
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Polyamine receptors containing anthracene as fluorescent probes for ketoprofen in H2O/EtOH solution†
Giammarco Maria
Romano‡
 a,
Liviana
Mummolo‡
b,
Matteo
Savastano
a,
Paola
Paoli
c,
Patrizia
Rossi
c,
Luca
Prodi
*b and
Andrea
Bencini
*a
a,
Liviana
Mummolo‡
b,
Matteo
Savastano
a,
Paola
Paoli
c,
Patrizia
Rossi
c,
Luca
Prodi
*b and
Andrea
Bencini
*a
aDipartimento di Chimica ‘Ugo Schiff’, Università di Firenze, Via della Lastruccia 3, 50019-Sesto Fiorentino, Firenze, Italy. E-mail: andrea.bencini@unifi.it
bDipartimento di Chimica “Giacomo Ciamician”, Università degli studi di Bologna, Via Selmi 2, 40126 Bologna, Italy. E-mail: luca.prodi@unibo.it
cDepartment of Industrial Engineering, Università di Firenze, Via S. Marta 3, Florence, I-50139, Italy
First published on 18th May 2022
Abstract
Triamine receptors containing anthracene units are able to bind and sense ketoprofen via fluorescence enhancement in a H2O/EtOH 50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :50 (Vol:Vol) mixture exploiting their protonation features, which are tuned by the interaction with the analyte.
:50 (Vol:Vol) mixture exploiting their protonation features, which are tuned by the interaction with the analyte.
Non-steroidal anti-inflammatory drugs (NSAIDs) are among the most worldwide used pharmaceuticals. Their chemical stability and widespread human consumption and veterinary use, accompanied by lack of efficient methodologies for removal, storage, and disposal of waste, lead to their increasing presence in the aquatic environment.1 So far, they have been classified as emerging pollutants, due to their recently proved ecotoxicity.2,3 Moreover, their release in the environment is still often poorly regulated and monitored.2 In this panorama, there is an effective challenge to obtain economically viable, highly sensitive and rapid response sensors to protect human health and ecosystems. Fluorescent sensors have been proven to satisfy these requirements.4
Nanostructured assemblies have been used for optical signalling of NSAIDs, including polyethyleneimine-passivated Cd/S quantum dots (QDs)5 or CdSe/ZnS QDs capped with N-acetyl-L-cysteine methyl ester,6 carbon dot-containing imprinted polymers,7 chitosan-stabilized Ag nanoparticles (NPs),8 hybrid organic-Ag NP assemblies,9,10 arrays of monolayered Au NPs,11 poly(p-aryleneethynylene) polymers,12 or hydrogel-embedded chemosensors.13 Less attention has been paid to fluorescent small molecules that are able to detect NSAIDs. NSAIDs present some common structural features, being normally composed of a carboxylic group linked to an aromatic portion. Binding and sensing of carboxylic acids are normally achieved by using receptors containing hydrogen bonding sites14 and it result difficult, especially in aqueous media, where solvation efficiently competes with the binding process.14 Examples are limited to cinchona alkaloids15 or calixpyrrole-based16 chemosensors and a BINOL-containing macrocycle,17 which are able to optically signal NSAIDs in non-aqueous solvents.
Receptors L1 and L2, obtained by using a well-known procedure for terminal alkylation of linear polyamines,18 contain a diethylen- or a dipropylen-triamine unit, two ‘classic’ ligands used in coordination chemistry, linked at their extremities via methylene bridges, to the 9-position of an anthracene unit, probably the most exploited fluorophore (Scheme 1). To test these simple receptors as NSAID probes, we chose ketoprofen (KP), one of the most common NSAIDs, which does not feature fluorescence emission.
