DOI:
10.1039/D0SC01017K
(Minireview)
Chem. Sci., 2020,
11, 7553-7561
Synthesis of azetidines by aza Paternò–Büchi reactions
Received
20th February 2020
, Accepted 6th May 2020
First published on 12th June 2020
Abstract
The [2 + 2] photocycloaddition reaction between an imine and an alkene component, the aza Paternò–Büchi reaction, is one of the most efficient ways to synthesize functionalized azetidines. However, the application of the aza Paternò–Büchi reaction has been met with limited success due to the inherent challenges associated with this approach. This review covers the current scope and limitations of reported examples of aza Paternò–Büchi reactions in organic synthesis. An outlook is provided, which highlights recent improvements and the discovery of new reaction protocols that have overcome some long-standing challenges within this field of research.
Introduction
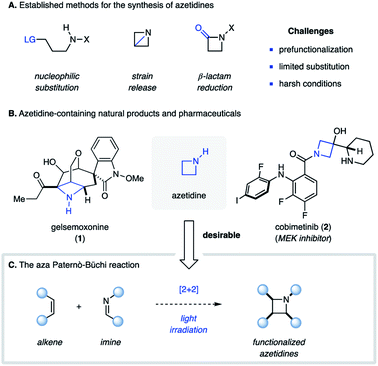
The photochemical [2 + 2] cycloaddition reaction is a powerful tool for the construction of four-membered carbocyclic and heterocyclic organic compounds.1 This approach can be considered among the most efficient methods for the rapid assembly of small rings as it typically proceeds in a single synthetic operation with high regio- and stereoselectivity. As a result, there exist numerous examples in the literature, in which [2 + 2] photocycloaddition reactions have been used for the synthesis of complex natural products.2 The most commonly employed type is the photocycloaddition between two alkene components to afford cyclobutanes, in which upon excitation of one of the reaction partners two new carbon–carbon bonds are formed.3 Similarly, excitation of a carbonyl compound in the presence of an alkene can result in a [2 + 2] photocycloaddition reaction to provide an oxetane as product, known as the Paternò–Büchi reaction.4 In contrast, the corresponding aza Paternò–Büchi reaction, in which an imine and alkene compound react under photochemical conditions to form an azetidine, is far less developed. Azetidines exist in numerous natural products (e.g.1)5 and studies have shown that incorporation of azetidines into pharmaceutically relevant scaffolds (e.g.2) can result in improved pharmacokinetic properties as well as metabolic stability.6 Established methods for the synthesis of azetidines rely on nucleophilic substitution reactions with nitrogen nucleophiles, the reduction of β-lactams7 or the ring-opening of highly strained azabicyclobutanes (Fig. 1A).8 Considering that most of these approaches require prefunctionalized starting materials and involve multistep sequences, aza Paternò–Büchi reactions represent a highly efficient and direct strategy for the synthesis of azetidines (Fig. 1C).
 |
| | Fig. 1 Synthetic methods to access azetidines. | |
Historical context
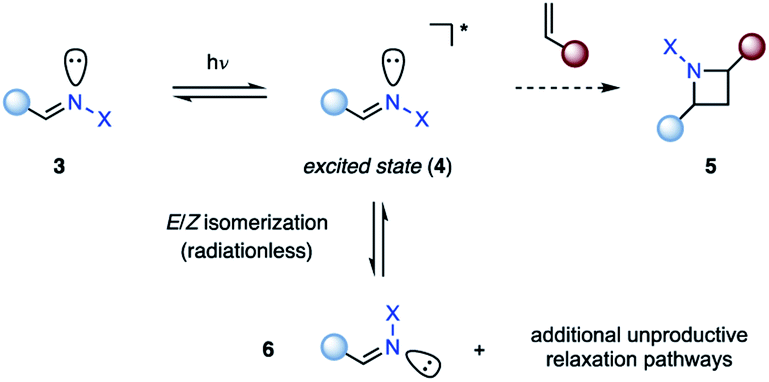
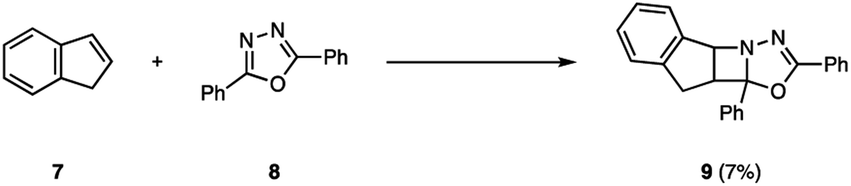
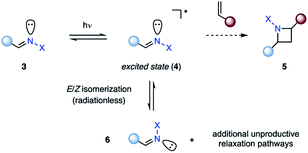
The photochemical behavior of excited state imines is in stark contrast to that of alkenes or carbonyls. In addition to relaxation pathways such as fragmentation, photoreduction or rearrangement reactions,9 the excited state of imines (4) is susceptible to radiationless decay back to the ground state (3, 6) via rotation around the carbon–nitrogen π-bond, rendering it traditionally unreactive in [2 + 2] photocycloadditions (5) (Fig. 2).10 The first successful example of an aza Paternò–Büchi reaction was reported by Tsuge in 1968, who utilized 1,3,4-oxadiazoles (8) to undergo an intermolecular [2 + 2] photocycloaddition with indene (7) providing 9 in 7% yield under ultraviolet (UV) light irradiation (Fig. 3).11 The addition of catalytic amounts of iodine was necessary to prevent 9 from reacting with a second equivalent of indene. Additionally, the reaction protocol was later found compatible with heteroaromatic compounds such as furan.12,13 Since this discovery, multiple research groups have reported new imine scaffolds that are reactive in [2 + 2] photocycloaddition reactions. However, the synthetic utility of the corresponding azetidine photocycloadducts is often limited. Importantly, photochemical methods that afford azetidine products, which allow for further synthetic modifications, have only recently been developed.
 |
| | Fig. 2 Isomerization of imines as competing relaxation pathway. | |
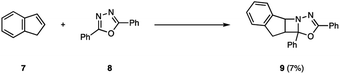 |
| | Fig. 3 First successful example of an aza Paternò–Büchi reaction. | |
Mechanistic considerations
While the general reactivity of excited state imines has been discussed previously in several excellent reviews,9 their behavior in [2 + 2] photocycloaddition reactions has not yet been extensively summarized.
