
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEfficient in vivo synthesis of lasso peptide pseudomycoidin proceeds in the absence of both the leader and the leader peptidase†
Tatyana
Zyubko‡
ab,
Marina
Serebryakova‡
acd,
Julia
Andreeva
ad,
Mikhail
Metelev§
 abd,
Guy
Lippens
e,
Svetlana
Dubiley
*ad and
Konstantin
Severinov
*af
abd,
Guy
Lippens
e,
Svetlana
Dubiley
*ad and
Konstantin
Severinov
*af
aCenter for Life Sciences, Skolkovo Institute of Science and Technology, 3 Nobel str., 143025 Moscow, Russia. E-mail: svetlana.dubiley@gmail.com
bPeter the Great St. Petersburg Polytechnic University, St. Petersburg, 195251, Russia
cA.N. Belozersky Institute of Physico-Chemical Biology, Lomonosov Moscow State University, Leninskie Gory 1, Bldg. 40, Moscow 119991, Russia
dInstitute of Gene Biology, Russian Academy of Science, 34/5 Vavilov str., 119334 Moscow, Russia
eToulouse Biotechnology Institute (TBI), CNRS, INRA, INSA, UPS 135 Avenue de Rangueil, 31077 Toulouse, France
fWaksman Institute for Microbiology, 190 Frelinghuysen Road, Piscataway, New Jersey 08854-8020, USA. E-mail: severik@waksman.rutgers.edu
First published on 30th August 2019
Abstract
Bacterial lasso peptides are made from linear ribosomally synthesized precursors by specific cleavage at the leader–core junction site of the precursor by a dedicated protease recognizing the leader, followed by cyclisation of the newly formed N-terminus of the core part with a side chain of the internal aspartic or glutamic residue catalyzed by a macrolactam synthetase. The resulting structure has a tail that is threaded and fixed inside the cycle formed. Here, we characterize a new lasso peptide, pseudomycoidin, encoded by Bacillus pseudomycoides DSM 12442. The most surprising and unique feature of pseudomycoidin is that it can be produced in vivo from the ribosomally synthesized core part by a macrolactam synthetase, in the absence of the leader protease. The minimalism of the pseudomycoidin synthesis system makes it a powerful model to generate pseudomycoidin-based lasso-peptide libraries and to study the poorly understood process of lasso formation. We detected two additional pseudomycoidin modifications: phosphorylation of a terminal residue that was previously observed in another lasso peptide, followed by glycosylation, which was not observed heretofore. We speculate that these bulky C-terminal modifications may help maintain the threaded lasso topology of the compound synthesized by the macrolactam synthetase.
Introduction
Bacterial lasso peptides are members of the RiPP (ribosomally synthesized post-translationally modified peptide) group of natural products. Lasso peptides characterized to date are 15–30 amino acids long and contain a ring formed by an amide bond between the backbone amine of the N-terminal residue and the side chain of the glutamic or aspartic residue at positions 7, 8 or 9, and a C-terminal tail. The tail is kept trapped within the ring either by disulfide bonds formed between side chains of cysteines in the ring and the tail (in class I, III, and IV lasso peptides) or by bulky side chains of C-terminal amino acids that strut both sides of the ring (class II).1Lasso peptide biosynthetic operons are widespread in diverse bacteria and archaea and contain one or several A genes coding for lasso peptide precursors and B and C genes coding for enzymes responsible for lasso peptide formation.1,2 The product of the B gene is a bifunctional protein that in some clusters is encoded by two separate genes, B1 and B2.1 Lasso peptide precursors comprise the N-terminal leader and the C-terminal core parts. The B1 protein, alone or in the context of fused B protein, recognizes the leader part and recruits B2, a cysteine protease, to hydrolyze the peptide bond at the leader–core junction. The core part, once formed, interacts with the C enzyme that catalyzes the formation of the ring.3
First lasso peptides were isolated in the course of functional screening of environmental bacteria for compounds with medically relevant functions ranging from inhibition of fusion of the HIV virus with eukaryotic cells4,5 to inhibition of human glucagon receptors.6 However, the biological role of these compounds in natural products is poorly understood. Most lasso peptide clusters encode export pumps, suggesting that the compounds are produced outside and may target other bacteria of the same or different species. Lasso peptides produced by Gram-negative bacteria, including the prototypical microcin J25, capistruin, acinetodin, and klebsidin, specifically target RNA polymerase of sensitive cells.7–10 Several other lasso peptides apparently target the ATP-dependent protease ClpC1P1P2,11 prolyl endopeptidase,12 and cell wall biosynthesis,13 though mechanisms remain undefined. For many other lasso peptides, no biological function that could benefit the producer is yet apparent. In addition to B and C enzymes, some lasso peptide clusters contain genes encoding enzymes that introduce additional tailoring modifications in the lasso scaffold, such as phosphorylation, methylation, or acetylation of the C-terminal residue,11,14,15 or its epimerization into a D-amino acid.16
In this work, we characterize pseudomycoidin – a new lasso peptide encoded by Bacillus pseudomycoides DSM 12442. We show that pseudomycoidin is modified by C-terminal phosphorylation followed by the previously unencountered mono and/or diglycosylation. Bioinformatics analysis suggests that homologs of B. pseudomycoides DSM 12442 genes required for pseudomycoidin glycosylation are common in lasso peptide gene clusters of diverse bacteria suggesting that this modification is widespread. We hypothesize that terminal phosphorylation followed by the introduction of bulky glucosyl modification may serve as a lock helping to stabilize the tail in the ring of wild-type pseudomycoidin. Uniquely, the B1/B2 enzyme and the leader part of the precursor are dispensable for the formation of the pseudomycoidin lasso. The minimal (core peptide and C enzyme) requirements for pseudomycoidin synthesis should make it an attractive model system to study lasso peptide synthesis and activity.
