
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRecent advances and perspectives of Ir-based anode catalysts in PEM water electrolysis
Chunyan
Wang
and
Ligang
Feng
 *
*
School of Chemistry and Chemical Engineering, Yangzhou University, Yangzhou, 225002, P. R. China. E-mail: ligang.feng@yzu.edu.cn
First published on 16th November 2023
Abstract
Proton exchange membrane water electrolysis (PEMWE) is a promising sustainable hydrogen production technology that can be effectively coupled with intermittent renewable energy. Currently, iridium (Ir) based catalysts are used that can well balance catalytic activity and stability in water oxidation. Herein, our attention is directed to the recent progress of Ir-based catalysts employed in PEMWE. We first briefly outline the basic working principle of PEMWE, key components, and their functions in the devices. Then, the latest progress of Ir-based anode catalysts and their practical applications in PEMWE are introduced in detail from the aspects of Ir-based single metals, Ir-based alloys, Ir-based oxides, and some supported Ir-based catalysts. Finally, the current problems and challenges faced by Ir-based anode catalysts in future development are commented on. It is concluded that the intrinsic catalytic activity can be significantly improved through precise structural design, morphology control, and support selection. Due to the strong corrosion under acidic conditions, the anti-dissolution of Ir active species should be carefully considered for catalyst fabrication in the future. Hopefully, the current efforts can help understand the current state of Ir-based anode catalysts and develop novel and effective catalysts for application in practical PEMWE.
 Chunyan Wang | Chunyan Wang received her bachelor's degree in chemical engineering and technology from Xi’an University of Science and Technology in 2021. She is currently pursuing a master's degree at the School of Chemistry and Chemical Engineering, Yangzhou University. Her research interests focus on the design and synthesis of nanostructured catalysts for electrochemical energy conversion, including water electrolysis and small molecule oxidation. |
 Ligang Feng | Prof. Ligang Feng received his PhD degree from Changchun Institute of Applied Chemistry (CIAC), Chinese Academy of Sciences in 2012 and worked briefly at CIAC and Ecole Polytechnique Fédérale de Lausanne (EPFL) and Chalmers University of Technology from 2012 to 2016. He started the professorship in April 2016 at Yangzhou University. His research interests focus on energy conversion and storage, particularly novel and cost-effective catalyst materials from earth-abundant materials and their application in fuel cells and water splitting. |
1. Introduction
Fossil energy consumption accompanied by the emission of greenhouse gases such as carbon dioxide has exacerbated the energy crisis and caused serious environmental pollution.1–3 Therefore, it is necessary to optimize the energy structure dominated by non-renewable carbon-based fossil fuels.4–6 Hydrogen (H2) has the advantages of high calorific value, high energy density, and being environmentally friendly, and is considered an ideal energy carrier to alleviate energy and climate problems.7,8 Hydrogen can be generated by water electrolysis technology driven by renewable energy built using a clean energy system to realize a green hydrogen cycle.9,10 At present, there are mainly two types of water electrolysis technology used in industry: alkaline water electrolysis (AWE) and proton exchange membrane water electrolysis (PEMWE).11 However, AWE requires a long start-up preparation and presents a slow response, which makes it difficult to adapt to frequent changes in electricity generated by renewable energy (such as solar energy and wind energy).12,13 In contrast, PEMWE has the advantage of a fast response under highly dynamic operating conditions, making it suitable for integration with intermittent renewable energy sources.14–16 Therefore, PEMWE attracts unprecedented attention in green hydrogen production and is considered a more promising water electrolysis technology for the future.Although PEMWE has developed rapidly in recent decades, it still faces numerous challenges for large-scale industrial applications.17,18 Specifically, the acidic environment severely limits the choice of materials for internal components that should be more corrosion-resistant.19,20 In particular, it is urgent to develop more effective anode catalysts for oxygen evolution reactions (OER) that undergo harsh oxidation conditions.21,22 The current electrodes that can be used as PEMWE anode catalysts mainly rely on iridium (Ir), ruthenium (Ru)-based, and other noble metal catalysts or their oxides.23,24 Among the catalysts, Ru-based catalysts have slightly higher OER activity but suffer from rapid dissolution due to the formation of RuO42− in OER potential regions.25 Ir-based catalysts are the most suitable anodic electrocatalysts in PEMWE because they can trade-off electrochemical activity and stability to a certain extent.20,26 However, Ir-based materials still face some problems, such as the high price and low abundance of Ir, and the excessively high and unstable working potential resulting from the PEMWE when coupled with intermittent renewable energy will also affect the durability of the Ir-based electrocatalyst.27,28 Therefore, developing Ir-based catalysts with cost-effectiveness and excellent stability has become a consensus for the commercialization of PEMWE.29,30 For example, supported Ir-based catalysts such as Ir@WOx,31 IrO2@TiN1+x,32 and Ir/Au/CP33 were prepared by introducing support materials with a large specific surface area, good conductivity, and corrosion resistance.34 It can evenly disperse the Ir/IrO2 active material on its surface, reduce the catalyst loading to improve the utilization of precious metals, enhance the conductivity in the catalyst layer, and provide stable support for the active metal.10,35 Ir@WOx can reach a current density of 2.2 A cm−2 when the cell voltage is 2.0 V, and its mass activity is 120 A mgIr−1, which is 15.3 times that of commercial Ir black.31 The IrO2@TiN1+x catalyst also showed excellent activity when used as the anode in PEM single cells.32 It only required a cell voltage of 1.916 V to drive 2 A cm−2, which was 57 mV and 130 mV lower than the cell voltages of IrO2@TiN and IrO2 as the anode, respectively, and the IrO2@TiN1+x electrode showed very good stability for 100 h at 1 A cm−2. Its excellent performance was attributed to the fact that the TiN support with Ti defects on the surface provides abundant nucleation sites for Ir ions, allowing sub-2 nm IrO2 nanoparticles to be evenly dispersed on the surface of the TiN1+x support, increasing the number of active sites and stability.
Considering the current exciting and inspiring achievements of Ir-based anode catalysts applied in the PEMWE, it would be timely required to review and summarize the efforts in this important field to provide references for relevant researchers. To the best of our knowledge, some relevant reviews have been completed mainly focused on reports of Ir and Ru-based catalysts in acidic OER, while there are very few summaries of their practical applications in PEMWE.14,27,36–38 Actually, this issue is very important to comprehensively understand the current state of the Ir-based catalysts development for practical applications. Therefore, in this review, we first briefly introduce the working principle of PEMWE and the key components and their functions in the devices. Combined with some typical case reports, the practical application of high-efficiency Ir-based catalysts in PEMWE such as Ir-based single metals, alloys, oxides, and supported catalysts was carefully summarized in detail. The development prospects and challenges of Ir-based catalysts in practical applications of PEMWE are also discussed and proposed. The intrinsic catalytic activity could be improved by precise structural design, morphology control, and support selection, while the high anti-corrosion ability of the novel Ir-based catalysts that can be applied in the real device should be mainly developed in the future. The current effort would help new readers and learners design more efficient Ir-based electrocatalysts for future energy conversion and storage devices.
2. PEMWE working principle
Generally, a PEMWE device is mainly composed of a proton exchange membrane (PEM), catalyst layer (cathode and anode electrocatalyst), gas diffusion layer (GDL)/porous transport layer (PTL), and bipolar plates (BPs) (Fig. 1a and b).39 In the electrolyzer design, the membrane electrode assembly (MEA) is the core component of PEMWE, accounting for 24% of the cost of the PEMWE stack, and largely determines the efficiency of water electrolysis (Fig. 1c).40,41 The MEA includes the anode and cathode sandwiched by the PEM. PEM separates the two half-cells, transports protons, and prevents the mix of the product gas during the reaction to ensure gas purity and safety.42–44 Among them, an ideal PEM should have excellent proton conductivity, low expansion ratio, low gas permeability, low cost, and good durability.45 Currently, the most commonly used commercial PEMs are perfluorosulfonic acid polymer membranes, mainly including Nafion, Aciplex, and Flemion.42–44 Among them, Nafion series PEMs (such as Nafion 117, 115, and 112, where the numbers representing different equivalent weights and thicknesses) have good chemical and mechanical stability at high current densities and are considered to be the most representative PEMs.46 The catalyst layer is composed of electrocatalysts and proton conductive ionomers, which mainly determine the performance of PEMWE.47 GDL is located between the catalytic layer and BPs, which plays a key role in supporting the catalyst layer and stabilizing the electrode structure.48–50 BPs are located on the outer layer of the PEMWE device to encapsulate the two half cells, separate each electrolyzer in the stack, and provide electrical contact and thermal conduction between each electrolyzer in the stack.51–53 However, due to the strong acidic environment of the PEMWE device, high corrosion-resistant titanium (Ti)-based metals and even the electroplate noble metal like Pt/Au coated on their surface are generally used to fabricate both GDL and BPs to meet the requirements of durability and high conductivity.54–56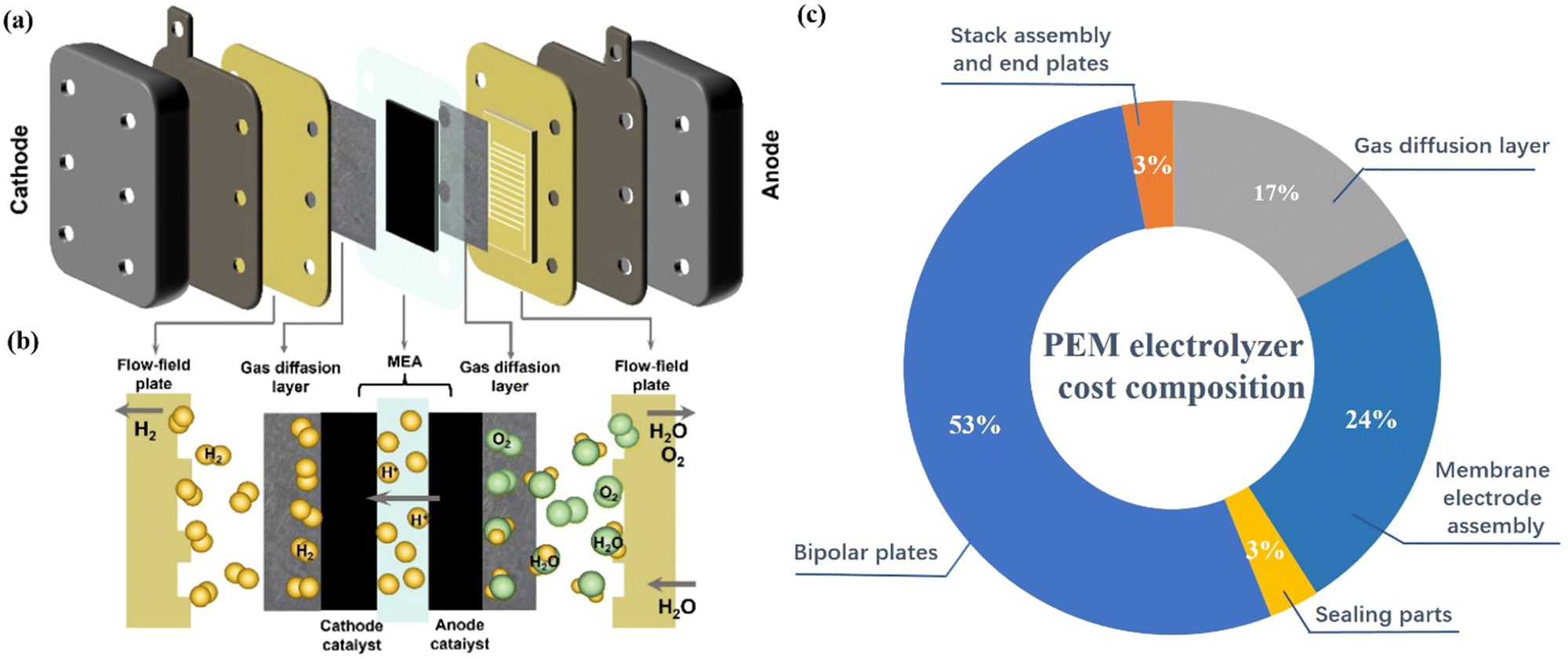 | ||
| Fig. 1 (a) Schematic illustration and (b) the working principle of PEM water electrolysis. Reproduced with permission from ref. 12. Copyright 2022, Tsinghua University Press. (c) The cost components of a PEM water electrolyzer. | ||
This additional process and high material costs make GDL and BPs account for 17% and 51% of the total cost of PEMWE devices respectively.53,57,58 In addition, the PEM water electrolyzer also requires some sealing parts to prevent gas and water leakage and other stack components, which account for 3% of the total cost of the electrolyzer.
The typical working principle of PEMWE for water electrolysis is as follows: Water is supplied on the anode side of the cell, then flows sequentially through the channels of BPs and GDL, and finally reaches the anode catalyst layer.59 The water molecules then break down into oxygen, protons, and electrons, and the resulting oxygen escapes from the anode surface of the PEMWE system (2H2O → O2 + 4H+ + 4e−). The generated electrons are transported to the surface of the cathode through the external power supply circuit, and the protons pass through the PEM to the cathode catalyst layer and combine with the electrons on the cathode surface to generate hydrogen (4H+ + 4e− → 2H2).12,15 Based on the understanding of the principles of PEMWE technology, it can be found that the catalyst layer is the core site for energy conversion through electrochemical reactions.60,61 The development of low-cost, high-performance catalysts is still very crucial to promote the large-scale application of PEMWE.62,63 At present, the hydrogen evolution reaction on the cathode side of PEMWE generally uses Pt-based catalysts, such as Pt/C catalysts with a Pt mass fraction of 20–60% and precious metal loading of 0.1–0.5 mgPt cm−2.64,65 The anode catalyst for the OER in PEMWE faces slow kinetics due to the four-electron process, and it is easily deactivated by corrosion at the high overpotential under the acidic conditions. Therefore, it is very important to develop highly active anode electrocatalysts and optimize their catalytic activity to maintain high performance in hydrogen production.
