
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAccelerated microdroplet synthesis of benzimidazoles by nucleophilic addition to protonated carboxylic acids†
Pallab
Basuri
a,
L. Edwin
Gonzalez
b,
Nicolás M.
Morato
 b,
Thalappil
Pradeep
*a and
R. Graham
Cooks
*b
b,
Thalappil
Pradeep
*a and
R. Graham
Cooks
*b
aDST Unit of Nanoscience (DST UNS), Thematic Unit of Excellence (TUE), Department of Chemistry, Indian Institute of Technology Madras, Chennai 600036, India
bDepartment of Chemistry, Purdue University, West Lafayette, Indiana 47907, USA. E-mail: cooks@purdue.edu
First published on 14th July 2020
Abstract
We report a metal-free novel route for the accelerated synthesis of benzimidazole and its derivatives in the ambient atmosphere. The synthetic procedure involves 1,2-aromatic diamines and alkyl or aryl carboxylic acids reacting in electrostatically charged microdroplets generated using a nano-electrospray (nESI) ion source. The reactions are accelerated by orders of magnitude in comparison to the bulk. No other acid, base or catalyst is used. Online analysis of the microdroplet accelerated reaction products is performed by mass spectrometry. We provide evidence for an acid catalyzed reaction mechanism based on identification of the intermediate arylamides. Their dehydration to give benzimidazoles occurs in a subsequent thermally enhanced step. It is suggested that the extraordinary acidity at the droplet surface allows the carboxylic acid to function as a C-centered electrophile. Comparisons of this methodology with data from thin film and bulk synthesis lead to the proposal of three key steps in the reaction: (i) formation of an unusual reagent (protonated carboxylic acid) because of the extraordinary conditions at the droplet interface, (ii) accelerated bimolecular reaction because of limited solvation at the interface and (iii) thermally assisted elimination of water. Eleven examples are shown as evidence of the scope of this chemistry. The accelerated synthesis has been scaled-up to establish the substituent-dependence and to isolate products for NMR characterization.
Introduction
Carbon–nitrogen (C–N) coupling chemistry1 is of comparable or even greater importance than is carbon–carbon coupling2 especially in the context of pharmaceutically active small molecules.3 These reactions involve in situ conversion of precatalysts (often ligated Pd) to the active catalyst and activation of the amine by base – typically an organic base, so permitting reaction in a homogeneous liquid phase.4 An enormous number of catalysts has been explored and C–N bond formation is currently possible under relatively mild conditions using Pd, Cu and other organometallic catalysts. Conceptually, an alternative to nucleophilic substitution at an aryl or alkyl (pseudo) halide is the acid catalyzed nucleophilic substitution at a carbonyl carbon, effectively amine addition to an acylium ion. Examples of accelerated droplet chemistry involving somewhat related mechanisms are known, in particular the acid catalyzed reaction of ketones with amines to form imines.5–13 It seems likely that the extraordinarily high acidity at the surface of aqueous droplets drives these rapid reactions, even in cases like this study where the solvent is nominally non-aqueous. We explore C–N coupling of amines using protonated carboxylic acids as reagents (clearly counter-intuitive entities, given that neutral carboxylic acids are normally proton donors not acceptors). The addition/water elimination product, the substituted amide, is not our primary focus here; rather we are most interested in systems in which the presence of an ortho amino group allows further reaction to give a cyclic benzimidazole.Benzimidazoles are an important class of heterocyclic compounds due to their wide application as active pharmaceutical moieties. Albendazole, mebendazole, triclabendazole, droperidol and pimozide are examples of drugs containing a benzimidazole scaffold.14 These drugs are used to treat cancers15 and ulcers,16 as well as fungal,17 viral18 and parasitic infections.19 The parent compound also serves as a precursor for the synthesis of vitamin B12.20 Derivatives of benzimidazoles are used industrially as ultraviolet filters and pigments.21,22 Conventionally, benzimidazole synthesis requires heating 1,2-phenylenediamine (PDA) with concentrated carboxylic acid for hours at high temperature, followed by the addition of a strong Lewis base to obtain the product.23,24 Recently, aldehydes and alcohols have been used as additives to facilitate the bulk reaction.25,26 Moreover, the reaction can be performed at a milder temperature by adding catalysts such as cobalt,27 palladium,28 copper29 and even boranes.30 Despite the use of a metal catalyst, the reaction generally takes 6 to 12 hours to accomplish. Reactions involving radical pathways under UV-radiation are faster but require a radical generator such as rose bengal.31 Other strategies have included the use of microwave irradiation in presence of triphenyl phosphite.32 Nevertheless, in terms of sustainable synthesis, reactions that occur rapidly under mild conditions remain highly desirable.
In this study, we demonstrate a metal-free synthetic strategy in which the rate of the benzimidazole synthesis is accelerated and occurs under ambient conditions inside charged microdroplets during their brief time of flight in the open air either into a mass spectrometer or, in scaled-up experiments (see below), onto a droplet collector. These electrosprayed microdroplets behave like micro/nano-reactors which undergo rapid desolvation and coulombic fission while finally releasing unsolvated product ions into the vacuum of the mass spectrometer.
Examples of reactions known to be accelerated in the microdroplet environment include Suzuki coupling,33 Fisher indole synthesis,10 Katritzky transamination,34 Claisen–Schmidt condensation,35 Eschenmoser coupling,35 Dakin reaction,36 Baeyer–Villiger oxidation,36N-alkylation of indoles,37 Combes reaction,12 Pomeranz–Fritsch synthesis,12 and cycloaddition reactions.38 These microdroplet accelerated reactions can be performed under ambient conditions using nESI10 or, in larger volumes, using electrosonic spray ionization (ESSI).12 Levitated Leidenfrost droplets13 allow milligram quantities of compounds to be synthesized in times on the order of minutes. Here, we use both nESI and ESSI, as well as mass spectral fragmentation patterns (MS/MS experiments) to generate and identify reaction intermediates and products, supplemented by isotope labeling and pH dependence experiments.