Results and discussion
Heterologous expression of putative lasso peptide biosynthetic psm operons from Bacillus pseudomycoides DSM 12442 in E. coli
A bioinformatics search identified a likely lasso peptide biosynthesis cluster in the Bacillus pseudomycoides DSM 12442 genome (Fig. 1A). The cluster is similar to the previously characterized Paenibacillus dendritiformis C454 lasso peptide paeninodin pade cluster.14 The B. pseudomycoides DSM 12442 cluster, which we will refer to as psm, contains a psmC gene coding for a homolog of known lasso peptide macrolactam synthetase, a putative precursor peptide gene (psmA), a psmK gene, encoding a histidine-containing protein kinase family protein (HprK), two genes coding for split B enzyme homologs (psmB1 and psmB2), and the ABC-type transporter gene psmD. The psm cluster also contains an additional gene, psmN, located between psmB2 and psmD. Such a gene is absent in the P. dendritiformis C454 cluster. The product of psmN is annotated as a putative uncharacterized NTP transferase. Similar genes have been found at the same location in several other putative lasso peptide clusters.14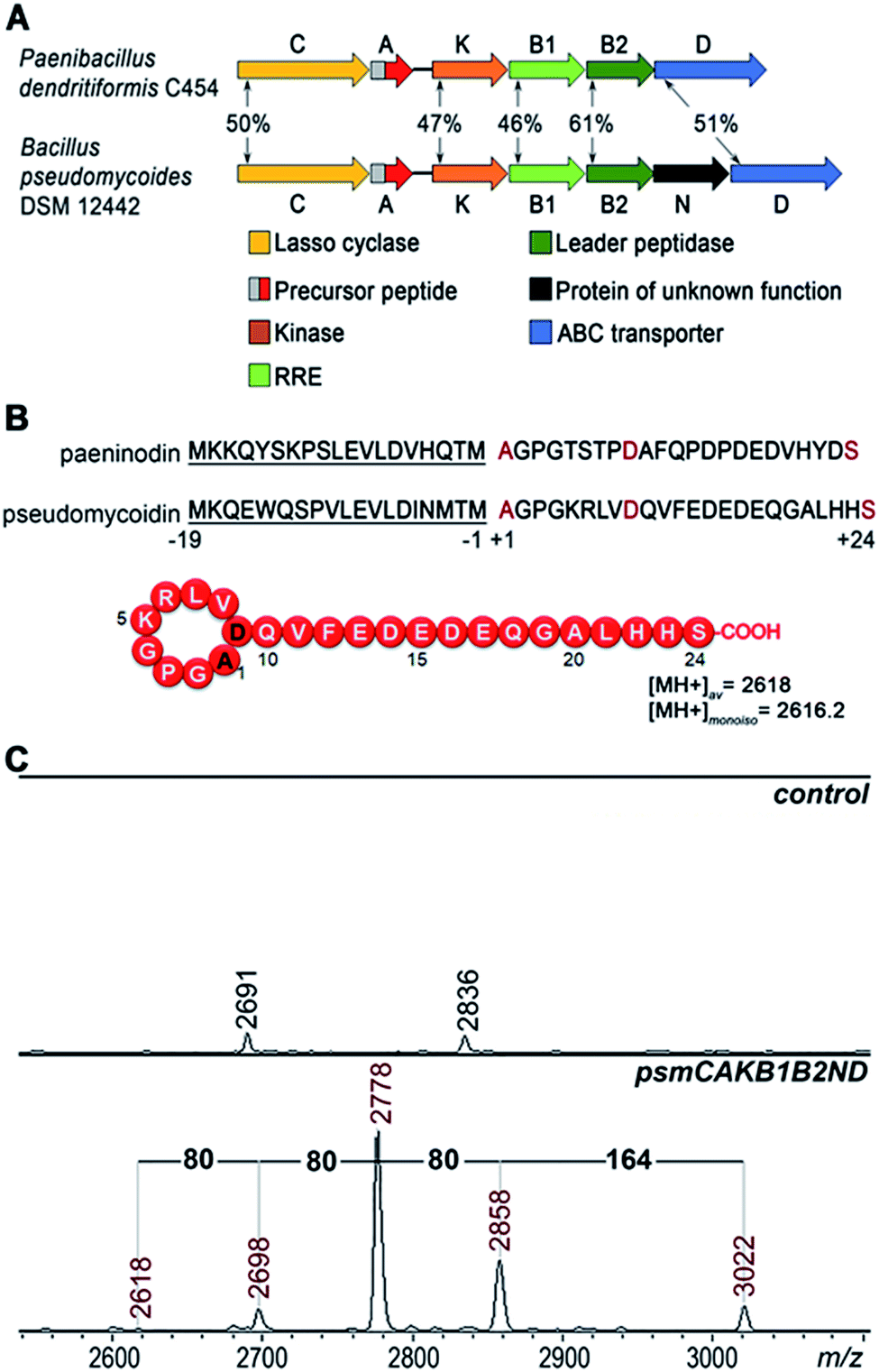 | ||
| Fig. 1 Heterologous expression of the B. pseudomycoides DSM 12442 lasso peptide psm gene cluster in E. coli. (A) Comparison of lasso peptide gene clusters from P. dendritiformis C454 and B. pseudomycoides DSM 12442. Genes are indicated by arrows (letters indicate gene names, i.e., “B” stands for either “padeB” or “psmB”), and homologous genes are indicated by same colors. Numbers indicate the percentage of identity between homologous protein sequences. Known or predicted gene product functions are listed at the bottom. RRE – RiPP precursor peptide recognition element. (B) At the top, the precursor of P. dendritiformis C454 paeninodin is aligned with the corrected B. pseudomycoides DSM 12442 pseudomycoidin precursor sequence. Leader parts are underlined. In the core parts, the N-terminal alanine and the inner aspartate that form the ring and C-terminal serine that becomes phosphorylated in paeninodin are indicated with red-color font. The predicted structure of pseudomycoidin is shown at the bottom with average and monoisotopic [MH+] of a molecule with the structure shown as indicated. (C) MALDI-MS spectra of E. coli cells harboring control (upper panel) and psm cluster (lower panel) expression plasmids. Average m/z values of difference mass-peaks are highlighted with red-color font (see text for details). | ||
Based on similarity to the pade cluster, one could expect that the psm cluster products may include lasso peptides with or without terminal phosphorylation and, possibly, with some additional unknown modifications introduced by PsmN. The predicted sequence of PsmA ends with an asparagine residue as opposed to C-terminal serine of paeninodin precursor PadeA, which is subject to phosphorylation.14,17 We resequenced the psmA gene and found a single nucleotide difference with the published sequence at the last codon. The correct sequence in fact codes for a PsmA peptide with a terminal serine. The expected average [MH+] of the lasso peptide formed from PsmA, if one assumes processing at the leader–core junction based on similarity to paeninodin, is 2618 Da (Fig. 1B). When B. pseudomycoides DSM 12442 cells and the cultured medium were analyzed by mass spectrometry, no peaks matching expected lasso-peptide cluster products – with or without terminal phosphates – were observed. We therefore introduced the psm cluster in a heterologous E. coli host. The psmCA genes were cloned on one expression plasmid under the control of a T5-lac promoter, while the remaining genes were cloned on a compatible plasmid under the control of an arabinose-inducible Para promoter. As can be seen from Fig. 1C, five difference mass peaks were present in the potential area of interest of the mass spectrum of cells carrying plasmids with psm genes compared to control (cells carrying empty plasmid vectors). One peak, with average m/z = 2618, matched the expected mass of a lasso peptide formed from the PsmA precursor. Three peaks, with average m/z = 2698, 2778, and 2858, differed from the m/z = 2618 peak and from each other by 80 atomic unit increments, a value matching a phosphate residue. The fifth difference peak had an m/z value of 3022 and could not be matched to any known lasso peptide modification.