3. Ir-based anode catalysts
The operation conditions of the PEMWE requires high anti-oxidation and corrosion resistance of the catalyst on the anode side. At present, the anode catalysts of Ir black and IrO2, etc. are mainly used in the PEM electrolyzers, and the amount of Ir metal is often on the order of several mg cm−2.63 Due to the scarcity of Ir metal, the amount of Ir in the catalyst must be reduced. Therefore, the development of low-Ir catalysts with high activity and high stability has become an important research field. To easily compare the performance of some typical catalysts used in the practical PEMWE, the composition of the MEA, catalyst loading and the performance and stability of these catalysts are compared in Table 1. It was found that most of the Ir-based catalysts reported so far for actual PEMWE were supported catalysts (such as lrO2@TiO2,66 IrO2/Sb–SnO2,67 and MWNT/PBI/IrO268), which can generally reach a current density of 1–2 A cm−2 between 1.6–1.8 V and can be stably maintained for hundreds of hours. In this section, we provided a comprehensive overview of the latest progress in the application of Ir-based anode catalysts in practical PEMWE that can be classified as Ir-based single metals, alloys, oxides, and supported catalysts.| Anode catalyst | Membrane | Ir/IrO2 loading (mg cm−2) | Current density (A cm−2)@cell voltage (V) | Stability (h)@current density (A cm−2) | Ref. |
|---|---|---|---|---|---|
| Ir@WOxNRs | Nafon 115 | 0.14 | 2.2 A cm−2@2.0 V | 1030 h@0.5 A cm−2 | 31 |
| Ir AC/NN | Nafon 212 | 1.0 | 3 A cm−2@1.82 V | 90 h@3 A cm−2 | 74 |
| npIrx-NS | Nafon 117 | 0.06 | 2.4 A cm−2@1.98 V | — | 75 |
| Nanoprous IrNS | Nafion 117 | 0.28 | 3 A cm−2@1.65 V | 90 h@1.8 A cm−2 | 76 |
| 30-layered WP 1200-Ir | Nafon 212 | 0.045 | 5.2 A cm−2@2.0 V | — | 77 |
| Nanosize IrOx | Nafon 117 | 0.08 | 1.8 A cm−2@1.86 V | 4500 h@1.8 A cm−2 | 78 |
| IrRu/T90G10 | Nafon 212 | 1.0 | 1 A cm−2@1.56 V | — | 79 |
| IrO2-ND/ATO | Nafon 212 | 1.0 | 1.5 A cm−2@1.8 V | — | 80 |
| Ir0.7Ru0.3Ox | Aquivion | 0.34 | 1 A cm−2@1.62 V | 1000 h@3 A cm−2 | 81 |
| IrO2 nanoneedles | Nafon 117 | 4.0 | 1 A cm−2@1.80 V | 200 h@2 A cm−2 | 82 |
| Ir0.7Ru0.3O2 | Nafon 115 | 1.8 | 1 A cm−2@1.656 V | 48 h@1 A cm−2 | 83 |
| Ir0.7Ru0.3Ox(EC) | Nafon 212 | 1.0 | 1 A cm−2@1.69 V | 400 h@1 A cm−2 | 84 |
| F108-Ir0.6Sn0.4O2 | Nafon 115 | 0.88 | 1 A cm−2@1.621 V | 200 h@0.5 A cm−2 | 85 |
| lrO2@TiO2 | Nafon 212 | 0.4 | 1 A cm−2@1.67 V | 150 h@1 A cm−2 | 66 |
| NPG/IrO2 | Nafon 212 | 0.086 | 2 A cm−2@1.728 V | 250 h@0.25 A cm−2 | 86 |
| IrO2-ITO | Nafon 115 | 1.8 | 1 A cm−2@1.74 V | — | 87 |
| IrO2/SBA-15 | Nafon 115 | 1.5 | 1 A cm−2@1.64 V | 370 h@0.5 A cm−2 | 88 |
| IrO2/Sb–SnO2 | Nafon 212 | 0.75 | 2 A cm−2@1.62 V | 646 h@0.5 A cm−2 | 67 |
| MWNT/PBI/IrO2 | Nafon 117 | 0.5 | 1 A cm−2@1.6 V | 100 h@0.3 A cm−2 | 68 |
| Sr2CaIrO6 | Nafion 212 | 0.4 | 2 A![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) cm−2@1.81 V cm−2@1.81 V |
450 h@2 A cm−2 | 89 |
| Ir–IrOx | Nafion 117 | 1.5 | 1 A cm−2@1.67 V | 100 h@1 A cm−2 | 90 |
| Fe2N@EIROF | Nafion 115 | 0.036 | 4.5 A cm−2@1.9 V | 120 h@2 A cm−2 | 91 |
| RuO2@IrOx | Nafon 117 | 2.5 | 1 A cm−2@1.683 V | 300 h@1 A cm−2 | 92 |
| IrOx/Nb4N5 | Nafion 115 | 1.2 | 1 A cm−2@1.52 V | 150 h@1 A cm−2 | 93 |
| IrO2/ATO | Nafon 115 | 1.2 | 1.625 A cm−2@1.8 V | — | 94 |
3.1. Ir-based single metal catalysts
Ir as the active metal in catalysis reactions has received special attention because research on single metal Ir would be helpful for the intrinsic activity improvement and novel hybrid catalyst design and fabrication. By reducing the particle size of Ir metal particles through structural design, more surface active sites can be generated due to the largely increased proportion of surface atoms to the total number of nanoparticles, thereby enhancing the catalytic activity.20,69,70 Morphology engineering is an effective way to improve the OER catalytic activity and Ir utilization of Ir metals.71 For example, ultrafine metal Ir catalysts (UF-Ir/IrOx) with an average particle size of 1.6 nm were prepared as OER catalysts through a surfactant-free hydrothermal reaction.72 They required a low overpotential of 299 mV to drive at 10 A cm−2 in 0.5 M H2SO4. When assembled in the MEA using UF-Ir/IrOx and commercial Pt/C as the anode and cathode catalysts, and Nafion 117 membrane by hot pressing, the PEMWE device showed cell voltages of 1.70 and 1.88 V and achieved current densities of 100 and 300 mA cm−2 at room temperature. Very high operation stability was found during the 400 h operation at 50 mA cm−2, demonstrating the excellent stability of UF-Ir/IrOx. The nanoclusters are a transition state between traditional nanoparticles and single atoms with the number of atoms varying from a few to hundreds, which showed much higher atom utilization.73 A low overpotential of 296 mV was reported to drive the current density of 10 mA cm−2 for the Ir atomic clusters (AC) anchored on the ultrathin rutile IrO2 nanoneedles (NN) in the acidic electrolyte (Ir AC/NN), and the catalyst can be fabricated using a polyol method (Fig. 2a).74 Better kinetics and improved conductivity were found in this system during the OER process, where the robustness of IrO2 NNs stabilized the surface Ir atomic clusters, and the crystalline IrO2 NN framework facilitated electron transfer to the surface Ir ACs (Fig. 2b). The anode and cathode catalyst layers prepared by Ir AC/NN catalyst ink on Ti felt and 40 wt% commercial Pt/C on carbon paper were employed to fabricate the MEA using the Nafion 212 film as PEM, and the device exhibited a current density of 3 A cm−2 at a cell voltage of 1.82 V. Its activity and stability were significantly better than those of IrO2 NN and IrO2 particles alone, and it can stably operate for more than 90 hours resulting from the good synergy between Ir AC and IrO2 NN (Fig. 2c). Similar to Ir AC/NN, an Ir–IrO2 catalyst with a heterogeneous interface was prepared by Adams and NaBH4 co-reduction method where Ir nanoclusters were highly dispersed on the IrO2 particle surfaces.95 Ir nanoclusters captured oxygen atoms from IrO2 after heat treatment, causing surface oxygen vacancy generation. It was reported that this catalyst only required an overpotential of 329 mV to reach 10 mA cm−2, and its mass activity was 1851 A gIrO2−1 at a potential of 1.6 V vs. RHE, which was approximately 2.4 times that of commercial IrO2 (779 A gIrO2−1). When integrated into the actual PEMWE, the cell voltage was only 1.74 V to reach 1 A cm−2, lower than that of 1.94 V for the commercial IrO2 catalyst; the stability of Ir–IrO2 was also much better than that of commercial IrO2 as shown in the stability test at a constant current density of 300 mA cm−2. The largely improved OER activity and stability were attributed to the combined effects of the oxygen vacancy-rich surface and the dual-site mechanism of the Ir–IrO2 heterostructure interface.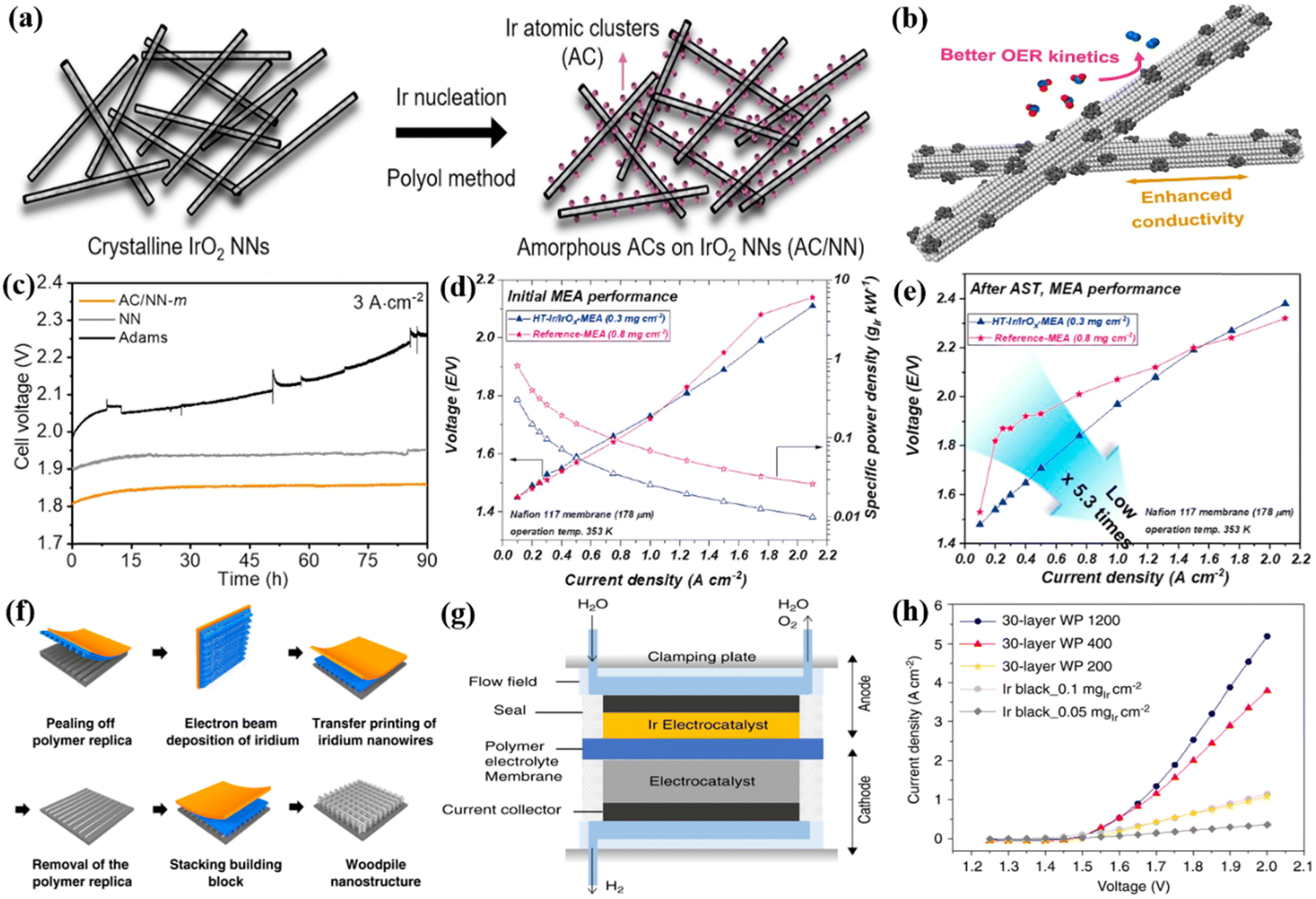 | ||
| Fig. 2 (a) A scheme of the synthetic process and (b) the outstanding properties and catalytic performance of amorphous Ir AC/NN catalysts. (c) Static durability test curves at 3 A cm−2 for 90 h of Ir AC/NN, IrO2 NN, and Adams IrO2. Reproduced with permission from ref. 74. Copyright 2022, Elsevier B.V. (d) Initial MEA performance and (e) the performances after the accelerated stress test (AST) of HT-Ir/IrOx-MEA and reference MEA. Reproduced with permission from ref. 97. Copyright 2021, Elsevier B.V. (f) Fabrication procedure of woodpile-structured Ir. (g) Schematic of PEMWE cells. (h) The performance curves for several catalysts in PEMWE. Reproduced with permission from ref. 77. Copyright 2020, Springer Nature. | ||
To some extent, the nanostructure of Ir influences the performance of the overall PEM electrolyzer because a suitable nanostructure will increase the electrochemical surface area and mass transfer capacity.96 A solution reduction technique to adjust the ratio of Ir oxidation states was used to form assembled Ir/IrOx OER catalysts with 2D layered structures and a controlled Ir oxidation state ratio.97 Its oxidation state gradually decreased from the shell layer to the core layer, which overcame the trade-off between the activity and stability of the OER. Specifically, the highly active surface oxide layer provided high activity through the lattice oxygen mechanism, and the highly stable internal nuclear layer maintained long-term stability through the adsorption oxygen mechanism. The Ir/IrOx was heat-treated in an N2 environment for 20 minutes at 398 K to produce HT-Ir/IrOx, and the HT-Ir/IrOx and unheated Ir/IrOx were assembled into the PEMWE device for the practical application performance comparison. The device using HT-Ir/IrOx MEA showed a cell performance of 1.73 V at 1.0 A cm−2 at 353 K (Fig. 2d). Furthermore, excellent catalytic stability was also demonstrated where the cell voltage of the HT-Ir/IrOx-MEA was only marginally increased by 70 mV after 48 h of stability test at 1 A cm−2, lower than that of the unheated Ir/IrOx MEA increased by 370 mV (Fig. 2e). To increase the surface area, porous Ir nanosheets (npIrx-NS) were fabricated by de-alloying NiIr alloy precursors and the porous nanosheets showed a thickness of 100 nm and a pore size of 5 nm.75 The high current density of 2.4 A cm−2 at 80 °C with a cell voltage of 1.98 V was achieved in the PEM electrolyzer constructed by npIrx-NS with an ultra-low noble metal loading of 0.06 mgIr cm−2 as the anode catalyst layer. Three-dimensional (3D) woodpile (WP)-structured Ir catalysts (nanowire periods of P1200, P400, and P200 nm) were prepared by stacking highly ordered Ir nanowire arrays via solvent-assisted nanotransfer printing technology (Fig. 2f).77 The influence of different layers and nanowire periods on catalytic activity was studied in a single cell (Fig. 2g), the current density of the single cell using WP 1200 as the anode catalyst was increased by increasing the number of stacked layers from 10 to 30 layers, and the 30-layer WP 1200 with an Ir loading of 45 μg cm−2 required the cell voltage of be 2.0 V to show the current density of 5.2 A cm−2, about 4.3 times that of the Ir black catalyst (Ir loading: 50 μg cm−2) (Fig. 2h). The multilayer stacking structure effectively increased the number of exposed active sites, and the mass transfer and removal of gas bubbles from the catalyst layer was also improved with the open and ordered features of the 3D WP structure, therefore, the improved performance in the PEM electrolyzer was observed.