In addition to being the focus for accelerated organic reactions as discussed here, sprays have also seen use in materials preparation, sometimes on a commercial scale as in the case of electrospinning.39 Other examples include fabrication of silver nanobrushes,40 metallic palladium nanosheets,41 high entropy alloys,42 dandelion-like CuO–Ag hybrid hierarchical nanostructures,43 hybrid perovskite quantum nanostructures,44 and multi-metal 3D printed microstructures.45
Results and discussion
Droplet reactions and MS analysis
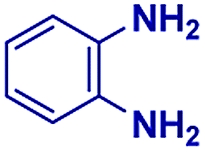
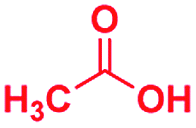
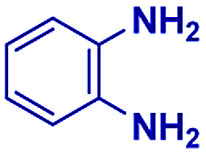
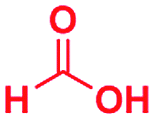
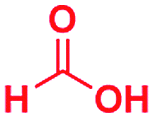
Scheme 1 displays the reaction conditions used in conventional23,24 and microdroplet benzimidazole synthesis. The droplet reaction was performed by electrospraying a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 molar ratio of o-aryl diamine and carboxylic acid at 2–2.5 kV applied potential using nESI emitters of 5 μm tip diameter. The solvent was methanol and the concentration of each reactant was 8 mM. Collision-induced dissociation (CID) with He as collision gas was used to record MS/MS spectra for the characterization of products and intermediates. The scope of the microdroplet synthesis was assessed using seven diamines (1,2-phenylenediamine (PDA), 4-methyl-1,2-phenylenediamine, 4,5-dimethyl-1,2-phenylenediamine, 4-nitro-1,2-phenylenediamine, 4-chloro-1,2-phenylenediamine, 4-methoxy-1,2-phenylenediamine and 1,2-diaminonaphthalene) and five carboxylic acids (formic acid (FA), acetic acid (AA), trifluoroacetic acid (TFAA), propanoic acid (PA) and benzoic acid (BA)).
:1 molar ratio of o-aryl diamine and carboxylic acid at 2–2.5 kV applied potential using nESI emitters of 5 μm tip diameter. The solvent was methanol and the concentration of each reactant was 8 mM. Collision-induced dissociation (CID) with He as collision gas was used to record MS/MS spectra for the characterization of products and intermediates. The scope of the microdroplet synthesis was assessed using seven diamines (1,2-phenylenediamine (PDA), 4-methyl-1,2-phenylenediamine, 4,5-dimethyl-1,2-phenylenediamine, 4-nitro-1,2-phenylenediamine, 4-chloro-1,2-phenylenediamine, 4-methoxy-1,2-phenylenediamine and 1,2-diaminonaphthalene) and five carboxylic acids (formic acid (FA), acetic acid (AA), trifluoroacetic acid (TFAA), propanoic acid (PA) and benzoic acid (BA)).
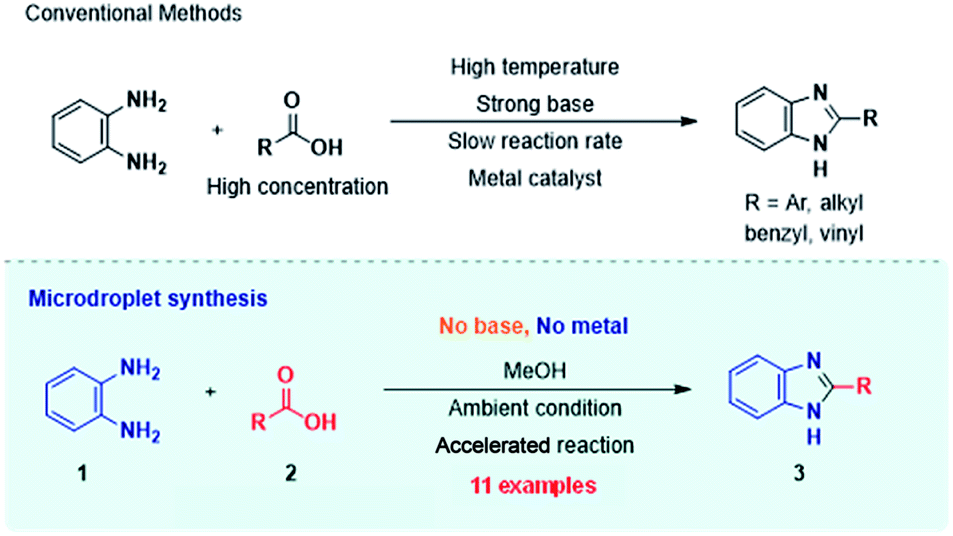 | ||
| Scheme 1 Synthesis of benzimidazole and its derivatives by conventional methods22,23 (above) and microdroplet synthesis (below). | ||
Fig. 1 compares the mass spectrum of PDA recorded with and without addition of formic acid. There is no benzimidazole product without the acid, the only signal being that at m/z 109 corresponding to protonated PDA. The inset of Fig. 1A schematically illustrates the procedure used for microdroplet synthesis. The MS/MS spectrum of the ion at m/z 109 shows a characteristic neutral loss of ammonia (ESI, Fig. S1A†) confirming that the peak corresponds to protonated PDA. The ion at m/z 119 observed when the reaction mixture was electrosprayed, fragmented by loss of a HCN molecule during CID (Fig. S1B†). This strongly suggests that the ion corresponds to protonated benzimidazole formed as a result of the reaction between PDA and FA. We verified this assignment by comparison with the MS/MS spectrum of the authentic compound (Fig. S2B†). In addition to the product, m/z 119, the mass spectrum of the reaction mixture shows a peak at m/z 137, which corresponds to a hydrated intermediate. This ion might be the formamide (i.e.2c in Scheme 2) in acyclic or cyclized form, each including two tautomers. This mass-selected ion dissociates under CID to give a major peak at m/z 109 and a minor peak at m/z 119 (Fig. S1C†) consistent with both the acyclic formamide (which fragments back to starting material) and with the cyclic product (which undergoes dehydration to the benzimidazole).
