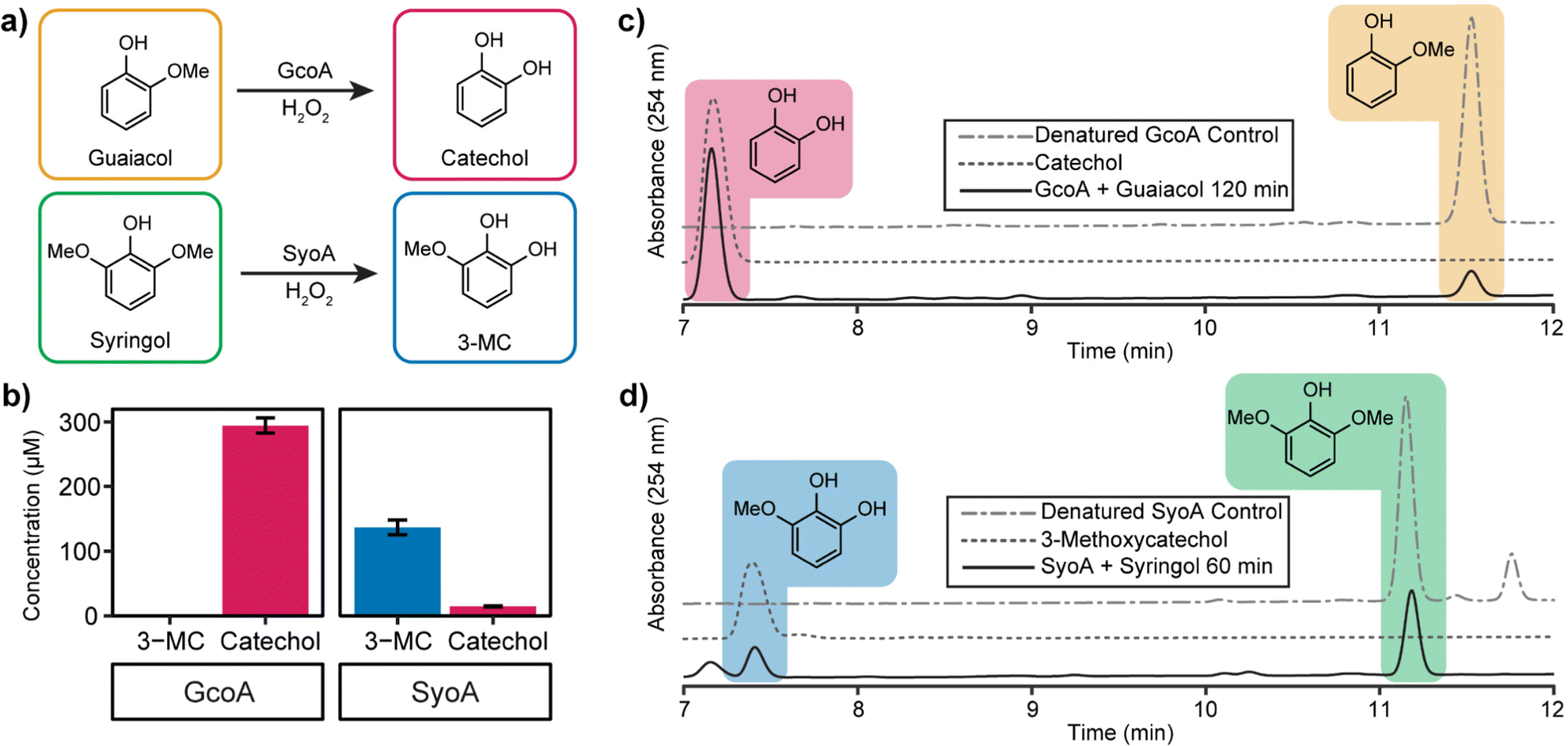 | ||
| Fig. 4 Optimised in vitro hydrogen peroxide turnovers demonstrate O-demethylase activity of GcoA and SyoA. (a) Scheme showing the O-demethylation of guaiacol and syringol by GcoA or SyoA in the presence of hydrogen peroxide. (b) Concentration of demethylated guaiacol and syringol aromatic products produced over 2 hours (guaiacol) and 1 hour (syringol). HPLC analysis of the hydrogen peroxide driven demethylation of (c) guaiacol by GcoA and (d) syringol by SyoA. Error bars indicate the SD from the mean of replicates n = 3. | ||
The H2O2-driven SyoA reaction with guaiacol yielded a small amount of catechol (15 ± 1 μM, TTN 15) after 120 min (Fig. 4b). In contrast, HPLC analysis of the reaction with SyoA and syringol revealed formation of a peak corresponding to the retention time of an authentic sample of 3-MC. Approximately 27% (137 ± 12 μM, TTN 137) of syringol was converted to 3-MC by SyoA after 60 min (Fig. 4a, b and d). Furthermore, we observed no evidence that 3-MC was further demethylated to pyrogallol by SyoA (Fig. S8, ESI†). The kcat and KM of syringol oxidation by SyoA were 6.3 ± 0.2 min−1 and 253 ± 27 μM (Fig. S9, ESI†).