Photocycloadditions from the excited singlet state
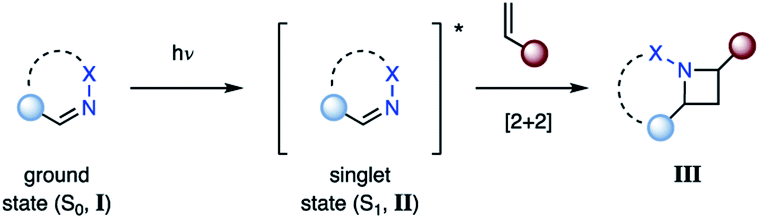
The most direct reaction path for photocycloaddition represents the direct excitation of an imine (I) from its ground state (S0) to the short-lived singlet state (S1, II) (Fig. 4). If an alkene is present, cycloaddition can occur to provide the corresponding azetidine product (III). This step proceeds likely in a concerted fashion, although the existence of singlet exciplexes has been suggested in some cases.14 As a result, the singlet state reaction can provide high levels of stereoselectivity. This approach is not viable for aliphatic imines due to the poor absorbance at higher wavelengths (λ > 250 nm). Thus, imines with adjacent conjugating groups are typically employed. For example, benzophenone imine absorbs strongly at 260 nm, likely due to a π–π* transition, while a weaker absorption at 340 nm is attributed to a n–π* transition.15,16 The specific excited states participating in aza Paternò–Büchi reactions have only been reported for a limited number of substrates. For example, Koch suggested that the excited state reactions of keto imino ethers occur from low-energy π–π* states, while unreactive compounds possess low-energy n–π* states.17 A limitation of the aza Paternò–Büchi reaction via singlet state imines is the short singlet state lifetime that often results in low concentrations of the excited state imine in solution. As a result, extended reaction times are typically necessary.
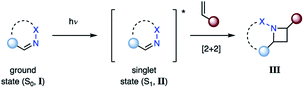 |
| | Fig. 4 Aza Paternò–Büchi reaction proceeding via a singlet state imine. | |
Photocycloadditions from the triplet excited state
In contrast, population of the triplet state (T1, IV) can be achieved via intersystem crossing (ISC) from II (Fig. 5), which is typically several orders of magnitudes longer lived than the corresponding singlet state.18 This intermediate can then efficiently undergo cycloaddition with an alkene. It is important to note that triplet state cycloaddition reactions are thought to proceed in a stepwise fashion via a 1,4-biradical intermediate (V) that converts to azetidine III upon intersystem crossing to the ground state. The stereoselectivity of this triplet state process often displays characteristic differences from the singlet state reaction, considering that biradical V can freely rotate around the carbon–carbon bond. Depending on the lifetime of V, the stereoselectivity is typically influenced by the steric environment in proximity to the biradical. In order for the triplet state reaction I → III to occur efficiently, it is important that the rate of intersystem crossing outcompetes other deactivation processes of the excited state II.
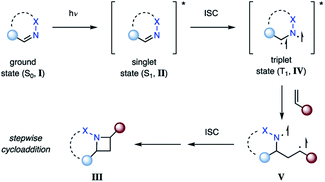 |
| | Fig. 5 Aza Paternò–Büchi reaction proceeding via a triplet state imine. | |
Alternatively, the triplet state IV can be populated via transfer of the excited state from a sensitizer to I (Fig. 6). This process is referred to as triplet energy transfer or triplet sensitization, and typically utilizes a photosensitizer with a high intersystem crossing rate close to unity and a sufficiently long triplet state lifetime.19 Triplet energy transfer proceeds via a simultaneous intermolecular two-electron transfer process between the triplet state photosensitizer and the ground state substrate (Dexter energy transfer).20 In order for this process to proceed, the photosensitizer and substrate have to be in proximity to provide sufficient electronic coupling. Additionally, the energy transfer typically has to be energetically favored. This is given when the triplet energy (ET) of the substrate is smaller than the triplet energy of the sensitizer, resulting in a formally exergonic energy transfer process. Utilizing triplet energy transfer to access the triplet excited state of imines (IV) has two main advantages: (1) it allows for high population of the triplet state IV, even for imines with inefficient intersystem crossing; (2) the photosensitizer often absorbs at higher wavelengths with an increased absorbance coefficient. Thus, the reaction can be carried out at higher wavelengths and with increased selectivity, as the photosensitizer is the major species in solution being excited.
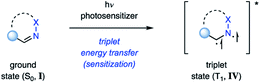 |
| | Fig. 6 Accessing triplet state imines via triplet energy transfer. | |
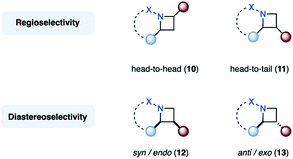
Finally, the nomenclature used within this review to describe the regiochemistry of azetidines will be used as follows. Regioisomers, for which the nitrogen is in a 1,2-relationship with the substituent of the highest priority originating from the alkene component (red), are referred to as head-to-head (10). In contrast, head-to-tail regioisomers (11) feature a 1,3-relationship (Fig. 7, top). Diastereomers, in which the alkene substituent (red) is on the same face of the azetidine ring as the imine substituent (blue) (12), are denoted as syn, or endo if the azetidine features a second ring. The corresponding other diastereomer (13) will be referred to as anti, or exo in the case of a bicyclic azetidine (Fig. 7, bottom).
 |
| | Fig. 7 Nomenclature used within this review to describe regio- and stereochemistry. | |
Scope & limitations
This review aims to provide an overview of reports that have utilized aza Paternò–Büchi reactions since Tsuge's initial report in 1968. Specific emphasis will be placed on both the scope and the limitations of current reaction protocols.
Intermolecular [2 + 2] photocycloadditions
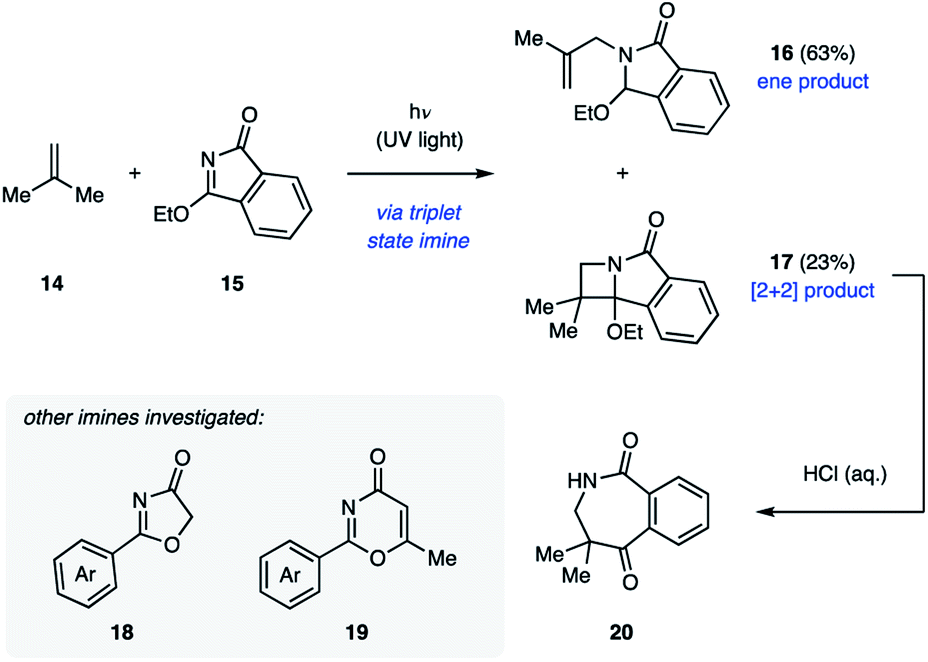
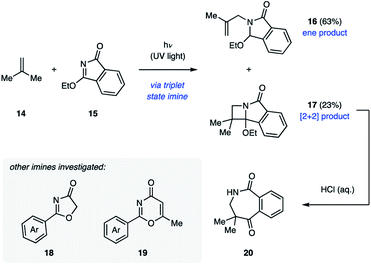
The majority of examples of aza Paternò–Büchi reactions represent intermolecular [2 + 2] photocycloadditions, in which irradiation with UV light results in excitation of an imine component that subsequently undergoes cycloaddition with an alkene. Due to the aforementioned competing E/Z isomerization of imines, most imines reported in the literature are cyclic to prevent this undesired relaxation pathway.21 The Koch group disclosed their work in pursuit of azetidines through [2 + 2] cycloadditions in 1972. Initial efforts focused on the ability of 3-ethoxyisoindolone (15) to undergo aza Paternò–Büchi reactions with a variety of sterically and electronically differentiated alkenes under UV light irradiation (Fig. 8).22 Unactivated alkenes proved to be effective reaction partners, providing the azetidine containing products in 26–40% yield. With alkenes such as isobutylene (14), a non-productive photochemical ene reaction (16) competed with the desired [2 + 2] cycloaddition (17).18 Cyclic and electron-rich alkenes were also competent reaction partners resulting in the desired azetidine products in moderate yields. Under acidic aqueous conditions these products undergo ring-opening to give seven-membered benzoazepinediones (20) in high yields.