E. coli cells that expressed psm cluster products grew well and did not inhibit the growth of several Gram-positive and Gram-negative bacteria tested. A similar result was earlier obtained with paeninodin, which was also devoid of antibacterial activity.14
Mutational analysis of the psm cluster
A set of expression plasmids carrying deletions of psm genes was prepared and introduced in E. coli and the products were analyzed. Deletion of psmK resulted in accumulation of the m/z = 2618 peak only (Fig. 2). The amplitude of its signal was strongly increased compared to the mass spectrum of cells with full clusters. The MS-MS fragmentation spectrum of this peak is consistent with it being a core PsmA peptide with a cycle between Ala1 and Glu.9 We will refer to this compound as pseudomycoidin (Fig. S1†).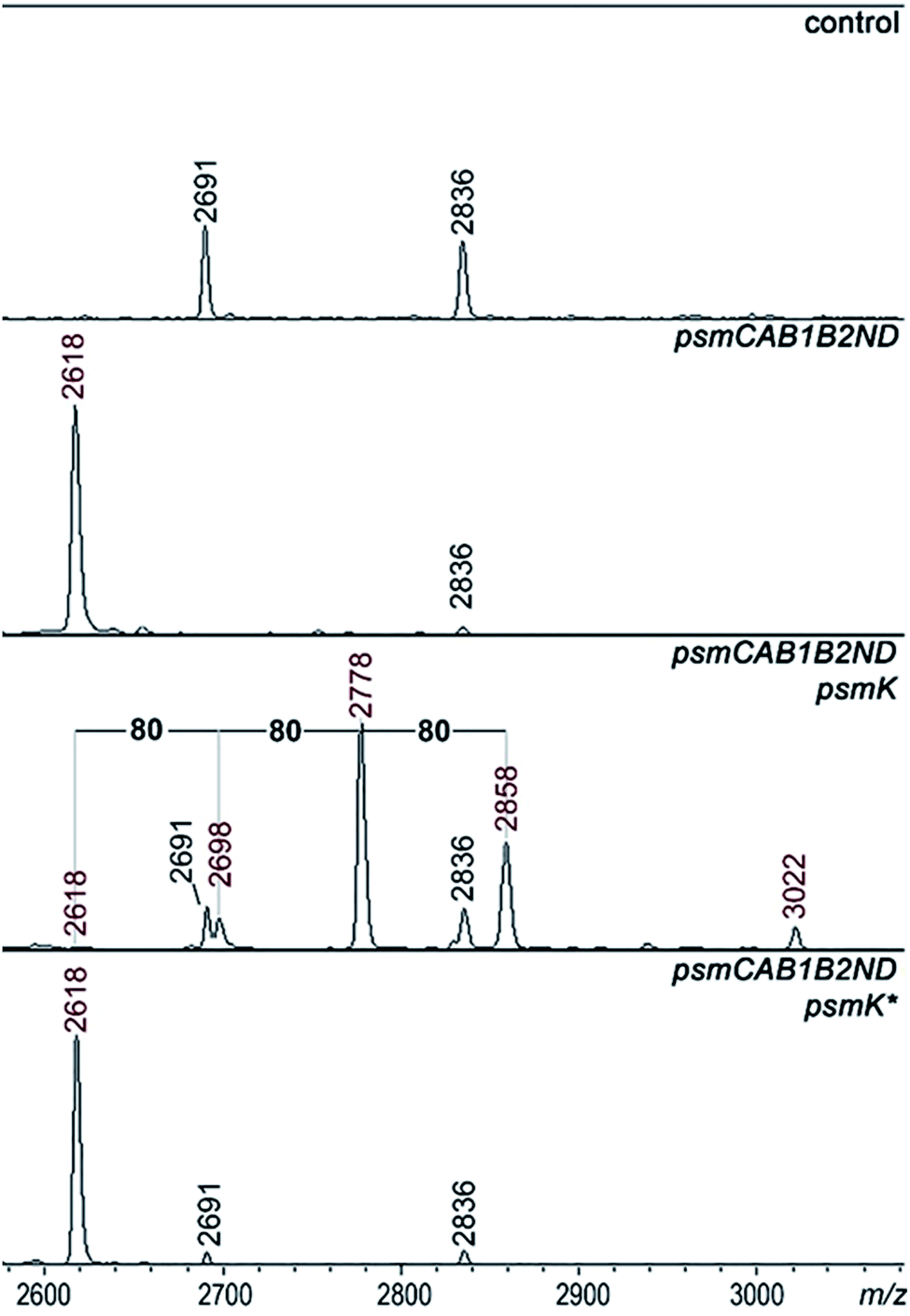 | ||
| Fig. 2 Identification of psm cluster products produced in the presence of the PsmK kinase. MALDI-MS spectra of E. coli cells harboring control empty vector plasmids (upper panel), plasmids expressing the psm cluster without the psmK gene (psmCAB1B2ND) or cells expressing the mutated cluster and harboring an additional plasmid expressing wild-type or D158A D159A double mutant psmK (psmK*). Mass-peaks that are common to both control and experimental cells are indicated by black-color font. The average m/z values of mass-peaks of psm cluster products are highlighted with red-color font (see text for details). | ||
The disappearance of m/z = 2698, 2778, and 2858 peaks in cells lacking psmK is consistent with them being, respectively, the products of mono-, di-, and triphosphorylation of pseudomycoidin. When these cells were supplemented with a compatible psmK expression plasmid, the 2778 and 2858 peaks reappeared. MS-MS analysis of the m/z = 2698 and m/z = 2778 peaks indicates that the +80 Da orthophosphate and +160 Da pyrophosphate are located at the C-terminal serine residue of pseudomycoidin, consistent with the PadeK-dependent phosphorylation of paeninodin, for which the side chain of the terminal Ser was shown to be a substrate.14 Expression of PsmK that contained D158A and D159A substitutions in the predicted metal binding site did not restore production of phosphorylated pseudomycoidin (Fig. 2).
Analysis of mass spectra of producing cells in the area corresponding to full-sized PsmA revealed multiple phosphorylated PsmA in cells carrying intact psmK. The result thus shows that PsmA precursors can be phosphorylated and phosphorylation may occur prior to cyclisation (Fig. S2†).
Deletion of psmN led to disappearance of the m/z = 3022 peak while the other four pseudomycoidin peaks remained unchanged. When the psmN gene was introduced into a separate plasmid, the 3022 peak reappeared and became more prominent. In addition, new peaks with m/z = 2940 and m/z = 2860 were observed. The mass difference between monophosphorylated pseudomycoidin (m/z = 2698) and the m/z = 2860 peak is 162 Da. The difference between m/z = 2860 and m/z = 3022 peaks is also 162 Da. In addition, the difference between diphosphorylated pseudomycoidin (m/z = 2778) and the m/z = 2940 peak is also 162 Da. It thus appears that PsmN is required for installation of a 162 Da modification onto phosphorylated forms of pseudomycoidin.
Cellular extracts prepared from cultures expressing the psm cluster without psmN or the same cultures harboring the additional psmN expression plasmid were prepared, treated with calf intestinal alkaline phosphatase (CIAP) and analyzed by mass spectrometry (Fig. S3†). In extracts lacking PsmN, CIAP treatment resulted, as expected, in accumulation of unphosphorylated pseudomycoidin (m/z = 2618) and decreased signals from phosphorylated forms. In cells expressing PsmN, the diphosphorylated pseudomycoidin peak disappeared, and the m/z = 2618 signal increased, but the m/z = 2860 and 3022 peaks remained. The results thus suggest that modification(s) in m/z = 2860 and 3022 compounds prevents the removal of terminal phosphate residues.
MS-MS fragmentation spectra of compounds from monoisotopic m/z = 2858.3 and m/z 3020.3 peaks were obtained (Fig. S4†). As a control, fragmentation of monoisotopic m/z = 2856.3 triphosphorylated pseudomycoidin was also performed. This resulted in an expected fragmentation pattern with peaks matching the successive loss of the phosphate groups from the C-terminal serine residue. The fragmentation of the m/z = 2858.3 compound revealed a loss of a 162 Da unit, followed by the loss of phosphate. The fragmentation of the m/z = 3020.3 compound proceeded similarly – through the sequential loss of 324 (2 × 162) and 98 Da from the C-terminal serine residue of the lasso peptide.