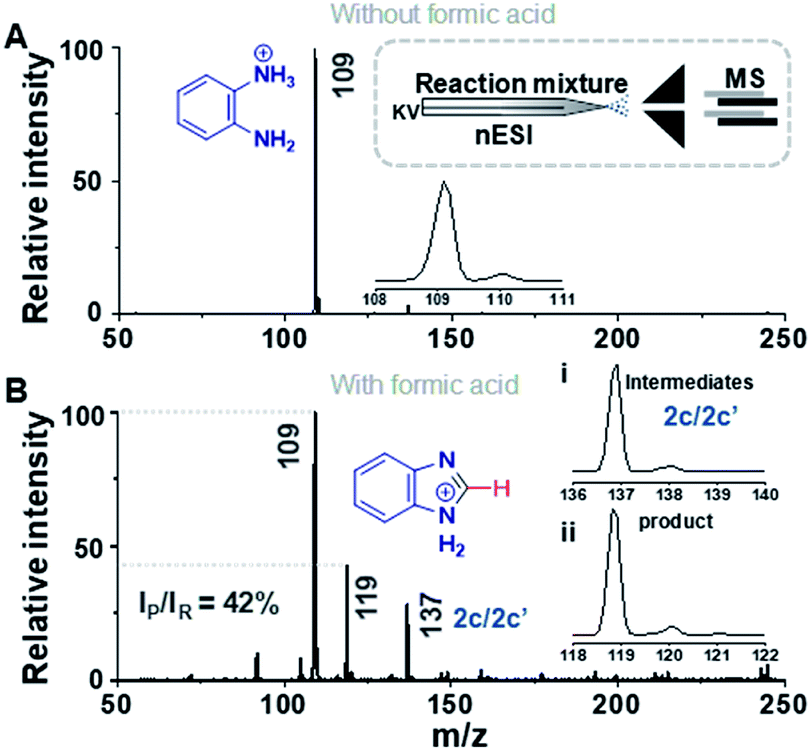 | ||
| Fig. 1 Microdroplet synthesis of benzimidazole. (A) Mass spectrum of PDA in methanol without addition of formic acid. Insets show the isotopic distribution of the peak at m/z 109 and a schematic illustration of the setup. (B) Mass spectrum of PDA with FA at 1:1 molar ratio in methanol. Insets show the isotopic distributions of the peaks at (i) m/z 137 and (ii) m/z 119. IP and IR refer to the intensity of signals for product and reactant, respectively. | ||
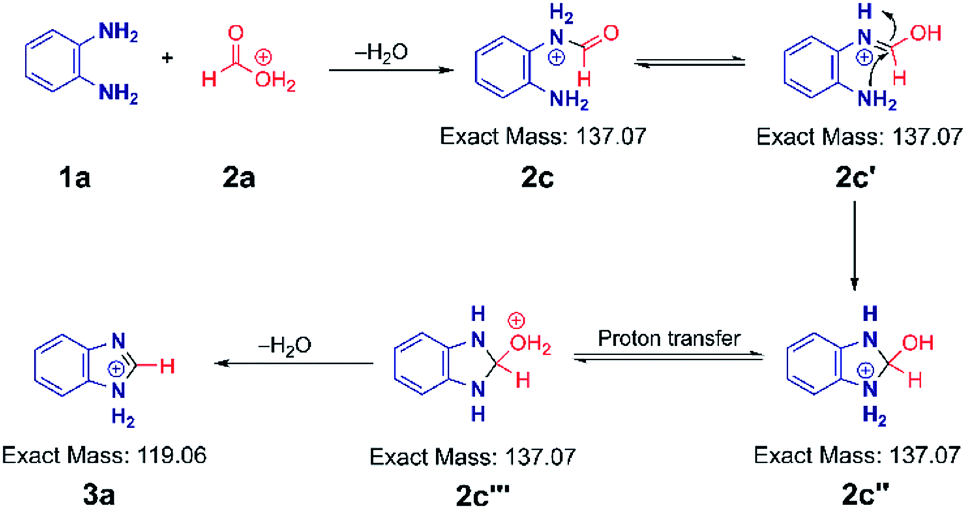 | ||
| Scheme 2 Proposed mechanism for the acid-catalysed benzimidazole synthesis in charged microdroplets. A similar mechanism can be written starting from the more stable carbonyl protonated form of the acid. | ||
A similar electrospray reaction was conducted between PDA and AA. This reaction produces 2-methylbenzimidazole, seen as the protonated form at m/z 133 in the full mass spectrum (Fig. S3A†), and confirmed by comparison of its MS/MS spectrum (Fig. S3B†) with that of the authentic compound (Fig. S4B†). The peak at m/z 151 in the full mass spectrum is assigned to the formation of the intermediate amide. Fragmentation of this ion shows a major peak at m/z 109, which corresponds to the starting reagent and, most significantly, it shows neutral loss of a water molecule to form the protonated reaction product at m/z 133 (Fig. S3C†).
To confirm that these reactions do not occur upon immediate mixing of the reagents in methanol, UV-vis and 1H NMR spectra of the authentic products and the reaction mixtures of the benzimidazole and 2-methylbenzimidazole synthesis were obtained. 1H NMR experiments were performed in deuterated chloroform for benzimidazole and the reaction mixture used to generate it, while deuterated methanol was used for 2-methylbenzimidazole and its reaction mixture. The mixtures of reagents were kept for 10 minutes at room temperature before recording the spectra. The UV-vis spectrum of the PDA/FA mixture (Fig. S5†) shows an expected small shift compared to the PDA spectrum upon addition of formic acid, which protonates the amine. However, the spectral signatures corresponding to the product were not observed in the UV-vis spectrum of the reaction mixture or in the 1H NMR spectra of these solutions (Fig. S6–S9†). Analogous results were obtained for the reaction between PDA and AA (Fig. S10–S14†). These results confirm that the reagents do not react in bulk.
Rough estimates of the yields of the microdroplet synthesis reactions were obtained by measuring the conversion ratio (CR): the ratio of the intensity between the product (P) and the sum of the intensities of the starting material (SM), intermediate (I), and product, viz., [P]/([SM] + [P] + [I]).11 Note that this estimate does not correct for differences in ionization energy so more accurate yields were obtained by collecting and purifying product (see below). The conversion ratios for the benzimidazole and 2-methylbenzimidazole syntheses were 27% and 3%, respectively (measured with MS inlet capillary temperature at 50 °C). This difference in the conversion ratio is surprisingly large but consistent with the expected lower electrophilicity of the methyl substituted carbon in AA vs. the unsubstituted carbon of FA. We noticed a change in the conversion ratios as a function of the mass spectrometer inlet capillary temperature as is now discussed.
Temperature effects
Studies of the Pomeranz–Fritsch and the Combes reactions showed that the temperature of the MS inlet can play an important role, with increasing inlet temperatures having opposite effects on the rate of these two reactions.46 In the benzimidazole synthesis, a significant increase in the product yield was observed upon increasing the inlet temperature. Both the FA and the AA reactions showed maximum conversion ratios at higher, albeit different, temperatures (Fig. 2A and B). In the case of PDA/FA a maximum was observed at 300 °C while for PDA/AA the maximum was reached at 350 °C. It was also noted in both cases, that the intensity of the intermediate peak decreases and almost disappears as the temperature is raised above 200 °C. Mass spectra of the reaction mixtures for the benzimidazole and 2-methylbenzimidazole syntheses at eight different temperatures ranging from 50 to 400 °C are shown in Fig. S15.† Based on the temperature dependence for PDA/FA (Fig. S15A†) the relative abundance of the product peak at m/z 119 increases from 50 to 180 °C and it becomes the base peak at 200 °C, decreasing beyond this point. The intermediate peak at m/z 137 also decreases sharply in this temperature range (Fig. S16†). The PDA/AA reaction shows a similar trend but over a higher temperature range (270 to 400 °C) and with a sharper increase in product signal, m/z 133 (Fig. S15B†). These observations also show that ionization efficiency decreases with increasing temperature so, in spite of the increase in product ions through conversion to reagents to products, there is a net decrease at high temperature.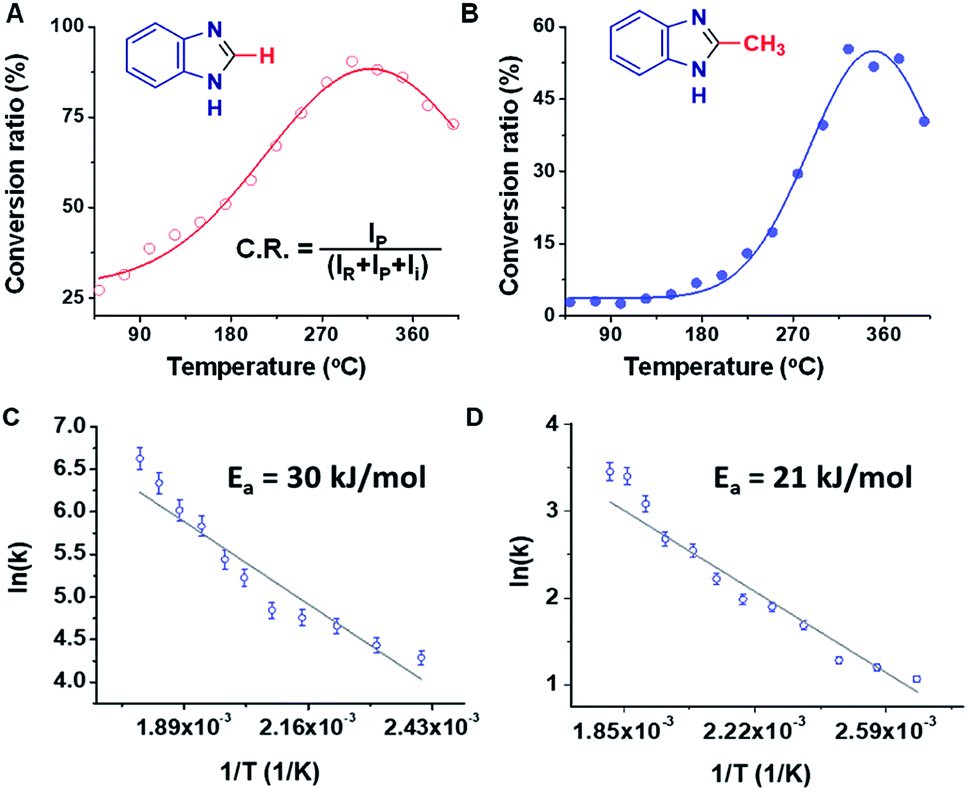 | ||