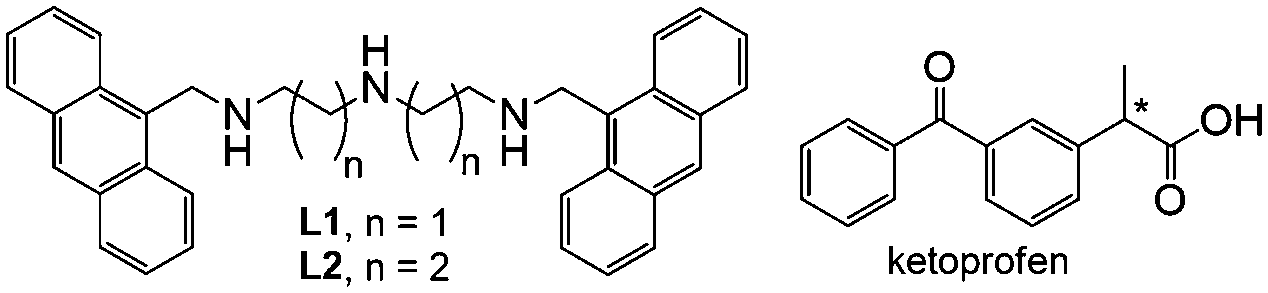 | ||
| Scheme 1 | ||
L1 and L2 protonate in aqueous media even at neutral pH to give polyammonium cations that are able to interact with the carboxylic group of NSAIDs, including KP, normally in its anionic form at pH 7, via charge–charge and H-bonding interactions. The anthracene units can undergo hydrophobic and/or π-stacking interactions with the aromatic section of KP. Indeed, potentiometric titrations in the H2O/EtOH 50:50 (Vol:Vol) mixture – the ligands being not sufficiently soluble in pure water under the conditions of potentiometric measurements – pointed out that the receptors bind up to three or two protons in the case of L1 and L2, respectively (Table S1 and Fig. S9, ESI†). At neutral pH, L1 is mainly in its [HL1]+ form, while in the case of L2 the not-protonated amine is present in solution together with [HL2]+ in relevant percentages. The basicity of L1 is in agreement with the drop observed moving from dien to its benzyl (logK1 = 9.4) and naphthyl (logK1 = 8.38) terminal bis-derivatives.19L2 is less basic than L1 in its first protonation step, likely due to the increased N⋯N distance that prevents stabilizing intramolecular H-bonding interactions between vicinal nitrogens. The third protonation step of L2 is likely to occur outside the investigated pH range (below pH 2.5).
K) of the L1 and L2 adducts with KP at 298.1 ± 0.1 K, 0.1 M NMe4Cl (H2O/EtOH 50:50 v/v)
| Equuilibrium | L1 | L2 |
|---|---|---|
| L + KP− = [L(KP)]− | 3.76(7) | 2.95(4) |
| HL+ + KP− = [HL(KP)] | 3.59(5) | 2.47(7) |
| H2L2+ + KP− = [H2L(KP)]+ | 3.6(1) | 2.98(4) |
| H2L2+ + HKP = [H2L(HKP)]2+ | 3.36(5) | — |
| [H2L(KP)]+ + KP− = [H2L(KP)2] | 3.1(1) | — |
| [H2L(HKP)]2+ + KP− = [H2L(HKP)(KP)]+ | 3.4(1) | — |
The polyammonium cations of both receptors form 1:1 adducts with KP in its anionic form, while in the case of L1 the formation of 1:2 complexes is also observed (Table 1). The adduct formation is likely due to a subtle balance between hydrophobic forces and salt bridging. Their stability is not particularly dependent on the receptor charge, suggesting that hydrophobic interactions play a relevant role in the formation of adducts. Both neutral ligands bind KP, probably thanks to hydrophobic effects and/or stacking forces and H-bonding. The association process is likely entropically guided by the effect of the solvent. First protonation of L1 and L2 does not favour KP binding (logK = 3.59 for L1 and 2.47 for L2) with respect to the neutral receptor (logK = 3.76 for L1 and 2.95 for L2): the extra stabilization due to salt-bridging comes at the price of a reduced hydrophobic effect. The second protonation, instead, leads to a small increase in stability, attributable to increased charge–charge and H-bonding interactions. This balance can be visualized from the plots of conditional stability constants vs. pH (Fig. S11, ESI†),20 which point out, for each ligand, a similar binding ability between pH 5 and 9. L1 functions as a better KP binder, as also evidenced by the BC50 values,21i.e., the total receptor concentration for which the molar ratio between the bound and total substrate concentrations assumes a 0.5 value (BC50 = 7.14 × 10−4 M (L1) and 2.01 × 10−3 M (L2) for [KP] = 0.001 M and pH 7). L2 shows a less gathered overall structure and a longer distance between the anthracene units, which would reduce the hydrophobic interactions with KP. In the case of L1, the constants for the addition of the second anion are slightly lower than those for the binding of the first one, as expected considering the lower positive charge of the 1:1 adduct with respect to the ‘free’ receptor.