 |
| | Fig. 8 Aza Paternò–Büchi reactions relying on 3-ethoxyisoindolone 15 (Koch and coworkers). | |
Koch and coworkers also discovered that 2-phenyl-2-oxazolin-4-one (18) can be used in place of 15 in otherwise analogous transformations (Fig. 8).23 This heterocycle can engage in [2 + 2] cycloaddition reactions with electron-rich alkenes and heterocycles in moderate yields. Additionally, electronically differentiated aromatic substituents are well tolerated. A large excess of the alkene component was found necessary to prevent Norrish type I cleavage. Furthermore, the carbon-nitrogen double bond of 5-methyl-2-phenyl-1,3-oxazin-4-one (19) was shown to react with dimethoxyethylene in a [2 + 2] cycloaddition to form the corresponding azetidine in 78% yield.24 Upon thermal retro-Diels–Alder fragmentation and subsequent hydrolysis, these compounds give rise to β-amino ketones.
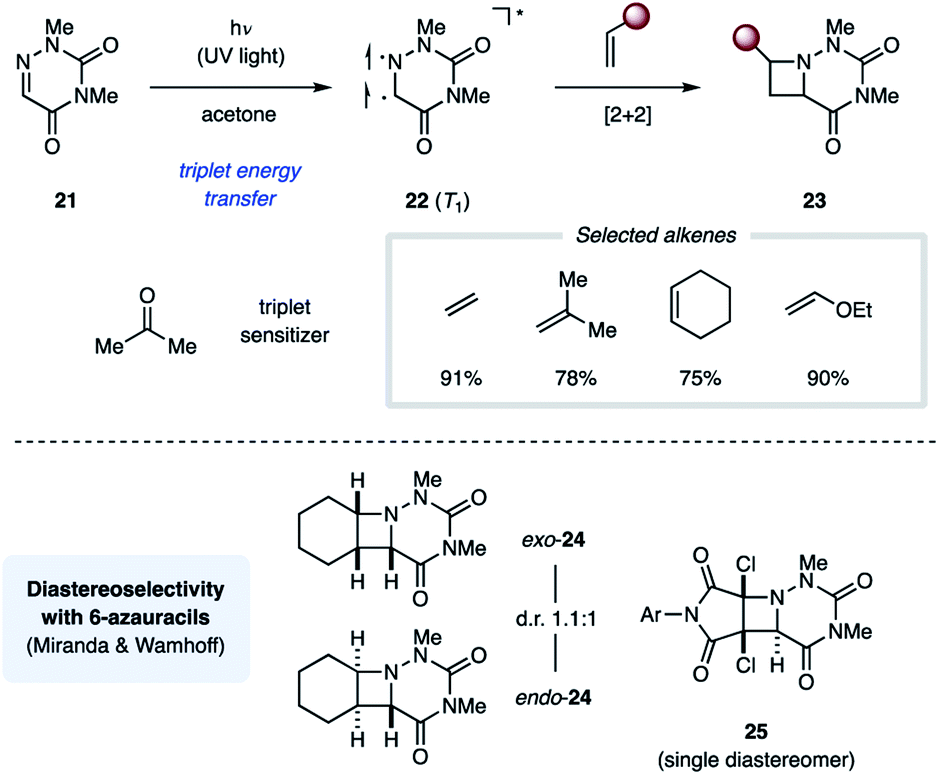
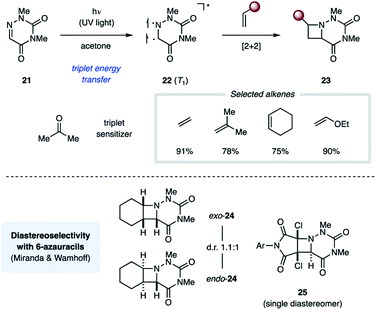
The Swenton group published a series of reports on photochemical [2 + 2] cycloaddition reactions relying on 1,3-dimethyl-6-azauracil (21). In the presence of acetone (ET = 79 kcal mol−1) as a triplet sensitizer, UV light irradiation of 21 resulted in efficient [2 + 2] cycloaddition with ethyl vinyl ether to yield the corresponding azetidine-containing product (23) in 90% overall yield (Fig. 9).25 In addition to electron-rich alkenes, both cyclic and acyclic unactivated alkenes work comparably well in this transformation.26 All reactions yield a mixture of endo and exo diastereomers with no significant stereoselectivity. The authors propose that this reaction proceeds through a triplet state uracil (22). Furthermore, the uracil ring of 23 could be cleaved under basic or reducing conditions to reveal the corresponding N-aminoazetidines.27 While the relative stereochemical configuration of the azetidine products was not assigned by Swenton, the Miranda group completed the stereochemical assignment of the azetidine products 24 from the cycloaddition reaction between 21 and cyclohexene, one of the compounds Swenton and coworkers initially reported.28 Similarly, Wamhoff and coworkers found that the cycloaddition reaction between 21 and dihalomaleamides proceeded in excellent stereoselectivity, giving exclusively the endo diastereomer (25).29
 |
| | Fig. 9 Aza Paternò–Büchi reaction relying on 6-azauracils (21) (Swenton and coworkers). | |
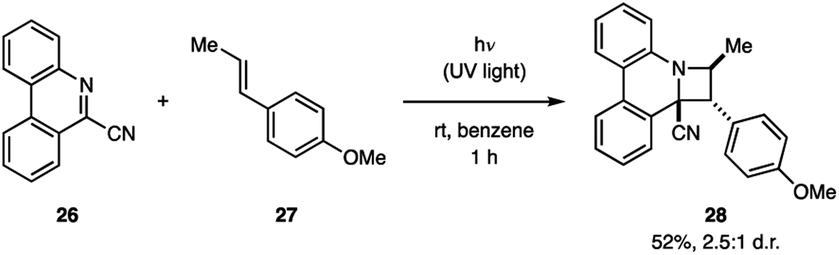
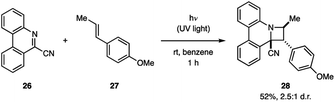
The Ohta group demonstrated that 9-cyanophenanthridine (26) undergoes a [2 + 2] cycloaddition with anethole (27) under irradiation with UV light (Fig. 10).30 The product 28 was obtained in 52% yield as a 2.5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of endo/exo diastereomers. Fluorescence spectroscopy of 26 in the presence of 27 shows a new emission pattern indicative of exciplex formation.14a Furthermore, the 26–27 exciplex emission was attenuated by acrylonitrile, a known singlet state quencher, but not by the triplet quencher trans-1,3-pentadiene, indicating that the cycloaddition occurs from the singlet state. Additionally, this hypothesis was supported by the fact that product formation was suppressed by the addition of acrylonitrile, but not trans-1,3-pentadiene.