3.2. Ir-based alloy catalysts
Combining Ir metal with other metal elements to prepare Ir-based alloy catalysts is an effective and direct strategy to reduce the amount of Ir and improve catalytic performance.98,99 The interface coordination environment of the active center could be modulated by some measures like alloying, multi-component construction, crystal structure mixing, etc., and the electronic structure could be modified resulting from the synergistic effect between different noble metal components that could optimize the binding energy between the intermediate and the active center.100,101 Similar to Ir, Ru is also active for acidic OER, therefore, IrRu alloys have been developed for the OER.102 For example, IrRu alloys supported on TiC could form an Ir-rich surface during the OER process that can protect Ru from further dissolution and increase catalytic stability. The IrRu alloy with an Ir:Ru ratio of 1:4 required only a low OER overpotential of 230 mV to drive a current density of 10 mA cm−2. When this catalyst was supported on TiC as an anode catalyst for PEMWE, it could drive a current density of 1 A cm−2 at a cell voltage of 1.48 V. The rapid dissolution and oxidation of Ru were found during the OER process via the valence state and coordination during the OER process via the valence state and coordination environment analysis probed by X-ray absorption spectra (XAS), and the active and protective Ir oxide shells were in situ formed on the surface of the IrRu alloy (Fig. 3a). In other words, the Ir-rich surface works not only as an active species for reactions but also prevents Ru in the core from further oxidation and leaching; and the electronic structure of the Ir shell was optimized by the core with high Ru content, which allowed the Ir1Ru4/TiC catalyst to deliver a current of 1 A at a cost of only 3.3 USD. Similarly, a core–shell electrocatalyst (IrRux@Ir) with an IrRux alloy core and an Ir-rich shell was prepared through a CO-induced phase separation strategy.103 An overpotential of 288 mV was required to reach 10 mA cm−2, and the MEA composed of IrRu@Ir showed a current density of 1 A cm−2 at the cell voltage of 1.83 ± 0.02 V and a very stable water electrolysis performance for nearly 400 h (Fig. 3c). The authors also attributed the performance to the strong electronic interaction between the IrRux core and the Ir-rich shell and the protection of the Ir-rich shell over the internal Ru atoms from loss during the long-term electrolysis process.
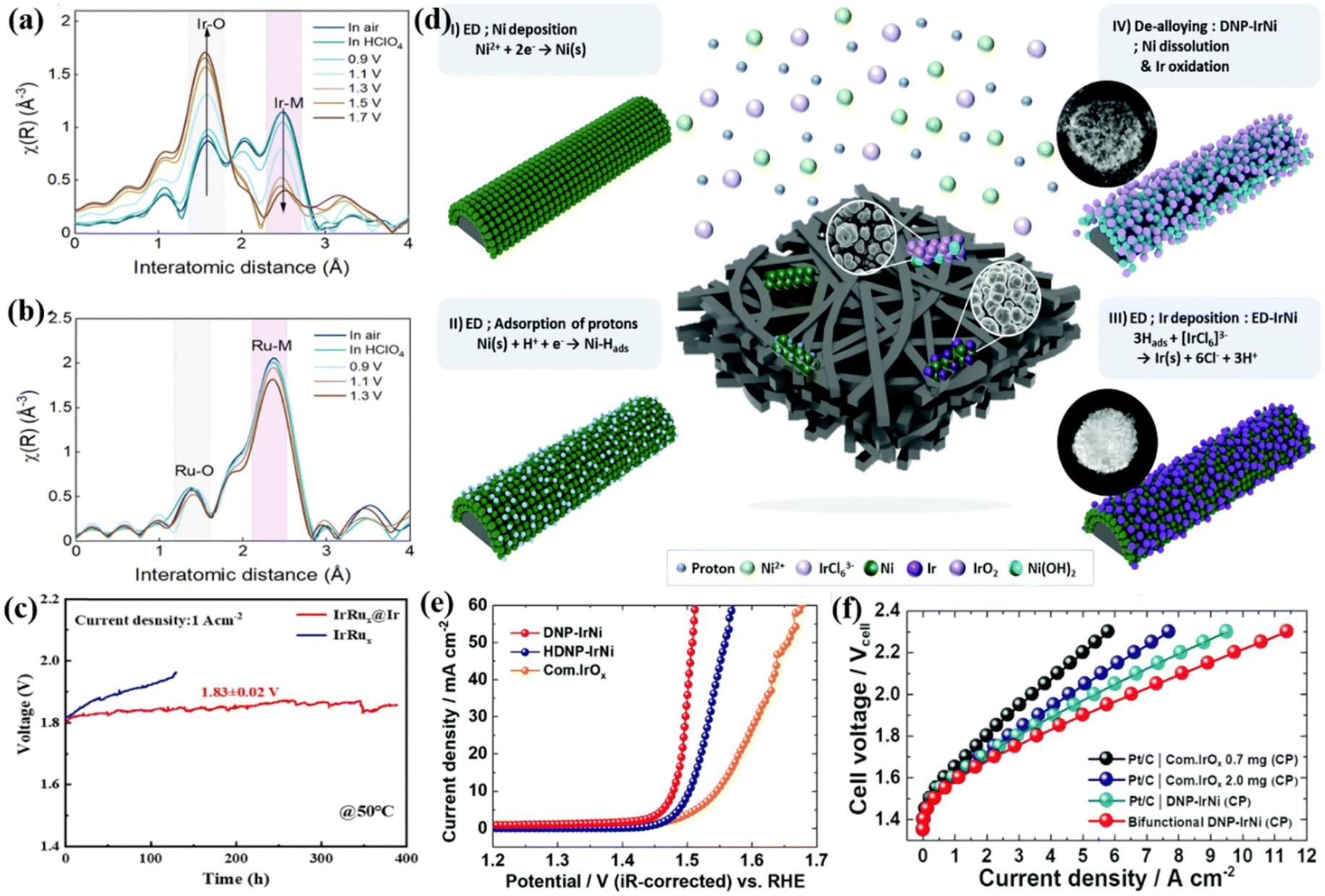 | ||
| Fig. 3 (a) Ir and (b) Ru of in situ Fourier transformed extended X-ray absorption fine structure (EXAFS) spectra for Ir1Ru4/TiC. Reproduced with permission from ref. 103. Copyright 2022, The Royal Society of Chemistry. (c) Stability test of PEMWE with IrRux@Ir and IrRux catalysts as the anode at 1 A cm−2, respectively. Reproduced with permission from ref. 115. Copyright 2022, Elsevier B.V. (d) Schematic illustration of the experimental process for 3D DNP-IrNi. (e) OER polarization curves of DNP-IrNi and HDNP-IrNi. (f) Single PEMWE cell polarization curves combined with DNP-IrNi catalyst and carbon paper. Reproduced with permission from ref. 108. Copyright 2022, The Royal Society of Chemistry. | ||
To further reduce the cost of catalysts, some researchers have tried to design Ir-based nanostructures by alloying Ir with cheap transition metals (such as Fe, Co, and Ni).104,105 Due to the leaching and dissolution of the cheap transition metals in the alloy, the dealloy of the catalyst could further increase the surface and modify the adsorption property.106,107 For instance, a de-alloyed self-supporting nanoporous IrNi (DNP-IrNi) catalyst was prepared through adsorbed H-induced co-electrodeposition to form an electrodeposited IrNi (ED-IrNi) precursor and a subsequent electrochemical dealloying method (Fig. 3d).108 They exhibited an OER overpotential of 248 mV at 10 mA cm−2 in acidic electrolytes, with very good catalytic stability (Fig. 3e). The DNP-IrNi with the Ir loading of 0.67 mg cm−2 coated on highly conductive carbon paper (CP) was measured in a real device of PEMWE electrolyzer, and a superior performance of 1.88 A cm−2 was obtained at the cell voltage of 1.7 V, much higher than that of 0.7 mgIr cm−2 for commercial IrOx (1.35 A cm−2) and 2.0 mgIr cm−2 for the commercial IrOx (1.79 A cm−2) (Fig. 3f). Moreover, at a cell voltage of 2.0 V, the electrolier showed a current density as high as 6.5 A cm−2. During a 100 h stability test at 2 A cm−2, the degradation rate was only 1.58 mV h−1 verifying the excellent stability of DNP-IrNi. The authors discussed the origin of the good performance resulting from the dandelion spore structure of DNP-IrNi, and some features would contribute to the high performance such as enlarged surface area, enhanced binding strength to oxygenated intermediates, controlled Ir4+ oxidation state, and anti-corrosion ability. A series of IrCo, IrNi and IrCoNi catalysts with hollow and porous structures were controllably synthesized by etching IrM (M = Co, Ni) solid metal nanocrystals using Fe3+ ions.109 Among them, IrCoNi porous hollow nanocrystals (PHNC) required the lowest overpotential of 303 mV to reach a current density of 10 mA cm−2 in 0.1 M HClO4. Density functional theory (DFT) calculations revealed that alloying Ir with 3d transition metals can weaken the adsorption of oxygen-based intermediates by appropriately regulating the d-band center. In addition, nanoscale high-entropy alloys (HEAs) containing multiple metal elements with ultra-low Ir loading have been designed to improve the stability and catalytic activity in acidic OERs.110 For example, FeCoNiIrRu HEA nanoparticles were deposited on carbon nanofibers (CNFs) through electrospinning and high-temperature carbonization methods to obtain an FeCoNiIrRu/CNFs catalyst which showed excellent acidic OER performance.104 The hysteretic diffusion effect of HEA effectively prevented the leaching and dissolution of metals, allowing the catalyst to exhibit high durability. In-situ Raman spectroscopy and DFT calculations revealed that alloying Ir with 3d transition metal elements not only reduced the adsorption of the key oxygen-containing intermediate *OH but also promoted the conversion of *OOH and the generation of O2. In summary, the Ir alloy with 3d transition metals can effectively adjust the Gibbs free energy of key oxygen-containing intermediates in the OER process, thereby improving the OER activity. While it should be noted however, that the Ir alloy employed in the PEMWE device is still used very rarely, probably due to the low stability and the easy leaching of the transition metals during catalysis, and thus some more elaborate design is still required to overcome this issue.