To investigate the receptor ability to optically signal KP, we focused our attention on the neutral pH region, performing titrations by adding to solutions of L1 and L2, buffered at pH 7 with TRIS, increasing amounts of KP, which is in its deprotonated anionic form at pH 7 (the pKa of KP is 5.31 under our experimental conditions, Table S1, ESI†). Under these conditions, both L1 and L2 show a typical structured band of anthracene derivatives both in their absorption and fluorescence spectra (Fig. S14 and S15, ESI†). Two excited state lifetimes can be obtained for both L1 (1.0 and 6.5 ns; Φ = 0.083) and L2 (1.2 and 6.3 ns; Φ = 0.061), being the shorter component the most abundant one in both cases (ca. 10:1). These data are compatible with the photoinduced electron transfer (PET) process between the non-protonated amines and the anthracene units (see below).22,23 Both ligands have a tail in the fluorescence spectrum in the 450–600 nm range, more pronounced in L1 than in L2, probably due to the shorter distance between the aromatic units. It is noteworthy that at pH 7, the ligand flexibility is reduced by partial protonation of the triamine chain. Excimer emission is not particularly intense and can be attributed to the formation of minor percentages of excimer conformers. By far stronger excimer and consequent reduction of monomer emission are observed for genuine interactions between two anthracene fragments.24
The addition of KP induces up to a 3-fold increase of the emission of the anthracene band (see Fig. 1 for L1). Interestingly, emission at 414 nm increases almost linearly up to the addition of 1 equiv. of KP (inset of Fig. 1); a linear increase, but with a higher slope, is also observed afterwards up to a 2:1 KP:L1 molar ratio, to achieve an almost constant value for molar ratios greater than 3, suggesting the successive formation of complexes with 1:1 and 2:1 stoichiometry between KP and L1. Analysis of the spectral data with the HYPSPEC25 program leads to estimating apparent binding constants at pH 7.0 (ligand speciation not analytically considered) for successive additions of one KP anion to L1 (logK1 = 5.7) and to its 1:1 complex (logK2 = 5.5). The almost equal constant values (errors are ca. 10% on K values) suggest that the higher slope observed for the increase in emission upon addition of more than 1 equiv. of KP cannot be attributed to the increased stability of the 1:2 complex, but to a stronger emission of the latter with respect to the 1:1 adduct.
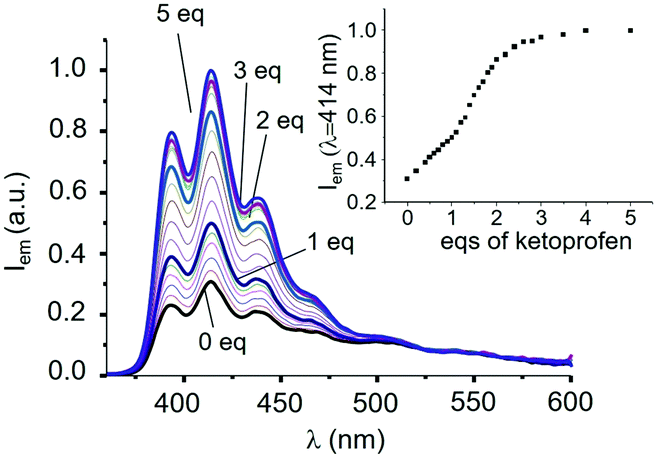 | ||
| Fig. 1 Fluorescence emission spectra of L1 at pH 7 (0.001 M TRIS buffer) in H2O/EtOH 50:50 (Vol:Vol) and (inset) emission intensity at 414 nm in the presence of increasing amounts of KP (λEXC = 340 nm, [L1] = 1 × 10−5 M, 298 K). | ||
Considering time-resolved emission measurements, the shorter excited state lifetime undergoes a two-fold elongation up to 2.1 ns, while the other experiences a modest change to 7.1 ns. Besides, the 450–600 nm tail becomes less prominent (Fig. S17, ESI†). L2 shows a similar behaviour upon KP binding (Fig. S15, ESI†), although in this case the emission enhancement is less marked. Apparent constants of 5.4 and 4.9 log units can be estimated for the addition of a single KP anion to L2 and its 1:1 complex, respectively. The values obtained from fluorescence titrations are higher than the ones obtained by potentiometry, which showed the formation of seemingly less stable complexes with L1 and L2. The comparison between constants obtained from potentiometric and fluorimetric measurements at a fixed pH value should take into account the different conditions used, including the presence, in the former, of a 0.1 M NMe4Cl ionic medium, in which Cl− can compete with KP binding thanks to salt bridging contacts with the polyamine chain (most likely, the stability constants of the 1:2 complexes with L2 are actually too low to be detected under these conditions). The constants derived from fluorimetric titrations at fixed pH in the absence of ionic medium are thus higher and obtained under conditions closer to the ones of possible applications.