| Fig. 2 Effect of inlet temperature of the mass spectrometer on ion intensity, showing conversion ratio (CR) vs. temperature plot for synthesis of (A) benzimidazole and (B) 2-methylbenzimidazole. The equation inset in (A) is used to calculate conversion ratio, where R, P, and I refer to reagent, product and intermediate, respectively. Plots (C) and (D) display temperature-dependent rates plotted against 1/T and give activation energies for the reactions leading to benzimidazole and 2-methylbenzimidazole, respectively. | ||
The temperature data are ascribed to removal of water shifting the dehydration/rehydration equilibrium to favor product formation. Although the effect of temperature is expected to be different for the different reactions because of their different energy requirements, the large difference between the product peak maxima (180 °C for benzimidazole but 350 °C for 2-methyl benzimidazole) is noteworthy. A plot of the inverse temperature dependence of these two reactions allows an estimation of activation energies. This is done in Fig. 2C and D, from which the values are 30 kJ mol−1vs. 21 kJ mol−1.
Mechanism (Scheme 2)
Based on the above data, we speculate that the reaction must go through an acid catalyzed pathway, but not one driven by protonation of the amine. It is known that the proton concentration at the surface of positively charged electrosprayed aqueous droplets is much higher than in the bulk.47–49 So we propose that a very high proton concentration at the surface of a droplet containing water will protonate neutral formic acid to create the electrophilic carbon (2a) which is subject to nucleophilic attack by the lone electron pair of the amine. (We observed protonated formic and trifluoracetic acid in the mass spectra (Fig. S17A and B†). This was confirmed by an MS/MS experiment (Fig. S17C†) on protonated TFAA). Subsequent loss of water results in the formation of a formamide intermediate (2c). Intramolecular nucleophilic attack at the carbon in the enol form of the intermediate (2c′) by the lone pair of the adjacent nitrogen forms the five-membered cyclic intermediate (2c′′). Finally, dehydration of its tautomer (2c′′′) leads to protonated benzimidazole (3a) as the final product. The intermediates 2c, 2c′, 2c′′ and 2c′′′ are indistinguishable by mass/charge ratio, all occurring at m/z 137 (Fig. 1B). The MS/MS spectrum of m/z 137 (Fig. S1C†) shows that this ion can either fragment back to protonated PDA (m/z 109) by loss of CO or lose water to form the benzimidazole product (m/z 119). These two processes give us confidence in the fact that the ion m/z 137 encompasses several isomers, as illustrated in Scheme 2. An analogous intermediate, m/z 151, is noted for the PDA/AA reaction and its fragmentation pattern (Fig. S3B†) confirms this.The mechanism illustrated in Scheme 2 was also tested using formic-d acid (DCOOH). We expected isotopically labeled intermediates and product according to the proposed reaction pathway and observed the deuterated benzimidazole at m/z 120 and the deuterated intermediate at m/z 138 (Fig. S18A†). Fragmentation of the m/z 138 peak gives the protonated reagent (m/z 109) as well as the deuterated product (m/z 120). Analogously, isotopically labeled 2-methylbenzimidazole at m/z 136 and the intermediate at m/z 154 were observed with CD3COOH (Fig. S18B†). These results agree with expectations and support the suggestion that reaction occurs through a protonated carboxylic acid pathway.
The fact that acids catalyze the formation of benzimidazole is well known.50 We suggest that the extraordinary acidity of microdroplets catalyzes the reaction on the milli- or micro-second time scale. Only in a formal sense (acid is not added to the reaction mixture) could one say that the droplet reaction is not acid catalyzed. More evidence for acid catalysis is shown below in connection with changes in solvent to increase the proportion of water.