3.3. Ir-based oxide catalysts
Ir-based metal oxides are the most extensively studied acidic OER catalysts due to their excellent flexibility in nanostructure, chemical composition, and crystal structure.111–113 Among them, commercial IrO2 with high activity and reliable performance is generally employed as the benchmark catalyst for acidic OER performance evaluation. Two different MEAs were prepared by spray-coating Ir-black (3 nm) and IrO2 (5 nm) onto the Aquivion membrane to evaluate their performance in PEMWE.114 At the beginning of the 1000 h durability test, although the average grain size of Ir-black was smaller, its initial catalytic activity was significantly lower than that of IrO2 (Fig. 4a). During subsequent operation at high current density (1 A cm−2), the cell voltage of Ir-black MEA showed a negative potential change with time, while the cell voltage of IrO2 MEA continued to increase with a degradation rate of approximately 12 μV h−1. The abnormal behavior of Ir-black was the result of the oxidation of metal Ir to IrOx. The hydrated IrO2 with a high oxidation state generated on the surface after long-term OER was beneficial to reducing the OER overpotential. In addition, the authors also explored the performance of Ir-black MEA and IrO2 MEA in PEMWE at different temperatures. By increasing the cell temperature, the cell potential gradually decreases, and the electrolysis efficiency increases. However, it should be noted that by further increasing the temperatures above 80 °C, in addition to the greater energy input, the stability of the catalyst and PEM will be reduced which will cause catalyst leaching and gas product mixing. Therefore, the effect of temperature on electrolysis rate, energy consumption, material stability and product purity is considered in practical applications, so that the temperature is controlled at 80 °C to obtain better comprehensive effects. IrOx/Nafion catalyst layers constructed by depositing well-dispersed IrOx particles on Nafion membranes showed excellent stability.78 With an ultra-low Ir loading of 0.08 mg cm−2, stable operation was maintained for more than 4500 h in an MEA electrolyzer at 1.8 A cm−2 and 80 °C. In addition to IrO2 nanoparticles, different nanostructures of IrO2 were also reported for OER performance study.116 For example, ultrafine IrO2 long nanoneedles (NN-L) with a diameter of about 2 nm were obtained through the molten salt synthesis method, and they were introduced into a single cell of a PEM water electrolyzer as an anode by comparing to the amorphous IrO2 nanoparticles.82 It was found that the IrO2 NN-L electrolyzer drove a current density of 2 A cm−2 at a cell voltage of 1.55 V and ran stably for more than 200 h. The conductivity, activity, and durability of IrO2 NN-L were found to be much better than amorphous IrO2 nanoparticles. Furthermore, hierarchical sea urchin-like IrO2 nanostructures were synthesized using a surfactant-free wet chemical route.117 By monitoring the morphology evolution of the product after different hydrothermal reaction times, it was found that the Ir precursor first transformed into nanospherical IrO2 with a rough surface, and then grew needle-like structures on the surface. The crystallinity of IrO2 was subsequently adjusted by controlling the hydrothermal temperature, and the sample obtained at 180 °C exhibited the lowest overpotential of 260 mV at 10 mA cm−2.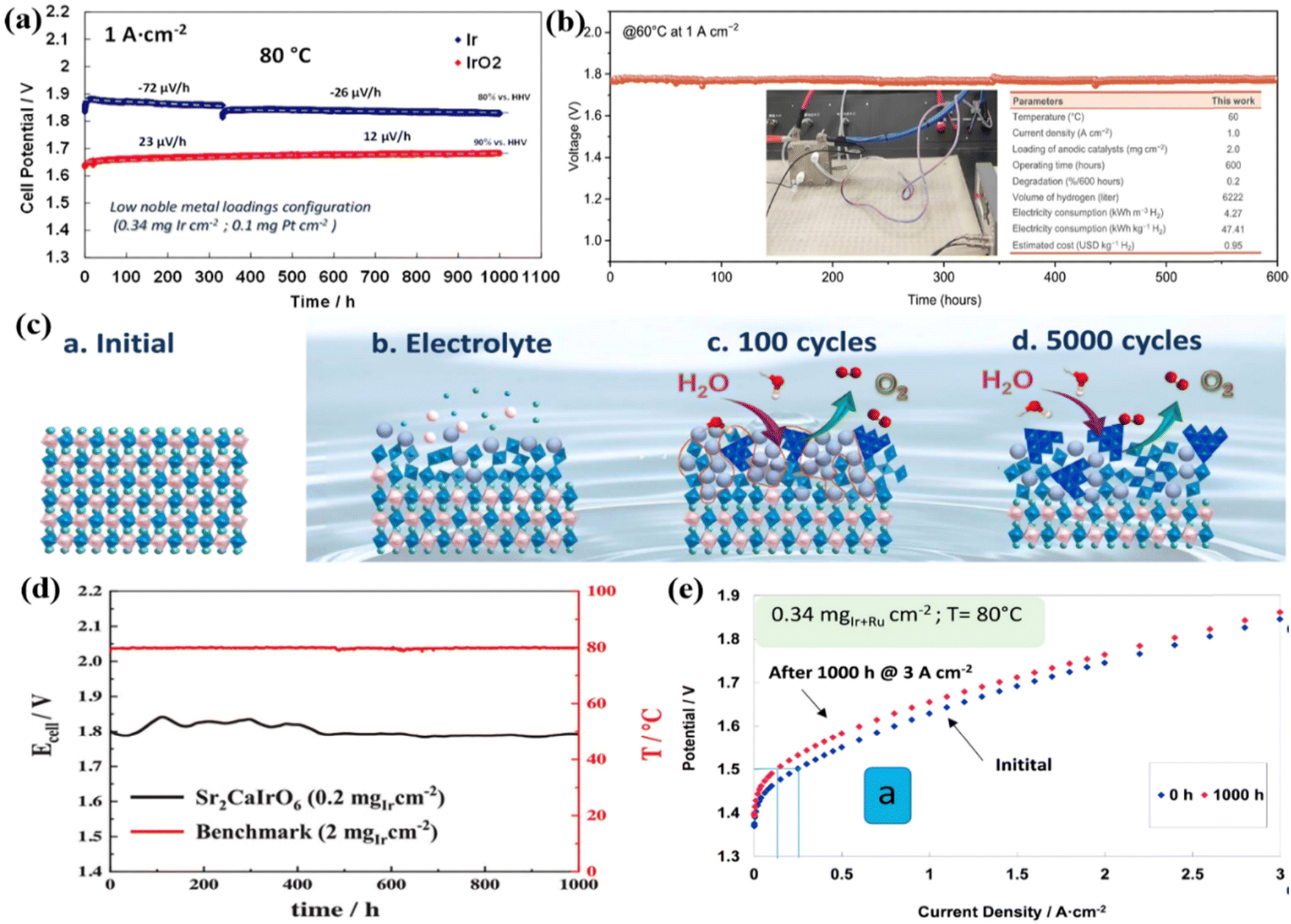 | ||
| Fig. 4 (a) The durability tests of Ir-black MEA and IrO2 MEA in the water electrolysis cell at 1 A cm−2 and 80 °C. Reproduced with permission from ref. 114. Copyright 2017, Elsevier B.V. (b) Chronopotentiometric curve of the PEMWE device using IrOx·nH2O. Reproduced with permission from ref. 123. Copyright 2023, American Association for the Advancement of Science. (c) Illustration of fresh Sr2CaIrO6, immersed in the electrolyte, after 100 OER CV cycles and after 5000 CV cycles. Reproduced with permission from ref. 89. Copyright 2022, Springer Nature. (d) Cell voltage and temperature evolution over time (1000 h) at a constant 2 A cm−2. Reproduced with permission from ref. 126. Copyright 2023, Wiley-VCH GmbH. (e) Polarization curves for MEA before and after durability tests at 3 A cm−2 for 1000 h at 80 °C. Reproduced with permission from ref. 81. Copyright 2017, Elsevier Ltd. | ||
The different crystal structures or the different atomic arrangements would influence the physical and chemical properties, as a result, resulting in different catalytic performances.118 Specific crystal structures with unique electronic structures and chemical environments can greatly promote the intrinsic activity of Ir-based catalysts.119,120 Generally, the amorphous or low crystallinity IrO2 exhibits much higher catalytic activity than the crystallized rutile IrO2 structure, and some more reports based on the amorphous or low crystallinity IrO2 were reported. For instance, the N/C doped amorphous IrO2 showed a larger active surface area and oxygen-defective characteristics, which was conducive to the OER.121 A low cell voltage of 1.774 V was obtained at 1.5 A cm−2 compared with the crystal IrO2 (1.847 V at 1.5 A cm−2), and it can run stably for more than 200 h without evident performance decay when operated in the PEMWE device. An Ir–IrOx catalyst with an amorphous IrOx surface and Ir skeleton was successfully synthesized via a facile complexation method.90 Through relevant physical characterization, it was verified that changes in the complex and calcination temperature can control the ratio of the amorphous surface and crystal skeleton, and it was found that as the surface content of amorphous oxide increases, the OER activity showed a volcanic trend. When the complex was citric acid (CA) and the calcination temperature was 375 °C, the Ir–IrOx-CA-375 catalyst had the lowest overpotential of 228 mV at 10 mA cm−2. Its excellent performance was attributed to the appropriate ratio of a highly active amorphous IrOx surface to stable Ir skeleton, which was beneficial to overcoming the trade-off between OER activity and stability of Ir-based catalysts. The MEA was constructed using 40 wt% Pt/C as the cathode and Nafion 117 membrane as the PEM, and the synthesized Ir–IrOx catalyst and the commercial IrO2 catalyst as the anode for comparison. The cell voltage of 1.67 V at 1 A cm−2 was obtained for the Ir–IrOx electrode, which was 54 mV lower than that assembled using commercial IrO2; the cell voltage remained almost constant during 100 h operation when tested at 1 A cm−2 and 80 °C. Low-crystallinity iridium molybdenum oxide (IrMoOx) nanofibers prepared using an electrospinning-calcination strategy also showed good OER activity in acidic electrolytes.122 They required an overpotential of 267 mV to drive 10 mA cm−2, lower than that of bare IrOx (333 mV), MoOx (almost no OER activity), and the benchmark commercial IrO2 catalyst (330 mV). The synergistic effect between IrOx and MoOx components and their unique heterostructure with low crystallinity provided more structural defects, generated more active sites and accelerated electrolyte ion diffusion. Furthermore, a lattice water-assisted short-range ordered iridium oxide (IrOx·nH2O) showed the combined advantage of the crystal IrO2 and the amorphous IrOx.123 The catalyst was prepared by incorporating lattice water into IrO2, which had abundant edge-sharing IrO6 octahedra, and during the reaction, the lattice water continued to participate in oxygen exchange causing very good stability in the OER for 5700 h (∼8 months). In PEMWE applications, IrOx·nH2O served as an anode catalyst that could deliver a cell voltage of 1.77 V at 1 A cm−2 and maintain stability for 600 h (Fig. 4b).
To further increase the intrinsic activity of the Ir sites, and maintain good stability, some mixed Ir-based oxides with special crystal structures were developed for the OER.124,125 Specifically, a series of double perovskite oxides such as Sr2CaIrO6, Sr2MgIrO6, and Sr2ZnIrO6 synthesized using the sol–gel method showed excellent performance in acidic OERs.89 Sr2CaIrO6 had the most excellent OER activity, which could reach 10 mA cm−2 with only a voltage of 1.48 V vs. RHE. The researchers deeply studied the structural evolution of Sr2CaIrO6 during the OER process and found that the surface of the perovskite was severely reconstructed during immersion in the electrolyte and subsequent OER cycles (Fig. 4c). Specifically, after immersing the Sr2CaIrO6 perovskite in an acidic electrolyte, the surface began to rebuild. Subsequently, the Ca and Sr cations on the catalyst surface that underwent 100 and 5000 OER cyclic voltammetry (CV) cycles between 1.2 and 1.7 V vs. RHE rapidly dissolved, causing H3O+ molecules to fill the gaps and form a stable surface with short-range ordered edge-sharing IrO6 octahedrons. The dissolution of alkaline cations forms an Ir-rich phase outer layer on the catalyst surface to provide high performance and maintain high stability. The rapid removal of basic cations led to the filling of the voids by H3O+ molecules, forming a stable surface with a short-range order of edge-sharing IrO6 octahedrons. The formation of an outer layer of an Ir-rich phase would be more stable and active for the OER in which the Ir atoms were in the 3+/4+ oxidation state. Sr2CaIrO6 was sprayed on the Nafion 212 membrane as an anode catalyst (0.4 mgIr cm−2) in a PEMWE single cell, and the cell voltage of 2.4 V was required to drive an ultra-high peak current of 6 A cm−2 at 80 °C with no significant mass transport limitation or rapid degradation process. When reducing the Ir loading to 0.2 mgIr cm−2 in PEMWE,126 a cell voltage of 1.78 V was found to reach a current of 2 A cm−2 at an operating temperature of 80 °C and remained stable for more than 1000 h (Fig. 4d). After the 1000 h stability test, a sponge-type morphology composed of small nanodomains of Ir–O–H was formed due to the rapid leaching of Ca and Sr, which resulted in a high OER performance.
The introduction of other active or inert components into IrO2 is considered to be one of the effective strategies to reduce the Ir content.127 For example, the Ir0.6Sn0.4O2 catalyst prepared by introducing Sn into IrO2 through sodium borohydride metal reduction and the subsequent annealing process showed excellent catalytic performance in single-cell tests.128 It could drive a current of 2.0 A cm−2 at a cell voltage of 1.96 V with a low Ir loading of 0.294 mgIr cm−2. Its excellent performance was attributed to the addition of Sn species which not only improved the utilization of Ir but also enhanced the electrochemical activity by changing the electronic structure. To reveal the electronic structure modification of Ir influenced by Sn species, the Ir0.83Sn0.17O2 catalyst was successfully synthesized by incorporating Sn into the IrO2 lattice through an ammonia-induced pore formation method without changing the stable IrO2 rutile phase.129 It only required an overpotential of 284 mV to reach 10 mA cm−2, which was 38 mV lower than commercial IrO2. Its high activity was attributed to electron transfer from Ir to Sn through the local structure of Ir–O–Sn by enhancing the Ir–O covalent bond, which in turn activated the lattice oxygen to participate in the OER. Furthermore, the ternary Ir0.7W0.2Sn0.1Ox nanocatalyst synthesized via a sol–gel strategy also showed similar results.130 That is, the introduction of Sn species was conducive to charge redistribution, limiting the valence state of the Ir active center to a stable state below 4+, and avoiding excessive oxidation of Ir. The oxides of Ir and Ru could also be combined to construct Ir–Ru solid solution catalysts (Ir0.7Ru0.3Ox) with surface-enriched Ir oxides through high-temperature annealing.81 As an anode catalyst for PEMWE, it could achieve a current density of 3 A cm−2 at a cell voltage of 1.8 V at 80 °C with a low Ir loading of 0.34 mgIr cm−2. However, there was a large potential increase after 1000 h operation at 3 A cm−2 (Fig. 4e). In other words, the stability of this kind of oxide was not satisfactory, and it should be further improved in future work.