:1 mixture of endo/exo diastereomers. Fluorescence spectroscopy of 26 in the presence of 27 shows a new emission pattern indicative of exciplex formation.14a Furthermore, the 26–27 exciplex emission was attenuated by acrylonitrile, a known singlet state quencher, but not by the triplet quencher trans-1,3-pentadiene, indicating that the cycloaddition occurs from the singlet state. Additionally, this hypothesis was supported by the fact that product formation was suppressed by the addition of acrylonitrile, but not trans-1,3-pentadiene.
 |
| | Fig. 10 Synthesis of azetidine 28 from phenanthridine 26 (Ohta and coworkers). | |
The Nishio group reported that quinoxaline-2(1H)-ones (29) are competent reagents to participate in aza Paternò–Büchi reactions with electron-deficient alkenes (Fig. 11).31 The alkene substrate scope included several differentially substituted methacrylates and acrylonitriles (30, 32) with yields ranging from 32% to >99%. In all cases, the isolated products were head-to-head regioisomers. Steric properties of both the imine and the alkene component were observed to dramatically impact the diastereoselectivity of the reaction. For example, utilizing 32 instead of 30 as the alkene partner, provided the respective azetidine product 33 as a single diastereomer. Furthermore, Nishio and coworkers developed related transformations, in which the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N bond of 1,4-benzoxazin-2-ones,32 pteridine-2,4,7-drione,33 and tetrahydroquinoxalin-2(1H)-ones were engaged in [2 + 2] photocycloaddition reactions.34
N bond of 1,4-benzoxazin-2-ones,32 pteridine-2,4,7-drione,33 and tetrahydroquinoxalin-2(1H)-ones were engaged in [2 + 2] photocycloaddition reactions.34
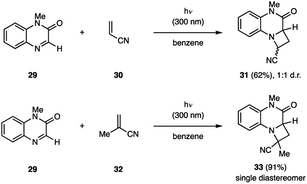 |
| | Fig. 11 Aza Paternò–Büchi reaction with quinoxaline-2(1H)-ones (29) (Nishio and coworkers). | |
During the course of their studies on 3-aryl-2-isoxazolines, the Mukai group discovered a novel [2 + 2] photocycloaddition, representing the first example of an oxime undergoing an aza Paternò–Büchi reaction (Fig. 12).35 Irradiation of isoxazoline 34 with UV light afforded 4% yield of the [2 + 2] cycloadduct between the substrate and the solvent benzene. Yields were significantly improved when using heteroaromatic or styrenyl alkenes, such as furan, thiophene or indene (7), providing azetidine products such as 36 in 75% yield.14b,c In contrast, cyclopentadiene, pyrrole and N-methylpyrrole failed to undergo this transformation. Evaluation of a number of different 3-aryl-2-isoxaolines revealed that an electron-withdrawing substituent on the aromatic ring was necessary for successful cycloaddition. The authors propose that the observed reactivity and diastereoselectivity can be accounted for by the existence of a singlet state exciplex (35).
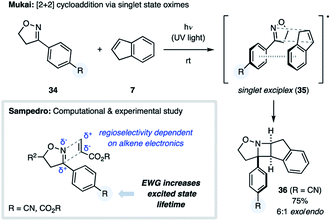 |
| | Fig. 12 The reactivity of aromatic oximes (34) in aza Paternò–Büchi reaction (Mukai and Sampedro). | |
This mechanism was further corroborated by Sampedro and coworkers, who studied the [2 + 2] photocycloaddition between aromatic oximes and activated alkenes computationally as well as experimentally (Fig. 12).14d,e Their computational investigations suggested that an electron-withdrawing substituent on the aryl ring is essential for prolonging the lifetime of the singlet excited state oxime allowing for the desired reactivity. The high regioselectivity of this process was attributed to the partial charges of both the oxime and alkene components, which was supported by the observation that alkenes featuring electron-donating or -withdrawing substituents provided opposite regioselectivity.
The Maruoka group overcame the requirement for cyclic imines by demonstrating that N-(arylsulfonyl)imines (37) undergo [2 + 2] photocycloadditions with styrenyl alkenes (38) such as styrene or benzofuran when irradiated with 365 nm UV light, providing protected azetidines (40–42) in 20–83% yield (Fig. 13).14f Importantly, these reactions were found to be stereospecific, which was attributed to the reaction proceeding via a singlet state exciplex (39). As a result of this sandwich-type intermediate the aryl substituent of both the imine and the alkene are on the same face of the azetidine ring.
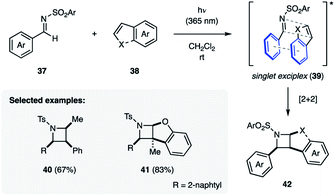 |
| | Fig. 13 Synthesis of protected azetidines from N-(arylsulfonyl)imines (37) (Maruoka and coworkers). | |
Intramolecular [2 + 2] photocycloadditions
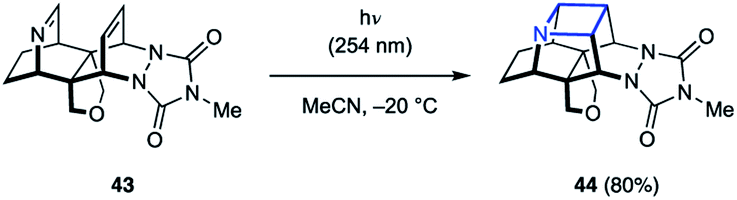
While the majority of examples of aza Paternò–Büchi reactions are intermolecular, azetidines can also be formed through intramolecular [2 + 2] cycloaddition reactions. During their synthesis and investigation of proximate bichromophoric systems, the Prinzbach group used an aza Paternò–Büchi reaction to access azetidine scaffold 44 (Fig. 14).36 Relying on either direct or acetone-sensitized excitation, the desired azetidine (44) was isolated in 80–85% yield. Notably, the imine 43 used in this report did not require any conjugating group for successful reactivity.