The ESI mass spectra of solutions of L1 and L2 in the presence of 2 equiv. of KP also confirm the formation of 1:1 and 1:2 adducts (Fig. S18–S23, ESI†). Significant 1:1 adduct peaks corresponding to [L1·KP + 2H]2+ or [L2·KP + 2H]2+ and to doubly charged 1:2 adducts are detected. The latter are less intense, likely due to the lower stability of 1:2 adducts.
The limit of detection (LOD) obtained with fluorescence experiments was 0.21 and 0.28 μM for L1 and L2, respectively. Somewhat higher LODs, 0.35 μM for L1 and 0.40 μM for L2, were found by using tap water in the H2O/EtOH (50:50 v/v) mixture. It is noteworthy that the emission enhancement is also observed in pure water at pH 7, although, in this case, the response of the probe is less intense (Fig. S26, ESI†).
The emission increase in the presence of KP may depend on the interactions between the aromatic units of both receptors and substrate and/or by salt bridging between the protonated polyamine chain and the carboxylate group of KP. To elucidate this point, we performed 1H NMR titrations in the CD3OD/D2O mixture (L1 and L2 are not sufficiently soluble in the EtOH/H2O mixture at the concentrations of NMR measurements). The addition of increasing amounts of KP to a solution of L1 at pH 7 gives rise to a progressive downfield shift of the resonance of CH2 (2AL) and to a simultaneous upfield shift of the CH2 (3AL) signal (Fig. 2a), strongly suggesting that host–guest binding takes place via salt bridging contacts between the carboxylate group of KP and the monoprotonated triamine group of L1. In the [HL1]+ species, the proton is localized on the central amine group of the polyamine chain, more basic than the ‘benzylic’ amines, adjacent to the aromatic units,19,22 as also demonstrated by the analysis of the pH dependence of the 1H NMR signals of L1 and L2 (Fig. S27, ESI†). Since in aliphatic amines protonation is accompanied by a downfield shift of the signal of the adjacent methylene unit,19 the shifts in opposite directions of the resonances 2AL and 3AL in L1 can be reasonably ascribed to a proton transfer process of the acidic proton from the central nitrogen to the benzylic amine group upon KP binding. A similar behaviour is also observed for the signal of the CH2 groups of L2 adjacent to central (4AL) and the benzylic amine groups (2AL), respectively (Fig. 2b). Proton transfer to the closer benzylic amine groups upon KP binding is expected to make the PET process less efficient, justifying the observed fluorescence enhancement. An additional, but definitely minor, element leading to the fluorescence increase is the observed decrease of the efficiency of the deactivation channel leading to the formation of the excimer upon complexation.