3.4. Supported Ir-based catalysts
Support materials have a considerable influence on the stability and activity of active metals through metal-support interactions.131,132 The good supports serviced the roles in effectively dispersing noble metals and preventing the active sites aggregation, as a result, the catalytic ability and stability of the catalysts could be increased.34,133 Metal oxides with high corrosion resistance are generally used as the main substrate in catalyst construction.134,135 For example, antimony-doped tin oxide (ATO) was synthesized using a synergistic sol–gel and hydrothermal technique, and the short rod-like Ir nano dendrites (Ir-ND) were subsequentially deposited onto the ATO substrate as anode catalysts for water splitting in PEMWE.80 The Ir-ND/ATO catalyst exhibited remarkable performance, surpassing the commercial Ir catalysts by over two-fold at practical current densities of 1.5 A cm−2 with a cell voltage of 1.80 V (Fig. 5a). Additionally, the mass activity was ca. 693 A gIr−1 cm−2 at 1.6 V, which was 2.3 times that of colloidal IrO2 (300 A gIr−1 cm−2). By loading metal Ir nanoparticles on Nb2O5−x, the Ir/Nb2O5−x catalyst was obtained where Ir atoms were closely arranged along the [001] crystal plane of Nb2O5−x, to form a thick quasi-Ir shell around 1 nm.136 The MEA was prepared using a Nafion 115 membrane as the PEM, Ir/Nb2O5−x catalyst as the anode, and commercial Pt/C as the cathode, and when tested at 80 °C, the electrolyzer showed a cell voltage of 1.839 V to drive 3 A cm−2, and it also showed very good stability during the 2000 h operation at 2 A cm−2, with no significant performance degradation (Fig. 5b). In the gradient increasing current density (from 2 to 6 A cm−2) stability test, very good results were also found, where the cell voltage at 1.937, 2.026, and 2.123 V could stably run for 150 h at the current density of 4, 5, and 6 A cm−2, respectively (Fig. 5c). Because of the unique chemical environment of Ir at the interface, namely, the Ir lattice beyond the grain boundaries and extending to Nb2O5−x, the close contact between Ir and Nb2O5−x accelerated the dynamic migration of oxygen species by reducing the electron transfer resistance (Fig. 5d).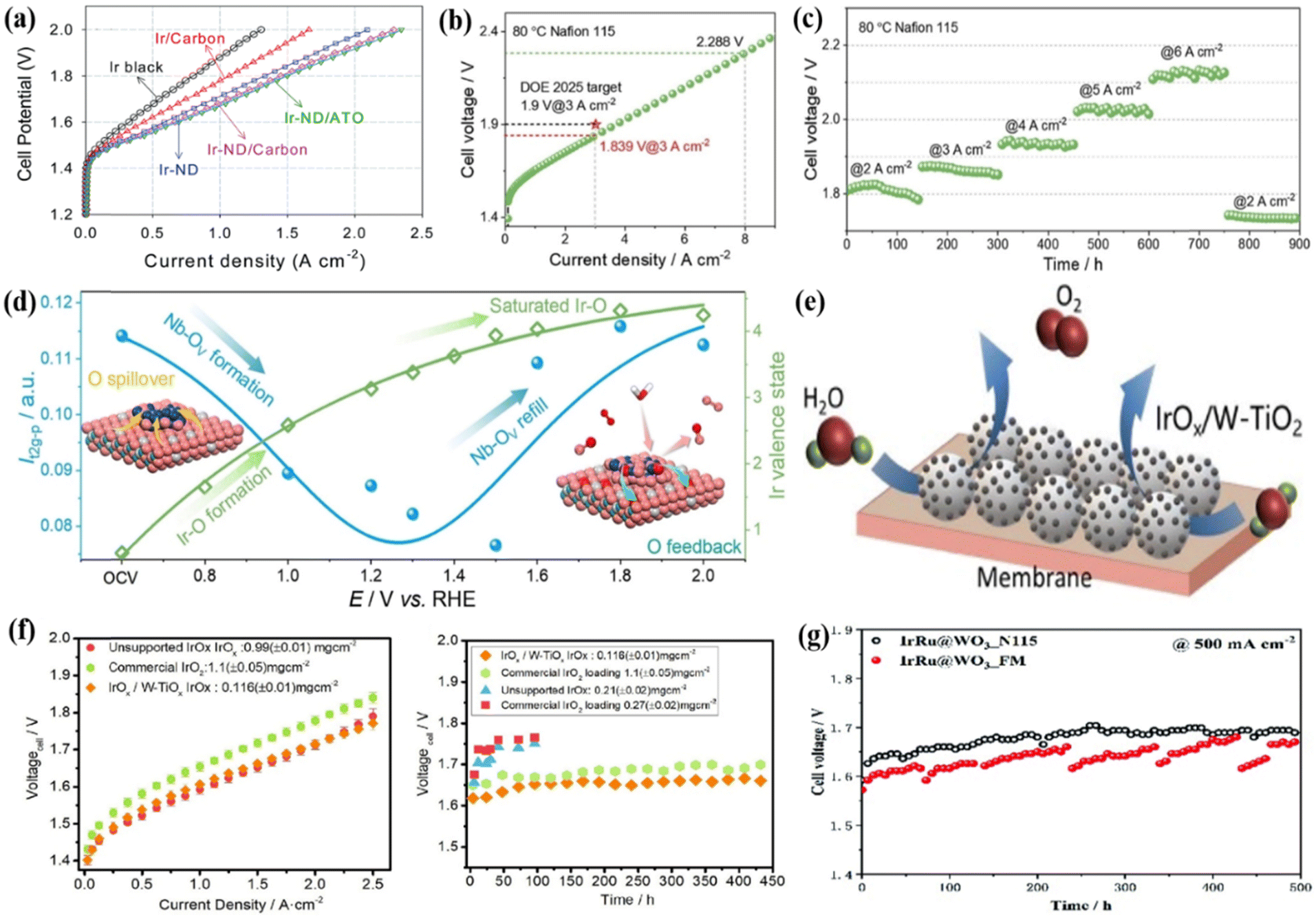 | ||
| Fig. 5 (a) Single cell performance of Ir-ND catalysts with a catalyst loading of 1.0 mg cm−2 at 80 °C. Reproduced with permission from ref. 80. Copyright 2017, Elsevier B.V. (b) Polarization curve of the PEMWE device with Ir/Nb2O5−x as the anode catalyst and (c) stability curves of the electrolyzer at gradient increasing current densities of 2, 3, 4, 5, and 6 Acm−2. (d) An illustration of the dynamic interface effect and the variation of the Ir valence state of Ir/Nb2O5−x. Reproduced with permission from ref. 136. Copyright 2022, Wiley-VCH GmbH. (e) An illustration of the MEA with an anode catalyst layer. (f) Polarization curves and durability test curves of the PEMWE device with different MEAs at 80 °C. Reproduced with permission from ref. 140. Copyright 2021, Elsevier Ltd. (g) The single cell voltage curves of IrRu@WO3 during stability testing at 80 °C. Reproduced with permission from ref. 141. Copyright 2022, The Royal Society of Chemistry. | ||
TiO2 is more anti-corrosive under acidic conditions, and Ir-based metals or oxides supported on TiO2 have been intensively studied for the OER in the PEMWE technique.137 Three different types of TiO2 materials TiO2-MC (only macropores), TiO2-MS (high surface area connecting mesopores and macropores), and TiO2-P25 (primary particles TiO2 nanopowder with a diameter of 25 nm) were used to support the Ir-based catalyst.138 Among them, Ir/TiO2-MC could drive 1.75 A cm−2 at a cell voltage of 1.8 V with a low Ir loading of 0.27 mgIr cm−2, showing the best performance in PEMWE due to the high electrochemical surface area resulting from the Ir interconnected mesoporous structure induced by the macroporous structure in TiO2-MC. TiO2 doped by N element supported IrOx could also enhance the OER performance, which required an overpotential of only 270 mV to provide 10 mA cm−2 in acidic electrolyte.139 When it was used as an anode catalyst in PEMWE, the single cell had a voltage of only 1.672 V at 1 A cm−2 and 1.761 V at 2 A cm−2. The N-doping process increased the conductivity of TiO2, improved the dispersion/stabilization of IrOx and weakened the oxidative dissolution of Ir substances, as a result, the performance could be increased compared to the un-doped support. Similarly, the IrOx/W-TiO2 catalyst was prepared by uniformly distributing amorphous IrOx on W-TiO2 (Fig. 5e).140 In the single-cell performance test, the anode catalyst layer constructed by IrOx/W-TiO2 with Ir loading of 0.2 mgIr cm−2 only required a cell voltage of 1.602 V to drive 1 A cm−2 (Fig. 5f). The activity and stability were significantly better than unloaded IrOx and commercial IrO2, further demonstrating the positive role of the supporting material.
An ordered array with a defective Ir layer with a thickness of ca.68 nm electrodeposited on the exterior of WOx nanorods (WOx NRs) was fabricated by 100-cycle CV scanning yielding an efficient Ir@WOx NRs-100 catalyst.31 Ir@WOx NRs-100 (144 mgIr cm−2) and commercial Ir black (0.5 mgIr cm−2) were sprayed on the Nafion 115 membrane and Pt/C (0.4 mg cm−2) was used as the cathode catalyst to construct MEA for PEMWE. The Ir@WOx NRs-100 showed good single-cell performances (2.2 A cm−2@2.0 V) with very good stability for 1030 h stability at 0.5 mA cm−2 at 80 °C. The outstanding stability of WOx NRs and good dispersion of Ir coating as well as the efficient mass transportation would contribute to the high performance. Ir-based alloys were also supported on metal oxide supports to better balance the activity and stability in the acidic medium for the OER. For example, IrRu particle-coated WO3 nanoarray electrodes (IrRu@WO3) were fabricated using the electrodeposition method, which exhibited an OER overpotential of 245 mV at 10 mA cm−2.141 This heterogeneous alloy structure could avoid the formation of soluble and over-oxidized Ru species by forming a rich Ir shell and an adjustable electronic structure, thereby maintaining high activity and stability. In addition, using IrRu@WO3 as an anode catalyst for PEMWE also showed good single-cell performance. Specifically, the electrolyzer showed a current density of 4.5 A cm−2 requiring a cell potential of 2.13 V with an Ir loading of 0.115 mg cm−2 in the MEA, outperforming the commercial Ir black-based electrode. The single cell could operate for 500 h at 80 °C to drive a stable current density of 0.5 A cm−2, and the cell voltage reached the highest value in the first 200 h (voltage growth rate: 1.96 μV h−1) and then became stable at 1.68 V for 200–500 h (Fig. 5g). This result demonstrated that the IrRu@WO3 nanostructured electrode is very promising for application in the PEMWE.
Although these corrosion-resistant metal oxides have good corrosion resistance, they have poor electrical conductivity. Carbon-based supports with high electrical conductivity are also largely studied under acid conditions for the OER, though they are easily corroded at a high oxidation potential.142,143 Therefore, some researchers have tried to combine metal oxides with good stability and carbon supports with high conductivity as supports for Ir-based catalysts in acidic environments. For example, TiO2 nanoparticles decorated on wrinkled reduced graphene oxide (rGO) were prepared by ultrasonic spray pyrolysis as a support to mitigate carbon corrosion and provide high conductivity (Fig. 6a).79 Subsequently, the IrRu/T90G10 (weight ratio of TiO2:rGO is 90:10) electrocatalyst was prepared by anchoring IrRu alloy nanoparticles onto a TiO2–rGO (TG) support and it exhibited high acidic OER activity with an overpotential of 254 mV to drive the current density of 10 mA cm−2 compared to commercial Ir black (340 mV), unsupported IrRuOx (372 mV), and IrRu/rGO (325 mV). Using IrRu/T90G10 as an anode catalyst, Nafion 212 membrane, and commercial Pt/C (46.5 wt%) as the cathode catalyst, the MEA was prepared and integrated into the single-cell electrolyzer for water electrolysis, which showed a cell voltage of 1.56 V at 80 °C to provide 1.0 A cm−2 (Fig. 6b). The mass activity based on noble metal (1.242 A mg−1 for IrRu/T90G10) at 1.6 V was much higher than that of commercial IrO2. The high performance was attributed to the TG support effectively dispersing IrRu nanoparticles and inducing the electronic structure modulation of Ir by lowering its d band center; in addition, the aggregation and oxidative dissolution of IrRu species were depressed in the acidic media. TiC-based support combined with the advantages of high corrosion resistance and high conductivity was reported to be a good support material for water electrolysis.144 Ir catalyst was coated on the TiC support surface to form an Ir film by magnetron sputtering, which was hot pressed on the PEM of Nafion 115 as the anode side of MEA.145 The loading of Ir in the catalyst layer had a significant impact on the performance of PEMWE. Specifically, cell voltages of 1.74 V (approximately 80 μg cm−2 Ir), 1.72 V (approximately 160 μg cm−2 Ir), and 1.71 V (approximately 240 μg cm−2 Ir) were required to drive a current density of 1 A cm−2 at 80 °C.