Thin film reactions
Our view of the reaction mechanism is that as the droplets evaporate, the increasingly high acidity of the droplet surface allows protonation of the carboxylic acid followed by nucleophilic attack by an amino group and dehydration, before cyclization and then a second dehydration event. It seems likely that the dehydration of the intermediate is the slow step in the reaction, hence the effect of the inlet temperature. If these arguments are correct, then it should be possible to observe the accelerated reaction entirely at ambient temperature (keeping the MS inlet at the lowest possible temperature of 50 °C). To test this claim, a thin film of reaction mixture in methanol was held at ambient temperature in the open lab. The mixture contained a trace (2.5 ppm) of a non-volatile acid, p-toluenesulfonic acid. This was added as a result of earlier ambient temperature accelerated thin film reactions11 in which traces of nonvolatile additives allowed continuous long-term reaction involving elimination of water or other small molecules.51 The nonvolatile acid helps to create and maintain a micro/nano-thin film. Kinetic data were recorded by MS (Fig. S19†) to follow benzimidazole and 2-methylbenzimidazole formation in this thin film experiment. In parallel with earlier studies on the base-catalyzed Katritzky transamination, we see that the reaction does not occur during an initial period of some minutes (while most of the methanol is evaporating) but it then increases in rate before plateauing after 6 hours, with 120% and 16% product-to-reagent peak intensity ratios for benzimidazole and 2-methylbenzimidazole, respectively. Clearly, the reactions are accelerated in the thin film compared to bulk, and increasingly so as the film becomes thinner with continued methanol evaporation. However, the reaction rate is moderate in comparison to the ambient droplet phase synthesis (using ESSI as discussed below) and small in comparison to the elevated temperature nESI microdroplet synthesis already discussed. The large differences in rate associated with simple methyl substitution are also replicated. These data support the conclusions on the reaction mechanism already outlined.Solvent effects
The mechanistic arguments also need to include consideration of the solvent system, especially because we invoke a highly acidic environment at the droplet surface. Experiments done by varying the percentage of water in methanol over the range 1–50% showed that a low percentage of water (1–10%) favors the reaction (Fig. S20†). The relative product plus intermediate ion intensity increases from 62 to 95% over this range. However, at higher water concentrations the relative intensity of the product plus intermediate peaks decreases significantly to 52–54% and when the reaction is performed in dry ACN we observe no product at all (Fig. S21†). These results are consistent with a balance between reagent solubility, methanol evaporation and formation of a superacid surface layer of water in the almost fully evaporated microdroplets. This clearly aligns with literature data47–49 on the high acidity of the surfaces of aqueous droplets while extending the concept of superacid formation in microdroplets to organic/aqueous mixed solvents. We imagine that water, being a stronger base than methanol, takes the charge and occupies the surface of a small methanol droplet thereby protonating neutral formic acid and initiating the benzimidazole synthesis. The final (dehydration) step, however, is impeded in aqueous solutions.pH dependence
We assessed the effect of decreasing the pH of the reaction mixture by adding equimolar concentrations of a strong inorganic acid, HCl. In the case of the microdroplet reaction of PDA/FA, addition of HCl does not cause significant changes in the mass spectrum of the reaction mixture (Fig. S22†). This lack of observable effect of pH reduction is consistent with the suggestion that the superacid at the surface of positively-charged water-containing droplets is responsible for protonating the carboxylic acid. The addition of HCl will reduce the pH in the core of the droplet, without changing the protonation of the carboxylic acid at the surface.Acceleration factors
The apparent acceleration factor (AAF) of a reaction can be calculated by comparing the product to starting material intensity ratio in the microdroplet reaction to the value in the bulk reaction carried out for the same period of time, viz. ([P]/[SM])droplet/([P]/[SM])bulk.33 Alternatively, it can be measured by recording the ratio of the time required to reach the same conversion ratio for both the conditions. To measure the AAF we performed a bulk reaction using an equimolar ratio of PDA and AA in methanol at ambient temperature. The AAF was calculated from the kinetic data as plotted in Fig. S23.† This simple method of approximating the apparent acceleration factor (not the true acceleration factor which is the ratio of rate constants) shows that the microdroplet reaction is accelerated by a very large factor (almost 109). This is not intended as a quantitative measure because the role of the inlet temperature in driving the reaction has not been considered.We compared high temperature microdroplet data with bulk reaction data obtained by refluxing the reaction mixture in methanol for 24 hours. Comparison was made between product formation in this bulk reaction (analysed using inlet temperature of 50 °C to minimize droplet reactions during analysis), with that in microdroplet reactions using 200 °C inlet temperature (Fig. S24A†). The data show that the relative intensity of the product peak at m/z 133 is significantly lower (ca. ×5) in the bulk than in the microdroplet experiment in spite of the difference of many orders of magnitude in reaction time. This difference becomes even more significant with higher inlet temperatures, which provide exponentially larger reaction acceleration factors (Fig. S24B†). The apparent acceleration factor at 50 °C inlet temperature was 1.1 × 108 and it increased to 6.6 × 108 at 350 °C. This is just one example of a very high AAF, others are shown later in the article. Factors of similar magnitude have been reported for the Biginelli reaction where similar temperature effects are associated with the last step of a reaction.52