The 162 and 324 Da mass differences match the addition of one (C6H10O5) or two hexose residues (C12H20O10), respectively. High-resolution mass spectrometry of unphosphorylated and monophosphorylated pseudomycoidin resulted in monoisotopic m/z values of 2616.2431 and 2696.2118, respectively, which are in 0.5 ppm accordance with the calculated values for brutto formulae of expected compounds (2616.2439 and 2696.2104, respectively). An m/z value of 3020.3175 was obtained for the major peak observed in cells expressing PsmN. This is in 0.5 ppm accordance with calculated values for a brutto formula of monophosphorylated pseudomycoidin with two hexose residues added (3020.3162). Indeed, the mass difference between m/z = 3020.3162 and m/z = 2696.2104 is 324.1057, which matches within 0.5 ppm a dihexose residue C12H20O10 (324.1056).
PsmN belongs to a family of uncharacterized nucleotidyltransferases and has no closely related proteins with a known function. Protein modeling using Phyre2 server (http://www.sbg.bio.ic.ac.uk/phyre2/html/) reveals limited structural homology between PsmN and 2′′-aminoglycoside nucleotidyltransferases (PDB 5cfs; PDB 4xje). This allowed us to identify possible catalytic metal binding amino acids in PsmN. A PsmN D134A D136A double mutant was inactive when overexpressed in cells bearing the psm cluster with the psmN deleted (Fig. 3A, lower panel), i.e., no hexose-modified pseudomycoidin peaks were observed.
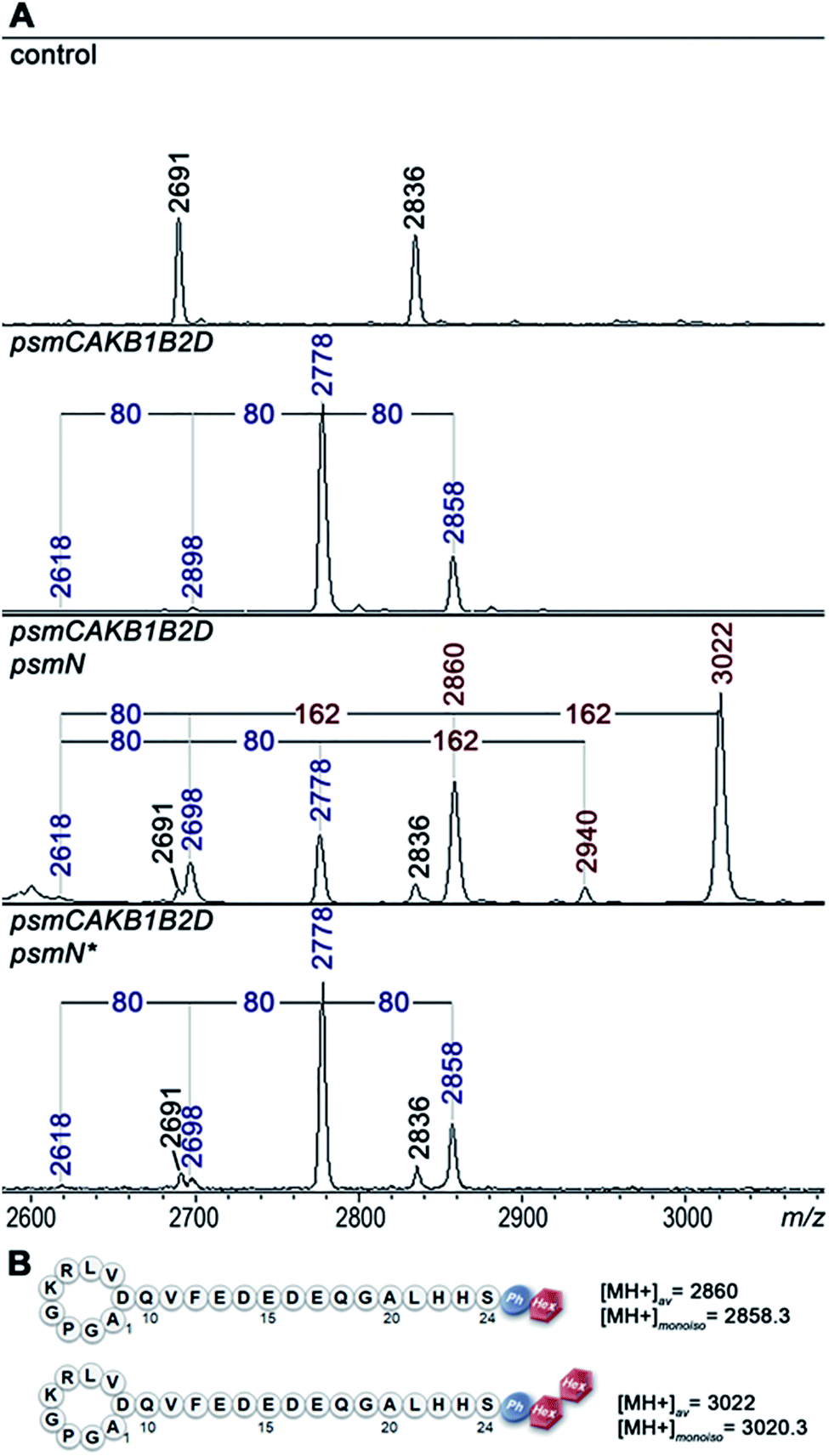 | ||
| Fig. 3 PsmN is required for an additional posttranslational modification of phosphorylated pseudomycoidin. (A) MALDI-MS analysis of E. coli BL21(DE3) cells containing control plasmids, or plasmids expressing the psm cluster with psmN deleted, with or without additional plasmid expressing wild-type, or mutant (psmN*, encodes the D134A D136A double mutant) psmN. The average m/z values of mass-peaks specific for cells carrying the psm genes are marked with blue font and for psmN-dependent peaks, brown color font. (B) Schematic structures of fully modified pseudomycoidins (see text for details). | ||
Overall, the data above show that in addition to variable numbers of phosphate groups attached to the C-terminal serine residue, pseudomycoidin can be decorated with an unidentified hexose(s) installed on the phosphate(s). PsmN is required for this modification.
Plasmid lacking psmB1 and psmB2 genes were also created. Surprisingly, cells carrying the psm cluster without these genes, which are essential for lasso peptide synthesis in all systems that have been investigated to date, continued to produce pseudomycoidin and its phosphorylated variants (Fig. S5†). Although both PsmB1 and PsmB2 are functional in vivo,18 the unexpected result suggested that in the absence of these two proteins their role is carried out by some host proteins. To confirm further functionality of PsmB1 and PsmB2, an in vitro experiment was performed using purified recombinant proteins and synthetic PsmA precursor peptides. As can be seen from Fig. S6,† the precursor peptide was specifically cleaved at the core–leader junction in the presence of both PsmB1 and PsmB2. No cleavage was observed when PsmB1 or PsmB2 were used separately (Fig. S6†). The PsmB1/PsmB2 cleavage was specific since the synthetic peptide corresponding to the precursor peptide encoded by a similar cluster from Paenibacillus polymyxa ATCC 842 (Fig. S7A†) was not cut despite having a leader sequence that is highly similar (14 out of 19 identical residues) to the PsmA leader sequence and an identical leader–core junction sequence (Fig. S7B†).