Control experiments with heat-denatured enzyme demonstrated that O-demethylation of guaiacol and syringol did not occur through reaction with H2O2 (Fig. 4c, d and Fig. S7a, b, ESI†). However, oxidation of syringol in the absence of enzyme to alternative metabolites could occur in the presence of H2O2 (Fig. S10, ESI†). Oxidation of the catechol and 3-MC metabolites appeared to occur with the formation of other oxidised species. Control reactions with catechol and 3-MC demonstrated that these breakdown products were generated from further oxidation of these reactive aromatics (Fig. S11, ESI†). Given these reactions also changed colour to red, we predict that the catechol products formed during the P450 mediated O-demethylation of guaiacol and syringol further oxidised to quinones and other hydroxylated products.31–33
In summary, we have characterised a new cytochrome P450 O-demethylase which displays specificity towards the LDAC syringol. To our knowledge, this is the first example of a cytochrome P450 that has native O-demethylase activity towards S-lignin LDACs. Additionally, we show that GcoA and SyoA are efficient peroxygenases and established a system to achieve H2O2-dependent O-demethylation of guaiacol and syringol. This work not only expands the toolkit for the enzymatic bioconversion of lignin, but also allows for a simple, cheap, and clean method for the O-demethylation of aromatics compared to traditional NAD(P)H-dependent systems. Biochemical and structural analysis to investigate the substrate range of SyoA for other S-lignin LDACs would be of interest.
This work was funded, in part, through Australian Research Council grant DP200102411 (to SGB and others). ACH was supported by a Research Training Program (RTP) scholarship from the Australian government and a Wine Australia top-up grant (Ph2002). FW was supported by the Ramsay Fellowship in Applied Science. We thank Prof. Kerry Wilkinson (University of Adelaide) for providing the guaiacol and syringol substrates.
Conflicts of interest
The authors declare no conflict of interest.References
- F. Weiland, M. Kohlstedt and C. Wittmann, Metab. Eng., 2022, 71, 13–41 CrossRef CAS PubMed.
- J. Becker and C. Wittmann, Biotechnol. Adv., 2019, 37, 107360 CrossRef CAS PubMed.
- R. Rinaldi, R. Jastrzebski, M. T. Clough, J. Ralph, M. Kennema, P. C. A. Bruijnincx and B. M. Weckhuysen, Angew. Chem., Int. Ed., 2016, 55, 8164–8215 CrossRef CAS PubMed.
- J. C. del Río, J. Rencoret, A. Gutiérrez, T. Elder, H. Kim and J. Ralph, ACS Sustainable Chem. Eng., 2020, 8, 4997–5012 CrossRef.
- C. Chio, M. Sain and W. Qin, Renewable Sustainable Energy Rev., 2019, 107, 232–249 CrossRef CAS.
- E. Erickson, A. Bleem, E. Kuatsjah, A. Z. Werner, J. L. DuBois, J. E. McGeehan, L. D. Eltis and G. T. Beckham, Nat. Catal., 2022, 5, 86–98 CrossRef CAS.
- C. C. Azubuike, M. N. Allemann and J. K. Michener, Curr. Opin. Microbiol., 2022, 65, 64–72 CrossRef CAS PubMed.
- S. Notonier, A. Z. Werner, E. Kuatsjah, L. Dumalo, P. E. Abraham, E. A. Hatmaker, C. B. Hoyt, A. Amore, K. J. Ramirez, S. P. Woodworth, D. M. Klingeman, R. J. Giannone, A. M. Guss, R. L. Hettich, L. D. Eltis, C. W. Johnson and G. T. Beckham, Metab. Eng., 2021, 65, 111–122 CrossRef CAS PubMed.
- D. Salvachúa, C. W. Johnson, C. A. Singer, H. Rohrer, D. J. Peterson, B. A. Black, A. Knapp and G. T. Beckham, Green Chem., 2018, 20, 5007–5019 RSC.
- M. E. Wolf, D. J. Hinchen, J. L. DuBois, J. E. McGeehan and L. D. Eltis, Curr. Opin. Biotechnol, 2022, 73, 43–50 CrossRef CAS PubMed.
- A. Greule, J. E. Stok, J. J. De Voss and M. J. Cryle, Nat. Prod. Rep., 2018, 35, 757–791 RSC.
- S. J. B. Mallinson, M. M. Machovina, R. L. Silveira, M. Garcia-Borràs, N. Gallup, C. W. Johnson, M. D. Allen, M. S. Skaf, M. F. Crowley, E. L. Neidle, K. N. Houk, G. T. Beckham, J. L. DuBois and J. E. McGeehan, Nat. Commun., 2018, 9, 2487 CrossRef PubMed.