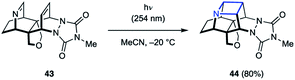 |
| | Fig. 14 First example of an intramolecular aza Paternò–Büchi reaction (Prinzbach and coworkers). | |
In 2017, Sivaguru and coworkers disclosed their efforts towards the synthesis of chiral azetidines (47) through an atroposelective [2 + 2] photocycloaddition (Fig. 15).37 The authors demonstrated that the need for cyclic imines can be circumvented by instead engaging an excited state alkene in the aza Paternò–Büchi reaction. Utilizing enamides (45) as the alkene equivalent, the triplet excited state could be accessed via sensitization using 30–100 mol% xanthone (46, ET = 74 kcal mol−1). With this reaction design, the CN bond of several oximes and hydrazones could be engaged in the cycloaddition reaction in good yields. Importantly, the authors observed no racemization under the reaction conditions, isolating the desired products in excellent enantiomeric excess. Unfortunately, high catalyst loadings were necessary for non-stabilized imines, which was attributed to undesired quenching of the imine component via an unproductive electron-transfer process. Extensive mechanistic experiments were carried out that corroborated the proposed mechanism proceeding via a triplet manifold. Specifically, the authors proposed a stepwise cycloaddition, in which the triplet enamide first undergoes carbon–carbon bond formation with the imine. Based on the observed scrambling of the imine E/Z ratio at low conversion, the authors conclude that this step is likely reversible. The resulting 1,4-biradical intermediate subsequently undergoes carbon–nitrogen bond formation upon intersystem crossing. Based on the stepwise nature of this process, the stereochemical configuration of the CN bond in the starting material is inconsequential to the stereochemistry of the product, as both the E- and Z-isomer convert to the same product diastereomer.
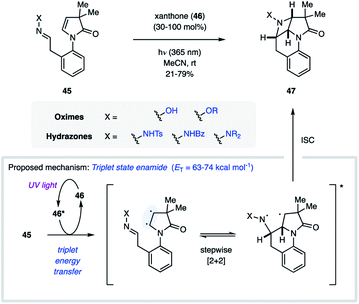 |
| | Fig. 15 Intramolecular aza Paternò–Büchi reaction relying on alkene activation via triplet energy transfer (Sivaguru and coworkers). | |
Photocycloadditions with alkynes
The [2 + 2] photocycloaddition reaction with alkynes affords four-membered rings that contain a double bond. Specifically, the reaction between an imine and an alkyne provides 2-azetines as photocycloadducts. Although the products represent interesting scaffolds,38 accessing these via aza Paternò–Büchi reactions has been met with limited success. Mei-Gong and coworkers reported that 6-azauracil 49 undergoes [2 + 2] photocycloaddition with alkynes such as propargyl alcohol (48) (Fig. 16).39 In addition to azetine 50, the authors also isolated diazabicycle 51, resulting from the reaction of 50 with uracil 49, and azetidine 52, formed from addition of the ethanol solvent to azetine 50. To date, this report represents the only example in which alkynes successfully participated in aza Paternò–Büchi reactions.
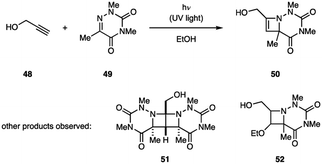 |
| | Fig. 16 Example of an aza Paternò–Büchi reaction involving alkynes (Mei-Gong and coworkers). | |
Synthetic applications
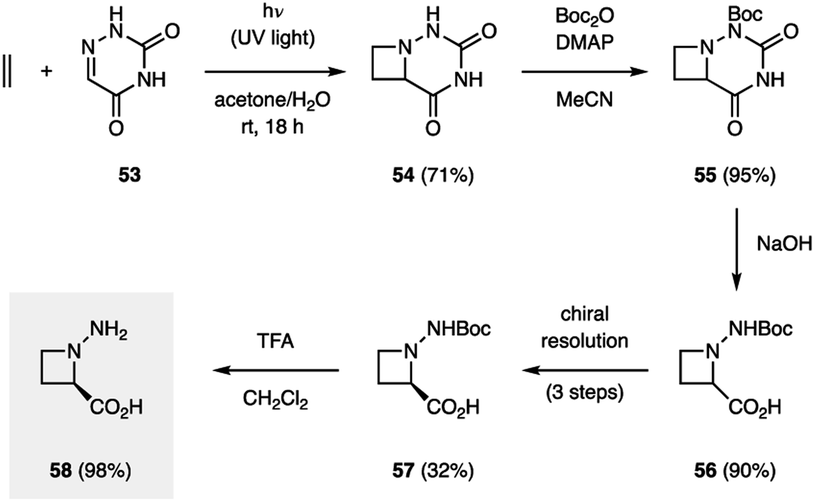
There only exist a few examples of aza Paternò–Büchi reactions in the context of target-directed synthesis. In contrast to the Paternò–Büchi reaction, which has found numerous applications in organic synthesis, only a small number of imine scaffolds have been reported to successfully participate in aza Paternò–Büchi reactions, typically providing azetidine scaffolds that limit further synthetic modifications. The Aitken group utilized an aza Paternò–Büchi reaction for the synthesis of N-aminoazetidinecarboxylic acid 58 (Fig. 17).40 Utilizing conditions developed by the Swenton group, azetidine 54 was accessed by irradiating 6-azauracil (53) and ethylene with UV light in the presence of acetone as a photosensitizer. A subsequent synthetic sequence involving Boc-protection, hydrolysis, chiral resolution and deprotection afforded 58 in high yield. The authors further studied 58 as an analogue to aminocyclobutanecarboxylic acids (ACBCs), which have been used as building blocks in the preparation of peptidomimetics. The resulting oligomers are recognized for their pharmacological activities and their ability to self-organize into complex molecular architectures. When a single residue of 58 was used as replacement for an ACBC, a change in helical structure was observed, demonstrating the utility of this cyclic β-amino acid.41
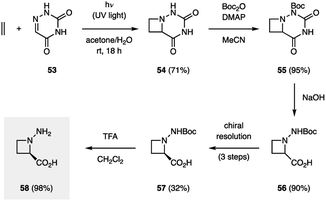 |
| | Fig. 17 Synthesis of aminoazetidines (58) (Aitken and coworkers). | |
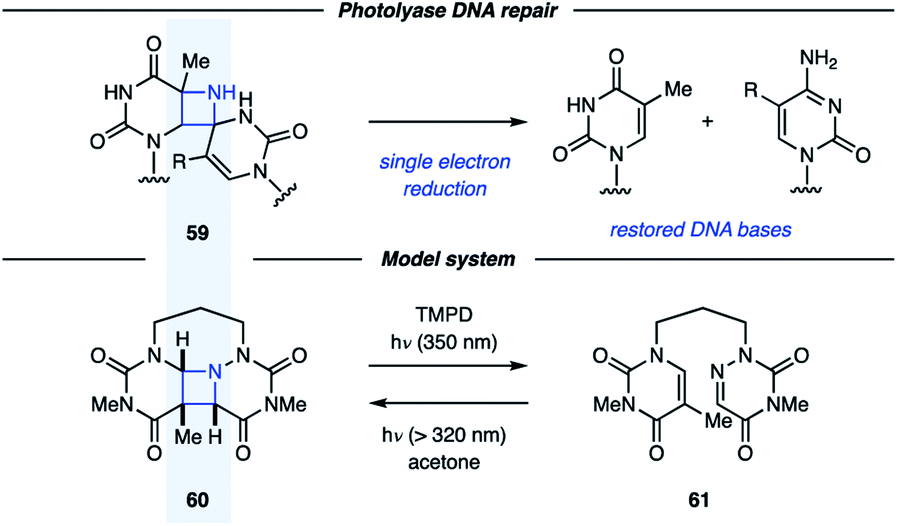