To further investigate Psm synthesis in the absence of PsmB1 and PsmB2 enzymes, we concentrated on analysis of a simplified psmCAD cluster lacking the psmK and psmN genes. E. coli cells harboring such a cluster produced pseudomycoidin without the phosphate or hexose modifications (Fig. 4). To test whether the ability to produce a lasso peptide without the universally conserved B1 and B2 enzymes is a specific feature of the heterologous production host, we introduced the psmCAD cluster into B. subtilis, an organism that is phylogenetically closer to the native producer than E. coli. As can be seen from Fig. S8,† cells harboring the psm expression plasmid produced correctly the processed linear core peptide of PsmA and the cyclized form, i.e., pseudomycoidin. We therefore conclude that PsmB1 and PsmB2 though functional and specific to the pseudomycoidin propeptide PsmA are not required for pseudomycoidin production either in E. coli or in B. subtilis.
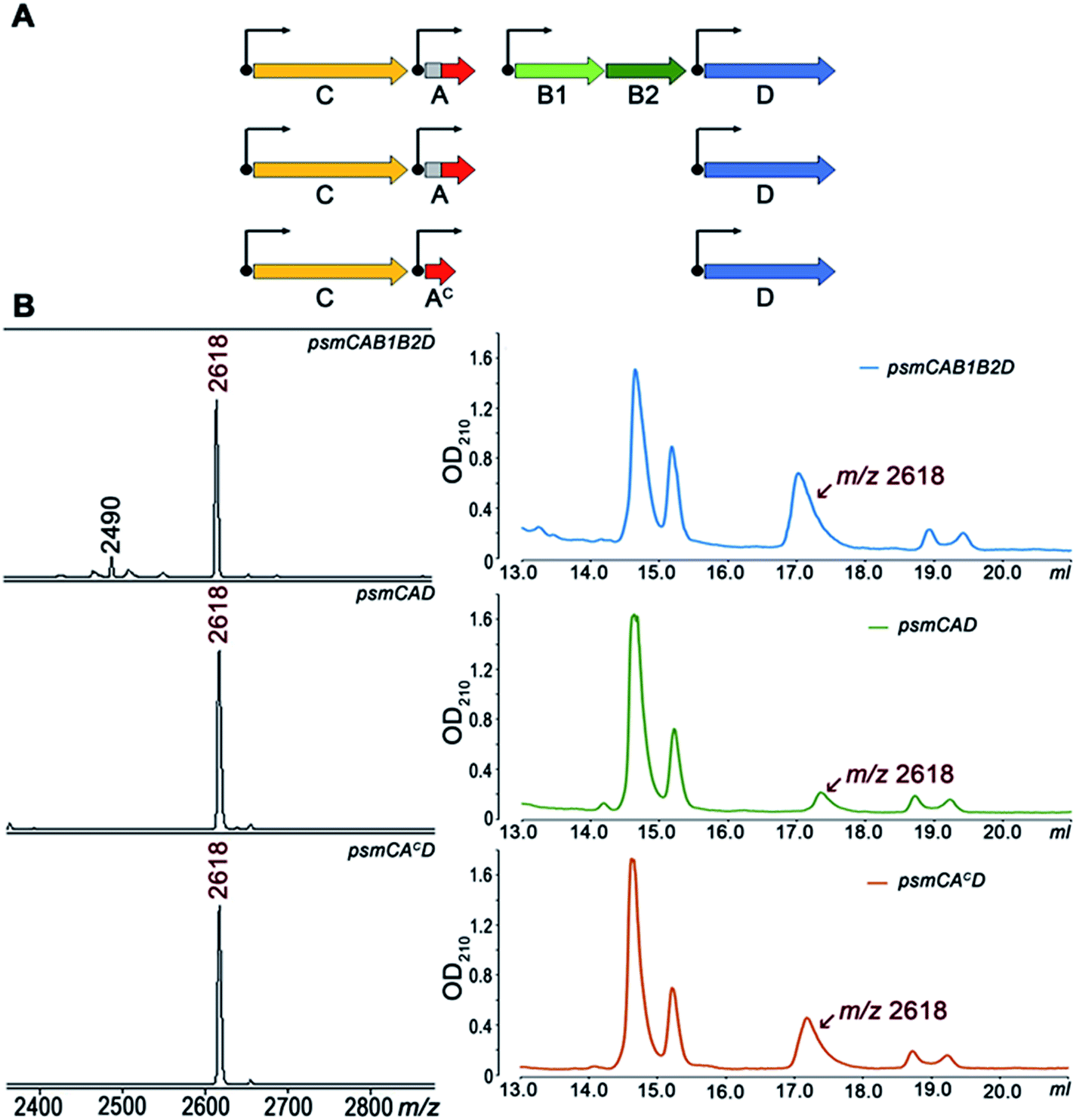 | ||
| Fig. 4 PsmB1/PsmB2 proteins are dispensable for pseudomycoidin lasso structure formation. (A) Three-gene arrangements used for pseudomycoidin production in E. coli. (B) MALDI MS spectra of the cells harboring the indicated sets of the psm genes (left panels) and HPLC traces of fractionation of corresponding conditioned media (right panels). HPLC fractions containing the lasso peptide (average m/z = 2618) were identified by MALDI MS. | ||
To determine if the PsmA leader sequence is needed for the cyclisation reaction catalyzed by PsmC, we next created a pseudomycoidin expression system that encoded the core peptide of PsmA and PsmAc, without the leader part. While the recombinant core peptide was produced with N-terminal formyl-methionine incorporated during translation initiation, we expected this residue to be efficiently removed by the E. coli methionine aminopeptidase since the first amino acid of the core peptide is Ala, the preferred substrate for this enzyme.19 Analysis of E. coli cells expressing psmCAcD genes revealed robust production of pseudomycoidin. The yield was higher than that in cells expressing psmCAD, suggesting that removal of the leader in the absence of PsmB1 and PsmB2 is a limiting step in production. Overall, we conclude that production of lasso peptide pseudomycoidin requires neither the leader part of the precursor peptide nor the universally conserved B1 and B2 enzymes.
To check whether the core part of PsmA can form a lasso in the absence of PsmC, plasmids expressing psmAB1B2D and psmAD were tested. Cells lacking PsmC produced only linear PsmA and fragments thereof. Thus, PsmC is strictly required for pseudomycoidin lasso structure formation.
We also tested pseudomycoidin production in the absence of the putative PsmD transporter by cells expressing psmCA and psmCAc. Pseudomycoidin was detected inside the cells in both cases, indicating that PsmD is not required for its synthesis. We conclude that a gene encoding the core part of the PsmA peptide and the lasso peptide macrolactam synthetase psmC gene are sufficient for production of pseudomycoidin. We attempted in vitro synthesis of pseudomycoidin from a chemically synthesized PsmA core peptide using recombinant PsmC, alone or in the presence of PsmB1/B2 proteins. No cyclisation was observed; however, the PsmC enzyme was very poorly expressed and likely misfolded (data not shown).