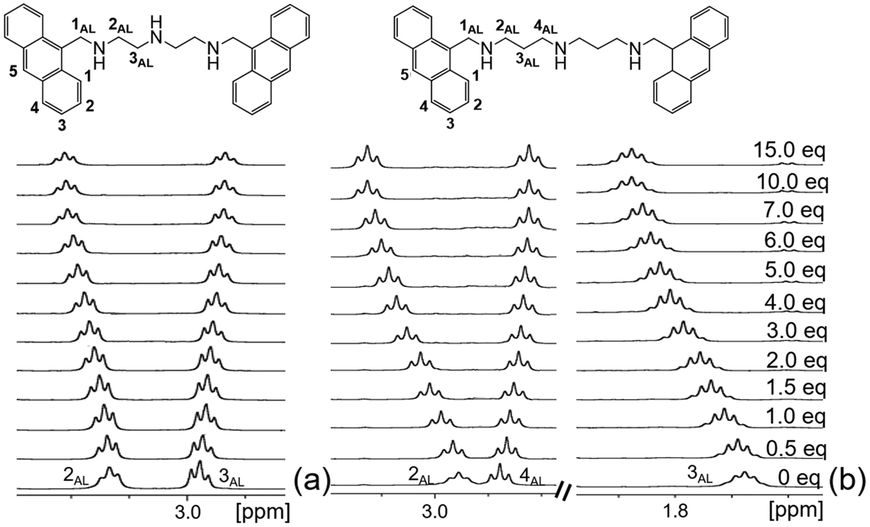 | ||
| Fig. 2 Aliphatic signals of the 1H NMR spectra of L1 (a) and L2 (b) at pH 7 in the presence of increasing amounts of KP. The signal of 1AL is superimposed to the HOD resonance and cannot be confidently monitored (D2O/CD3OD 30:70) (Vol:Vol, [L1] = 1 × 10−2 M, 298 K, for L1, J2AL,3AL = 6 Hz; for L2, J2AL,3AL 6 Hz; J3AL,4AL = 7 Hz; J2AL,4AL cannot be confidently estimated). | ||
The 1H NMR titrations point out that the anthracene and KP signals are not affected by the host–guest interaction (Fig. S28 and S29, ESI†), indicating that the aromatic units are likely not directly involved in substrate binding via π-stacking.
Taken as a whole, these results suggest that charge–charge and H-bonding interactions play the major role in KP sensing by both protonated receptors at pH 7. Hydrophobic effects are not significantly involved in KP sensing, although they contribute to complex stabilization. No interaction with L1 or L2 is detected by potentiometric and fluorimetric measurements with carboxylate anions with smaller hydrophobic units, including acetate, benzoate and ibuprofen (Fig. S30 and S31, ESI†), suggesting that the overall hydrophobic character of L1 and L2 contributes to stabilize the adducts with KP, containing a diphenylmethane moiety with respect to less hydrophobic carboxylates. The sensing ability is not particularly affected by relevant interfering agents, in particular of biological interest, including metal cations (Na+, K+ Mg2+, Ca2+, Fe2+, and Fe3+), inorganic anions (Cl−, Br−, HPO4−, SO42−, NO3−, and ClO4−) and amino acids (glycine, phenylalanine, and cysteine) (Fig. S32–S36, ESI†). Only Cu2+ and Zn2+ appreciably interfere, the former quenching the emission of both L1 and L2, in the presence and in the absence of KP (due to its affinity for polyamines and its paramagnetic nature) and the latter enhancing the emission, as often observed for Zn2+ polyamine complexes.23 Original emission of the KP adducts is restored by sequestering the metals with a strong chelator N,N,N′,N′-tetrakis(2-pyridylmethyl)ethylenediamine (TPEN).
KP adduct stabilization by salt bridging is also supported by the crystal structure of the [H2L2](KP)2·0.75EtOH·2.75H2O complex. Fig. 3 shows the asymmetric unit, containing the KPA and KPB anions, together with carboxylate groups of the symmetry related KPA′ and KPB′ KP units. The non-symmetry related KPA and KPB anions are bound to the ligand via 2 salt-bridges each (KPA: N2⋯O1A, N3⋯O2A′; KPB: N2⋯O1B, N3⋯O1B′). KPB is further stabilized by an additional H-bond (N1⋯O3B), between the not-protonated amino group and KPB carbonyl function. KPB simultaneously interacts with two amino/ammonium groups supports the lower affinity obtained for a second substrate and the 1:2 host–guest stoichiometry found in solution, although at the solid-state it is realized via a larger 2:4 adduct (Fig. S38, ESI†). Of note, KPB forms a bifurcated salt-bridge, generally less stable than standard ones (average salt-bridge length: KPA, 2.69 ± 0.04 Å, vs. KPB, 2.73 ± 0.08 Å), maintaining its carbonyl-amine H-bond and overcoming the energy loss.26 This structure demonstrates that anion coordination influences acidic proton localization within the polyamine backbone. In [H2L2](KP)2 the two acidic protons are localized on the central and one benzylic nitrogen, an unexpected positioning considering that in protonated polyamines acidic proton distribution is normally regulated by minimization of electrostatic repulsion. In this case, interaction of one KP unit with a central ammonium group allows for the formation of a further stabilizing H-bond between the carbonyl oxygen of KP and the not-protonated amine group, which, in turn, would be lost in the case of interaction of KP with two ammonium groups of L2.
