
Atomically miniaturized bi-phase IrOx/Ir catalysts loaded on N-doped carbon nanotubes for high-performance Li–CO2 batteries†

Abstract
Li–CO2 batteries are bifunctional, spontaneously storing energy and fixing environmental CO2 without external electricity. Identifying efficient catalysts that can accelerate the reversible formation and decomposition of the insulating carbonate products formed on the electrode remains challenging. To overcome this limitation, we atomically dispersed IrOx/Ir bi-phase particles as single-atom catalysts (SACs) on nitrogen-doped carbon nanotubes (NCNTs) and introduced them to facilitate a reversible Li–CO2 reaction with low overpotential and stable cycle performance for 120 cycles. The IrOx/Ir SACs were successfully minimized to an atomic scale of 4 Å and formed unique surface oxides via dangling bonds on the atomic Ir catalyst, enhancing the surface catalytic activities. The N sites doped on the carbon nanotubes increased the electronic conductivity and provided favorable nucleation sites for Ir loading. The Li–CO2 cells employing IrOx/Ir SACs loaded on NCNTs exhibited improved cell performance, reduced polarization, lower charge transfer resistance, and higher stable cyclability compared to cells employing larger-sized Ir particles on NCNTs. The reversible Li–CO2 reaction mechanism facilitated by the IrOx/Ir SAC-loaded NCNT catalyst is explained through density functional theory (DFT) calculations that demonstrated that the bond of SACs with (Li+ + e−) is strong and forms products, whereas the bond with 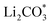 is weak and evolves products reversibly. This strategy to atomically minimize noble metal catalysts may facilitate the realization of high-performance and economical Li–CO2 batteries to achieve carbon-negativity targets.
is weak and evolves products reversibly. This strategy to atomically minimize noble metal catalysts may facilitate the realization of high-performance and economical Li–CO2 batteries to achieve carbon-negativity targets.

- This article is part of the themed collection: Journal of Materials Chemistry A Emerging Investigators
 Please wait while we load your content...
Please wait while we load your content...