- M. M. Fetherolf, D. J. Levy-Booth, L. E. Navas, J. Liu, J. C. Grigg, A. Wilson, R. Katahira, G. T. Beckham, W. W. Mohn and L. D. Eltis, Proc. Natl. Acad. Sci. U. S. A., 2020, 117, 25771–25778 CrossRef CAS PubMed.
- Y. Jiang, C. Wang, N. Ma, J. Chen, C. Liu, F. Wang, J. Xu and Z. Cong, Catal. Sci. Technol., 2020, 10, 1219–1223 RSC.
- A. Bleem, E. Kuatsjah, G. N. Presley, D. J. Hinchen, M. Zahn, D. C. Garcia, W. E. Michener, G. König, K. Tornesakis, M. N. Allemann, R. J. Giannone, J. E. McGeehan, G. T. Beckham and J. K. Michener, Chem. Catal., 2022, 2, 1–23 CrossRef.
- T. Abe, E. Masai, K. Miyauchi, Y. Katayama and M. Fukuda, J. Bacteriol., 2005, 187, 2030–2037 CrossRef CAS PubMed.
- D. P. Brink, K. Ravi, G. Lidén and M. F. Gorwa-Grauslund, Appl. Microbiol. Biotechnol., 2019, 103, 3979–4002 CrossRef CAS PubMed.
- M. M. Machovina, S. J. B. Mallinson, B. C. Knott, A. W. Meyers, M. Garcia-Borràs, L. Bu, J. E. Gado, A. Oliver, G. P. Schmidt, D. J. Hinchen, M. F. Crowley, C. W. Johnson, E. L. Neidle, C. M. Payne, K. N. Houk, G. T. Beckham, J. E. McGeehan and J. L. DuBois, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 13970–13976 CrossRef CAS PubMed.
- E. Masai, M. Sasaki, Y. Minakawa, T. Abe, T. Sonoki, K. Miyauchi, Y. Katayama and M. Fukuda, J. Bacteriol., 2004, 186, 2757–2765 CrossRef CAS PubMed.
- J. H. Cecil, D. C. Garcia, R. J. Giannone and J. K. Michener, Appl. Environ. Microbiol., 2018, 84, e01185-18 CrossRef PubMed.
- J. M. Klenk, M.-P. Fischer, P. Dubiel, M. Sharma, B. Rowlinson, G. Grogan and B. Hauer, J. Biochem., 2019, 166, 51–66 CrossRef CAS PubMed.
- D. Holtmann, M. W. Fraaije, I. W. C. E. Arends, D. J. Opperman and F. Hollmann, Chem. Commun., 2014, 50, 13180–13200 RSC.
- S. Nagano and T. L. Poulos, J. Biol. Chem., 2005, 280, 31659–31663 CrossRef CAS PubMed.
- M. Gajhede, D. J. Schuller, A. Henriksen, A. T. Smith and T. L. Poulos, Nat. Struct. Biol., 1997, 4, 1032–1038 CrossRef CAS PubMed.
- M. Hofrichter and R. Ullrich, Curr. Opin. Chem. Biol., 2014, 19, 116–125 CrossRef CAS PubMed.
- X. Huang and J. T. Groves, Chem. Rev., 2018, 118, 2491–2553 CrossRef CAS PubMed.
- M. N. Podgorski, J. S. Harbort, J. H. Z. Lee, G. T. H. Nguyen, J. B. Bruning, W. A. Donald, P. V. Bernhardt, J. R. Harmer and S. G. Bell, ACS Catal., 2022, 12, 1614–1625 CrossRef CAS.
- O. Shoji, T. Fujishiro, K. Nishio, Y. Kano, H. Kimoto, S.-C. Chien, H. Onoda, A. Muramatsu, S. Tanaka, A. Hori, H. Sugimoto, Y. Shiro and Y. Watanabe, Catal. Sci. Technol., 2016, 6, 5806–5811 RSC.
- P. C. Cirino and F. H. Arnold, Angew. Chem., Int. Ed., 2003, 42, 3299–3301 CrossRef CAS PubMed.
- A. Greule, T. Izoré, D. Machell, M. H. Hansen, M. Schoppet, J. J. De Voss, L. K. Charkoudian, R. B. Schittenhelm, J. R. Harmer and M. J. Cryle, Front. Chem., 2022, 10, 868240 CrossRef CAS PubMed.
- J. Lasovsky, J. Hrbac, D. Sichertova and P. Bednar, Luminescence, 2007, 22, 501–506 CrossRef CAS PubMed.
- J. Su, L. Yang, R. N. Liu and H. Lin, Chin. J. Catal., 2014, 35, 622–630 CrossRef CAS.
- R. Ma, Y. Xu and X. Zhang, ChemSusChem, 2015, 8, 24–51 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2cc04698a |
| This journal is © The Royal Society of Chemistry 2022 |