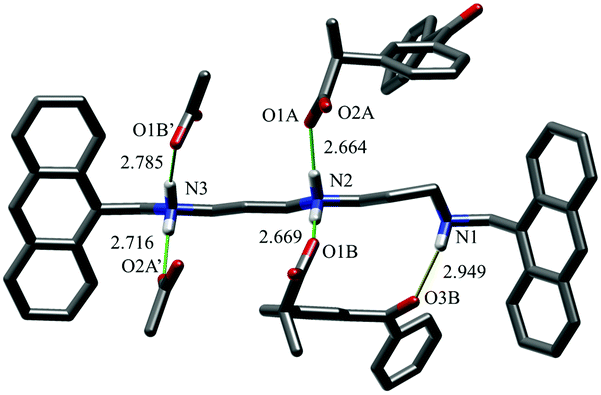 | ||
| Fig. 3 Main interactions in the crystal structure of the complex. Asymmetric unit, comprising one ligand molecule and 2 KP anions, KPA and KPB, is completely shown. Carboxylate groups of symmetry-related KPA and KPB have been added to show a complete H-bond environment of the [H2L2]2+ cation. Salt-bridges are depicted in green H-bonds are in yellow. | ||
These results demonstrate that simple molecular receptors, obtained by straightforward coupling of a linear polyamine with common fluorogenic units, can be used for binding and sensing – with some selectivity in the fluorescence response – of elusive analytes, such as NSAIDs, exploiting the proton binding features of these receptors.
Financial support from the Italian Ministry for University and Research (PRIN 2017EKCS35 project) is gratefully acknowledged. We thank Paolo Neviani for having performed mass spectrometry.
Conflicts of interest
There are no conflicts to declare.Notes and references
- (a) P. Izadi, P. Izadi, R. Salem, S. A. Papry, S. Magdouli, R. Pulicharla and S. K. Brar, Environ. Pollut., 2020, 267, 115370 CrossRef CAS PubMed; (b) A. Jurado, E. Vázquez-Suñé and E. Pujades, Water, 2021, 13, 72 CrossRef.
- Non-Steroidal Anti-Inflammatory Drugs in Water, ed. L. M. Gómez-Oliván, Springer International Publishing, 2020 Search PubMed.
- (a) N. Patel, M. Z.-A. Khan, S. Shahane, D. Rai, D. Chauhan, C. Kant and V. Chaudhary, Pollution, 2020, 99–113 CAS; (b) J. Wilkinson, P. S. Hooda, J. Barker, S. Barton and J. Swinden, Environ. Pollut., 2017, 231, 954–970 CrossRef CAS PubMed.
- (a) P. Gale, T. Gunnlaugsson and S. Kubik, Supramolecular chemistry of anionic species themed issue Anion recognition in water, Chem. Soc. Rev., 2010, 39 CAS; (b) P. A. Gale and C. Caltagirone, Coord. Chem. Rev., 2018, 354, 2–27 CrossRef CAS; (c) W. Laxman, T. Anushree, K. Pawan, S. Yong, P. Samandhan, D. Akash and H. Ki, Trends Anal. Chem., 2017, 97, 458–467 CrossRef.
- S. Chen, S. Zhou, J. Fu, S. Tang, X. Wu, P. Zhao and Z. Zhang, Anal. Methods, 2021, 13, 2836–2846 RSC.
- T. Delgado-Pérez, L. M. Bouchet, M. de la Guardia, R. E. Galian and J. Pérez-Prieto, Chem. – Eur. J., 2013, 19, 11068–11076 CrossRef PubMed.
- B. Shikha, K. Kaur, S. Maheshwari and A. Malik., J. Fluoresc., 2018, 29, 145–154 Search PubMed.