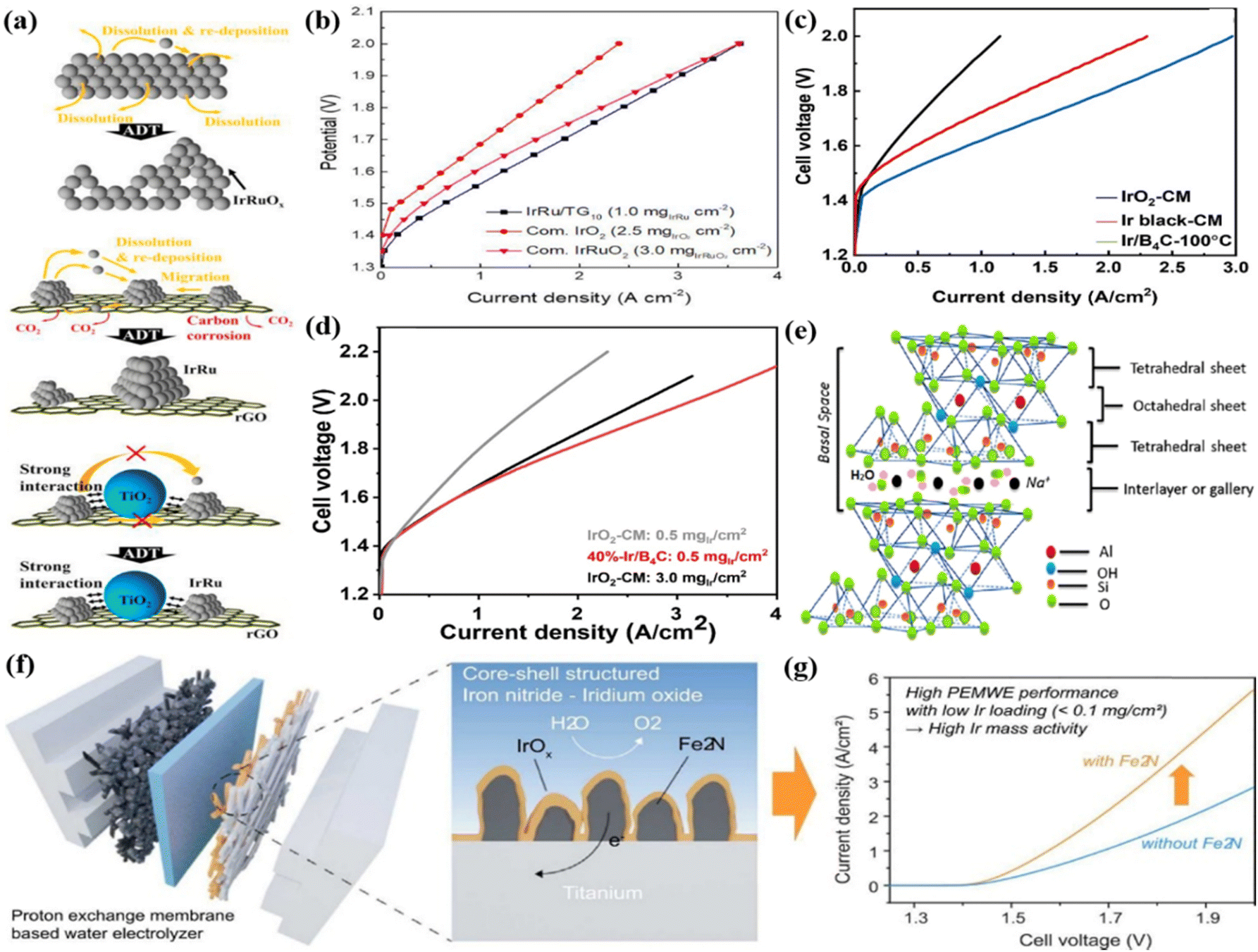 | ||
| Fig. 6 (a) The degradation mechanisms occurring during the accelerated durability tests of IrRuOx, IrRu/rGO, and IrRu/T90G10 catalyst and (b) the performance curves for IrRu/T90G10 with three conventional MEAs. Reproduced with permission from ref. 79. Copyright 2021, Elsevier B.V. (c) MEA Performance of Ir/B4C-100 °C (Ir loading: 0.5 mgIr cm−2). (d) The single-cell performance of 40%-Ir/B4C and commercial IrO2 catalysts. Reproduced with permission from ref. 147. Copyright 2023, Elsevier Ltd. (e) An illustration of the montmorillonite structure. Reproduced with permission from ref. 148. Copyright 2018, Elsevier Ltd. (f) Conventional catalyst-coated MEA for a water electrolyzer and fabrication of Fe2N@EIROF core–shell structured Ti GDL. (g) MEA performance of Fe2N@EIROF/Ti and EIROF/Ti in PEMWE single cells. Reproduced with permission from ref. 91. Copyright 2023, Elsevier B.V. | ||
A series of Ir/B4C catalysts with different reduction temperatures (30–100 °C) were prepared by loading Ir nanoparticles on the boron carbide (B4C) support through a wet impregnation method.146 Among them, Ir/B4C-100 °C showed the highest OER activity and single-cell performance among all the catalysts measured by a PEMWE. Specifically, it required a current density of 1.98 A cm−2 to reach 1.8 V, whereas those with two commercial catalysts (Ir black and IrO2) exhibited values of 1.36 and 0.692 A cm−2 at 1.8 V, respectively (Fig. 6c). The enhanced activity was associated with the well-dispersed Ir nanoparticles on the support and the high amount of Ir(III) and OH species formed on the surface; the high reduction temperature produced a strong metal-support interaction that inhibited the oxidative dissolution of Ir(III) and the aggregation of Ir substances. Subsequently, a series of Ir/B4C catalysts with different iridium contents (10–60 wt%) were prepared by changing the Ir content from 10 wt% to 60 wt%.147 Due to its high electrochemical surface area and suitable Ir particle size, 40%-Ir/B4C showed the highest catalytic activity and excellent durability. A membrane electrode assembly (MEA) was prepared using it as an anode catalyst, commercial Nafion 115 as a membrane, and commercial Pt/C as a cathode electrocatalyst to evaluate the catalyst performance in PEM electrolyzers. The PEMWE using 40%-Ir/B4C with an Ir loading of 0.5 mg cm−2 showed a current density of 1.89 A cm−2 at 1.8 V, while the IrO2 electrode with an Ir loading of 3.0 mg cm−2 showed a current density of 1.01 A cm−2 at 1.8 V (Fig. 6d).
In addition, some support materials with high conductivity and good chemical stability were also used to support Ir-based catalysts. For example, the high surface area natural mineral montmorillonite (MMT) with a layered structure was used as a carbon-free catalytic support to support IrO2 for acidic OER (Fig. 6e).148 By integrating into the PEMWE, a catalyst loading of 0.5 mgIr cm−2 ensured sustainable oxygen evolution at 80 °C with a current density of 2 A cm−2 at 1.6 V. Moreover, no obvious performance degradation was observed under the conditions of higher temperature and prolonged potential cycling. A Fe2N support was employed to support the iridium-oxide film as a Fe2N@EIROF core–shell structure for application in PEMWE (Fig. 6f).91 When evaluated in PMEWE, it can reach a current density of 4.5 A cm−2 at a cell voltage of 1.9 V with an Ir loading of 0.036 mgIr cm−2, and achieve a mass activity of 103 A mgIr−1 (Fig. 6g). The significantly enhanced performance compared with PEMWE without Fe2N support was due to the higher surface area and roughness of the porous Fe2N nanostructure, which provided ion and material transport paths for water electrolysis.
4 Conclusion and perspective
Here, we provide a systematic and comprehensive review of the catalytic performance of Ir-based catalysts in PEMWE, aiming to summarize the latest progress of Ir-based catalysts and promote the development of efficient catalysts. We showed the working principle of PEMWE and summarized the latest progress of Ir-based catalysts in PEMWE from four aspects: Ir-based single metals, alloys, oxides, and supported catalysts. By summarizing relevant reports, it was found that morphology engineering, the synergistic effect of alloys, and amorphous substances can change the electronic structure of Ir-based catalysts and the binding energy of reaction intermediates, and can improve the intrinsic activity of the catalyst. Supports can be used to increase the number of exposed active sites, and strong metal-support interactions can also improve the long-term stability of Ir-based catalysts in acidic electrolytes. Efficient and stable Ir-based electrocatalysts that meet commercial standards should continue to be developed, and their degradation mechanisms should be explored to improve their stability. In short, future research needs to comprehensively improve catalytic efficiency, reduce costs, and optimize the catalyst activity in the catalyst layer. Despite the impressive progress in the development of Ir-based anode catalysts, the large-scale production of hydrogen via PEMWE still faces the challenge of achieving a balance between performance and cost, e.g. operating current density greater than 2 A cm−2@1.8 V@80 °C, lifetime greater than 3 thousand hours, overpotential growth rate less than 2%, and Ir loading less than 0.3 mg cm−2.12,149 To achieve these goals, the activity and durability of the anode catalyst in practical PEMWE must be further improved (Fig. 7).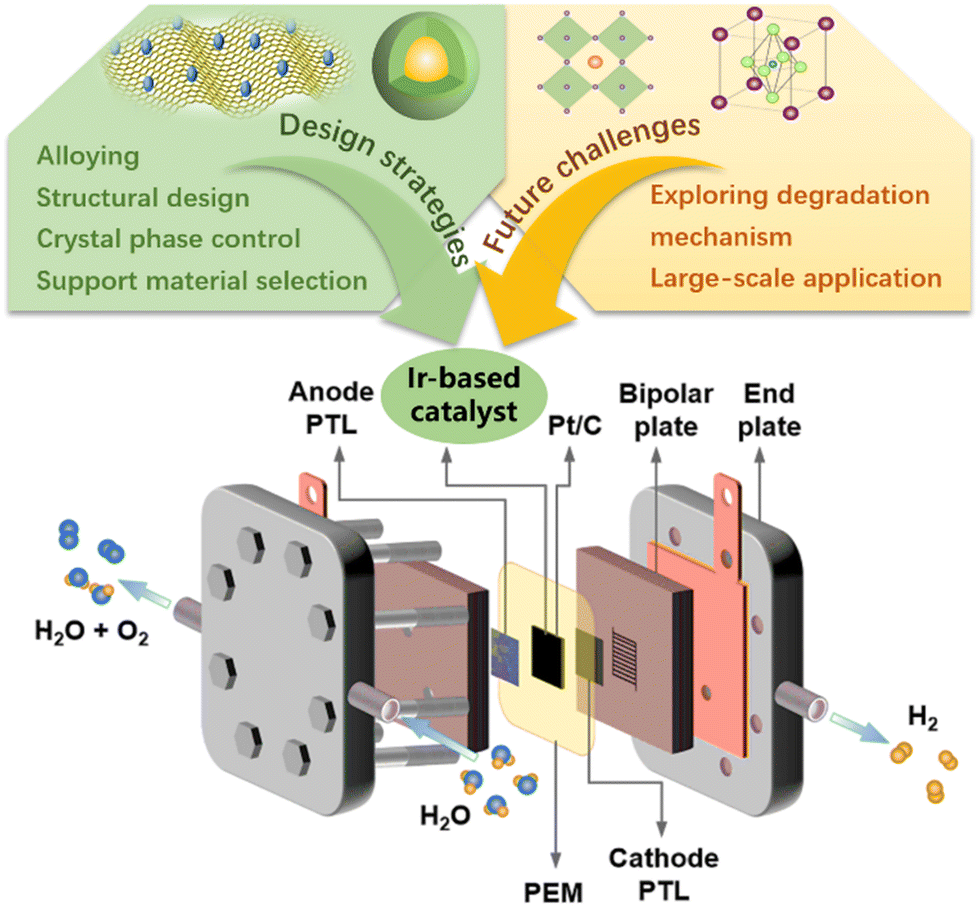 | ||
| Fig. 7 The design strategies and future challenges in developing Ir-based catalysts for practical PEMWE. | ||
The harsh operating conditions of PEMWE (i.e., strongly acidic environment and high oxidation potential) limit the selection of catalyst materials. The high price and low reserves of the precious metal Ir seriously hinder the large-scale application of PEM water electrolysis for hydrogen production. Therefore, without sacrificing activity and stability, reducing the amount of noble metal Ir used in electrocatalysts (≤0.3 mg cm−2) and improving the utilization efficiency of noble metals are reasonable ways to find ideal catalysts at this stage. To balance the relationship between cost, activity and stability, the design strategy of ideal Ir-based catalysts can be summarized as follows: (a) to prepare catalysts with special surface morphology, smaller size, and hollow/porous structures through surface engineering. This can obtain a larger specific surface area to increase the contact area between the electrode and the electrolyte and accelerate the transfer of electrons and reaction intermediates. (b) Introducing non-noble metals or cheap precious metal components can dilute the content of precious metals in the electrocatalysts which reduces the costs in practical application. It can also adjust the electronic structure and local environment of the catalyst to enhance the intrinsic activity and ensure catalytic stability. (c) The development of supports with high conductivity and large surface area enables good dispersion of active metal through metal (oxide)-support interaction. It is beneficial to maximize the number of exposed active sites and improve atomic utilization. Overall, in future electrocatalyst research, it is necessary to significantly improve the intrinsic catalytic activity by considering factors affecting catalyst performance such as structure design, morphology control, and support selection; focus on the performance of electrocatalysts under practical PEMWE operating conditions and feasibility of large-scale production.
Since the anode catalyst of PEMWE needs to have a long service life in a highly acidic environment and ampere-level high current density, it is very important and challenging to improve the long-term stability of the catalyst during operation. At present, most researchers believe that the OER mainly proceeds through the adsorbed oxygen mechanism on the surface of highly crystalline IrO2, while more oxygen atoms from the electrocatalyst lattice can participate in the OER on amorphous IrOx through the lattice oxygen mechanism. The lattice oxygen mechanism leads to high OER activity but poor durability due to the rapid dissolution of Ir species, which tends to collapse the catalyst structure. Therefore, it is necessary to use advanced in situ operational techniques (such as X-ray photoelectron spectroscopy, XAS, Raman spectroscopy, etc.) to monitor real-time changes in valence state, chemical composition, and structure/surface dissolution to explore the real catalytic active sites in the reaction, surface evolution/reconstruction behavior, clarifying the reaction mechanism more clearly to guide the construction of highly stable catalysts. Currently, the four-electron transfer process of Ir-based electrocatalysts in acidic OER is accompanied by the redox of Ir or O sites, in which IrO2 can be dissolved in the form of Ir(III) and Ir (>IV) species. This will not only lead to the change in the catalyst composition but also affect the atomic rearrangement and crystal plane adjustment of the catalyst, which in turn leads to deactivation of the active sites of the catalyst. Therefore, how to avoid the dissolution of active species and increase the capability of Ir active sites should be carefully studied in the future and more attention might be directed toward Ir state monitoring in realistic devices.
Author contributions
Chunyan Wang: conceptualization, writing – draft, creating the graphics, investigation, reviewing and editing; Ligang Feng: writing – review & editing, formal analysis and funding acquisition.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (21972124 and 22272148), and a project funded by the Priority Academic Program Development of Jiangsu Higher Education Institution was also appreciated by the authors.Notes and references
- F. Zhou, L. Zhang, J. Li, Q. Wang, Y. Chen, H. Chen, G. Lu, G. Chen, H. Jin, S. Wang and J. Wang, Eng. Rep., 2021, 3, e12437 CrossRef CAS.
- Y. Zhou, Q. Wang, X. Tian, J. Chang and L. Feng, J. Energy Chem., 2022, 75, 46–54 CrossRef CAS.
- C. Liu, F. Yang, A. Schechter and L. Feng, Adv. Sens. Energy Mater., 2023, 2, 100055 CrossRef.
- L. She, G. Zhao, T. Ma, J. Chen, W. Sun and H. Pan, Adv. Funct. Mater., 2022, 32, 2108465 CrossRef CAS.
- M. Kim, J. Park, M. Wang, Q. Wang, M. J. Kim, J. Y. Kim, H. S. Cho, C. H. Kim, Z. Feng, B. H. Kim and S. W. Lee, Appl. Catal., B, 2022, 302, 120834 CrossRef CAS.
- S. Wang, J. Zhu, X. Wu and L. Feng, Chin. Chem. Lett., 2022, 33, 1105–1109 CrossRef CAS.
- R. Liu, Z. Xu, F. Li, F. Chen, J. Yu, Y. Yan, Y. Chen and B. Y. Xia, Chem. Soc. Rev., 2023, 52, 5652–5683 RSC.
- Y. Zhou, Y. Kuang, G. Hu, X. Wang and L. Feng, Mater. Today Phys., 2022, 27, 100831 CrossRef CAS.