In addition to being attractive targets in synthesis, azetidines have been proposed as intermediates along the pathway for DNA (6–4) photoproduct repair.42 Unfortunately, the instability of these proposed azetidines (59) has made isolation and characterization difficult. Miranda and coworkers studied azetidine 60 as a model system for the proposed azetidine intermediates in the photolyase repair mechanism (Fig. 18).43 In the presence of acetone as a photosensitizer, UV light irradiation resulted in conversion of 61 to azetidine 60. This process could be reversed under irradiation with 350 nm light in the presence of a photocatalyst with an oxidation potential close to that of FADH−*, the proposed single-electron reductant in vivo. This result indicates that single-electron reduction of dimeric azetidines can result in cycloreversion, further promoting the understanding of the repair mechanism involving photolyases. In a subsequent study, the Miranda group reported that the azetidine fragmentation could be achieved by both single-electron reduction and oxidation.44
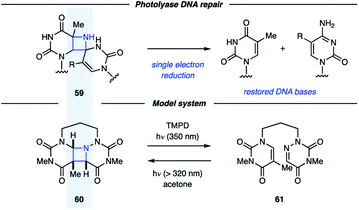 |
| | Fig. 18 Model system for photolyase DNA repair (Miranda and coworkers). | |
Outlook
While [2 + 2] photocycloadditions continue to grow among the most prominent reactions for the construction of cyclobutanes and oxetanes, the applications of the aza Paternò–Büchi reaction for the synthesis of azetidines remain limited. Current protocols require imine reagents that provide photocycloadducts with limited synthetic utility, and the use of UV light to access the excited state species. Recently, visible light triplet energy transfer has emerged as a prominent tool in organic synthesis to access triplet excited state species under mild conditions. A vast number of visible light photocatalysts have been developed over the years that were successfully used in transformations such as isomerization, cycloaddition or bond dissociation reactions.45
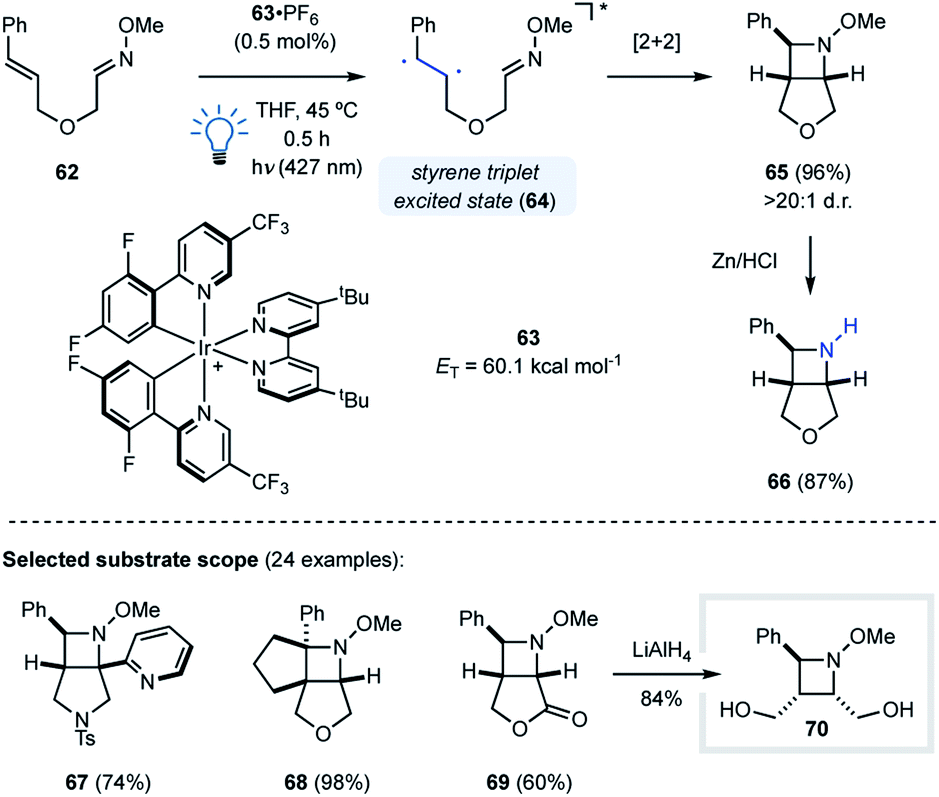
Our group utilized this design principle for the development of an intramolecular visible light-mediated aza Paternò–Büchi reaction (Fig. 19).46 Similar to the pioneering work by Sivaguru and coworkers,37 the developed aza Paternò–Büchi reaction protocol relies on activation of the alkene component via triplet energy transfer. Activated alkenes such as styrenes or dienes possess triplet energies that are accessible with visible light photocatalysts, which was previously utilized for the synthesis of cyclobutanes by Yoon and coworkers.47,48 With 63·PF6 (ET = 60.1 kcal mol−1) as the photocatalyst, triplet energy transfer to the styrene moiety of 62 resulted in triplet excited 64 that subsequently underwent intramolecular [2 + 2] cycloaddition with the adjacent oxime to form bicyclic azetidine 65 in 96% yield and >20:1 d.r. (Fig. 19). Mechanistic experiments were carried out that were in agreement with a mechanism relying on a triplet state styrene. This mode of activation proceeds under very mild conditions, as demonstrated by the high functional group tolerance of this transformation (e.g.67–69). Furthermore, utilizing oximes as imine equivalents was found beneficial based on their superior stability towards hydrolysis, and the N–O bond in the azetidine products can be readily cleaved to obtain free azetidines (66) in high yields.
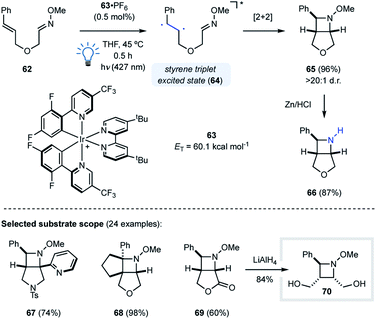 |
| | Fig. 19 Development of a visible light-mediated intramolecular aza Paternò–Büchi reaction (Schindler and coworkers). | |
Conclusions
In summary, this review has provided an overview of aza Paternò–Büchi reactions used in organic synthesis. Since its first discovery, several research groups have developed new reagents that have enabled the construction of novel azetidine-containing frameworks. However, there are still major challenges that have to be overcome to improve the general synthetic utility of this transformation. Most examples presented herein require UV light irradiation, are limited in scope or do not allow for further synthetic applications, which currently represents challenges for the use of the aza Paternò–Büchi reaction in both synthetic and industrial applications. Furthermore, the competing E/Z isomerization of excited state imines provides a challenge for designing new, acyclic imine reagents, thus, further development is required to overcome this obstacle. Nevertheless, recent reports have shown that utilizing emerging synthetic tools, such as visible light triplet energy transfer catalysis, can overcome some of the aforementioned challenges. Therefore, we consider this area of research to hold great promise for the synthesis of complex azetidine-containing scaffolds, and the development of improved protocols for aza Paternò–Büchi reactions will arise with the application and discovery of new photocatalytic methods.