Scope of ambient droplet reaction
The results of a systematic study of the scope of the microdroplet synthesis of benzimidazoles was undertaken, now using ambient temperatures accessed by ESSI. This procedure not only allows the entire reaction to occur at ambient temperature but it also allows ready scale up to several hundred mg. MS and NMR were used to characterize and quantify the reaction products which were collected on a surface rather than being directly transferred into the MS. The results are summarized in Table 1, which gives the experimental conversion ratio (CR), the apparent acceleration factor (AAF) and the experimentally determined yield, as measured after product isolation by flash chromatography.|
|
||||||
|---|---|---|---|---|---|---|
| Entry | Diamine 1 | Acid 2 | Product 3 | AAF × 109 | C.R. | Yield% |
|
a The reactions were performed using a home built ESSI source. Each diamine (200 mg) was used in a 1:1 molar ratio with the acid in methanol. The flow rate and the gas pressure used for droplet deposition were 10 μL min−1 and 30 psi. The ESSI experiment was done under ambient conditions (in contrast to the nESI data reported above, which used an inlet temperature of 50 °C unless otherwise indicated). In a few cases the CR and the yield trends differ, likely due to poor product separation by flash chromatography.
|
||||||
| 1 |
|
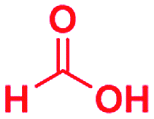
|
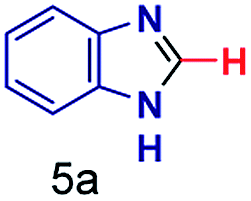
|
0.83 | 27 | 72 |
| 2 |
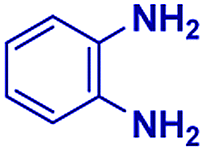
|
|
|
0.74 | 3 | 16 |
| 3 |
|
|
|
0.11 | 18 | 22 |
| 4 |
|

|
|
25 | 1 | 10 |
| 5 |
|
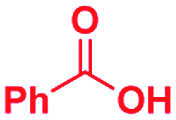
|
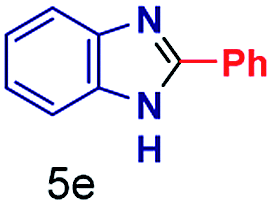
|
0.02 | 5 | 5 |
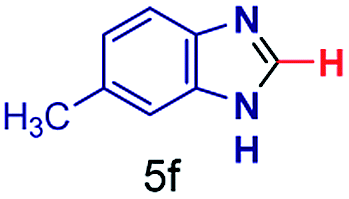
| 6 |
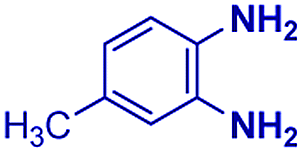
|

|
|
0.25 | 37 | 93 |
| 7 |
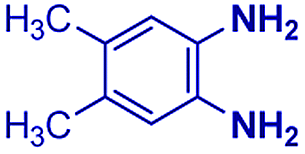
|
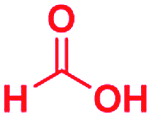
|
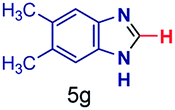
|
4.2 | 3 | 38 |
| 8 |
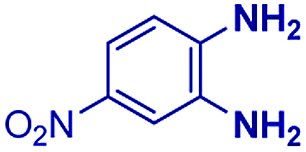
|
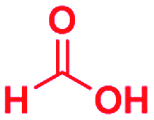
|
|
No reaction | No reaction | No reaction |
| 9 |
|
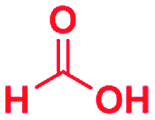
|

|
0.59 | 14 | 60 |
| 10 |
|
|
|
0.04 | 36 | 67 |
| 11 |
|
|
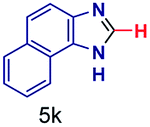
|
200 | 6 | 29 |
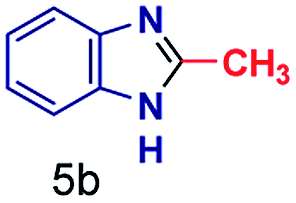
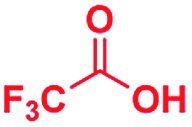
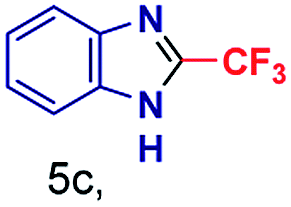
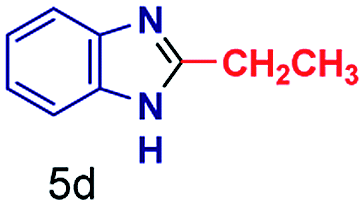
The mass spectra of the isolated products typically show a single peak due to the protonated product with no trace of reagents or intermediates, as typified by benzimidazole (Fig. S25†). The 1H NMR of this isolated product shown in Fig. S26† also demonstrates moderately high purity. We found the actual yield of the product of benzimidazole synthesis to be 72% (entry 1, Table 1). However, the yield was reduced to 16% in the case of 2-methylbenzimidazole (entry 2, Table 1) presumably due to the inductive effect of the methyl group in the carboxylic acid, which weakens the electrophilic center of the acid. MS and NMR spectra are presented in Fig. S27 and S28.† Product isolation using flash chromatography was not always effective in separating all the products as reflected in these mass and NMR spectra. Other 2-substituted benzimidazoles were synthetized by varying the structures of the carboxylic acids. An electron withdrawing group such as CF3 in the carboxylic acid increases the yield to 22% (entry 3, Table 1). The ethyl substituted benzimidazole (entry 4, Table 1) has a 10% yield, similar to the methyl substituted case. Correspondingly, the phenyl substituent on the carboxylic acid provides a lower yield (5%) due to resonance electron donation.
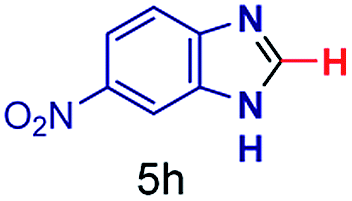
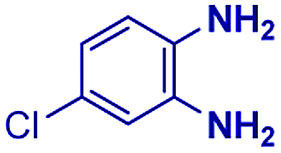
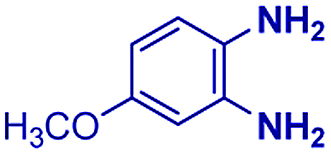
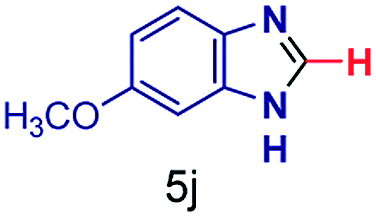
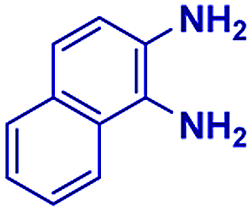
The reaction scope was further extended by performing reactions between formic acid and several substituted o-aryl diamines, specifically: 4-methyl-1,2-phenylenediamine, 4,5-dimethyl-1,2-phenylenediamine, 4-nitro-1,2-phenylenediamine, 4-chloro-1,2-phenylenediamine, 4-methoxy-1,2-phenylenediamine and 1,2-diaminonaphthalene, to synthesize 4-methylbenzimidazole (5f), 4,5-dimethylbenzimidazole (5g), 4-nitrobenzimidazole (5h), 4-chlorobenzimidazole (5i), 4-methoxybenzimidazole (5j), and naphth[1,2]imidazole (5k), respectively (entries 6–11 in Table 1). It is observed that for the first two cases the inductive effect facilitates product formation leading to 93 and 38% product yields, respectively. We suspect that a steric effect reduces the product yield in the case of the dimethyl substituted amine. However, the conjugated strong electron withdrawing group (nitro-) results in no product formation (entry 8, Table 1), while the chloro- and the methoxy-substituted products (entry 9 and 10, Table 1) have yields of 60% and 67%, respectively. We also extended the method to the synthesis naphth[1,2]imidazole (entry 11, Table 1) and observed product in 29% of yield, with a very high AAF of the order of 2 × 1011. 1H NMR and mass spectra of the isolated products are available in the ESI.†
There is a rough correlation between the experimentally observed isolated yields and the experimentally estimated acceleration factors (as seen in Table 1). There is also rough agreement with the electron donating/withdrawing character of the diamine and the carboxylic acid (with some exceptions potentially due to steric effects). The AAF for the benzimidazole synthesis (0.83 × 109) is on the same order of other reactions with substituted amines and carboxylic acids. However, the increase of AAF in case of the reactions of PDA/PA (2.5 × 1010), 4,5-dimethyl PDA/FA (4.2 × 109) and naphthalene-1,2-diamine/FA (2 × 1011) may be due to their low reactivity in bulk because of steric effects. The high yield (and conversion ratio) for the methoxyphenylenediamine (entry 10, Table 1) is consistent with the expected high reactivity of the amine due the electron donation by the oxygen of the methoxyl group. These relationships provide strong evidence for the nucleophilic attack at the carboxylic acid carbon which must be ascribed to the extraordinary acidity at the droplet (and thin film) interfaces.