- S. Wang, T. Shen, C. Yang, G. Luo and D. Wang, ACS Catal., 2023, 13, 8670–8691 CrossRef CAS.
- J. Li, S. Wang, J. Chang and L. Feng, Adv. Powder Mater., 2022, 1, 100030 CrossRef.
- Q. Feng, X. Z. Yuan, G. Liu, B. Wei, Z. Zhang, H. Li and H. Wang, J. Power Sources, 2017, 366, 33–55 CrossRef CAS.
- K. Zhang, X. Liang, L. Wang, K. Sun, Y. Wang, Z. Xie, Q. Wu, X. Bai, M. S. Hamdy, H. Chen and X. Zou, Nano Res. Energy, 2022, 1, e9120032 CrossRef.
- N. Wang, D. Ma, S. Zhou, M. Hu, X. Li, X. Wu and Q. Zhu, Chin. Chem. Lett., 2023, 34, 107788 CrossRef CAS.
- Q. Wu, Y. Wang, K. Zhang, Z. Xie, K. Sun, W. An, X. Liang and X. Zou, Mater. Chem. Front., 2023, 7, 1025–1045 RSC.
- S. S. Kumar and H. Lim, Sustainable Energy Fuels, 2023, 7, 3560–3583 RSC.
- Z. Wang, P. Wang, H. Zhang, W. Tian, Y. Xu, X. Li, L. Wang and H. Wang, J. Mater. Chem. A, 2021, 9, 18576–18581 RSC.
- Y. Liu, X. Liang, H. Chen, R. Gao, L. Shi, L. Yang and X. Zou, Chin. J. Catal., 2021, 42, 1054–1077 CrossRef CAS.
- M. Liu, S. Liu, Q. Mao, S. Yin, Z. Wang, Y. Xu, X. Li, L. Wang and H. Wang, J. Mater. Chem. A, 2022, 10, 2021–2026 RSC.
- L. An, C. Wei, M. Lu, H. Liu, Y. Chen, G. G. Scherer, A. C. Fisher, P. Xi, Z. J. Xu and C. Yan, Adv. Mater., 2021, 33, 2006328 CrossRef CAS.
- C. Wang, F. Yang and L. Feng, Nanoscale Horiz., 2023, 8, 1174–1193 RSC.
- H. J. Song, H. Yoon, B. Ju and D. W. Kim, Adv. Energy Mater., 2021, 11, 2002428 CrossRef CAS.
- R. Yang, X. Shi, Y. Wang, J. Jin, H. Liu, J. Yin, Y. Zhao and P. Xi, Chin. Chem. Lett., 2022, 33, 4930–4935 CrossRef CAS.
- C. Wang, A. Schechter and L. Feng, Nano Res. Energy, 2023, 2, e9120056 CrossRef.
- L. E. CamachoForero, F. GodínezSalomón, G. RamosSánchez, C. P. Rhodes and P. B. Balbuena, J. Catal., 2022, 408, 64–80 CrossRef CAS.
- H. Jin, S. Choi, G. J. Bang, T. Kwon, H. S. Kim, S. J. Lee, Y. Hong, D. W. Lee, H. S. Park, H. Baik, Y. Jung, S. J. Yoo and K. Lee, Energy Environ. Sci., 2022, 15, 1119–1130 RSC.
- L. Li, P. Wang, Z. Cheng, Q. Shao and X. Huang, Nano Res., 2022, 15, 1087–1093 CrossRef CAS.
- H. Wu, Y. Wang, Z. Shi, X. Wang, J. Yang, M. Xiao, J. Ge, W. Xing and C. Liu, J. Mater. Chem. A, 2022, 10, 13170–13189 RSC.
- C. Yang, X. Zhang, Q. An, M. Liu, W. Zhou, Y. Li, F. Hu, Q. Liu and H. Su, J. Energy Chem., 2023, 78, 374–380 CrossRef CAS.
- L. Li, P. Wang, Q. Shao and X. Huang, Adv. Mater., 2021, 33, 2004243 CrossRef CAS.
- R. Li, H. Wang, F. Hu, K. C. Chan, X. Liu, Z. Lu, J. Wang, Z. Li, L. Zeng, Y. Li, X. Wu and Y. Xiong, Nat. Commun., 2021, 12, 3540 CrossRef CAS.
- G. Jiang, H. Yu, Y. Li, D. Yao, J. Chi, S. Sun and Z. Shao, ACS Appl. Mater. Interfaces, 2021, 13, 15073–15082 CrossRef CAS.
- S. Wang, H. Lv, S. Bi, T. Li, Y. Sun, W. Ji, C. Feng and C. Zhang, Mater. Chem. Front., 2021, 5, 8047–8055 RSC.
- H. Kim, J. Kim, J. Kim, G. H. Han, W. Guo, S. Hong, H. S. Park, H. W. Jang, S. Y. Kim and S. H. Ahn, Appl. Catal., B, 2021, 283, 119596 CrossRef CAS.
- W. Qiao, X. Huang and L. Feng, Chin. J. Struct. Chem., 2022, 41, 2207016 CAS.
- D. Zhou, M. He, Y. Ding, J. Yu, K. Fan and L. Sun, ACS Appl. Energy Mater., 2022, 5, 970–980 CrossRef CAS.
- H. Yu, J. Ke and Q. Shao, Small, 2023, 2304307, DOI:10.1002/smll.202304307.
- N. Yu, Z. J. Zhang, Y. M. Chai and B. Dong, Mater. Chem. Front., 2023, 7, 4236–4258 RSC.
- Z. Chen, X. Duan, W. Wei, S. Wang and B. Ni, Nano Energy, 2020, 78, 105270 CrossRef CAS.
- M. Bühler, P. Holzapfel, D. McLaughlin and S. Thiele, J. Electrochem. Soc., 2019, 166, F1070 CrossRef.
- Q. Feng, Z. Zhang, H. Huang, K. Yao, J. Fan, L. Zeng, M. C. Williams, H. Li and H. Wang, Chem. Eng. J., 2020, 395, 124428 CrossRef CAS.
- P. Holzapfel, M. Bühler, C. Van Pham, F. Hegge, T. Böhm, D. McLaughlin, M. Breitwieser and S. Thiele, Electrochem. Commun., 2020, 110, 106640 CrossRef CAS.
- C. Klose, T. Saatkamp, A. Münchinger, L. Bohn, G. Titvinidze, M. Breitwieser, K. D. Kreuer and S. Vierrath, Adv. Energy Mater., 2020, 10, 1903995 CrossRef CAS.
- J. E. Park, J. Kim, J. Han, K. Kim, S. Park, S. Kim, H. S. Park, Y. H. Cho, J. C. Lee and Y. E. Sung, J. Membr. Sci., 2021, 620, 118871 CrossRef CAS.
- A. Kusoglu and A. Z. Weber, Chem. Rev., 2017, 117, 987–1104 CrossRef CAS PubMed.
- B. Tian, Y. Li, Y. Liu, F. Ning, X. Dan, Q. Wen, L. He, C. He, M. Shen and X. Zhou, Nano Lett., 2023, 23, 6474–6481 CrossRef CAS PubMed.
- P. C. Okonkwo, I. Ben Belgacem, W. Emori and P. C. Uzoma, Int. J. Hydrogen Energy, 2021, 46, 27956–27973 CrossRef CAS.
- T. A. M. Suter, K. Smith, J. Hack, L. Rasha, Z. Rana, G. M. A. Angel, P. R. Shearing, T. S. Miller and D. J. L. Brett, Adv. Energy Mater., 2021, 11, 2101025 CrossRef CAS.
- Z. Kang, J. Mo, G. Yang, S. T. Retterer, D. A. Cullen, T. J. Toops, J. B. Green Jr, M. M. Mench and F. Zhang, Energy Environ. Sci., 2017, 10, 166–175 RSC.
- T. Li, K. Wang, J. Wang, Y. Liu, Y. Han, Z. Xu, G. Lin and Y. Liu, Renewable Energy, 2021, 179, 2086–2093 CrossRef CAS.
- K. Zhou, T. Li, Y. Han, J. Wang, J. Chen and K. Wang, RSC Adv., 2021, 11, 2010–2019 RSC.
- P. Shirvanian and F. van Berkel, Electrochem. Commun., 2020, 114, 106704 CrossRef CAS.
- R. Liu, Q. Jia, B. Zhang, Z. Lai and L. Chen, Int. J. Hydrogen Energy, 2022, 47, 22915–22937 CrossRef CAS.
- K. Xiong, W. Wu, S. Wang and L. Zhang, Appl. Energy, 2021, 301, 117443 CrossRef CAS.
- B. G. Abraham and R. Chetty, Int. J. Hydrogen Energy, 2021, 46, 6845–6856 CrossRef CAS.
- Y. Liu, S. Huang, D. Wang, H. Zhang, D. Shan, S. Peng, G. Shen, L. Wang and X. Wang, ACS Appl. Mater. Interfaces, 2022, 14, 15728–15735 CrossRef CAS.
- N. F. Asri, T. Husaini, A. B. Sulong, E. H. Majlan and W. R. W. Daud, Int. J. Hydrogen Energy, 2017, 42, 9135–9148 CrossRef CAS.
- Q. Feng, X. Yuan, G. Liu, B. Wei, Z. Zhang, H. Li and H. Wang, J. Power Sources, 2017, 366, 33–55 CrossRef CAS.
- S. Shimpalee, V. Lilavivat, H. McCrabb, Y. Khunatorn, H. K. Lee, W. K. Lee and J. W. Weidner, Int. J. Hydrogen Energy, 2016, 41, 13688–13696 CrossRef CAS.
- M. Maier, K. Smith, J. Dodwell, G. Hinds, P. R. Shearing and D. J. L. Brett, Int. J. Hydrogen Energy, 2022, 47, 30–56 CrossRef CAS.
- Y. Sun, S. Polani, F. Luo, S. Ott, P. Strasser and F. Dionigi, Nat. Commun., 2021, 12, 5984 CrossRef CAS.
- X. Peng, P. Satjaritanun, Z. Taie, L. Wiles, A. Keane, C. Capuano, I. V. Zenyuk and N. Danilovic, Adv. Sci., 2021, 8, 2102950 CrossRef CAS.
- Z. Taie, X. Peng, D. Kulkarni, I. V. Zenyuk, A. Z. Weber, C. Hagen and N. Danilovic, ACS Appl. Mater. Interfaces, 2020, 12, 52701–52712 CrossRef CAS.
- M. Clapp, C. M. Zalitis and M. Ryan, Catal. Today, 2023, 420, 114140 CrossRef CAS.
- X. Ren, Y. Wang, A. Liu, Z. Zhang, Q. Lv and B. Liu, J. Mater. Chem. A, 2020, 8, 24284–24306 RSC.
- S. Liu, S. Hua, R. Lin, H. Wang, X. Cai and W. Ji, Energy, 2022, 253, 124201 CrossRef CAS.
- C. V. Pham, M. Bühler, J. Knöppel, M. Bierling, D. Seeberger, D. Escalera López, K. J. J. Mayrhofer, S. Cherevko and S. Thiele, Appl. Catal., B, 2020, 269, 118762 CrossRef CAS.
- G. Liu, J. Xu, Y. Wang and X. Wang, J. Mater. Chem. A, 2015, 3, 20791–20800 RSC.
- T. Fujigaya, Y. Shi, J. Yang, H. Li, K. Ito and N. Nakashima, J. Mater. Chem. A, 2017, 5, 10584–10590 RSC.
- L. Wang, H. Liu, J. Zhuang and D. Wang, Small Sci., 2022, 2, 2200036 CrossRef CAS.
- T. Tang, Z. Wang and J. Guan, Coord. Chem. Rev., 2023, 492, 215288 CrossRef CAS.
- T. Kim, B. Kim, T. Kwon, H. Y. Kim, J. Y. Kim and K. Lee, Mater. Chem. Front., 2021, 5, 4445–4473 RSC.
- A. Chen, M. Deng, Z. Lu, Y. Lin and L. Chen, Mater. Chem. Front., 2023, 7, 4900–4907 RSC.
- C. Dong, Y. Li, D. Cheng, M. Zhang, J. Liu, Y. Wang, D. Xiao and D. Ma, ACS Catal., 2020, 10, 11011–11045 CrossRef CAS.
- J. Lim, G. Kang, J. W. Lee, S. S. Jeon, H. Jeon, P. W. Kang and H. Lee, J. Power Sources, 2022, 524, 231069 CrossRef CAS.
- S. Chatterjee, X. Peng, S. Intikhab, G. Zeng, N. N. Kariuki, D. J. Myers, N. Danilovic and J. Snyder, Adv. Energy Mater., 2021, 11, 2101438 CrossRef CAS.
- Z. Xie, L. Ding, S. Yu, W. Wang, C. B. Capuano, A. Keane, K. Ayers, D. A. Cullen, H. M. Meyer and F. Zhang, Appl. Catal., B, 2024, 341, 123298 CrossRef CAS.
- Y. J. Kim, A. Lim, J. M. Kim, D. Lim, K. H. Chae, E. N. Cho, H. J. Han, K. U. Jeon, M. Kim, G. H. Lee, G. R. Lee, H. S. Ahn, H. S. Park, H. Kim, J. Y. Kim and Y. S. Jung, Nat. Commun., 2020, 11, 4921 CrossRef CAS PubMed.
- H. Yu, N. Danilovic, Y. Wang, W. Willis, A. Poozhikunnath, L. Bonville, C. Capuano, K. Ayers and R. Maric, Appl. Catal., B, 2018, 239, 133–146 CrossRef CAS.
- I. G. Kim, A. Lim, J. H. Jang, K. Y. Lee, I. W. Nah and S. Park, J. Power Sources, 2021, 501, 230002 CrossRef CAS.
- H.-S. Oh, H. N. Nong, T. Reier, M. Gliech and P. Strasser, Chem. Sci., 2015, 6, 3321–3328 RSC.
- S. Siracusano, N. Hodnik, P. Jovanovic, F. Ruiz Zepeda, M. Šala, V. Baglio and A. S. Aricò, Nano Energy, 2017, 40, 618–632 CrossRef CAS.
- J. Lim, D. Park, S. S. Jeon, C. W. Roh, J. Choi, D. Yoon, M. Park, H. Jung and H. Lee, Adv. Funct. Mater., 2018, 28, 1704796 CrossRef.