Conflicts of interest
There are no conflicts to declare.
Abbreviations
| Boc2O | Di-tert-butyl dicarbonate |
| DMAP | 4-Dimethylaminopyridine |
| d.r. | Diastereomeric ratio |
|
E
T
| Triplet energy |
| EtOH | Ethanol |
| EWG | Electron-withdrawing group |
| ISC | Intersystem crossing |
| MeCN | Acetonitrile |
|
p-TsOH |
p-Toluenesulfonic acid |
| rt | Room temperature |
| r.r. | Regioisomer ratio |
| S0 | Singlet ground state |
| S1 | First singlet excited state |
| T1 | First triplet excited state |
| THF | Tetrahydrofuran |
| TFA | Tifluoroacetic acid |
| UV | Ultraviolet |
Acknowledgements
C. S. S. thanks the Alfred P. Sloan Foundation, the David and Lucile Packard Foundation, and the Camille and Henry Dreyfus Foundation for fellowships.
References
-
(a)
M. T. Crimmins, in Comprehensive Organic Synthesis, ed. B. M. Trost and I. Fleming, Elsevier, Oxford, 1st edn, 1991, vol. 5, ch. 2.3, pp. 123–150 Search PubMed;
(b)
J. A. Porco and S. L. Schreiber, in Comprehensive Organic Synthesis, ed. B. M. Trost and I. Fleming, Elsevier, Oxford, 1st edn, 1991, vol. 5, ch. 2.4, pp. 151–192 Search PubMed.
- D. Sarkar, N. Bera and S. Ghosh, Eur. J. Org. Chem., 2020, 1310–1326 CrossRef CAS.
- S. Poplata, A. Tröster, Y.-Q. Zou and T. Bach, Chem. Rev., 2016, 116, 9748–9815 CrossRef CAS PubMed.
-
(a) M. D'Auria, Photochem. Photobiol. Sci., 2019, 18, 2297–2362 RSC;
(b) M. D'Auria and R. Racioppi, Molecules, 2013, 18, 11384–11428 CrossRef PubMed.
- For examples of azetidine-containing natural products, see:
(a)
H. Yoda, M. Takahashi and T. Sengoku, in Heterocycles in Natural Product Synthesis, ed. K. C. Majumdar and S. K. Chattopadhyay, Wiley-VCH, Weinheim, 1st edn, 2011 Search PubMed;
(b) M. Kitajima, N. Kogure, K. Yamaguchi, H. Takayama and N. Aimi, Org. Lett., 2003, 5, 2075–2078 CrossRef CAS PubMed;
(c) T. Akihisa, S. Mafune, M. Ukiya, Y. Kimura, K. Yasukawa, T. Suzuki, H. Tokuda, N. Tanabe and T. Fukuoka, J. Nat. Prod., 2004, 67, 479–480 CrossRef CAS PubMed.
-
(a) P. V. Fish, A. D. Brown, E. Evrard and L. R. Roberts, Bioorg. Med. Chem. Lett., 2009, 19, 1871–1875 CrossRef CAS PubMed;
(b) A. Brown, T. B. Brown, A. Calabrese, D. Ellis, N. Puhalo, M. Ralph and L. Watson, Bioorg. Med. Chem. Lett., 2010, 20, 516–520 CrossRef CAS PubMed;
(c)
E. H. Kerns, and L. Di, Drug-Like Properties: Concepts, Structure Design and Methods, Academic Press, 1st edn, 2008, pp. 137–168 Search PubMed;
(d) D. J. S. Jean and C. Fotsch, J. Med. Chem., 2012, 55, 6002–6020 CrossRef PubMed;
(e) F. Lovering, J. Bikker and C. Humblet, J. Med. Chem., 2009, 52, 6752–6756 CrossRef CAS PubMed.
- For reviews on the synthesis of azetidines, see:
(a) A. Brandi, S. Cicchi and F. M. Cordero, Chem. Rev., 2008, 108, 3988–4035 CrossRef CAS PubMed;
(b) N. H. Cromwell and B. Phillips, Chem. Rev., 1979, 79, 331–358 CrossRef CAS;
(c) D. Antermite, L. Degennaro and R. Luisi, Org. Biomol. Chem., 2017, 15, 34–50 RSC.
- R. Gianatassio, J. M. Lopchuk, J. Wang, C.-M. Pan, L. R. Malins, L. Prieto, T. A. Brandt, M. R. Collins, G. M. Gallego, N. W. Sach, J. E. Spangler, H. Zhu, J. Zhu and P. S. Baran, Science, 2016, 351, 241–246 CrossRef CAS PubMed.
- For reviews on the photochemistry of imines, see:
(a) A. Padwa, Chem. Rev., 1977, 77, 37–68 CrossRef CAS;
(b) A. C. Pratt, Chem. Soc. Rev., 1977, 6, 63–81 RSC;
(c)
H. D. Roth, in PATAI's Chemistry of Functional Groups, ed. I. Marek, Wiley, Weinheim, 2010 Search PubMed.
- A. Padwa and F. Albrecht, J. Am. Chem. Soc., 1972, 94, 1000–1002 CrossRef CAS.
- O. Tsuge, M. Tashiro and K. Oe, Tetrahedron Lett., 1968, 9, 3971–3974 CrossRef.
- O. Tsuge, K. Oe and M. Tashiro, Tetrahedron, 1973, 29, 41–46 CrossRef CAS.
- K. Oe, M. Tashiro and O. Tsuge, J. Org. Chem., 1977, 42, 1496–1499 CrossRef CAS.
- For examples of aza Paternò–Büchi reactions proceeding via singlet exciplexes, see:
(a) S. Futamura, H. Ohta and Y. Kamiya, Bull. Chem. Soc. Jpn., 1982, 55, 2190–2194 CrossRef CAS;
(b) T. Kumagai, K. Shimizu, Y. Kawamura and T. Mukai, Chem. Lett., 1983, 12, 1357–1360 CrossRef;
(c) Y. Kawamura, T. Kumagai and T. Mukai, Chem. Lett., 1985, 14, 1937–1940 CrossRef;
(d) D. Sampedro, ChemPhysChem, 2006, 7, 2456–2459 CrossRef CAS PubMed;
(e) D. Sampedro, A. Soldevilla, P. J. Campos, R. Ruiz and M. A. Rodríguez, J. Org. Chem., 2008, 73, 8331–8336 CrossRef CAS PubMed;
(f) R. Sakamoto, T. Inada, S. Sakurai and K. Maruoka, Org. Lett., 2016, 18, 6252–6255 CrossRef CAS PubMed.