Experimental
Online reaction monitoring
Online monitoring was carried out using a nESI source in which the reagents were mixed prior to being sprayed. The nESI tip (5 μm diameter) was held 1 cm away from the inlet of the mass spectrometer. The applied potential for spray generation was kept at 2–2.5 kV. The capillary and tube lens voltages were set to 50 and 100 V, respectively. For the temperature dependence study, the capillary temperature was initially set to 50 °C but later increased to a final temperature of 400 °C. Unless otherwise noted, the mass spectrometric studies were performed at 50 °C inlet temperature. All mass spectra were recorded by averaging 4 microscans of 100 ms scan times. A linear ion trap was used for all MS studies, and CID with He as collision gas was used for MS/MS analysis.Thin film synthesis
Thin film reactions were performed by applying a thin film of the reactants onto the surface of parafilm. The parafilm was wrapped around a glass slide to prevent catalysis by the glass. The thin film of reaction mixture was produced by drop-casting 5 μL of mixture containing 2.5 ppm of p-toluenesulfonic acid onto the parafilm surface. The film was allowed to dry slowly in ambient air. Sampling was done by transferring the crude product into 10 μL of methanol and then quenching by diluting the sample ×10 in methanol before measuring by nESI. Measurements at different times were done using different thin films. All thin film reactions were performed in triplicate to verify reproducibility.Scaled-up reactions and product isolation
Scaling-up and product deposition used ambient ESSI. Scale-up was achieved by increasing the flux of droplets using a high flow rate of the solution in a fused silica capillary (150 μm of ID and 300 μm of OD). A flow rate of 10 μL min−1 was used with +3 kV applied potential and 30 psi nebulization N2 gas pressure. HPLC grade methanol was used for the preparation of all the reaction mixtures. The amount of starting material used for the deposition experiment was 200 mg in each case. The collected crude product mixture was dissolved in methanol to perform thin layer chromatography to allow optimization of the solvent system for better separation of product from the crude. With this information the crude reaction mixture was separated using normal-phase silica flash column chromatography in a Biotage automated flash chromatography column. A methanol and DCM solvent gradient was selected for column chromatography based on the Rf value of the reagent and the product. The fractionated solutions were characterized using nESI MS and 1H NMR. The fractions which contained the product were mixed and solvent was removed at reduced pressure to obtain isolated solid product. Yields of the microdroplet reactions were calculated using the mass of the recovered solid.Bulk synthesis
The bulk synthesis of 2-methylbenzimidazole was performed by mixing equimolar methanolic solutions of PDA and AA in a 500 mL two-neck round bottom flask fitted with a condenser. The final concentrations of each reagent in the mixture was 8 mM. The reaction mixture was then stirred and refluxed using a sand bath for continuous heating for 24 hours. The time dependent studies were carried out by removing 15 μL aliquots of sample at fixed time intervals. Bulk reactions were also carried out under ambient conditions and characterized in a similar way.Monitoring of time dependent mass spectra of the bulk reaction mixture was performed after dilution. The dilution was done to quench the reaction as well as to avoid microdroplet synthesis. Inlet temperature was also set to 50 °C and the tip of the nESI emitter was kept as close as possible to the inlet of the mass spectrometer to prevent reaction during bulk product analysis. The applied potential to generate the electrospray plume was 1 kV. ([P]/[SM])bulk data of the 24 hour sample was used to calculate the apparent acceleration factor.
Conclusions
It is commonplace knowledge that amines and carboxylic acids react in Brønsted fashion to produce a salt, while amines react with carboxylic acid halides by nucleophilic substitution to give amides. The corresponding nucleophilic substitution with an acid would require the carboxylic acid to be protonated to give the conjugate base which could then eliminate water. This situation would call for extraordinary conditions in which a very strong acid is present so that not only is the ionization of the acid to its conjugated base suppressed by the common ion effect, but the neutral acid itself is protonated so it can react as a carbon-centered Lewis acid. These conditions are satisfied in the benzimidazole chemistry examined in this study.Accelerated microdroplet synthesis of benzimidazole and its derivatives have been demonstrated under ambient conditions. The reaction involves aromatic-1,2-diamines and carboxylic acids in a metal free environment with no requirement of addition of base to complete the product formation. Online mass spectrometric monitoring enables detection of reaction intermediates and provides an understanding of the reaction mechanism. Ten examples are shown to illustrate the scope of the microdroplet synthesis.
The described mechanism of benzimidazole synthesis has three elements: (i) the formation of a highly reactive reagent because of the extraordinary conditions in at the interface (protonated of the carboxylic acid), (ii) the accelerated bimolecular reaction because of limited solvation at the interface and (iii) the thermally assisted elimination of water. Point (i) is supported by the effect of adding water to the methanol solvent. Point (ii) is suggested by analogy to other organic solvent droplet reactions where this mechanism is supported by experimental and recent computations.53 Point (iii) is shown directly by the experiments. Note that bimolecular reaction acceleration is seen in formation of the intermediate amide and that the thermal elimination of water is a subsequent and secondary factor. We think that many other droplet-accelerated reactions will contain some elements in common with this case.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
PB thanks IIT Madras and acknowledges a visiting doctoral fellowship from the Science and Engineering Board of India. RGC acknowledges support of the US National Science Foundation (1908057).References
- P. Ruiz-Castillo and S. L. Buchwald, Chem. Rev., 2016, 116, 12564–12649 CrossRef CAS PubMed.
- E. Negishi, Pure Appl. Chem., 1981, 53, 2333–2356 CAS.
- T. Cernak, K. D. Dykstra, S. Tyagarajan, P. Vachal and S. W. Krska, Chem. Soc. Rev., 2016, 45, 546–576 RSC.
- L. M. Baumgartner, J. M. Dennis, N. A. White, S. L. Buchwald and K. F. Jensen, Org. Process Res. Dev., 2019, 23, 1594–1601 CrossRef CAS.
- X. Yan, R. M. Bain and R. G. Cooks, Angew. Chem., Int. Ed., 2016, 55, 12960–12972 CrossRef CAS PubMed.
- S. T. Ayrton, R. G. Cooks and M. Pugia, Analyst, 2016, 141, 5389–5403 RSC.
- R. D. Espy, M. Wleklinski, X. Yan and R. G. Cooks, TrAC, Trends Anal. Chem., 2014, 57, 135–146 CrossRef CAS.
- A. K. Badu-Tawiah, D. I. Campbell and R. G. Cooks, J. Am. Soc. Mass Spectrom., 2012, 23, 1077–1084 CrossRef CAS PubMed.
- A. Saidykhan, Y. Nazir, W. H. C. Martin, R. T. Gallager and R. D. Bowen, Eur. J. Mass Spectrom., 2017, 24, 3–11 CrossRef PubMed.
- R. M. Bain, S. T. Ayrton and R. G. Cooks, J. Am. Soc. Mass Spectrom., 2017, 28, 1359–1364 CrossRef CAS PubMed.
- Z. Wei, X. Zhang, J. Wang, S. Zhang, X. Zhang and R. G. Cooks, Chem. Sci., 2018, 9, 7779–7786 RSC.
- S. Banerjee and R. N. Zare, Angew. Chem., Int. Ed., 2015, 54, 14795–14799 CrossRef CAS PubMed.
- R. M. Bain, C. J. Pulliam, F. Thery and R. G. Cooks, Angew. Chem., Int. Ed., 2016, 55, 10478–10482 CrossRef CAS PubMed.
- K. Anand and S. Wakode, Int. J. Chem. Stud., 2017, 5, 350–362 CAS.
- B. Chu, F. Liu, L. Li, C. Ding, K. Chen, Q. Sun, Z. Shen, Y. Tan, C. Tan and Y. Jiang, Cell Death Dis., 2015, 6, e1686 CrossRef CAS PubMed.
- A. Bettarello, Dig. Dis. Sci., 1985, 30, 36S–42S CrossRef CAS PubMed.
- Y.-B. Bai, A.-L. Zhang, J.-J. Tang and J.-M. Gao, J. Agric. Food Chem., 2013, 61, 2789–2795 CrossRef CAS PubMed.