- Z. L. Zhao, Q. Wang, X. Huang, Q. Feng, S. Gu, Z. Zhang, H. Xu, L. Zeng, M. Gu and H. Li, Energy Environ. Sci., 2020, 13, 5143–5151 RSC.
- L. Wang, V. A. Saveleva, S. Zafeiratos, E. R. Savinova, P. Lettenmeier, P. Gazdzicki, A. S. Gago and K. A. Friedrich, Nano Energy, 2017, 34, 385–391 CrossRef CAS.
- G. Li, H. Yu, D. Yang, J. Chi, X. Wang, S. Sun, Z. Shao and B. Yi, J. Power Sources, 2016, 325, 15–24 CrossRef CAS.
- Y. Zeng, X. Guo, Z. Shao, H. Yu, W. Song, Z. Wang, H. Zhang and B. Yi, J. Power Sources, 2017, 342, 947–955 CrossRef CAS.
- V. K. Puthiyapura, S. Pasupathi, H. Su, X. Liu, B. Pollet and K. Scott, Int. J. Hydrogen Energy, 2014, 39, 1905–1913 CrossRef CAS.
- G. Li, H. Yu, W. Song, M. Dou, Y. Li, Z. Shao and B. Yi, ChemSusChem, 2012, 5, 858–861 CrossRef CAS PubMed.
- M. Retuerto, L. Pascual, J. Torrero, M. A. Salam, Á. Tolosana-Moranchel, D. Gianolio, P. Ferrer, P. Kayser, V. Wilke, S. Stiber, V. Celorrio, M. Mokthar, D. G. Sanchez, A. S. Gago, K. A. Friedrich, M. A. Peña, J. A. Alonso and S. Rojas, Nat. Commun., 2022, 13, 7935 CrossRef CAS PubMed.
- F. Gu, L. Zheng, H. Wei, W. Mi, C. Zhang, Q. Su, W. Zhu and W. Lin, Appl. Surf. Sci., 2022, 606, 155008 CrossRef CAS.
- H. Y. Jeong, J. Oh, G. S. Yi, H. Y. Park, S. K. Cho, J. H. Jang, S. J. Yoo and H. S. Park, Appl. Catal., B, 2023, 330, 122596 CrossRef CAS.
- H. Lv, S. Wang, J. Li, C. Shao, W. Zhou, X. Shen, M. Xue and C. Zhang, Appl. Surf. Sci., 2020, 514, 145943 CrossRef CAS.
- X. Duan, H. Liu, W. Zhang, Q. Ma, Q. Xu, L. Khotseng and H. Su, Electrochim. Acta, 2023, 470, 143271 CrossRef CAS.
- V. K. Puthiyapura, M. Mamlouk, S. Pasupathi, B. G. Pollet and K. Scott, J. Power Sources, 2014, 269, 451–460 CrossRef CAS.
- G. Liu, F. Hou, X. Wang and B. Fang, Appl. Surf. Sci., 2023, 615, 156333 CrossRef CAS.
- T. Kwon, H. Hwang, Y. J. Sa, J. Park, H. Baik, S. H. Joo and K. Lee, Adv. Funct. Mater., 2017, 27, 1604688 CrossRef.
- S. W. Lee, C. Baik, D. Kim and C. Pak, J. Power Sources, 2021, 493, 229689 CrossRef CAS.
- H. Guo, Z. Fang, H. Li, D. Fernandez, G. Henkelman, S. M. Humphrey and G. Yu, ACS Nano, 2019, 13, 13225–13234 CrossRef CAS PubMed.
- L. Zhang, Q. Fan, K. Li, S. Zhang and X. Ma, Sustainable Energy Fuels, 2020, 4, 5417–5432 RSC.
- H. Hu, F. M. D. Kazim, Z. Ye, Y. Xie, Q. Zhang, K. Qu, J. Xu, W. Cai, S. Xiao and Z. Yang, J. Mater. Chem. A, 2020, 8, 20168–20174 RSC.
- H. Wang, Z. Chen, D. Wu, M. Cao, F. Sun, H. Zhang, H. You, W. Zhuang and R. Cao, J. Am. Chem. Soc., 2021, 143, 4639–4645 CrossRef CAS.
- T. Hrbek, P. Kúš, T. Košutová, K. Veltruská, T. N. Dinhová, M. Dopita, V. Matolín and I. Matolínová, Int. J. Hydrogen Energy, 2022, 47, 21033–21043 CrossRef CAS.
- J. Zhang, X. Cao, Y. Jiang, S. Hung, W. Liu, H. B. Yang, C. Xu, D. Li, T. Zhang, Y. Li, J. Li and B. Liu, Chem. Sci., 2022, 13, 12114–12121 RSC.
- H. Zhu, Z. Zhu, J. Hao, S. Sun, S. Lu, C. Wang, P. Ma, W. Dong and M. Du, Chem. Eng. J., 2022, 431, 133251 CrossRef CAS.
- W. Gao, Q. Xu, Z. Wang, M. Wang, X. Ren, G. Yuan and Q. Wang, Electrochim. Acta, 2020, 337, 135738 CrossRef CAS.
- J. Guan, X. Bai and T. Tang, Nano Res., 2022, 15, 818–837 CrossRef CAS.
- T. Tang, S. Li, J. Sun, Z. Wang and J. Guan, Nano Res., 2022, 15, 8714–8750 CrossRef CAS.
- K. Yeo, K. Lee, H. Kim, J. Lee and S. Kim, Energy Environ. Sci., 2022, 15, 3449–3461 RSC.
- J. Feng, F. Lv, W. Zhang, P. Li, K. Wang, C. Yang, B. Wang, Y. Yang, J. Zhou, F. Lin, G. Wang and S. Guo, Adv. Mater., 2017, 29, 1703798 CrossRef PubMed.
- J. Kwon, S. Sun, S. Choi, K. Lee, S. Jo, K. Park, Y. K. Kim, H. B. Park, H. Y. Park, J. H. Jang, H. Han, U. Paik and T. Song, Adv. Mater., 2023, 35, 2300091 CrossRef CAS PubMed.
- S. Shin, T. Kwon, K. Kim, M. Kim, M. H. Kim and Y. Lee, ACS Appl. Energy Mater., 2022, 5, 6146–6154 CrossRef CAS.
- M. A. Hubert, A. Gallo, Y. Liu, E. Valle, J. Sanchez, D. Sokaras, R. Sinclair, L. A. King and T. F. Jaramillo, J. Phys. Chem. C, 2022, 126, 1751–1760 CrossRef CAS.
- N. Liu, Z. Duan, Q. Zhang and J. Guan, Chem. Eng. J., 2021, 419, 129567 CrossRef CAS.
- S. Siracusano, V. Baglio, S. A. Grigoriev, L. Merlo, V. N. Fateev and A. S. Aricò, J. Power Sources, 2017, 366, 105–114 CrossRef CAS.
- Y. Zheng, F. Zhang, G. Wang, D. Lai, L. Zou, Q. Cheng, J. Li, Z. Zou and H. Yang, J. Power Sources, 2022, 528, 231189 CrossRef CAS.
- Z. Lu, Y. Shi, P. Gupta, X. Min, H. Tan, Z. Wang, C. Guo, Z. Zou, H. Yang, S. Mukerjee and C. Yan, Electrochim. Acta, 2020, 348, 136302 CrossRef CAS.
- Q. Deng, Y. Sun, J. Wang, S. Chang, M. Ji, Y. Qu, K. Zhang and B. Li, Dalton Trans., 2021, 50, 6083–6087 RSC.
- S. Lee, Y. J. Lee, G. Lee and A. Soon, Nat. Commun., 2022, 13, 3171 CrossRef CAS PubMed.
- W. Gou, Z. Xia, X. Tan, Q. Xue, F. Ye, S. Dai, M. Zhang, R. Si, Y. Zou, Y. Ma, J. C. Ho and Y. Qu, Nano Energy, 2022, 104, 107960 CrossRef CAS.
- J. Bak, T. G. Yun, J. S. An, H. B. Bae and S. Y. Chung, Energy Environ. Sci., 2022, 15, 610–620 RSC.
- H. Lv, Y. Sun, S. Wang, W. Ji, W. Zhou and C. Zhang, Int. J. Hydrogen Energy, 2023, 48, 16949–16957 CrossRef CAS.
- X. Chen, W. Li, N. Song, M. Zhong, S. Yan, J. Xu, W. Zhu, C. Wang and X. Lu, Chem. Eng. J., 2022, 440, 135851 CrossRef CAS.
- J. Xu, H. Jin, T. Lu, J. Li, Y. Liu, K. Davey, Y. Zheng and S. Qiao, Sci. Adv., 2023, 9, eadh1718 CrossRef CAS.
- L. Zhang, Y. Wang, Y. Wang, H. Liu, Q. Qin and X. Liu, ACS Sustainable Chem. Eng., 2022, 10, 10658–10665 CrossRef CAS.
- H. Li, H. Liu, Q. Qin and X. Liu, Inorg. Chem. Front., 2022, 9, 702–708 RSC.
- J. Torrero, T. Morawietz, D. García Sanchez, D. Galyamin, M. Retuerto, V. Martin-Diaconescu, S. Rojas, J. A. Alonso, A. S. Gago and K. A. Friedrich, Adv. Energy Mater., 2023, 13, 2204169 CrossRef CAS.
- J. Joo, Y. Park, J. Kim, T. Kwon, M. Jun, D. Ahn, H. Baik, J. H. Jang, J. Y. Kim and K. Lee, Small Methods, 2022, 6, 2101236 CrossRef CAS.
- G. Jiang, H. Yu, J. Hao, J. Chi, Z. Fan, D. Yao, B. Qin and Z. Shao, J. Energy Chem., 2019, 39, 23–28 CrossRef.
- H. Wu, Y. Wang, Z. Shi, D. Han, J. Yang, P. Wang, J. Ni, M. Xiao, C. Liu and W. Xing, J. Phys. Chem. C, 2023, 127, 12541–12547 CrossRef CAS.
- J. He, G. Fu, J. Zhang, P. Xu and J. Sun, Small, 2022, 18, 2203365 CrossRef CAS.
- J. Gao, H. Tao and B. Liu, Adv. Mater., 2021, 33, 2003786 CrossRef CAS.
- J. Wang, J. Kim, S. Choi, H. Wang and J. Lim, Small Methods, 2020, 4, 2000621 CrossRef CAS.
- A. P. Murthy, J. Madhavan and K. Murugan, J. Power Sources, 2018, 398, 9–26 CrossRef CAS.
- A. Martínez Séptimo, M. A. Valenzuela, P. Del Angel and R. D. G. González Huerta, Int. J. Hydrogen Energy, 2021, 46, 25918–25928 CrossRef.
- A. Hartig Weiss, M. Miller, H. Beyer, A. Schmitt, A. Siebel, A. T. S. Freiberg, H. A. Gasteiger and H. A. ElSayed, ACS Appl. Nano Mater., 2020, 3, 2185–2196 CrossRef CAS.
- Z. Shi, J. Li, J. Jiang, Y. Wang, X. Wang, Y. Li, L. Yang, Y. Chu, J. Bai, J. Yang, J. Ni, Y. Wang, L. Zhang, Z. Jiang, C. Liu, J. Ge and W. Xing, Angew. Chem., Int. Ed., 2022, 61, e202212341 CrossRef CAS PubMed.
- D. Böhm, M. Beetz, C. Gebauer, M. Bernt, J. Schröter, M. Kornherr, F. Zoller, T. Bein and D. Fattakhova-Rohlfing, Appl. Mater. Today, 2021, 24, 101134 CrossRef.
- J. Islam, B. S. Yoon, P. T. Thien, C. H. Ko and S. Kim, Catal. Today, 2024, 425, 114349 CrossRef CAS.
- G. Li, H. Jia, H. Liu, X. Yang and M. Lin, RSC Adv., 2022, 12, 28929–28936 RSC.
- X. Min, Y. Shi, Z. Lu, L. Shen, T. O. Ogundipe, P. Gupta, C. Wang, C. Guo, Z. Wang, H. Tan, S. Mukerjee and C. Yan, Electrochim. Acta, 2021, 385, 138391 CrossRef CAS.
- G. Jiang, H. Yu, D. Yao, Y. Li, J. Chi, H. Zhang and Z. Shao, J. Mater. Chem. A, 2022, 10, 11893–11903 RSC.
- S. Zaman, M. Wang, H. Liu, F. Sun, Y. Yu, J. Shui, M. Chen and H. Wang, Trends Chem., 2022, 4, 886–906 CrossRef CAS.
- R. Li, L. Luo, X. Ma, W. Wu, M. Wang and J. Zeng, J. Mater. Chem. A, 2022, 10, 5717–5742 RSC.
- P. Kúš, A. Ostroverkh, I. Khalakhan, R. Fiala, Y. Kosto, B. Šmíd, Y. Lobko, Y. Yakovlev, J. Nováková, I. Matolínová and V. Matolín, Int. J. Hydrogen Energy, 2019, 44, 16087–16098 CrossRef.
- P. Kúš, A. Ostroverkh, K. Ševčíková, I. Khalakhan, R. Fiala, T. Skála, N. Tsud and V. Matolin, Int. J. Hydrogen Energy, 2016, 41, 15124–15132 CrossRef.
- J. Islam, S. Kim, P. T. Thien, M. Kim, H. Cho, W. Cho, C. Kim, C. Lee and J. H. Lee, J. Power Sources, 2021, 512, 230506 CrossRef CAS.
- J. Islam, S. Kim, M. M. Rahman, P. T. Thien, M. Kim, H. Cho, C. Lee, J. H. Lee and S. Lee, Mater. Today Energy, 2023, 32, 101237 CrossRef CAS.
- I. Boshnakova, E. Lefterova and E. Slavcheva, Int. J. Hydrogen Energy, 2018, 43, 16897–16904 CrossRef CAS.
- Y. Chen, C. Liu, J. Xu, C. Xia, P. Wang, B. Y. Xia, Y. Yan and X. Wang, Small Struct., 2023, 4, 2200130 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2024 |