The product of the psm cluster produced in the absence of psmK and psmN is not a threaded lasso
We were next interested to determine whether pseudomycoidin is a threaded lasso peptide or a branched cycle. High temperature treatment has been shown to stimulate the release of the tail in some threaded lasso compounds.20 We compared chromatographic behavior of pseudomycoidin produced from full-sized PsmA or from its core part and incubated for 4 hours at 95 °C with untreated control. While heat treatment caused partial degradation of the peptide, no change in HPLC retention time for full-sized pseudomycoidin peak was observed (Fig. S9†). Since in studied systems chromatographic behavior of branched cycles and threaded lasso isomers is usually distinct,20 the result suggests that pseudomycoidin is either a branched cycle or a stable threaded lasso, such as microcin J25 (ref. 9) or capistruin.21We performed several additional tests that have been previously used to distinguish branched cycles from threaded lassos and to compare pseudomycoidin produced from full-sized or the core part of PsmA and in the presence or the absence of PsmB1/B2 proteins. To have a reference substrate with a known topology, we treated pseudomycoidin produced by cells harboring psmCAB1B2D with trypsin, which was found to cut inside the ring between after Lys5 and Arg6 residues (Fig. 1). The resulting molecule should be Y-shaped and its tail shall be free from any possible topological constrains. Equal amounts of trypsin-digested pseudomycoidin and intact compounds produced by cells with various combinations of psm genes were combined and treated with carboxypeptidase Y (Fig. S10†). The 18 Da difference between trypsin-digested and intact substrates allowed us to follow protease digestion of both substrates in the same reaction. Carboxypeptidase Y was expected to cleave from C-termini of both intact and trypsin digested peptides. It was also expected to cleave amino acids from a C-terminus that was formed after the opening of the ring in the trypsin digested substrate; however, mass peaks corresponding to this activity were minor and did not interfere with analysis (Fig. S10†). Cleavage of threaded lasso substrates but not of trypsin-treated ones was expected to be limited by topology. As can be seen from Fig. S10,† digestion with carboxypeptidase led to accumulation of products ending at His,23 Ala,20 Gly,19 Gln,18 and Gln.10 Peaks corresponding to each of these intermediates were split, with a characteristic 18 Da difference indicating that they were produced from both intact and the Y-shaped pseudomycoidin. The absence of differences in carboxypeptidase digestion product accumulation is consistent with the branched cycle topology of pseudomycoidin. Compounds produced by cells harboring various combinations of psm genes appeared to be identical.
Another test included digestions with Glu-C protease (Fig. S11†). Under our conditions, this protease cleaves pseudomycoidin at positions 13, 15, and 17 in the tail. For a stable threaded lasso such as microcin J25, cleavage in the tail results in the formation of a rotaxane intermediate consisting of a ring and a linear tail trapped within it. HPLC analysis of Glu-C treated pseudomycoidin revealed the disappearance of intact compounds and the appearance of additional peaks. According to MALDI-MS, these peaks contained separate rings or tail products of Glu-C digestion (Fig. S11†) with no peaks containing both fragments, as would be expected for a rotaxane topology. Taken together, the results suggest that pseudomycoidin, once purified from cells harboring psmCAB1B2D or smaller sets of genes, is a branched cyclic peptide.
Introduction of a bulky residue at the tail of pseudomycoidin leads to threaded lasso topology
Given the fact that pseudomycoidin synthesis machinery is homologous to that of validated lasso peptides, the branched lasso topology may arise when the tail is withdrawn from the macrolactam cycle during purification. To test this possibility, a bulky phenylalanine residue was introduced instead of pseudomycoidin leucine 21 in the context of plasmid-borne psmCAB1B2D or psmCAcD clusters. Cells producing either of the clusters produced compounds with average m/z = 2652, an expected value for a pseudomycoidin lasso peptide carrying the L21F substitution. The mutant peptide produced by cells harboring either psmCAB1B2D or psmCAcD clusters eluted much earlier than wild-type pseudomycoidin (Fig. S12†). After incubation at 95 °C and rechromatography, the pseudomycoidin L21F peak became split, with a new peak eluting later, at a position similar to where the wild-type pseudomycoidin eluted (Fig. 5A and S12†). Mass-spectrometric analysis revealed that this later peak still had an m/z = 2652 and thus corresponded to chemically intact pseudomycoidin L21F. Material from each peak was next treated with carboxypeptidase Y. For peak B that appeared after thermal treatment (Fig. 5B), a pattern that was previously observed for wild-type pseudomycoidin and consistent with branched lasso topology was observed. In contrast, for the earlier eluting peak A, the cleavage rate was considerably slower, as only cleavage products corresponding to removal of C-terminal 4 amino acids were observed (Fig. 5B).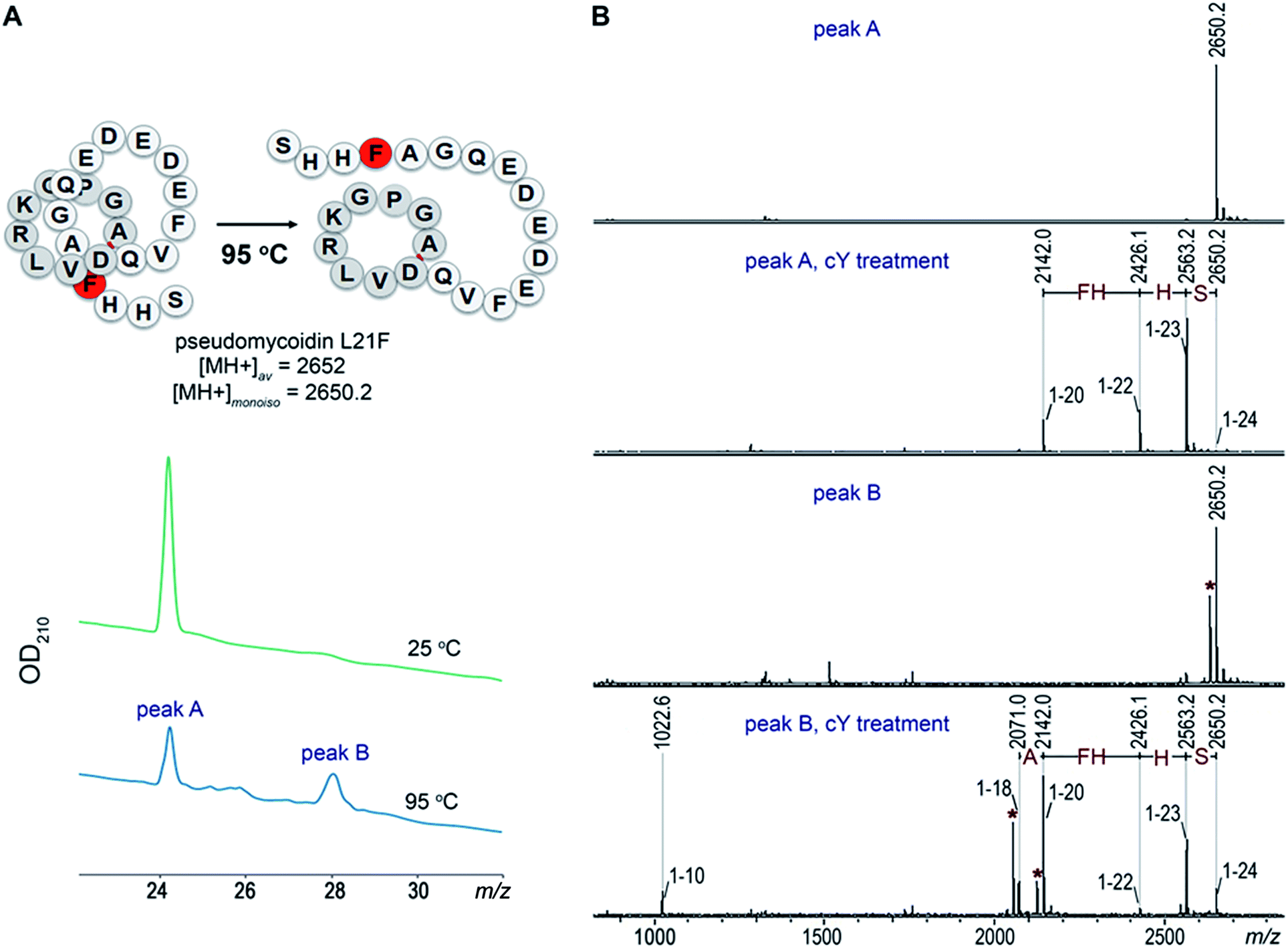 | ||
| Fig. 5 L21F mutant of pseudomycoidin is a threaded lasso peptide. An L21F mutant of pseudomycoidin was produced in the expression system, which encoded the mutant core peptide without the leader and the psmC and psmD genes. (A) HPLC traces of the pseudomycoidin L21F before (green) and after (blue) incubation at 95 °C for 2 h. (B) MALDI MS spectra of lasso peptides from the HPLC peaks A (two lower panels) and B (two top panels) before and after 80 min treatment with carboxypeptidase Y. The smallest proteolytic fragments identified (1–10) correspond to N-terminal macrocycle extended by one amino acid. Dehydrated fragments are marked with asterisk. | ||