-
G. Wettermark, in The Chemistry of the carbon-nitrogen double bond, ed. S. Patai, Wiley & Sons, 1970 Search PubMed.
- M. A. El-Bayoumi, M. El-Aasser and F. Abdel-Halim, J. Am. Chem. Soc., 1971, 93, 586–590 CrossRef CAS.
- T. H. Koch, J. A. Olesen and J. DeNiro, J. Org. Chem., 1975, 40, 14–19 CrossRef CAS.
- D. R. Anderson, J. S. Keute, T. H. Koch and R. H. Moseley, J. Am. Chem. Soc., 1977, 99, 6332–6340 CrossRef CAS.
- J. Zhao, W. Wu, J. Sun and S. Guo, Chem. Soc. Rev., 2013, 42, 5323–5351 RSC.
- D. L. A. Dexter, J. Chem. Phys., 1953, 21, 836–850 CrossRef CAS.
- For an example of an acyclic imine undergoing an aza Paternò–Büchi reaction, see: P. Margaretha, Helv. Chim. Acta, 1978, 61, 1025–1032 CrossRef CAS.
-
(a) T. H. Koch and K. H. Howard, Tetrahedron Lett., 1972, 13, 4035–4038 CrossRef;
(b) K. A Howard and T. H Koch, J. Am. Chem. Soc., 1975, 97, 7288–7298 CrossRef.
-
(a) T. H. Koch and R. M. Roderhorst, Tetrahedron Lett., 1972, 13, 4039–4042 CrossRef;
(b) T. H. Koch and R. M. Rodehorst, J. Am. Chem. Soc., 1975, 97, 7298–7304 CrossRef.
- T. H. Koch, R. H. Higgins and H. F. Schuster, Tetrahedron Lett., 1977, 18, 431–434 CrossRef.
- J. A. Hyatt and J. S. Swenton, J. Chem. Soc., Chem. Commun., 1972, 1144–1145 RSC.
- J. S. Swenton and J. A. Hyatt, J. Am. Chem. Soc., 1974, 96, 4879–4885 CrossRef CAS PubMed.
- J. S. Swenton and R. J. Balchunis, J. Heterocycl. Chem., 1974, 11, 917–920 CrossRef CAS.
- A. B. Fraga-Timiraos, G. M. Rodríguez-Muñiz, V. Peiro-Penalba, M. A. Miranda and V. Lhiaubet-Vallet, Molecules, 2016, 21, 1683 CrossRef PubMed.
-
(a) G. Szilagyi and H. Wamhoff, Angew. Chem., 1980, 92, 1066–1067 CrossRef CAS;
(b) G. Szilágyi and H. Wamhoff, Angew. Chem., 1983, 95, 156–157 CrossRef.
- S. Futamura, H. Ohta and Y. Kamiya, Chem. Lett., 1980, 9, 655–658 CrossRef.
- T. Nishio, J. Org. Chem., 1984, 49, 827–832 CrossRef CAS.
- T. Nishio and Y. Omote, J. Org. Chem., 1985, 50, 1370–1373 CrossRef CAS.
- T. Nishio, T. Nishiyama and Y. Omote, Liebigs Ann. Chem., 1988, 441–443 CrossRef CAS.
- T. Nishio, M. Kondo and Y. Omote, Helv. Chim. Acta, 1991, 74, 225–231 CrossRef CAS.
- T. Kumagai, K. Shimizu, Y. Kawamura and T. Mukai, Tetrahedron, 1981, 37, 3365–3376 CrossRef CAS.
-
(a) G. Fischer, H. Fritz and H. Prinzbach, Tetrahedron Lett., 1986, 27, 1269–1272 CrossRef CAS;
(b) W. Marterer, H. Prinzbach, G. Rihs, J. Wirz, J. Lecoultre and E. Heilbronner, Helv. Chim. Acta, 1988, 71, 1937–1965 CrossRef CAS;
(c) G. Fischer, H. Fritz, G. Rihs, D. Hunkler, K. Exner, L. Knothe and H. Prinzbach, Eur. J. Org. Chem., 2000, 743–762 CrossRef CAS.
- E. Kumarasamy, S. K. Kandappa, R. Raghunathan, S. Jockusch and J. Sivaguru, Angew. Chem., Int. Ed., 2017, 56, 7056–7061 CrossRef CAS PubMed.
- D. Didier, A. N. Baumann and M. Eisold, Tetrahedron Lett., 2018, 59, 3975–3987 CrossRef CAS.
- F. Mei-Gong, Z. Ya-Lin and F. Ping, Acta Chim. Sin., 1985, 43, 778–781 Search PubMed.
- V. Declerch and D. J. Aitken, J. Org. Chem., 2011, 76, 708–711 CrossRef PubMed.
- A. Altmayer-Henzien, V. Declerck, J. Farjon, D. Merlet, R. Guillot and D. J. Aitken, Angew. Chem., Int. Ed., 2015, 54, 10807–10810 CrossRef CAS PubMed.
- T. Carell, L. T. Burgdorf, L. M. Kundu and M. Cichon, Curr. Opin. Chem. Biol., 2001, 5, 491–498 CrossRef CAS PubMed.
- A. B. Fraga-Timiraos, V. Lhiaubet-Vallet and M. A. Miranda, Angew. Chem., Int. Ed., 2016, 55, 6037–6040 CrossRef CAS PubMed.
- A. B. Fraga-Timiraos, A. Francés-Monerris, G. M. Rodríguez-Muñiz, M. Navarrete-Miguel, M. A. Miranda, D. Roca-Sanjuán and V. Lhiaubet-Vallet, Chem.–Eur. J., 2018, 24, 15346–15354 CrossRef CAS PubMed.
- For reviews on visible light triplet energy transfer catalysis, see:
(a) F. Strieth-Kalthoff, M. J. James, M. Teders, L. Pitzer and F. Glorius, Chem. Soc. Rev., 2018, 47, 7190–7202 RSC;
(b) Q.-Q. Zhou, Y.-Q. Zou, L.-Q. Lu and W.-J. Xiao, Angew. Chem., Int. Ed., 2019, 58, 1586–1604 CrossRef CAS PubMed.
- M. R. Becker, A. D. Richardson and C. S. Schindler, Nat. Commun., 2019, 10, 5095, DOI:10.1038/s41467-019-13072-x.
- Z. Lu and T. P. Yoon, Angew. Chem., Int. Ed., 2012, 51, 10329–10332 CrossRef CAS.
- A. E. Hurtley, Z. Lu and T. P. Yoon, Angew. Chem., Int. Ed., 2014, 53, 8991–8994 CrossRef CAS.
Footnote |
| † These authors contributed equally. |
|
| This journal is © The Royal Society of Chemistry 2020 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence ,
Marc R.
Becker†
,
Marc R.
Becker†