- M. Tonelli, M. Simone, B. Tasso, F. Novelli, V. Boido, F. Sparatore, G. Paglietti, S. Pricl, G. Giliberti, S. Blois, C. Ibba, G. Sanna, R. Loddo and P. La Colla, Bioorg. Med. Chem., 2010, 18, 2937–2953 CrossRef CAS PubMed.
- L. H. Jaeger and F. A. Carvalho-Costa, BMC Vet. Res., 2017, 13, 358 CrossRef PubMed.
- M. J. Warren, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 4799 CrossRef CAS PubMed.
- A. Bino, A. Baldisserotto, E. Scalambra, V. Dissette, D. E. Vedaldi, A. Salvador, E. Durini, S. Manfredini and S. Vertuani, J. Enzyme Inhib. Med. Chem., 2017, 32, 527–537 CrossRef CAS PubMed.
- D. V. Dikshit, S. D. Deval and K. D. Deodhar, Dyes Pigm., 1985, 6, 39–46 CrossRef CAS.
- E. C. Wagner and W. H. Millett, Org. Synth., 1939, 19, 12–14 CrossRef CAS.
- P. N. Preston, Chem. Rev., 1974, 74, 279–314 CrossRef CAS.
- D. Mahesh, P. Sadhu and T. Punniyamurthy, J. Org. Chem., 2015, 80, 1644–1650 CrossRef CAS PubMed.
- K. Das, A. Mondal and D. Srimani, J. Org. Chem., 2018, 83, 9553–9560 CrossRef CAS PubMed.
- P. Daw, Y. Ben-David and D. Milstein, ACS Catal., 2017, 7, 7456–7460 CrossRef CAS.
- J. E. R. Sadig, R. Foster, F. Wakenhut and M. C. Willis, J. Org. Chem., 2012, 77, 9473–9486 CrossRef CAS PubMed.
- Y. Kim, M. R. Kumar, N. Park, Y. Heo and S. Lee, J. Org. Chem., 2011, 76, 9577–9583 CrossRef CAS PubMed.
- W. Cui, R. B. Kargbo, Z. Sajjadi-Hashemi, F. Ahmed and J. F. Gauuan, Synlett, 2012, 23, 247–250 CrossRef CAS.
- J. Kovvuri, B. Nagaraju, A. Kamal and A. K. Srivastava, ACS Comb. Sci., 2016, 18, 644–650 CrossRef CAS PubMed.
- S. Lin, Y. Isome, E. Stewart, J. Liu, D. Yohannes and L. Yu, Tetrahedron Lett., 2006, 47, 2883–2886 CrossRef CAS.
- P. W. Fedick, K. Iyer, Z. Wei, L. Avramova, G. O. Capek and R. G. Cooks, J. Am. Soc. Mass Spectrom., 2019, 30, 2144–2151 CrossRef CAS PubMed.
- R. L. Schrader, P. W. Fedick, T. F. Mehari and R. G. Cooks, J. Chem. Educ., 2019, 96, 360–365 CrossRef CAS.
- C. Liu, J. Li, H. Chen and R. N. Zare, Chem. Sci., 2019, 10, 9367–9373 RSC.
- D. Gao, F. Jin, J. K. Lee and R. N. Zare, Chem. Sci., 2019, 10, 10974–10978 RSC.
- E. Gnanamani, X. Yan and R. N. Zare, Angew. Chem., Int. Ed., 2020, 59, 3069–3072 CrossRef CAS PubMed.
- R. M. Bain, S. Sathyamoorthi and R. N. Zare, Angew. Chem., Int. Ed., 2017, 56, 15083–15087 CrossRef CAS PubMed.
- C.-L. Zhang and S.-H. Yu, Chem. Soc. Rev., 2014, 43, 4423–4448 RSC.
- D. Sarkar, M. K. Mahitha, A. Som, A. Li, M. Wleklinski, R. G. Cooks and T. Pradeep, Adv. Mater., 2016, 28, 2223–2228 CrossRef CAS PubMed.
- D. Sarkar, R. Singh, A. Som, C. K. Manju, M. A. Ganayee, R. Adhikari and T. Pradeep, J. Phys. Chem. C, 2018, 122, 17777–17783 CrossRef CAS.
- Y. Yang, B. Song, X. Ke, F. Xu, K. N. Bozhilov, L. Hu, R. Shahbazian-Yassar and M. R. Zachariah, Langmuir, 2020, 36, 1985–1992 CrossRef CAS PubMed.
- H. Fang, D. Wang, L. Yuan, X. Wu, H. Guo, H. Chen, K. Huang and S. Feng, New J. Chem., 2017, 41, 2878–2882 RSC.
- R. Naphade, S. Nagane, G. S. Shanker, R. Fernandes, D. Kothari, Y. Zhou, N. P. Padture and S. Ogale, ACS Appl. Mater. Interfaces, 2016, 8, 854–861 CrossRef CAS PubMed.
- A. Reiser, M. Lindén, P. Rohner, A. Marchand, H. Galinski, A. S. Sologubenko, J. M. Wheeler, R. Zenobi, D. Poulikakos and R. Spolenak, Nat. Commun., 2019, 10, 1853 CrossRef PubMed.
- S. Banerjee and R. N. Zare, J. Phys. Chem. A, 2019, 123, 7704–7709 CrossRef CAS PubMed.
- H. Wei, E. P. Vejerano, W. Leng, Q. Huang, M. R. Willner, L. C. Marr and P. J. Vikesland, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, 7272–7277 CrossRef CAS PubMed.
- J. L. Lee, D. Samanta, H. G. Nam and R. N. Zare, J. Am. Chem. Soc., 2019, 141, 10585–10589 CrossRef CAS PubMed.
- K. Luo, J. Li, Y. Cao, C. Liu, J. Ge, H. Chen and R. N. Zare, Chem. Sci., 2020, 11, 2558–2565 RSC.
- I. Mohammadpoor-Baltork, M. Moghadam, S. Tangestaninejad, V. Mirkhani, M. A. Zolfigol and S. F. Hojati, J. Iran. Chem. Soc., 2008, 5, S65–S70 CrossRef CAS.
- Z. Wei, M. Wleklinski, C. Ferreira and R. G. Cooks, Angew. Chem., Int. Ed., 2017, 56, 9386–9390 CrossRef CAS PubMed.
- N. Sahota, D. I. AbuSalim, M. L. Wang, C. J. Brown, Z. Zhang, T. J. El-Baba, S. Cook and D. E. Clemmer, Chem. Sci., 2019, 10, 4822–4827 RSC.
- N. Narendra, X. Chen, J. Wang, J. Charles, R. G. Cooks and T. Kubis, J. Phys. Chem. A, 2020, 124, 4984–4989 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: MS, MS/MS, 1H NMR and UV-vis spectra; kinetic plots; experimental diagrams. See DOI: 10.1039/d0sc02467h |
| This journal is © The Royal Society of Chemistry 2020 |