In order to confirm our findings by an independent approach, we recorded NMR spectra of wild-type and L21F pseudomycoidins purified from cells harboring psmCAB1B2D and psmCAcD gene sets, correspondingly, in an aqueous phosphate buffer. Homonuclear TOCSY and NOESY spectra were recorded at 800 MHz and led to a full assignment of both peptides (Fig. S13 and S14†). Importantly, we observed the characteristic NOE peak between the amide proton of Ala1 and the Hβ protons of Asp9 in both spectra (Fig. 6), confirming the cyclic nature of both peptides.22 However, upon closer comparison of the spectra, both the amide and methyl proton spectral regions of the wild-type pseudomycoidin (Fig. S13†) appear more compact than those of the L21F mutant (Fig. S14†). As an example, we show the methyl groups of V11 that resonate at 0.84 ppm in the spectrum regions of the wild type pseudomycoidin, whereas they shift to 0.3 ppm in the L21F mutant (Fig. 6). Another salient difference between both spectra is that while the amide protons of the wild type peptide are all sharp and of comparable intensity, several amide protons of the L21F mutant pseudomycoidin are severely broadened. This is the case for A1 and G2 and also for Q10 and F12 (Fig. S14†). Although a full conformational analysis by NMR is beyond the scope of the present article, these findings confirm the hypothesis of the C-terminus threading through the macrolactam ring when a bulky Phe occupies position 21. The tail is however not static, but slides back and forth through the ring, thereby leading to signal broadening.
 | ||
| Fig. 6 1D spectra (top) and homonuclear TOCSY/NOESY spectra (bottom) of wild type (left) and mutant L21F (right) pseudomycoidin. The amide and methyl proton regions of the wild type peptide spectrum are reduced when compared to those of the L21F mutant, and peaks are sharper in the former. The characteristic NOE contact between A1 HN and D9 Hb is circled in both NOESY spectra. | ||
Overall, these data are consistent with threaded lasso topology of pseudomycoidin L21F and suggest that the branched lasso topology of wild-type pseudomycoidin is derived from threaded lasso after the tail is withdrawn from the ring during purification. E. coli cells that produced pseudomycoidin L21F and cells producing wild-type pseudomycoidin grew and did not inhibit the growth of laboratory strains of E. coli B, B. subtilis 168, or Arthrobacter sp. FB24, suggesting that the lasso topology in itself is not sufficient for antibacterial activity.
Conclusions
The principal result of this work is the demonstration that a lasso peptide can be formed in vivo in the absence of B1/B2 enzymes that heretofore were thought to be a universal component of lasso peptide biosynthesis machinery. In systems that have been studied, the B1 protein recognizes the lasso peptide precursor leader sequence and then recruits the B2 protease to cleave at the leader–core junction and generate an N-terminus, which is used by the C macrolactam synthetase to form an amide bond with a side chain of a Glu or Asp residue in the core. In the case of pseudomycoidin, this requirement must be bypassed through involvement of a cellular protease(s) that cleaves at the leader–core junction. The necessary enzymes are present in either E. coli or B. subtilis since both surrogate hosts produce pseudomycoidin in the absence of B1/B2 proteins. Given the phylogenetic distance between E. coli and B. subtilis, it is likely that the PsmA precursor has a specific structure, which exposes the leader–core junction to proteolytic attack by divergent proteases. In systems that have been investigated, the B and C enzymes are thought to interact with the core part, once generated, being passed over to the macrolactam synthetase.23,24 In the case of pseudomycoidin, the situation must be different since the C protein must be able to convert the peptide part to a lasso in the absence of any additional factors, as exemplified by the efficient synthesis of pseudomycoidin in the presence of the PsmA core peptide and PsmC only. The idea of universality of requirement of the B1/B2 enzymes (sometimes fused into a single B protein) may have originated due to a bias during bioinformatics searches used to reveal lasso peptide clusters. In fact, we identified a putative lasso peptide cluster from a cyanobacterium Stanieria cyanosphaera PCC 7437 (core genes locus tags STA7437_RS03725 and STA7437_RS03720) that does not encode B proteins, while coding for an apparently functional macrolactam synthetase, a precursor peptide, and a PsmK kinase homolog. It is thus possible that many lasso peptide clusters were overlooked during prior searches.We here show that psmN is required for installation of unique mono and/or dihexose modification on phosphorylated pseudomycoidin. The exact nature of hexose modification of pseudomycoidin is beyond the scope of the present study but the results suggest that PsmN is either a glycosyl transferase of a novel kind, since it does not contain recognizable motifs characteristic of known enzymes of this class or, alternatively, is an enzyme that produces a substrate used by one of glucosyl transferases present in the E. coli host. Interestingly, the cyanobacterial cluster mentioned above encodes a clearly recognizable (unlike PsmN) glycosyltransferase that may perform a similar function. Be as it may, the psmN-like genes are common in many lasso peptide clusters and must play an evolutionary conserved essential function. It is possible that the bulky hexose residues appended to phosphorylated C termini of pseudomycoidin and related peptides may serve as a lock to maintain the threaded lasso conformation. If true, this will be an additional, third strategy for stabilizing this topology.