- M. Mabrouk, S. F. Hammad, A. A. Abdella and F. Mansour, Colloids Surf., A, 2021, 614, 126182 CrossRef CAS.
- A. Saini, M. Kaur, S. Mayank, A. Kuwar, N. Kaur and N. Singh, Mol. Syst. Des. Eng., 2020, 5, 1428–1436 RSC.
- A. Jouyban and E. Rahimpour, Talanta, 2020, 217, 218924501 CrossRef PubMed.
- X. Sun, P. Liu and F. Mancin, Analyst, 2018, 143, 5754–5763 RSC.
- J. Han, B. Wang, M. Bender, S. Kushida, K. Seehafer and U. H. F. Bunz, ACS Appl. Mater. Interfaces, 2017, 9, 790–797 CrossRef CAS PubMed.
- Y. Liu, T. Minami, R. Nishiyabu, Z. Wang and P. Anzenbacher, J. Am. Chem. Soc., 2013, 135, 7705–7712 CrossRef CAS PubMed.
- (a) K. Bowman-James and A. Bianchi, Enrique García-España, Anion Coordination Chemistry, Wiley-VCH, Weinheim, Germany, 2012 Search PubMed; (b) J. W. Steed and J. L. Atwood, Supramolecular Chemistry, Wiley-VCH, Weinheim, Germany, 2nd edn, 2009 Search PubMed.
- A. Akdeniz, L. Mosca, T. Minami and P. Anzenbacher, Chem. Commun., 2015, 51, 5770–5773 RSC.
- M. Pushina, P. Koutnik, R. Nishiyabu, T. Minami, S. Tsuyoshi and P. Anzenbacher Pavel, Jr, Chem. – Eur. J., 2018, 24, 4879–4884 CrossRef CAS PubMed.
- A. Akdeniz, T. Minami, S. Watanabe, M. Yokoyama, T. Ema and P. Anzenbacher, Chem. Sci., 2016, 13, 2016–2022 RSC.
- J. A. Sclafani, M. T. Maranto, T. M. Sisk and S. A. Van Arman, J. Org. Chem., 1996, 61, 3221–3222 CrossRef CAS PubMed.
- (a) A. Bencini, A. Bianchi, E. García-España, M. Micheloni and J. A. Ramírez, Coord. Chem. Rev., 1999, 188, 97–156 CrossRef CAS; (b) A. Bianchi, B. Escuder, E. García-España, S. V. Luis, V. Marcelino, J. F. Miravet and J. A. Ramírez, J. Chem. Soc., Perkin Trans. 2, 1994, 1253–1259 RSC.
- C. Bazzicalupi, A. Bianchi, C. Giorgi, M. Paz Clares and E. García-Espana, Coord. Chem. Rev., 2012, 256, 13–27 CrossRef CAS.
- A. Vacca, O. Francesconi and S. Roelens, Chem. Rec., 2012, 12, 544–566 CrossRef CAS PubMed.
- (a) S. Alves, F. Pina, M. T. Albelda, E. García-España, C. Soriano and S. V. Luis, Eur. J. Inorg. Chem., 2001, 405–412 CrossRef CAS; (b) J. Seixas de Melo, M. T. Albelda, P. Díaz, E. García-España, C. Lodeiro, S. Alves, J. C. Lima, F. Pina and C. Soriano, J. Chem. Soc., Perkin Trans. 2, 2002, 991–998 RSC; (c) C. Bazzicalupi, A. Bencini, A. Bianchi, C. Giorgi, V. Fusi, B. Valtancoli, M. A. Bernardo and F. Pina, Inorg. Chem., 1999, 38, 3806–3813 CrossRef CAS.
- B. Valeur, Molecular fluorescence principles and applications, Wiley-VCH, Weinheim, 2001 Search PubMed.
- D. Marquis, J.-P. Desvergne and H. Bouas-Laurent, J. Org. Chem., 1995, 60(24), 7984–7996 CrossRef CAS.
- P. Gans, A. Sabatini and A. Vacca, Talanta, 1996, 43, 1739–1753 CrossRef CAS PubMed.
- I. Rozas, I. Alkorta and J. Elguero, J. Phys. Chem. A, 1998, 102, 9925–9932 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 2116440. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d2cc01107g |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2022 |