The unique fold and exceptional stability make lasso peptides an attractive scaffold for development of non-natural bioactive compounds. Genetic engineering reveals that the core part of the precursor peptide can tolerate significant alterations without preventing recognition and processing by the biosynthesis machinery in vivo.23,25 The availability of the minimal pseudomycoidin in vivo system should make it straightforward to generate libraries of various pseudomycoidin derivatives and understand the determinants of PsmC specificity. These experiments are currently ongoing in our laboratory.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by NIH RO1 grant AI117270 (to KS and Satish A. Nair) and Skoltech institutional funds to KS, and the Russian Science Foundation grant RSF 19-14-00266 to SD. MALDI MS facility became available to us in the framework of the Moscow State University Development Program PNG 5.13. We are grateful to Dr I. Ivanov (Oncolead GmbH&Co.KG, Germany), Dr T. Artamonova and Dr M. Khodorkovsky of Peter the Great St. Petersburg Polytechnic University, St. Petersburg, Russia for their invaluable help. Access to the 800 MHz NMR spectrometer was provided by MetaToul (Toulouse Metabolomics & Fluxomics facilities, http://www.metatoul.fr) as part of the French National Infrastructure for Metabolomics and Fluxomics MetaboHUB-AR-11-INBS-0010 (http://www.metabohub.fr) and supported by the Région Midi-Pyrénées, the ERDF, the SICOVAL and the French Minister of Education & Research, who are gratefully acknowledged.References
- J. I. Tietz, C. J. Schwalen, P. S. Patel, T. Maxson, P. M. Blair, H.-C. Tai, U. I. Zakai and D. A. Mitchell, Nat. Chem. Biol., 2017, 13, 470–478 CrossRef CAS.
- M. O. Maksimov and A. J. Link, J. Ind. Microbiol. Biotechnol., 2014, 41, 333–344 CrossRef CAS.
- S. Duquesne, D. Destoumieux-Garzón, S. Zirah, C. Goulard, J. Peduzzi and S. Rebuffat, Chem. Biol., 2007, 14, 793–803 CrossRef CAS.
- P. F. Lin, H. Samanta, C. M. Bechtold, C. A. Deminie, A. K. Patick, M. Alam, K. Riccardi, R. E. Rose, R. J. White and R. J. Colonno, Antimicrob. Agents Chemother., 1996, 40, 133–138 CrossRef CAS.
- K. L. Constantine, M. S. Friedrichs, D. Detlefsen, M. Nishio, M. Tsunakawa, T. Furumai, H. Ohkuma, T. Oki, S. Hill, R. E. Bruccoleri, P. F. Lin and A. Mueller, J. Biomol. NMR, 1995, 5, 271–286 CrossRef CAS.
- T. A. Knappe, U. Linne, X. Xie and M. A. Marahiel, FEBS Lett., 2010, 584, 785–789 CrossRef CAS.
- M. A. Delgado, M. R. Rintoul, R. N. Farías and R. A. Salomón, J. Bacteriol., 2001, 183, 4543–4550 CrossRef CAS.
- J. Yuzenkova, M. Delgado, S. Nechaev, D. Savalia, V. Epshtein, I. Artsimovitch, R. A. Mooney, R. Landick, R. N. Farias, R. Salomon and K. Severinov, J. Biol. Chem., 2002, 277, 50867–50875 CrossRef CAS.
- K. Kuznedelov, E. Semenova, T. A. Knappe, D. Mukhamedyarov, A. Srivastava, S. Chatterjee, R. H. Ebright, M. A. Marahiel and K. Severinov, J. Mol. Biol., 2011, 412, 842–848 CrossRef CAS.
- M. Metelev, A. Arseniev, L. B. Bushin, K. Kuznedelov, T. O. Artamonova, R. Kondratenko, M. Khodorkovskii, M. R. Seyedsayamdost and K. Severinov, ACS Chem. Biol., 2017, 12, 814–824 CrossRef CAS.
- E. Gavrish, C. S. Sit, S. Cao, O. Kandror, A. Spoering, A. Peoples, L. Ling, A. Fetterman, D. Hughes, A. Bissell, H. Torrey, T. Akopian, A. Mueller, S. Epstein, A. Goldberg, J. Clardy and K. Lewis, Chem. Biol., 2014, 21, 509–518 CrossRef CAS.
- K. Kimura, F. Kanou, H. Takahashi, Y. Esumi, M. Uramoto and M. Yoshihama, J. Antibiot., 1997, 50, 373–378 CrossRef CAS.
- M. Daniel-Ivad, N. Hameed, S. Tan, R. Dhanjal, D. Socko, P. Pak, T. Gverzdys, M. A. Elliot and J. R. Nodwell, ACS Chem. Biol., 2017, 12, 628–634 CrossRef CAS.
- S. Zhu, J. D. Hegemann, C. D. Fage, M. Zimmermann, X. Xie, U. Linne and M. A. Marahiel, J. Biol. Chem., 2016, 291, 13662–13678 CrossRef CAS.
- C. Zong, W. L. Cheung-Lee, H. E. Elashal, M. Raj and A. J. Link, Chem. Commun., 2018, 54, 1339–1342 RSC.
- Z. Feng, Y. Ogasawara, S. Nomura and T. Dairi, ChemBioChem, 2018, 19, 2045–2048 CrossRef CAS.
- S. Zhu, C. D. Fage, J. D. Hegemann, D. Yan and M. A. Marahiel, FEBS Lett., 2016, 590, 3323–3334 CrossRef CAS.
- T. Sumida, S. Dubiley, B. Wilcox, K. Severinov and S. Tagami, ACS Chem. Biol., 2019, 14, 1619–1627 CrossRef CAS.
- Q. Xiao, F. Zhang, B. A. Nacev, J. O. Liu and D. Pei, Biochemistry, 2010, 49, 5588–5599 CrossRef CAS.
- M. Zimmermann, J. D. Hegemann, X. Xie and M. A. Marahiel, Chem. Biol., 2013, 20, 558–569 CrossRef CAS.
- T. A. Knappe, U. Linne, L. Robbel and M. A. Marahiel, Chem. Biol., 2009, 16, 1290–1298 CrossRef CAS.
- X. Xie and M. A. Marahiel, ChemBioChem, 2012, 13, 621–625 CrossRef CAS.
- K.-P. Yan, Y. Li, S. Zirah, C. Goulard, T. A. Knappe, M. A. Marahiel and S. Rebuffat, ChemBioChem, 2012, 13, 1046–1052 CrossRef CAS.
- W. L. Cheung, M. Y. Chen, M. O. Maksimov and A. J. Link, ACS Cent. Sci., 2016, 2, 702–709 CrossRef CAS.
- O. Pavlova, J. Mukhopadhyay, E. Sineva, R. H. Ebright and K. Severinov, J. Biol. Chem., 2008, 283, 25589–25595 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Supplementary Fig. S1–S14 and experimental procedures. See DOI: 10.1039/c9sc02370d |
| ‡ These authors contributed equally to this work. |
| § Present address: Department of Cell and Molecular Biology, Uppsala University, Uppsala, Sweden. |
| This journal is © The Royal Society of Chemistry 2019 |